

Background: Estrogen receptor α (ERα) activity in breast cancer cells is increased by Pin1, an isomerase that alters protein conformation.
Results: Pin1-mediated isomerization increases ERα DNA binding affinity.
Conclusion: Pin1 allosterically regulates ERα DNA binding directly via isomerization of phosphorylated receptor.
Significance: Pin1 regulation of ERα provides a framework for understanding regulation by intrinsically disordered domains on transcription factor function.
Keywords: DNA-protein interaction; estrogen receptor; intrinsically disordered protein; nuclear receptor; phosphorylation; prolyl isomerase; DNA-protein interaction, proline isomerization, SNAP (specificity and affinity for protein) microarray
Abstract
The transcriptional activity of estrogen receptor α (ERα), the key driver of breast cancer proliferation, is enhanced by multiple cellular interactions, including phosphorylation-dependent interaction with Pin1, a proline isomerase, which mediates cis-trans isomerization of the N-terminal Ser(P)118-Pro119 in the intrinsically disordered AF1 (activation function 1) domain of ERα. Because both ERα and Pin1 have multiple cellular partners, it is unclear how Pin1 assists in the regulation of ERα transactivation mechanisms and whether the functional effects of Pin1 on ERα signaling are direct or indirect. Here, we tested the specific action of Pin1 on an essential step in ERα transactivation, binding to specific DNA sites. DNA binding analysis demonstrates that stable overexpression of Pin1 increases endogenous ERα DNA binding activity when activated by estrogen but not by tamoxifen or EGF. Increased DNA binding affinity is a direct effect of Pin1 on ERα because it is observed in solution-based assays with purified components. Further, our data indicate that isomerization is required for Pin1-modulation of ERα-DNA interactions. In an unbiased in vitro DNA binding microarray with hundreds of thousands of permutations of ERα-binding elements, Pin1 selectively enhances the binding affinity of ERα to consensus DNA elements. These studies reveal that Pin1 isomerization of phosphorylated ERα can directly regulate the function of the adjacent DNA binding domain, and this interaction is further modulated by ligand binding in the ligand-binding domain, providing evidence for Pin1-dependent allosteric regulation of ERα function.
Introduction
ERα,3 a key driver of proliferation in breast cancer cells, is a member of the nuclear receptor (NR) superfamily of transcription factors and activates gene programs by interacting with transcriptional coregulators, components of the RNA polymerase II complex, and, importantly, DNA. ERα binds a palindromic DNA consensus sequence (5′-AGGTCANNNTGACCT-3′, where N is any nucleotide) known as an estrogen response element (ERE) (1–4). The centrally located DNA-binding domain (DBD) of ERα is flanked by two transactivation domains, AF1 in the N terminus and AF2 in the ligand-binding C-terminal domain (LBD) (5, 6). The ligand-dependent AF2 regulates ERα transcriptional function by acting as a hub for coregulator interactions, and these interactions can affect DNA binding capacity (7–9). On the other hand, AF1 resides in the N-terminal region of ERα and is predominantly governed via phosphorylation by multiple kinases (10–16). Although sequences at the N terminus are important for both ligand-dependent and ligand-independent activation of ERα, how signals originating from the N terminus translate into differences in ERα transactivation and DNA binding function remain poorly understood.
Historically, NRs were thought to be composed of semiautonomous protein domains, such as the LBD and DBD, that independently carried out functions for ligand binding and DNA interactions, respectively. However, recent crystal and solution structural studies of full-length NRs highlight the signaling convergence of several NR domains via interactive protein surfaces (17, 18). These structures have confirmed an ever-growing body of work, indicating that the AF1, DBD, and LBD/AF2 serve as hubs of allosterically mediated intraprotein communication for several NRs. This complex interplay incorporates kinase and protein-protein interactions via the AF1 domain, ligand binding and coregulator interactions at the LBD, and DNA interactions at the DBD.
The AF1 domain of ERα is an intrinsically disordered (ID) domain. Although ID domains lack a specific identifying motif or secondary structure, they play key roles in post-translational modifications, protein-protein interactions, and allosteric control (19–21). By virtue of their unstructured characteristics, ID domains are difficult to study, yet AF1 has been shown to provide an interaction surface for coregulator proteins (10, 22–25). An understanding of the interactions in ID domains could provide a mechanistic window to better understand the allosteric role of unstructured domains in transcriptional signaling (26).
Pin1 is a peptidylprolyl isomerase that binds to Ser(P)/Thr(P)-Pro through its WW domains and catalyzes the cis/trans-isomerization of the Ser(P)/Thr(P)-Pro bond through its prolyl isomerase domain (27–29). Phosphorylation-dependent isomerization by Pin1 affects diverse cellular processes, such as protein-protein interactions, subcellular localization, dephosphorylation, and transcription (30). We previously identified cis-trans isomerization of the Ser(P)118–Pro119 bond in AF1 by Pin1 as a novel post-translational modification regulating ERα conformation, protein stability, and transcriptional function (25, 31). Ser118 in the AF1 region of ERα is a major site of phosphorylation by proline-directed kinases in response to estrogen, growth factors, and anti-estrogens, such as tamoxifen (11–15). Ser118 phosphorylation (Ser(P)118) also plays an important role in ERα transcriptional function (12, 25, 32, 33).
Our studies indicate that high levels of Pin1 expression are associated with worse outcomes in ERα+ breast tumors as well as increased ERα signaling and transcriptional activity (25, 31). Pin1 is recruited to phosphorylated ERα at Ser118 in response to estrogen, tamoxifen, and EGF (25). Pin1 enhances ERα-mediated transcription induced by estrogen and tamoxifen and increases ERα dimerization. These previous studies also showed that the Pin1-mediated increase in ERα transcriptional activity was mediated through the AF1 domain (25). Collectively, these studies establish Pin1 as an AF1 binding partner that enhances ERα transcriptional function.
Multiple potential mechanisms have been proposed to describe Pin1 regulation of ERα transcriptional function, highlighting the complexity of Pin1 activity on ERα signaling. Like ERα, Pin1 binds multiple components of transcriptional complexes, including RNA polymerase II, histones, general transcription factors, and ERα coactivators and corepressors (34–38). Moreover, Pin1 alters signaling in breast cancer cells that can indirectly influence ERα activity (34, 36, 39–41). Hence, traditional cell-based approaches to elucidate Pin1 activity on ERα-mediated transcription are confounded by the potential for both direct and indirect effects. Therefore, to focus specifically on direct effects of Pin1 on ERα function, we examined how Pin1 binding and isomerization might affect ERα DNA binding activity. To enable direct analysis of DNA binding affinity and sequence specificity, we used purified proteins and an unbiased approach on the scale of 385,000 DNA sequences, providing a mechanistic readout of Pin1-mediated effects on ERα DNA binding in the process. Together, the data provide novel insights into the roles of phosphorylation and Pin1 on ERα transcriptional regulation and describe a new approach for refined definition of components within large transcription factor complexes.
Experimental Procedures
Cell Culture and Treatments
MCF7 cells and MCF7 cells stably overexpressing GFP-Pin1 and GFP (25) were maintained in DMEM (Life Technologies, Inc.), 10% fetal bovine serum (FBS; Hyclone), and 1% penicillin-streptomycin (Life Technologies) at 37 °C and 10% CO2. In experiments involving treatments with 0.1% EtOH (vehicle), 10 nm 17β-estradiol (E2; Steraloids, Inc.), or 100 nm 4-hydrotamoxifen (OHT), cells were placed in estrogen-depleted phenol-red free medium consisting of 10% dextran-charcoal-stripped FBS for 3 days prior to the addition of hormone or vehicle. For EGF treatments, the medium was changed to phenol red-free DMEM or Opti-MEM (Life Technologies) overnight prior to treatment. The treatments were carried out for the times indicated in the figure legends.
Western Blot and Antibodies
Western blot was performed as described (25). ERα antibody (Santa Cruz Biotechnology, Inc.) was used to detect purified and endogenous ERα. Antibody directed against Ser(P)118 ERα (Cell Signaling) was used to detect Ser(P)118-ERα. Pin1 antibody (Epitome) was used to detect purified and endogenous Pin1. Detection of actin with an anti-actin antibody (Sigma) was used as loading control.
Electrophoretic Mobility Shift Assay (EMSA)
ERE and mutated ERE (mERE) probes listed in Table 1 were annealed as 32-mer sense (0.5 μg) and 13-mer antisense (0.2 μg) in solution containing 10× Klenow buffer in 100 μl of H2O at 90 °C and slow cooled to room temperature. An extension reaction was then performed to fill the overhang in a 200-μl reaction containing 100 μl of annealed oligonucleotide, 4 μl of 10 mm dNTP (2.5 mm concentration of each: dATP, dCTP, dGTP, and dTTP), 10 μl of 10× Klenow buffer, 5 μl of Klenow polymerase (New England Biolabs), and H2O. This mixture was incubated at 16 °C overnight. Unincorporated dNTPs were removed by passing the mixture through a G-50 column twice. To end-label probes with radioactive 32P, 20 ng of reannealed oligonucleotides were mixed with 2 μl of 10× T3 PNK buffer, 1 μl of 0.1 m DTT, 1 μl of T4 PNK (New England Biolabs), and the final volume was brought up to 18 μl by adding H2O. Two μl of [γ-32P]ATP (6,000 Ci/mmol) was then added to the reaction and incubated for 4 h at 37 °C. Unincorporated 32P was removed by passing the reaction through a G-50 column twice. Afterward, TE buffer (10 mm Tris-Cl, pH 8.0, 1 mm EDTA) was added to the purified reaction to a final volume of 50–100 μl and stored at −20 °C. University of Wisconsin (Madison, WI) radioactive safety guidelines were strictly followed during these procedures.
TABLE 1.
