

Background: Acyl carrier protein (ACP) is required for fatty acid synthesis.
Results: Exchange of domains between an ACP that is nonfunctional in Escherichia coli and E. coli ACP results in a functional protein in one direction but not the reverse.
Conclusion: Incompatibility of ACP domains can reside in both helices I and II.
Significance: The modular elements of ACPs are not swappable.
Keywords: acyl carrier protein (ACP), bacteria, bacterial metabolism, enzyme, Escherichia coli (E. coli), lipid
Abstract
Prior work showed that expression of acyl carrier proteins (ACPs) of a diverse set of bacteria replaced the function of Escherichia coli ACP in lipid biosynthesis. However, the AcpAs of Lactococcus lactis and Enterococcus faecalis were inactive. Both failed to support growth of an E. coli acpP mutant strain. This defect seemed likely because of the helix II sequences of the two AcpAs, which differed markedly from those of the proteins that supported growth. To test this premise, chimeric ACPs were constructed in which L. lactis helix II replaced helix II of E. coli AcpP and vice versa. Expression of the AcpP protein L. lactis AcpA helix II allowed weak growth, whereas the L. lactis AcpA-derived protein that contained E. coli AcpP helix II failed to support growth of the E. coli mutant strain. Replacement of the L. lactis AcpA helix II residues in this protein showed that substitution of valine for the phenylalanine residue four residues downstream of the phosphopanthetheine-modified serine gave robust growth and allowed modification by the endogenous AcpS phosphopantetheinyl transferase (rather than the promiscuous Sfp transferase required to modify the L. lactis AcpA and the chimera of L. lactis AcpA helix II in AcpP). Further chimera constructs showed that the lack of function of the L. lactis AcpA-derived protein containing E. coli AcpP helix II was due to incompatibility of L. lactis AcpA helix I with the downstream elements of AcpP. Therefore, the origins of ACP incompatibility can reside in either helix I or in helix II.
Introduction
Acyl carrier proteins (ACPs)2 are highly conserved proteins that play essential roles both in the synthesis of fatty acids and in transfer of completed fatty acyl chains into complex lipids (e.g. phospholipids) (1–3). ACP function requires modification of the protein by transfer of a 4′-phosphopantetheine moiety from CoA to a conserved serine residue. The phosphopantetheine thiol tethers the starting materials and lipid synthetic intermediates as their thioesters. ACPs are small highly soluble proteins composed of four α-helices. The helices form a bundle that act as a hydrophobic sheath that sequesters the acyl chains and their thioesters from solvent. Escherichia coli ACP, the most thoroughly studied member of this protein family, is a small (77 residues; molecular weight, 8,860), very acidic (pI of 4.1), and extremely soluble protein. In prior work, we reported that ACP species encoded by 11 diverse bacteria plus that of the apicoplast of the malarial protozoan parasite Plasmodium falciparum functionally replaced E. coli ACP in vivo (4). This generality was unforeseen because E. coli ACP interacts with 21 different lipid metabolism enzymes (5), virtually all of which are essential for growth. Moreover, the successful ACPs have markedly diverse sequences (see Fig. 1).
FIGURE 1.

ACP Structure and Alignments. A, the structure of the decanoyl thioester of E. coli ACP modified from PDB file 2FAE using Chimera (25). The 4′-phosphopantetheine prosthetic group and acyl chain are stick models with CPK coloring. The helices are numbered. B, alignments of the ACPs of various bacteria and that of P. falciparum with E. coli ACP. Red asterisks denote the two ACPs that failed to complement growth of the conditional acpP mutant strain of E. coli (4). The bacteria were E. coli, Vibrio harveyi, Nitrosomonas europea, Agrobacterium tumefaciens, Bacteroides thetaiotaomicron, Deinococcus radiodurans, Helicobacter pylori, Aquifex aeolicus, E. faecalis, L. lactis, and B. subtilis. The apicoplast ACP of the P. falciparum malaria parasite was also found to support growth (4).
However, there were two exceptions to this tolerance. Expression of the ACP proteins of Lactococcus lactis and Enterococcus faecalis (ACP is called AcpA in these bacteria) failed to support E. coli growth (4). This failure was not due to a dichotomy between Gram-positive and Gram-negative bacteria because the ACP of another Gram-positive bacterium, Bacillus subtilis, was fully functional in E. coli. The lack of function of the L. lactis and E. faecalis AcpAs could be explained by the failure of these two highly similar ACPs to be modified by the E. coli AcpS 4′-phosphopantetheinyl transferase. However, upon co-expression with the promiscuous B. subtilis Sfp 4′-phosphopantetheinyl transferase, both AcpA proteins were modified but remained unable to restore growth to a conditionally lethal acpP mutant strain of E. coli (4). These findings were puzzling because we had previously observed that genes from both of these closely related bacteria could replace the functions of E. coli fatty acid genes in vivo (6–8). Hence, these L. lactis and E. faecalis enzymes functioned with E. coli ACP, whereas neither the L. lactis ACP nor the E. faecalis ACP could functionally replace the E. coli protein.
The first ACP-enzyme complex structure was that of an enzyme-product complex: ACP complexed to B. subtilis AcpS, the enzyme that transfers the phosphopantetheinyl moiety from CoA to Ser-36 of ACP (9). The structure showed that the enzyme interacted with ACP largely through contacts with ACP helix II, the helix that begins with Ser-36 (see Fig. 1). Moreover, the interactions are predominately hydrophilic in nature, with almost all being salt bridges between acidic residues of helix II and AcpS arginine residues. This finding gave rise to the concept that ACP-requiring enzymes have an arginine/lysine-rich “positive patch” and that ACP helix II would be the site of interaction with the patches. The AcpS-AcpP structure significantly predated the next high resolution structure of an enzyme-ACP complex and thus had a primacy that colored interpretations of enzyme-acyl-ACP interactions studied by indirect means (e.g. the activities of mutated enzymes measured in vitro). Although the positive patch concept has stood the test of four subsequent acyl-ACP-enzyme crystal structures (for review see Ref. 3), in some complexes hydrogen bonds that involve acidic residues of helices I and III and the connecting loops are observed together with helix II interactions.
Given these precedents, it seemed likely that the markedly different helix II sequences of the L. lactis and E. faecalis AcpAs (Fig. 1) relative to E. coli AcpP and the other functional nonnative ACPs, might explain their inability to function in E. coli. Hence, our first experiments tested the effects of swapping the helix II sequences between the L. lactis and E. coli ACPs by construction of genes encoding protein chimeras. These chimeras were then tested for their ability to support growth of an E. coli strain conditionally defective in AcpP synthesis. We report that the effects of helix II exchange between the E. coli and L. lactis ACPs are idiosyncratic. The chimeric E. coli protein that contained the L. lactis helix II weakly supported growth of the E. coli strain, whereas the inverse construct was completely inactive.
Experimental Procedures
Plasmid Constructions
The plasmids, strains, and oligonucleotides are given in Tables 1, 2, and 3, respectively. The parental E. coli acpP and L. lactis acpA genes were derived from plasmids pNRD25 (10) and pNRD73 (4), respectively. In general, the gene chimeras were constructed in a small high copy number vector under conditions where the genes were oriented such that they were not expressed to avoid possible toxicity because of high levels of ACP expression. Plasmid pNRD25 was modified to remove the EcoRI site just upstream of acpP and to introduce a PciI site overlapping the acpP initiation codon by replacing the BamHI-MluI with a synthetic DNA made by annealing oligonucleotides Pci-top and Pci-btm (the MluI site was preserved) to