Sequence for EMSA probes
| Probe name | Sequence |
|---|---|
| ERE sense | AGCTTCGCGGCGGTCACAGTGACCCTGGAGCG |
| ERE antisense | CGCTCCAGGTCAC |
| mERE sense | AGCTTCGAGGAGATCACAGTGATCTGGAGCG |
| mERE antisense | CGCTCCAGATCAC |
Cells were treated as indicated in the figure legends and harvested. Cells were pelleted and resuspended in 100 μl of buffer A (10 mm HEPES, pH 7.8, 10 mm KCl, 1.5 mm MgCl2) supplemented freshly with 0.5 mm PMSF, 10 μg/ml aprotinin, 0.1% Nonidet P-40, and 1 mm DTT. Cells were then incubated on ice for 30 min and spun down for 1 min at 13,000 rpm and 4 °C. The supernatant (cytoplasmic fraction) was moved to a new tube. For nuclei lysis, the pellet was dissolved in 50 μl of buffer C (20 mm HEPES, pH 7.8, 450 mm NaCl, 0.2 mm EDTA, 1.5 mm MgCl2, 25% glycerol) supplemented with 0.5 mm PMSF, 10 μg/ml aprotinin, and 0.5 mm DTT. The lysis reaction was incubated on ice for 30 min and spun down for 10 min at 13,000 rpm and 4 °C. Supernatants were transferred to new tubes, and protein concentrations were measured by Bradford assay (Bio-Rad).
The EMSA was performed using fresh nuclear extracts or purified proteins. For cell studies, 10 μg of nuclear extract was incubated with 2 μl of 5× binding buffer (375 mm NaCl, 75 mm Tris-Cl, pH 7.5, 7.5 mm EDTA, 20% glycerol, 100 μg/ml BSA), 1.5 mm DTT, 1 μl of poly(dI-dC) (Sigma), and H2O was added up to a volume of 9 μl on ice. ERα supershifts were performed with 2–4 μl of ERα antibody Ab712 (42). For in vitro EMSA, purified ERα (Life Technologies or Pierce) was used in the presence and absence of Pin1, Pin1 K63A, or juglone (Sigma) and incubated at 30 °C for 30 min. One μl of 32P-end-labeled probe was added to the reaction and further incubated at room temperature for 20 min. Afterward, samples were loaded onto a native 4% acrylamide gel composed of 8 ml of 30% acrylamide, 2% bis, 1.5 ml of TBE, 600 μl of 10% APS, and 60 μl of TEMED and electrophoresed at 200 V for 1.5 h in 0.25× TBE. The gel was then placed on Whatman paper, dried in a gel dryer (Bio-Rad) for 45 min, and exposed to x-ray film (Eastman Kodak Co.) at −80 °C for 1–7 days, depending upon the signal. Bands were quantified by exposing the dried gel to a phosphoscreen (Eastman Kodak Co.) and quantified by a PhosphorImager (GE Healthcare).
Specificity and Affinity for Protein (SNAP) Array
A custom-designed DNA array known as the SNAP array (Proteovista LLC) was synthesized using maskless array technology (Roche Nimblegen). ERE variations were tested within the sequence, 5′-GCGGTAGGTCA(N9)GCTGCGGAGCAGC(N9)′TGACCTACCGC-3′, where N is any nucleotide, N′ is the reverse complement, and the 15-mer core sequence is in boldface type. Control probes included response elements for androgen receptor and glucocorticoid receptor as well as mutated EREs. DNA hairpins were induced on an array, as described previously (43). A hybridization chamber was applied over the array, and the array was hydrated twice with deionized water. The array was then blocked with 2.5% milk for 1 h at room temperature on a rotator (2–3 rpm). After the blocking solution was removed from the array, the transcription factor binding buffer (Proteovista LLC) was used to rinse the array. Purified ERα (Life Technologies), preincubated with or without a 10-fold molar excess of Pin1, was applied to the array in buffer plus 1 μm E2, incubated for 1 h with rotation, and detected with an Alexa Fluor 647-labeled anti-ERα antibody (Santa Cruz Biotechnology). After incubation, the array was rinsed briefly, dried, and scanned using a GenePix 4000B scanner (Molecular Devices). Arrays were performed in duplicate. The data from the arrays were extracted using NimbleScan 2.4 (Roche Applied Science), and the data were processed as described previously (44, 45).
Fluorescence Polarization (FP)
SNAP microarray results were verified using individual DNA sequences in FP assays. Dilutions of ERα, either alone or with a 10-fold molar excess of Pin1 or Pin1 starting concentration, as noted in the legend, were preincubated at 30 °C for 20 min in a 96-well plate (Corning 3676). Fluorescein-labeled, hairpinned DNA (IDT Technologies; core sequences shown in Fig. 5) was added to a final concentration of 1 nm, and the plate was incubated for an additional 10 min at 30 °C. To avoid quenching of the fluorescein label by the 5′G of the array DNA probe, a T nucleotide was included at the 5′-end of the 53-mer DNA sequences (Seq1, Seq2, and Seq3). The mERE had the sequence, 5′-TGCGGTGAGACACAGGGTTTCGCTGCGGAGCAGCGAAACCCTGTGTCTCACCGC-3′, where the mutated ERE is in boldface type. The core sequence that was altered between different probes is underlined within the adjacent backbone separated by a GGA loop in the hairpin. FP was measured using a Safire2 plate reader (Tecan), and the data were analyzed by sigmoidal dose response (variable slope) using Prism (GraphPad) to yield the dissociation constant (Kd).
FIGURE 5.

Effect of Pin1 on ERα DNA binding using the comprehensive SNAP microarray. A, top, motif for estrogen-occupied ERα binding to all permutations of the core sequence, AGGTCAN9, in the 25-bp hairpin on the SNAP array. Bottom, SSL for estrogen-occupied ERα binding to all 262,144 (9-mer) DNA sequences. Each ring contains sequences with increasing mismatches from the seed motif composed of the sequences NNNTGACCT and TGACCTNNN. Left, motif and SSL for ERα. Right, motif and SSL for ERα + Pin1. B, the effect of Pin1 was determined by plotting normalized fluorescence intensity for ERα (Array1_noPin1) versus ERα + Pin1 (Array1_wtPin1) binding to different DNA probes from the groups listed in Table 2. A representative graph from two independent replicates is shown. Error bars, 1 S.D. C–E, determination of Kd values for estrogen-occupied ERα (filled circles) or ERα + Pin1 (open squares) for three DNA sequences (Seq1, Seq2, and Seq3; shown by arrows in Fig. 5A) from the SNAP array using FP. F, correlation between normalized SNAP intensity and Ka, relating fluorescence intensities on the SNAP arrays to affinity of ERα (filled circles) or ERα + Pin1 (open squares) for each DNA sequence.
Chromatin Immunoprecipitation (ChIP)
MCF7 cells overexpressing Pin1 or vector were treated with E2 for the indicated times periods, followed by chromatin cross-linking, lysis, fragmentation, immunoprecipitation, and quantitative real-time RT-PCR as described previously (33, 46). ERα was immunoprecipitated using ERα (HC-20, SC-543) antibody (Santa Cruz Biotechnology). Quantitative real-time RT-PCR was carried out with 1 μl of input or 4 μl of immunoprecipitated sample and 200 nm forward and reverse primers (primer sequences available upon request). Data are calculated as percentage of input by the ΔΔCt method.
Protein Purification
GST-tagged Pin1 and K63A Pin1-expressing plasmids (gifts from Dr. Kun Ping Lu, Harvard Medical School) were transformed into Escherichia coli BL21 (pLysS) cells (Novagen). Proteins were purified as described previously (29). To cleave the GST tag, proteins were incubated with thrombin (Sigma) solution overnight at 4 °C. The eluted fractions were combined and buffer-exchanged with 50 mm Tris-Cl, pH 8.0, 200 mm NaCl, and 10% glycerol using 10,000 kDa cut-off centrifugal filters (Millipore). The level of protein purity was assessed using SDS-PAGE stained with Coomassie Brilliant Blue (Bio-Rad).
In Vitro Dephosphorylation Assay
This assay was carried out as described previously with modifications (25). Purified ERα (Life Technologies) was incubated with 50 ng of purified trans-specific PP2A holoenzyme (a gift from Dr. Yongna Xing, University of Wisconsin (Madison, WI)) and 1 μg of purified BSA (control) or Pin1 in dephosphorylation buffer (50 mm Tris-Cl, pH 7.5, 50 mm NaCl, 50 μm MnCl2) at 30 °C for 0–90 min. The reaction was terminated with 2× SDS sample buffer at the indicated times, and phosphorylation was assessed by Western blot using Ser(P)118 ERα-specific antibody. Equivalent loading was confirmed by reprobing with ERα-specific antibody.
Statistical Analysis
Student's paired t tests and analysis of variance were performed using GraphPad Prism (GraphPad Software). p values of less than 0.05 were considered significant and are indicated by asterisks in the figures.
Results
Pin1 Enhances the DNA Binding Activity of ERα from MCF7 Breast Cancer Cells in the Presence of Estrogen
Our previous studies indicated that Pin1 catalyzed conformational shifts in the ERα N-terminal AF1 domain, leading to enhanced transcriptional activity (25). Phosphorylation can also functionally alter the DBD and LBD of ERα (16, 47). Thus, we hypothesized that Pin1-induced changes in AF1 may alter receptor transcriptional activity through other functional domains. We chose to focus on the DBD, which is immediately adjacent to the AF1 domain. ERα is maximally phosphorylated within 30 min by E2 treatment in our MCF7 model system (Fig. 1A). To evaluate the role of Pin1 in endogenous ERα DNA binding function, MCF7 breast cancer cells stably expressing GFP-Pin1 or GFP alone (vector) were stimulated with E2 for 30 min. EMSA with nuclear extracts showed that Pin1 overexpression increased ERα DNA binding activity in the presence of ligand by ∼5-fold (Fig. 1, B and C). The Pin1-induced increase in DNA binding affinity concurs with increased transcriptional activity of liganded ERα induced by Pin1 overexpression in MCF7 (25). Because Pin1 can regulate ERα protein abundance levels (31), Western blot analysis was performed to confirm that ERα protein levels were equivalent in lysates used in EMSAs (Fig. 1D). These data indicate that Pin1 can increase DNA binding independent of its activity on receptor protein levels. The increase in DNA binding in E2-treated cells implicates a role for ligand and the LBD. Hence, similar experiments were conducted in MCF7 cells treated with OHT (Fig. 1, E and F). In contrast to E2, Pin1 did not significantly enhance the ERα-DNA interaction in the presence of OHT. Together, these data indicate that Pin1 regulates ERα DNA binding in a ligand-specific manner, independent of effects on ERα protein levels.
FIGURE 1.

Pin1 overexpression increases ERα-ERE interaction in breast cancer cells. A, purified recombinant ERα (rERα; 0.075–0.3 pmol, 5–20 nm) and MCF-7 cell extracts treated with EtOH or 10 nm E2 (0–480 min) were separated by SDS-PAGE, and Western blot was performed for Ser(P)118-ERα, ERα, and actin using appropriate antibodies. B, nuclear extracts from MCF7 cells stably overexpressing GFP (vector (V)) or GFP-Pin1 (Pin1) treated without (−) and with 10 nm E2 (E2) were incubated with 32P-labeled ERE followed by gel electrophoresis and autoradiography. SS, ERα supershift using anti-ERα antibody. ERE represents unbound free probe. C, EMSA autoradiography was quantified using a PhosphorImager, and data are represented as means ± S.E. (error bars) for at least three independent experiments. *, p < 0.05 comparing E2-treated vector and Pin1-expressing cells. Data were normalized to vehicle-treated vector-expressing cells. D, Western blot analysis was performed for ERα and Pin1 on 10-μg nuclear extracts of E2-treated and untreated MCF7 cells stably overexpressing GFP (vector (V)) or GFP-Pin1 (Pin1) using appropriate antibodies. E, EMSA was performed as in B in vector- or Pin1-expressing cells treated with (+) or without (−) 100 nm OHT. F, EMSA for E was quantified as in C, and data are represented as means ± S.E. for at least three independent experiments. n.s., not significant comparing OHT-treated vector and Pin1-expressing cells. Data were normalized to vehicle-treated vector-expressing cells.
EGF activates ERα by kinase-mediated phosphorylation of the AF1 domain and promotes Pin1 binding to the receptor (12, 25, 48). To further explore the specificity of Pin1 regulation on ERα DNA binding, EMSA was conducted with EGF-treated cells. EGF-induced phosphorylation occurs on a different time scale than E2, and maximal Ser118 phosphorylation occurred within 5 min of stimulation (Fig. 2A). Thus, MCF7-GFP and MCF7-Pin1 GFP cells were treated with EGF for 5 min, and nuclear extracts were prepared. EMSAs showed that Pin1 increased DNA binding activity in the absence of stimulation (Fig. 2, B and C). However, EGF treatment resulted in no further enhancement of the Pin1 effect on ERα DNA binding function. Because previous studies showed that EGF treatment was sufficient to promote Pin1 binding to ERα (25), these data suggest that EGF-induced phosphorylation and recruitment of Pin1 to ERα is not sufficient for Pin1-mediated effects on ERα DNA binding activity.
FIGURE 2.

Phosphorylation by EGF is insufficient to enhance Pin1-mediated effects on ERα-DNA binding. A, Western blot of ERα phosphorylated at Ser118 in response to 100 ng/ml of EGF at the indicated time points. NS, nonspecific band for loading control. B, nuclear extracts (10 μg) from MCF7 cells stably overexpressing GFP (vector (V)) or GFP-Pin1 (Pin1) treated with (EGF) and without (−) 100 ng/ml EGF were incubated with ERE followed by gel electrophoresis and autoradiography. The ERα-ERE complex was competed with unlabeled ERE or mERE. SS, ERα supershift using anti-ERα antibody. ERE represents unbound free probe. Bands in the oval box were quantified. C, EMSA autoradiography (B) was quantified using a PhosphorImager, and data are represented as means ± S.E. (error bars) for at least three independent experiments. No statistically significant differences were observed (p > 0.05).
Pin1 cis/trans-Isomerase Activity Is Required to Increase ERα DNA Binding Function
To better define Pin1 activity on ERα DNA binding, solution-based DNA binding assays using purified components were conducted. We used commercially purified ERα that is phosphorylated at Ser118 (Fig. 1A). Recombinant ERα binds to a perfect ERE but not to the mERE in EMSAs, providing evidence of specific binding. Furthermore, competition binding with cold ERE and mERE confirmed that this interaction was specific to the wild-type response element (Fig. 3A). Purified recombinant Pin1 used in our assays is catalytically active and causes cis/trans-isomerization of the Ser(P)118–Pro119 bond of purified ERα, as determined by increased sensitivity to dephosphorylation by trans-specific PP2A (Fig. 3B) (25). FP using a DNA probe consisting of a consensus ERE was used to quantitatively determine the Kd of ERα DNA binding in the presence and absence of Pin1. The consensus ERE used for the FP assays consisted of the core AGGTCA-CCG-TGACCT flanked by constant regions that formed a hairpin DNA. Pin1 significantly improved the Kd of the ER-ERE interaction by at least 2-fold (4.0 ± 1.6 nm versus 2.0 ± 0.6 nm, p = 0.003), and a representative experiment is shown in Fig. 3C. Similar to findings with EMSAs, there was undetectable binding of ERα to mERE in FP experiments under all conditions tested.
FIGURE 3.

In vitro ERα interaction with ERE is increased in the presence of Pin1. A, recombinant purified ERα (300 nm) was incubated with 32P-labeled ERE, 32P-labeled mERE, or 100-fold unlabeled ERE, and/or unlabeled mERE, followed by gel electrophoresis and autoradiography. Free probe indicates unbound ERE and mERE. The indicated unlabeled EREs were used for competition (comp.) of the complex. B, top, model representing cis and trans conformations of the Ser(P)118–Pro119 bond. The schematic of ERα N-terminal domain (NTD) shown at the top represents purified ERα. A stick model shows the phosphorylated Ser(P)118–Pro119 bond in cis and trans conformations and potential catalysis by Pin1. PP2A selectively dephosphorylates the trans-isomer and was used as a biochemical tool to assess cis and trans isomers of the Ser(P)118–Pro119 bond of ERα. Bottom, in vitro dephosphorylation was performed as described under “Experimental Procedures” in the presence of BSA or Pin1. Reaction mixtures were incubated at 30 °C for the indicated length of time and immunoblotted for Ser(P)118-ERα, ERα, and Pin1 using appropriate antibodies. C, determination of Kd values for purified ERα (closed circle and square) or ERα + Pin1 (closed triangle and open square) for DNA sequences (ERE and mERE), with the same sequences as probes from the SNAP array, using FP. ND, not detectable Kd. Shown is a representative figure for at least three independent experiments. *, p < 0.05 between ERα and ERα + Pin1. D, in vitro EMSA was performed as in A with 200 nm ERα and different Pin1 concentrations (0–12 μm). E, determination of Kd values for ERα (filled circles) or ERα + Pin1 (2–20 μm) for DNA sequences using FP. Shown is a representative figure for at least three independent experiments. *, p < 0.05 between ERα and ERα + Pin1 (2 μm).
Dose-response curves indicated that Pin1 regulation of ERα DNA binding occurs within a broad range of Pin1 concentrations (Fig. 3, D and E). EMSA analysis indicated that Pin1 increased ERα binding affinity at doses ranging from 0.060 to 12 μm, with maximal binding occurring at 1.5 μm, and higher doses of Pin1 showed a diminution of ERα-DNA binding. In addition, a reproducible but unexplainable decrease in binding occurred at 600 nm Pin1. In agreement with the EMSA data, quantitative determination by FP indicated that although the addition of Pin1 enhanced the Kd of ERα binding in all cases, the effect was greater at lower doses of Pin1 (2 μm) relative to high doses (10 or 20 μm) (Fig. 3E). These results suggest that Pin1 activity on ERα is concentration-dependent, and based on our EMSA analysis, Pin1 exhibits a biphasic dose response (Fig. 3D).
Next, the requirement for isomerization by Pin1 on ERα DNA interactions was tested. To avoid potential saturation of binding and to best visualize the effect of Pin1, subsequent EMSA experiments used a suboptimal concentration of 100 nm purified ERα unless otherwise noted (Fig. 4A). Juglone is a specific pharmacological inhibitor of the catalytic activity of Pin1 in vitro (49). Quantification of the EMSA results showed that there was an ∼16-fold increase in DNA binding activity of purified ERα due to Pin1. This effect was completely abolished in the presence of juglone (Fig. 4, B and C). These results were recapitulated by FP, where juglone consistently blocked the effect of Pin1 on ERα DNA binding affinity (Fig. 4, D and E). The role of Pin1-mediated cis-trans-isomerization in ERα DNA binding function was further confirmed using a catalytically inactive (K63A) mutant Pin1 protein (50). Compared with wild-type Pin1, the K63A Pin1 mutant did not increase ERα DNA binding activity by either EMSA or FP (Fig. 4, F and G). These data provide evidence that Pin1 isomerase activity is necessary for Pin1 to enhance ERα binding to its response element.
FIGURE 4.
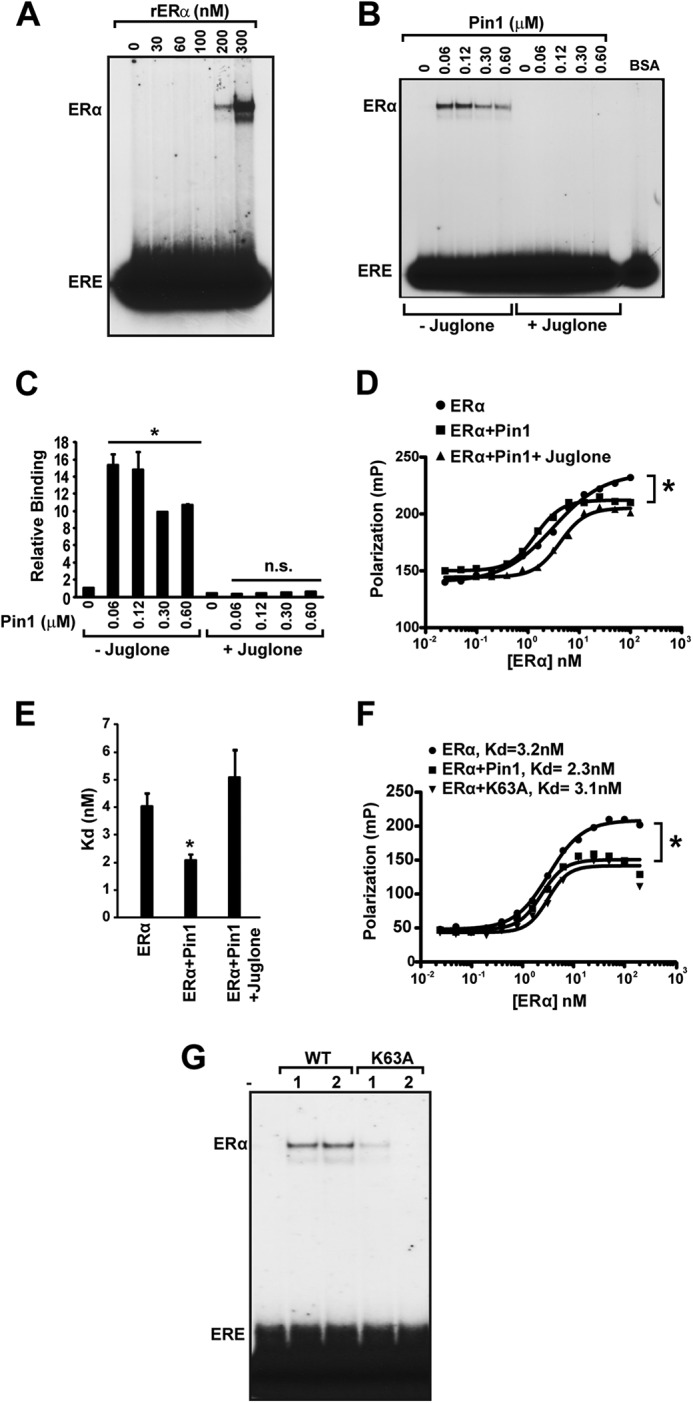
Pin1 isomerase function increases ERα binding affinity to ERE. A, to assess various levels of ERα-ERE binding, recombinant purified ERα (0–300 nm) was first incubated at 30 °C for 30 min and then incubated with 32P-labeled ERE, followed by gel electrophoresis and autoradiography. B, in vitro EMSA was performed as in (Fig. 3D) with 100 nm ERα and different Pin1 amounts (0–0.6 μm) in the presence and absence of Pin1 catalytic inhibitor, juglone (10 μm), and 100 ng of BSA. C, EMSA autoradiography in B was quantified using a PhosphorImager, and data are represented as means ± S.E. (error bars) for at least three independent experiments. *, p < 0.05; n.s., not significant comparing ERα and ERα + Pin1. D, determination of Kd values for unoccupied ERα (closed circles), ERα + Pin1 (closed squares), or ERα + Pin1 + juglone (closed triangles) for the ERE was assessed by FP. E, graph showing Kd values for ERα, ERα + Pin1, and ERα + Pin1 + juglone. Data are represented as means ± S.E. for at least three independent experiments. *, p < 0.05. F, determination of Kd values for unoccupied ERα (closed circles), ERα + Pin1 (closed squares), or ERα + K63A mutant Pin1 (closed triangles) for the ERE was assessed by FP, and a representative graph is shown. *, p < 0.05. G, in vitro EMSA was performed as in B in the presence and absence of 1 and 2 μm Pin1 (WT) or catalytic mutant K63A.
Specificity and Affinity Landscape of ERα DNA Binding Properties Regulated by Pin1
Because all of the DNA binding experiments in previous figures were performed with just two variations of the consensus EREs, we next wanted to unbiasedly explore the global effect of Pin1 on the DNA binding profile of ERα. For this, we utilized the high throughput DNA microarray, referred to as the SNAP array, which displays 385,000 double-stranded DNA sequences in unique spatially resolved features (44, 45). On the custom-designed SNAP array, duplex DNA probes were designed to contain 9 permuted bp, or 49 ERE variations, preceded by a constant ERα half-site. Each probe was additionally flanked by a constant 5-bp region on each end to form 25-bp B-form DNA after hairpinning (Table 2) (44, 45). Binding of purified recombinant ERα to the DNA was detected by fluorescence, and the comprehensive DNA binding specificity and affinity of ERα to all sequences assayed were analyzed using motif-finding algorithms (51, 52) and sequence specificity landscapes (SSLs; Fig. 5A) (44). Motif analysis of the top fluorescence intensity sequences indicated that ERα alone and ERα + Pin1 bound to the consensus ERE (Fig. 5A, top). However, when SSL analysis was employed to display the binding intensity from all 9-mer core sequences (underlined) using the dual core-motif AGGTCA-NNNTGACCT and AGGTCA-TGACCTNNN, Pin1-induced differences in ERα binding to DNA were revealed.
TABLE 2.
Each group contains different DNA sequences tested
DNA sequences (underlined) represent potential ERα binding sites, and additional sequences are the flanking, hairpin loop, or spacer sequences between half-sites. N, any nucleotide; N′, complement of N; GRE, glucocorticoid response elements; ARE, androgen response elements; ERR, estrogen-related receptor; IR, inverted repeat; DR, direct repeat.
| Group | Description | Sequence |
|---|---|---|
| A | ER half-site held constant; 3-bp spacing and second half-site varied | FLANK-AGGTCAN1–9-FLANK-GGA-FLANK-N9′–1′-TGACCT-FLANK |
| B | Perfect consensus with 5-mer spacer permuted | FLANK-AGGTCA-N1–5-TGACCT-FLANK-GGA-FLANK-AGGTCA-N5′–1′-TGACCT-FLANK |
| C | Perfect consensus with 4-mer spacer permuted | FLANK-AGGTCA-N1–4-TGACCT-FLANK-GGA-FLANK-AGGTCA-N4′–1′-TGACCT-FLANK |
| D | Perfect consensus with 3-mer spacer permuted | FLANK-AGGTCA-N1–3-TGACCT-FLANK-GGA-FLANK-AGGTCA-N3′–1′-TGACCT-FLANK |
| E | Perfect consensus with 2-mer spacer permuted | FLANK-AGGTCA-N1-2-TGACCT-FLANK-GGA-FLANK-AGGTCA-N2′–1′-TGACCT-FLANK |
| F | Perfect consensus with 1-mer spacer permuted | FLANK-AGGTCA-N-TGACCT-FLANK-GGA-FLANK-AGGTCA-N-TGACCT-FLANK |
| G | Perfect consensus with 0 mer spacer | FLANK-AGGTCATGACCT-FLANK-GGA-FLANK-AGGTCATGACCT-ACCGC |
| H | Randomized 8-mer ERE/GRE/ARE | FLANK-N1GN2N3CN4-GGC-N5GN6N7CN8-FLANK-GGA-FLANK-N8′GN7′N6′CN5′-GCC-N4′GN3′N2′CN1′-FLANK |
| I | ERE | FLANK-AGGTCA-CAG-TGACCT-FLANK-GGA-FLANK-AGGTCA-CTG-TGACCT-FLANK |
| J | mERE | FLANK-AGATCA-CAG-TGATCT-FLANK-GGA-FLANK-AGATCA-CTG-TGATCT-FLANK |
| K | mERE2 | FLANK-GAGACA-CAG-GGTTTC-FLANK-GGA-FLANK-GAAACC-CTG-TGTCTC-FLANK |
| L | ER positive control | FLANK-AGGTCA-GGC-TGACCT-FLANK-GGA-FLANK-AGGTCA-GCC-TGACCT-FLANK |
| M | ER negative control | FLANK-AGATCA-GGC-TGATCT-FLANK-GGA-FLANK-AGATCA-GCC-TGATCT-FLANK |
| N | GR positive control | FLANK-AGAACA-GGC-TGTTCT-FLANK-GGA-FLANK-AGAACA-GCC-TGTTCT-FLANK |
| O | GR negative control | FLANK-GAGACA-GGC-GGTTTC-FLANK-GGA-FLANK-GAAACC-GCC-TGTCTC-FLANK |
| P | AR positive control | FLANK-GGTACA-GGC-TGTTCT-FLANK-GGA-FLANK-AGAACA-GCC-TGTACC-FLANK |
| Q | AR negative control | FLANK-TGGTCA-GGC-AGTTCT-FLANK-GGA-FLANK-AGAACT-GCC-TGACCA-FLANK |
| R | ERRγ positive control | FLANK-TCAAGGTCA-FLANK-GGA-FLANK-TGACCTTGA-FLANK |
| S | ERRγ negative control | FLANK-CAGAGGTCA-FLANK-GGA-FLANK-TGACCTCTG-FLANK |
| T | ERE IRO | FLANK-AGGTCA-TGACCT-FLANK-GGA-FLANK-AGGTCA-TGACCT-FLANK |
| U | ERE IR1 | FLANK-AGGTCA-C-TGACCT-FLANK-GGA-FLANK-AGGTCA-G-TGACCT-FLANK |
| V | ERE IR2 | FLANK-AGGTCA-CA-TGACCT-FLANK-GGA-FLANK-AGGTCA-TG-TGACCT-FLANK |
| W | ERE IR3 | FLANK-AGGTCA-CAC-TGACCT-FLANK-GGA-FLANK-AGGTCA-GTG-TGACCT-FLANK |
| X | ERE IR4 | FLANK-AGGTCA-CACA-TGACCT-FLANK-GGA-FLANK-AGGTCA-TGTG-TGACCT-FLANK |
| Y | ERE IR5 | FLANK-AGGTCA-CACAC-TGACCT-FLANK-GGA-FLANK-AGGTCA-GTGTG-TGACCT-FLANK |
| Z | ERE DR0 | FLANK-AGGTCAAGGTCA-FLANK-GGA-FLANK-TGACCTTGACCT-FLANK |
| AA | ERE DR1 | FLANK-AGGTCA-C-AGGTCA-FLANK-GGA-FLANK-TGACCT-G-TGACCT-FLANK |
| BB | ERE DR2 | FLANK-AGGTCA-CA-AGGTCA-FLANK-GGA-FLANK-TGACCT-TG-TGACCT-FLANK |
| CC | ERE DR3 | FLANK-AGGTCA-CAC-AGGTCA-FLANK-GGA-FLANK-TGACCT-GTG-TGACCT-FLANK |
| DD | ERE DR4 | FLANK-AGGTCA-CACA-AGGTCA-FLANK-GGA-FLANK-TGACCT-TGTG-TGACCT-FLANK |
| EE | ERE DR5 | FLANK-AGGTCA-CACAC-AGGTCA-FLANK-GGA-FLANK-TGACCT-GTGTG-TGACCT-FLANK |
| FF | ER or ERR half-site, 6-mer flanking | FLANK-N1N2N3-AGGTCA-N4N5N6-FLANK-GGA-FLANK-N6′N5′N4′-TGACCT-N3′N2′N1′-FLANK |
In SSLs, intensities for each DNA sequence from the array are plotted on a series of concentric rings, where each array sequence in the center ring contains a subsequence that perfectly matches the motif, sequences in the second ring contain a subsequence with one mismatch to the consensus, sequences in the third ring contain a subsequence with two mismatches to the consensus, and so forth. The height of the peak corresponds to intensity and is proportional to binding affinity (Ka). Results indicate that the affinity of ERα binding to DNA, in particular to DNA sequences bearing the consensus ERE AGGTCANNNTGACCT (Fig. 5A), were enhanced in the presence of Pin1. Pin1 affected ERα binding to the AGGTCA presented within the context of a palindromic sequence (Groups D, I, and L as examples; Table 2 and Fig. 5B) but not with a simple AGGTCA half-site (Groups R and FF; Table 2 and Fig. 5B). Moreover, DNA sequences bearing inverted repeats were preferred over direct repeats. These findings are observed in the SSLs, in which the sequences in the central ring (perfect match to the SSL consensus; shown by arrows) of the ERα + Pin1 landscape are of significantly higher intensity than the central ring of the ERα-alone landscape. Comparison of the SSLs showed several Pin1-induced high intensity peaks in the outer rings, and these sequences correspond to AGGTCA direct repeats of 0 and 3 spacers, which are known ERα targets (53). A ridge of higher intensity peaks radiating outward in the ERα + Pin1 landscape also contains sequences in which the motif degrades starting at the 3′-end. The first ring in the ridge contains NNNTGACCN, and the subsequent second ring includes NNNTGACNN. ERα plus Pin1 demonstrated preferences for the consensus ERE and 3-bp spacer, specifically the spacer CCG (labeled Seq1). Additional spacer analysis indicated that Pin1 also enhanced ERα interaction at inverted repeats with 0-, 2-, and 3-bp spacers.
To confirm the ERα signal enhancement on the SNAP array by Pin1, we used FP to quantify the effect of Pin1 on ERα binding to three different DNA sequences (Seq1, Seq2, and Seq3; 9-mer embedded within the 25-bp hairpin) (Fig. 5, C–E). As expected, the EREs bearing a consensus AGGTCANNNTGACCT resulted in the highest affinity of ERα. A plot of Ka versus normalized intensity yielded a linear correlation (r2 > 0.94), thereby allowing the intensity of every 9-mer sequence to be related to affinity by Ka (Fig. 5F). Consistent with our previous EMSA and FP results, the Ka plot also shows that Pin1 enhances the affinity of ERα for the three sequences analyzed by FP.
FP and SNAP analysis indicated that Pin1 does not form a stable interaction with the ERα-ERE complex but has a transient enzymatic interaction perhaps similar to other “allosteric cofactors” (54, 55). The FP curve for ERα in the presence of Pin1 reached a lower plateau than ERα alone. FP provides readout on both the shape and size of the ERα-DNA complex (56). Hence, the change in the ERα + Pin1 FP curve is consistent with Pin1 inducing a conformational change in the ERα protein (25).
Pin1 Impacts ERα Recruitment to Endogenous Estrogen Response Elements
We used a ChIP assay in MCF7 cells overexpressing Pin1 or vector to probe for ERα-DNA interaction at ERα binding sites on endogenous targets. Binding sites interrogated include the well characterized pS2 promoter, the ESR1 distal region (ENH1; −150 kb), the ESR1 promoter (+60), and a nonspecific intervening region (−811) (46) (Fig. 6, A–C). ERα enrichment on ENH1 and pS2 ERE sites were higher in Pin1-overexpressing cells (Fig. 6, A and B). In agreement with our SNAP data, these sites bear near perfect EREs, supporting the notion that Pin1 enhances ERα occupancy at the ERE sites most resembling a perfect palindromic consensus. In contrast, Pin1 had essentially no impact on the ERα binding site on the ESR1 proximal promoter region (+60) (Fig. 6C) and a nonspecific region (−811) (Fig. 6, A–C). The latter two sites lack a consensus ERE, reflected by the low percentage of input DNA bound, although the ESR1 promoter is a site of estrogen-regulated ERα binding (46). Although the effect of Pin1 on ERα occupancy on DNA in cells is not as robust as in in vitro studies, the trend is consistent with EMSA and FP findings that Pin1 regulates ERα interaction with EREs on target genes.
FIGURE 6.

Pin1 regulates ERα enrichment on its target genes. A–C, MCF7 cells expressing GFP or GFP-Pin1 were treated with 10 nm E2 for the indicated times, and ChIP analyses were performed to examine occupancy of ERα at regions of pS2 promoter (A), ESR1 ENH1 (B), ESR1 +60 promoter (C), and ESR1 −811 nonspecific region (A–C). Data are represented as means ± S.E. (error bars) for at least three independent experiments. No statistically significant differences were observed (p > 0.05).
Discussion
ERα and Pin1 are important regulatory molecules in cell division and growth control that have overlapping and distinct roles in cancer (57, 58). The growth-promoting activity of ERα is mediated through the transcriptional regulation of gene expression. The mechanisms governing ERα transactivation are well described, and hundreds of ERα protein partners have been invoked as part of the process (59). They include multiple chromatin-remodeling and chromatin-modifying enzymes as well as the multiprotein Mediator complex, which bridges ERα with the basal transcriptional machinery. Phosphorylated ERα comprises ∼48% of the total ERα in the presence of ligand (60), and gene expression analysis indicates that phosphorylated ERα regulates a subset of ERα target genes involved in cell growth (32). Pin1 was discovered as a critical regulator of mitosis and is overexpressed in a number of cancers, including breast, prostate, lung, and colon, among others relative to normal controls (27, 57, 61, 62). A large number of substrates have been reported for Pin1, including critical cancer oncogenes (e.g. c-Myc, cyclin D1, and p53), transcriptional co-regulators (e.g. SMRT and AIB1), chromatin, and components of the transcriptional machinery, such as RNA polymerase II (30, 63). Pin1 is also known to associate with several nuclear receptors, including glucorticoid receptor, PPARγ, Nur77, androgen receptor, and retinoic acid receptor (38, 64–66, 68). The broad activities of ERα and Pin1, coordinated through multiprotein complexes, make it difficult to dissect the specific activities of these partners in transcription and growth.
Our previous studies showed that Pin1 binds to the phosphorylated ERα, and Pin1 isomerase activity increases receptor transactivation function in both the presence and absence of ligand (25). However, given the extensive protein interactions of both ERα and Pin1, an understanding of the direct role of Pin1 in ERα transactivation was lacking. In this study, we provide evidence that Pin1 can directly increase ERα DNA binding affinity, providing a plausible explanation for increased ERα transcriptional function by Pin1 in estrogen-treated cells. Using both breast cancer cell lysates and purified components, our data indicate that the overexpression or addition of Pin1 is sufficient to increase ERα affinity for DNA binding sites. Pin1 isomerase activity is responsible for the resultant effect, suggesting that Pin1-induced conformational changes are involved.
These studies, combined with our previous studies (25), support a model in which Pin1-catalyzed isomerization induces conformational changes in the ERα N terminus, which in turn modulates the ability of ERα to dimerize, recognize DNA binding sites, and regulate transcription. Order of addition experiments as well as inhibition of Pin1 catalytic function indicate that Pin1 isomerase activity must occur prior to the effects on ERα-DNA binding affinity. These Pin1-mediated effects can act on unliganded ERα but are further enhanced for estrogen-bound ERα, which suggests that conformational changes from the N terminus are coordinated with ligand-induced changes in the LBD to modulate the activities of the DBD. Of interest, Pin1 enhances transactivation of ERα in the presence of OHT (25) but does not significantly increase the binding of OHT-occupied ERα to DNA. This finding points to context-specific control of ERα by Pin1 and indicates that Pin1 enhances tamoxifen-driven transcriptional activity through a separate mechanism. In either case, our data reveal that Pin1 regulation of ERα transcription involves the integrated activities of multiple functional domains.
Interestingly, Pin1 does not directly interact with the ERα DBD but rather binds to a phosphorylation site in the adjacent ID region (25). In addition to Ser118, Pin1 has also been reported to bind to Ser294 in the disordered hinge region immediately downstream of the DBD (69). Changes in DNA binding activity coordinated by ID regions have been observed in other transcription factors. For example, the ID regions in Cys2-His2 zinc finger transcription factors lock the DNA binding motif into conformations that contribute to tighter and specific DNA interactions (70, 71). Moreover, ID regions can also coordinate hydrophobic and electrostatic interactions that shape the interface of the DBD and DNA, as has been shown for Max and Ets factors (72, 73). ID regions are mostly enriched with proline residues, and proline isomerization has been shown to kink the protein backbone which functions as a molecular switch to regulate the structure of nearby domains (74–76). Therefore, allosteric regulation of the ERα DBD through extended conformational changes originating in the AF1 domain is plausible, although the lack of effect of EGF, which induces phosphorylation and Pin1 binding but does not affect ERα DNA binding, suggests additional complexities. Given the high proportion of ID regions in transcription factors that are common sites of phosphorylation, it is possible that the effects of Pin1 on DNA binding observed here on ERα may be more generally applied and be a model for the role of ID domain regulation in transcription factor function.
Pin1 may also regulate ERα DNA binding via promoting interdomain interactions. As mentioned previously, the importance of ligand in modulating Pin1 activity implicates coordinated regulation of the N-terminal AF1 and the C-terminal LBD on the DBD. This finding is in agreement with other results, which indicate that the N and C termini of receptors coordinately regulate receptor activity (77–79). Other receptor binding partners have been identified that enhance allosteric interactions among functional domains. Direct p300 interaction with the N-terminal A/B region of ERα not only bridges the N and C termini of ERα but also enhances receptor transactivation through ligand-induced synergism between AF1 and AF2 (80). Similarly, Dutertre and Smith (24) showed the importance of AF1 in recruiting SRC1 (steroid receptor coactivator-1) and CBP (CREB-binding protein) in mediating interaction and transcriptional synergism between AF1 and AF2 domains. Previous studies have also suggested that AF1 coregulator binding may have long range effects through changes in structure. Like Pin1, TBP binds to the N-terminal AF1 domains of ERα as well as the glucorticoid receptor and progesterone receptor (10, 81–83). In the case of glucorticoid receptor, TBP binding induces the transition to an ordered structure that facilitates interaction with the coactivator SRC1 to enhance ligand activated transcription. Recent studies of progesterone receptor using hydrogen-deuterium exchange have shown that TBP binding to progesterone receptor AF1 domain stabilized the AF2 domain, supporting the notion that coregulator binding at the AF1 domain may facilitate physical interactions between the AF1 and AF2 to alter transcriptional responses (83). NMR analysis of the ERα N terminus showed that, unlike TBP, Pin1 binding and isomerization of ERα does not induce an ordered structure in the N terminus (25). Pin1 did, however, increase ERα dimerization (25), which is necessary for receptor DNA binding function. We propose that Pin1 probably plays multiple roles in directing ERα DNA binding function by inducing conformational changes that are transmitted to both the DBD and dimer interface and that can be further modulated by ligand interactions in the LBD.
The SNAP-SSL analysis provided an unprecedented view into the effects of Pin1 on the ERα-ERE interactions. For our analysis, we employed the SNAP technology which, unlike ChIP-sequencing, can distinguish direct ERα-DNA interactions from indirect/protein-tethered interactions with DNA and also overcomes complications associated with Pin1 interactions with basal transcriptional machinery. Moreover, although ChIP-sequencing is an established and powerful technique, it requires next generation sequencing expertise, takes days or weeks to complete depending on the number of lanes needed, generates complicated data, is particular to a cell type or condition, is labor-intensive, and includes DNA amplification (84–86). SNAP arrays, however, can be performed with purified proteins or lysates with high throughput. SNAP arrays were particularly well suited for our analysis because we wanted to pinpoint the specific biochemical effects of Pin1 on the ERα-ERE interaction without the input of other cellular proteins. The DNA probes examined on the SNAP array contained the core sequence AGGTCA-9-mer embedded within a 25-bp hairpin, allowing for an unbiased analysis of the 3-mer spacer and 6-mer half site (49 or 262,144 permutations). Motif searching indicated an ERα preference for the consensus TGACCT at the second half-site in the presence or absence of Pin1.
SNAP-SSL analysis of all 9-mer permutations adjacent to the AGGTCA sequence further uncovered additional Pin1-induced effects on ERα-DNA binding specificity and affinity. The most apparent effect of Pin1 was an enhancement of ERα affinity for the consensus ERE, but Pin1 caused ERα to be recruited to other ERα binding sites as well, including inverted repeats of zero, two, and three spacers as well as AGGTCA direct repeats. The SNAP data indicated that ERE spacer sizes of 0- and 3-mer are preferred (Groups T and W; Table 2 and Fig. 5B), as has been observed previously (53). In addition, enhanced ERα interaction at an inverted repeat is consistent with ERα dimer binding, and our previous findings indicated that Pin1 enhances ERα dimerization (25). Data showing that ERα + Pin1 consistently preferred the CCG spacer within the context of the inverted repeat suggest that Pin1 may also alter ERα-DNA interactions directly via subtle spacer sequence preferences.
To further support Pin1 effects on ERα specificity, we performed a differential binding analysis of the full 262,144 SNAP sequences. In comparing ERα versus ERα + Pin1 affinities in this analysis, ERα + Pin1 is enriched for strong palindromic ERE sequences, whereas ERα alone is enriched for sequences with a nearly unidentifiable degenerate second half-site. The ability of ERα to recognize a strong half-site with a highly degenerate second half-site has been observed in genome-wide ERα assays (67, 87). Our comparative analysis of Pin1 enhancement of ERα binding at sites that best resemble the consensus ERE is further supported by our ChIP findings.
Identification of direct actions of Pin1 on ERα transactivation were facilitated by in vitro approaches, which revealed activities that would not be uncovered in the complex environments of the cell. The studies reported here indicate that although Pin1 may regulate multiple factors in the transcriptional process, at least one component of Pin1 isomerase activity on ERα is through the direct regulation of receptor DNA binding activity. Although Pin1 is not required for ERα binding to the DNA, these results suggest that one of the outcomes of Pin1 binding is to tune the receptor function and contribute to the specificity of gene regulation by phosphorylated forms of the receptor. Importantly, the identification of Pin1 as an ID region-binding protein that changes ERα interactions with DNA opens the door to future structural studies to pursue the allosteric transmission of signal from an important yet poorly understood ID region of the ERα to other functional domains of the receptor. Such insight would contribute to an understanding of how communication between ID functional domains of nuclear receptors and other Pin1-regulated transcription factors coordinate DNA binding functions and transcriptional activation, offering novel avenues to target and control Pin1 interactions in disease.
Acknowledgments
We thank McArdle Laboratories for Cancer Research for support of this project. We also thank the University of Wisconsin Carbone Comprehensive Cancer Center for use of its shared services to complete this research. We thank Dr. Kun Ping Lu (Harvard Medical School) for Pin1 reagents and Dr. Yongna Xing (University of Wisconsin, Madison, WI) for purified PP2A holoenzyme.
This work was supported, in whole or in part, by National Institutes of Health Grants CA159578 (to E. T. A.) and T32 CA009135 (to P. R.). This work was also supported by Department of Defense Concept Award W81XWH-09-1-0581 (to M. S. O.). M. S. O. is a founder of Proteovista LLC.
- ERα
- estrogen receptor-α
- NR
- nuclear receptor
- DBD
- DNA-binding domain
- LBD
- ligand-binding domain
- ERE
- estrogen response element
- mERE
- mutated ERE
- E2
- 17β-estradiol
- OHT
- 4-hydroxytamoxifen
- ID
- intrinsically disordered
- SNAP
- specificity and affinity for protein
- FP
- fluorescence polarization
- SSL
- sequence specificity landscape
- AF
- activation function 1 and 2, respectively
- Seq
- sequence
- TBP
- TATA-binding protein
- CREB
- cAMP-response element-binding protein.
References
- 1. Klein-Hitpass L., Ryffel G. U., Heitlinger E., Cato A. C. (1988) A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res. 16, 647–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Klein-Hitpass L., Tsai S. Y., Greene G. L., Clark J. H., Tsai M. J., O'Malley B. W. (1989) Specific binding of estrogen receptor to the estrogen response element. Mol. Cell. Biol. 9, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schwabe J. W., Chapman L., Finch J. T., Rhodes D. (1993) The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 75, 567–578 [DOI] [PubMed] [Google Scholar]
- 4. Klinge C. M. (2001) Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 29, 2905–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kumar V., Green S., Stack G., Berry M., Jin J. R., Chambon P. (1987) Functional domains of the human estrogen receptor. Cell 51, 941–951 [DOI] [PubMed] [Google Scholar]
- 6. Tora L., White J., Brou C., Tasset D., Webster N., Scheer E., Chambon P. (1989) The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell 59, 477–487 [DOI] [PubMed] [Google Scholar]
- 7. Klinge C. M. (2000) Estrogen receptor interaction with co-activators and co-repressors. Steroids 65, 227–251 [DOI] [PubMed] [Google Scholar]
- 8. Métivier R., Penot G., Hübner M. R., Reid G., Brand H., Kos M., Gannon F. (2003) Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115, 751–763 [DOI] [PubMed] [Google Scholar]
- 9. Shang Y., Hu X., DiRenzo J., Lazar M. A., Brown M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852 [DOI] [PubMed] [Google Scholar]
- 10. Wärnmark A., Wikström A., Wright A. P., Gustafsson J. A., Härd T. (2001) The N-terminal regions of estrogen receptor α and β are unstructured in vitro and show different TBP binding properties. J. Biol. Chem. 276, 45939–45944 [DOI] [PubMed] [Google Scholar]
- 11. Chen D., Riedl T., Washbrook E., Pace P. E., Coombes R. C., Egly J. M., Ali S. (2000) Activation of estrogen receptor α by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol. Cell 6, 127–137 [PubMed] [Google Scholar]
- 12. Kato S., Endoh H., Masuhiro Y., Kitamoto T., Uchiyama S., Sasaki H., Masushige S., Gotoh Y., Nishida E., Kawashima H., Metzger D., Chambon P. (1995) Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 270, 1491–1494 [DOI] [PubMed] [Google Scholar]
- 13. Medunjanin S., Hermani A., De Servi B., Grisouard J., Rincke G., Mayer D. (2005) Glycogen synthase kinase-3 interacts with and phosphorylates estrogen receptor α and is involved in the regulation of receptor activity. J. Biol. Chem. 280, 33006–33014 [DOI] [PubMed] [Google Scholar]
- 14. Rogatsky I., Trowbridge J. M., Garabedian M. J. (1999) Potentiation of human estrogen receptor α transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin A-CDK2 complex. J. Biol. Chem. 274, 22296–22302 [DOI] [PubMed] [Google Scholar]
- 15. Thomas R. S., Sarwar N., Phoenix F., Coombes R. C., Ali S. (2008) Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-α activity. J. Mol. Endocrinol. 40, 173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Likhite V. S., Stossi F., Kim K., Katzenellenbogen B. S., Katzenellenbogen J. A. (2006) Kinase-specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, deoxyribonucleic acid, and coregulators associated with alterations in estrogen and tamoxifen activity. Mol. Endocrinol. 20, 3120–3132 [DOI] [PubMed] [Google Scholar]
- 17. Chandra V., Huang P., Hamuro Y., Raghuram S., Wang Y., Burris T. P., Rastinejad F. (2008) Structure of the intact PPAR-γ-RXR-nuclear receptor complex on DNA. Nature 456, 350–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chandra V., Huang P., Potluri N., Wu D., Kim Y., Rastinejad F. (2013) Multidomain integration in the structure of the HNF-4α nuclear receptor complex. Nature 495, 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He B., Wang K., Liu Y., Xue B., Uversky V. N., Dunker A. K. (2009) Predicting intrinsic disorder in proteins: an overview. Cell Res. 19, 929–949 [DOI] [PubMed] [Google Scholar]
- 20. Hilser V. J., Thompson E. B. (2011) Structural dynamics, intrinsic disorder, and allostery in nuclear receptors as transcription factors. J. Biol. Chem. 286, 39675–39682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu J., Perumal N. B., Oldfield C. J., Su E. W., Uversky V. N., Dunker A. K. (2006) Intrinsic disorder in transcription factors. Biochemistry 45, 6873–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Endoh H., Maruyama K., Masuhiro Y., Kobayashi Y., Goto M., Tai H., Yanagisawa J., Metzger D., Hashimoto S., Kato S. (1999) Purification and identification of p68 RNA helicase acting as a transcriptional coactivator specific for the activation function 1 of human estrogen receptor α. Mol. Cell. Biol. 19, 5363–5372 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Watanabe M., Yanagisawa J., Kitagawa H., Takeyama K., Ogawa S., Arao Y., Suzawa M., Kobayashi Y., Yano T., Yoshikawa H., Masuhiro Y., Kato S. (2001) A subfamily of RNA-binding DEAD-box proteins acts as an estrogen receptor α coactivator through the N-terminal activation domain (AF-1) with an RNA coactivator, SRA. EMBO J. 20, 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24. Dutertre M., Smith C. L. (2003) Ligand-independent interactions of p160/steroid receptor coactivators and CREB-binding protein (CBP) with estrogen receptor-α: regulation by phosphorylation sites in the A/B region depends on other receptor domains. Mol. Endocrinol. 17, 1296–1314 [DOI] [PubMed] [Google Scholar]
- 25. Rajbhandari P., Finn G., Solodin N. M., Singarapu K. K., Sahu S. C., Markley J. L., Kadunc K. J., Ellison-Zelski S. J., Kariagina A., Haslam S. Z., Lu K. P., Alarid E. T. (2012) Regulation of estrogen receptor α N-terminus conformation and function by peptidyl prolyl isomerase Pin1. Mol. Cell. Biol. 32, 445–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simons S. S., Jr., Edwards D. P., Kumar R. (2014) Minireview: dynamic structures of nuclear hormone receptors: new promises and challenges. Mol. Endocrinol. 28, 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu K. P., Hanes S. D., Hunter T. (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380, 544–547 [DOI] [PubMed] [Google Scholar]
- 28. Verdecia M. A., Bowman M. E., Lu K. P., Hunter T., Noel J. P. (2000) Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Biol. 7, 639–643 [DOI] [PubMed] [Google Scholar]
- 29. Yaffe M. B., Schutkowski M., Shen M., Zhou X. Z., Stukenberg P. T., Rahfeld J. U., Xu J., Kuang J., Kirschner M. W., Fischer G., Cantley L. C., Lu K. P. (1997) Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278, 1957–1960 [DOI] [PubMed] [Google Scholar]
- 30. Wulf G., Finn G., Suizu F., Lu K. P. (2005) Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat. Cell Biol. 7, 435–441 [DOI] [PubMed] [Google Scholar]
- 31. Rajbhandari P., Schalper K. A., Solodin N. M., Ellison-Zelski S. J., Ping Lu K., Rimm D. L., Alarid E. T. (2014) Pin1 modulates ERα levels in breast cancer through inhibition of phosphorylation-dependent ubiquitination and degradation. Oncogene 33, 1438–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng J., Zhang C., Shapiro D. J. (2007) A functional serine 118 phosphorylation site in estrogen receptor-α is required for down-regulation of gene expression by 17β-estradiol and 4-hydroxytamoxifen. Endocrinology 148, 4634–4641 [DOI] [PubMed] [Google Scholar]
- 33. Valley C. C., Métivier R., Solodin N. M., Fowler A. M., Mashek M. T., Hill L., Alarid E. T. (2005) Differential regulation of estrogen-inducible proteolysis and transcription by the estrogen receptor α N terminus. Mol. Cell. Biol. 25, 5417–5428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stanya K. J., Liu Y., Means A. R., Kao H. Y. (2008) Cdk2 and Pin1 negatively regulate the transcriptional corepressor SMRT. J. Cell Biol. 183, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu Y. X., Manley J. L. (2007) Pin1 modulates RNA polymerase II activity during the transcription cycle. Genes Dev. 21, 2950–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yi P., Wu R. C., Sandquist J., Wong J., Tsai S. Y., Tsai M. J., Means A. R., O'Malley B. W. (2005) Peptidyl-prolyl isomerase 1 (Pin1) serves as a coactivator of steroid receptor by regulating the activity of phosphorylated steroid receptor coactivator 3 (SRC-3/AIB1). Mol. Cell. Biol. 25, 9687–9699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Farrell A. S., Pelz C., Wang X., Daniel C. J., Wang Z., Su Y., Janghorban M., Zhang X., Morgan C., Impey S., Sears R. C. (2013) Pin1 regulates the dynamics of c-Myc DNA binding to facilitate target gene regulation and oncogenesis. Mol. Cell. Biol. 33, 2930–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Poolman T. M., Farrow S. N., Matthews L., Loudon A. S., Ray D. W. (2013) Pin1 promotes GR transactivation by enhancing recruitment to target genes. Nucleic Acids Res. 41, 8515–8525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Khanal P., Yun H. J., Lim S. C., Ahn S. G., Yoon H. E., Kang K. W., Hong R., Choi H. S. (2012) Proyl isomerase Pin1 facilitates ubiquitin-mediated degradation of cyclin-dependent kinase 10 to induce tamoxifen resistance in breast cancer cells. Oncogene 31, 3845–3856 [DOI] [PubMed] [Google Scholar]
- 40. Liou Y. C., Ryo A., Huang H. K., Lu P. J., Bronson R., Fujimori F., Uchida T., Hunter T., Lu K. P. (2002) Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc. Natl. Acad. Sci. U.S.A. 99, 1335–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wulf G. M., Ryo A., Wulf G. G., Lee S. W., Niu T., Petkova V., Lu K. P. (2001) Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 20, 3459–3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Furlow J. D., Ahrens H., Mueller G. C., Gorski J. (1990) Antisera to a synthetic peptide recognize native and denatured rat estrogen receptors. Endocrinology 127, 1028–1032 [DOI] [PubMed] [Google Scholar]
- 43. Ozers M. S., Warren C. L., Ansari A. Z. (2009) Determining DNA sequence specificity of natural and artificial transcription factors by cognate site identifier analysis. Methods Mol. Biol. 544, 637–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carlson C. D., Warren C. L., Hauschild K. E., Ozers M. S., Qadir N., Bhimsaria D., Lee Y., Cerrina F., Ansari A. Z. (2010) Specificity landscapes of DNA binding molecules elucidate biological function. Proc. Natl. Acad. Sci. U.S.A. 107, 4544–4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Warren C. L., Kratochvil N. C., Hauschild K. E., Foister S., Brezinski M. L., Dervan P. B., Phillips G. N., Jr., Ansari A. Z. (2006) Defining the sequence-recognition profile of DNA-binding molecules. Proc. Natl. Acad. Sci. U.S.A. 103, 867–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ellison-Zelski S. J., Solodin N. M., Alarid E. T. (2009) Repression of ESR1 through actions of estrogen receptor α and Sin3A at the proximal promoter. Mol. Cell. Biol. 29, 4949–4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tzeng D. Z., Klinge C. M. (1996) Phosphorylation of purified estradiol-liganded estrogen receptor by casein kinase II increases estrogen response element binding but does not alter ligand stability. Biochem. Biophys. Res. Commun. 223, 554–560 [DOI] [PubMed] [Google Scholar]
- 48. Bunone G., Briand P. A., Miksicek R. J., Picard D. (1996) Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 15, 2174–2183 [PMC free article] [PubMed] [Google Scholar]
- 49. Hennig L., Christner C., Kipping M., Schelbert B., Rücknagel K. P., Grabley S., Küllertz G., Fischer G. (1998) Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 37, 5953–5960 [DOI] [PubMed] [Google Scholar]
- 50. Ryo A., Suizu F., Yoshida Y., Perrem K., Liou Y. C., Wulf G., Rottapel R., Yamaoka S., Lu K. P. (2003) Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 12, 1413–1426 [DOI] [PubMed] [Google Scholar]
- 51. Bailey T. L., Elkan C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 [PubMed] [Google Scholar]
- 52. Liu X. S., Brutlag D. L., Liu J. S. (2002) An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 20, 835–839 [DOI] [PubMed] [Google Scholar]
- 53. El Marzouk S., Gahattamaneni R., Joshi S. R., Scovell W. M. (2008) The plasticity of estrogen receptor-DNA complexes: binding affinity and specificity of estrogen receptors to estrogen response element half-sites separated by variant spacers. J. Steroid Biochem. Mol. Biol. 110, 186–195 [DOI] [PubMed] [Google Scholar]
- 54. Das D., Peterson R. C., Scovell W. M. (2004) High mobility group B proteins facilitate strong estrogen receptor binding to classical and half-site estrogen response elements and relax binding selectivity. Mol. Endocrinol. 18, 2616–2632 [DOI] [PubMed] [Google Scholar]
- 55. Wei G., Li A. G., Liu X. (2005) Insights into selective activation of p53 DNA binding by c-Abl. J. Biol. Chem. 280, 12271–12278 [DOI] [PubMed] [Google Scholar]
- 56. Ozers M. S., Hill J. J., Ervin K., Wood J. R., Nardulli A. M., Royer C. A., Gorski J. (1997) Equilibrium binding of estrogen receptor with DNA using fluorescence anisotropy. J. Biol. Chem. 272, 30405–30411 [DOI] [PubMed] [Google Scholar]
- 57. Lu Z., Hunter T. (2014) Prolyl isomerase Pin1 in cancer. Cell Res. 24, 1033–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sommer S., Fuqua S. A. (2001) Estrogen receptor and breast cancer. Semin. Cancer Biol. 11, 339–352 [DOI] [PubMed] [Google Scholar]
- 59. Malovannaya A., Lanz R. B., Jung S. Y., Bulynko Y., Le N. T., Chan D. W., Ding C., Shi Y., Yucer N., Krenciute G., Kim B. J., Li C., Chen R., Li W., Wang Y., O'Malley B. W., Qin J. (2011) Analysis of the human endogenous coregulator complexome. Cell 145, 787–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Atsriku C., Britton D. J., Held J. M., Schilling B., Scott G. K., Gibson B. W., Benz C. C., Baldwin M. A. (2009) Systematic mapping of posttranslational modifications in human estrogen receptor-α with emphasis on novel phosphorylation sites. Mol. Cell Proteomics 8, 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lu K. P., Suizu F., Zhou X. Z., Finn G., Lam P., Wulf G. (2006) Targeting carcinogenesis: a role for the prolyl isomerase Pin1? Mol. Carcinog. 45, 397–402 [DOI] [PubMed] [Google Scholar]
- 62. Wulf G., Ryo A., Liou Y. C., Lu K. P. (2003) The prolyl isomerase Pin1 in breast development and cancer. Breast Cancer Res. 5, 76–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lu K. P., Zhou X. Z. (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916 [DOI] [PubMed] [Google Scholar]
- 64. La Montagna R., Caligiuri I., Maranta P., Lucchetti C., Esposito L., Paggi M. G., Toffoli G., Rizzolio F., Giordano A. (2012) Androgen receptor serine 81 mediates Pin1 interaction and activity. Cell Cycle 11, 3415–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fujimoto Y., Shiraki T., Horiuchi Y., Waku T., Shigenaga A., Otaka A., Ikura T., Igarashi K., Aimoto S., Tate S., Morikawa K. (2010) Proline cis/trans-isomerase Pin1 regulates peroxisome proliferator-activated receptor γ activity through the direct binding to the activation function-1 domain. J. Biol. Chem. 285, 3126–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brondani V., Schefer Q., Hamy F., Klimkait T. (2005) The peptidyl-prolyl isomerase Pin1 regulates phospho-Ser77 retinoic acid receptor α stability. Biochem. Biophys. Res. Commun. 328, 6–13 [DOI] [PubMed] [Google Scholar]
- 67. Ross-Innes C. S., Stark R., Teschendorff A. E., Holmes K. A., Ali H. R., Dunning M. J., Brown G. D., Gojis O., Ellis I. O., Green A. R., Ali S., Chin S. F., Palmieri C., Caldas C., Carroll J. S. (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gianni M., Boldetti A., Guarnaccia V., Rambaldi A., Parrella E., Raska I., Jr., Rochette-Egly C., Del Sal G., Rustighi A., Terao M., Garattini E. (2009) Inhibition of the peptidyl-prolyl-isomerase Pin1 enhances the responses of acute myeloid leukemia cells to retinoic acid via stabilization of RARα and PML-RARα. Cancer Res. 69, 1016–1026 [DOI] [PubMed] [Google Scholar]
- 69. Lucchetti C., Caligiuri I., Toffoli G., Giordano A., Rizzolio F. (2013) The Prolyl isomerase Pin1 acts synergistically with CDK2 to regulate the basal activity of estrogen receptor α in breast cancer. PLoS One 8, e55355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Laity J. H., Chung J., Dyson H. J., Wright P. E. (2000) Alternative splicing of Wilms' tumor suppressor protein modulates DNA binding activity through isoform-specific DNA-induced conformational changes. Biochemistry 39, 5341–5348 [DOI] [PubMed] [Google Scholar]
- 71. Laity J. H., Dyson H. J., Wright P. E. (2000) DNA-induced α-helix capping in conserved linker sequences is a determinant of binding affinity in Cys(2)-His(2) zinc fingers. J. Mol. Biol. 295, 719–727 [DOI] [PubMed] [Google Scholar]
- 72. Pufall M. A., Lee G. M., Nelson M. L., Kang H. S., Velyvis A., Kay L. E., McIntosh L. P., Graves B. J. (2005) Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science 309, 142–145 [DOI] [PubMed] [Google Scholar]
- 73. Pursglove S. E., Fladvad M., Bellanda M., Moshref A., Henriksson M., Carey J., Sunnerhagen M. (2004) Biophysical properties of regions flanking the bHLH-Zip motif in the p22 Max protein. Biochem. Biophys. Res. Commun. 323, 750–759 [DOI] [PubMed] [Google Scholar]
- 74. Eckert B., Martin A., Balbach J., Schmid F. X. (2005) Prolyl isomerization as a molecular timer in phage infection. Nat. Struct. Mol. Biol. 12, 619–623 [DOI] [PubMed] [Google Scholar]
- 75. Lummis S. C., Beene D. L., Lee L. W., Lester H. A., Broadhurst R. W., Dougherty D. A. (2005) Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 438, 248–252 [DOI] [PubMed] [Google Scholar]
- 76. Sarkar P., Saleh T., Tzeng S. R., Birge R. B., Kalodimos C. G. (2011) Structural basis for regulation of the Crk signaling protein by a proline switch. Nat. Chem. Biol. 7, 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Benecke A., Chambon P., Gronemeyer H. (2000) Synergy between estrogen receptor alpha activation functions AF1 and AF2 mediated by transcription intermediary factor TIF2. EMBO Rep. 1, 151–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Métivier R., Penot G., Flouriot G., Pakdel F. (2001) Synergism between ERalpha transactivation function 1 (AF-1) and AF-2 mediated by steroid receptor coactivator protein-1: requirement for the AF-1 α-helical core and for a direct interaction between the N- and C-terminal domains. Mol. Endocrinol. 15, 1953–1970 [DOI] [PubMed] [Google Scholar]
- 79. Schaufele F., Carbonell X., Guerbadot M., Borngraeber S., Chapman M. S., Ma A. A., Miner J. N., Diamond M. I. (2005) The structural basis of androgen receptor activation: intramolecular and intermolecular amino-carboxy interactions. Proc. Natl. Acad. Sci. U.S.A. 102, 9802–9807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kobayashi Y., Kitamoto T., Masuhiro Y., Watanabe M., Kase T., Metzger D., Yanagisawa J., Kato S. (2000) p300 mediates functional synergism between AF-1 and AF-2 of estrogen receptor α and β by interacting directly with the N-terminal A/B domains. J. Biol. Chem. 275, 15645–15651 [DOI] [PubMed] [Google Scholar]
- 81. Khan S. H., Ling J., Kumar R. (2011) TBP binding-induced folding of the glucocorticoid receptor AF1 domain facilitates its interaction with steroid receptor coactivator-1. PLoS One 6, e21939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kumar R., Volk D. E., Li J., Lee J. C., Gorenstein D. G., Thompson E. B. (2004) TATA box binding protein induces structure in the recombinant glucocorticoid receptor AF1 domain. Proc. Natl. Acad. Sci. U.S.A. 101, 16425–16430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kumar R., Moure C. M., Khan S. H., Callaway C., Grimm S. L., Goswami D., Griffin P. R., Edwards D. P. (2013) Regulation of the structurally dynamic N-terminal domain of progesterone receptor by protein-induced folding. J. Biol. Chem. 288, 30285–30299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Johnson D. S., Mortazavi A., Myers R. M., Wold B. (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 [DOI] [PubMed] [Google Scholar]
- 85. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 86. Handel A. E., Sandve G. K., Disanto G., Handunnetthi L., Giovannoni G., Ramagopalan S. V. (2013) Integrating multiple oestrogen receptor α ChIP studies: overlap with disease susceptibility regions, DNase I hypersensitivity peaks and gene expression. BMC Med. Genomics 6, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lin C. Y., Vega V. B., Thomsen J. S., Zhang T., Kong S. L., Xie M., Chiu K. P., Lipovich L., Barnett D. H., Stossi F., Yeo A., George J., Kuznetsov V. A., Lee Y. K., Charn T. H., Palanisamy N., Miller L. D., Cheung E., Katzenellenbogen B. S., Ruan Y., Bourque G., Wei C. L., Liu E. T. (2007) Whole-genome cartography of estrogen receptor α binding sites. PLoS Genet. 3, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]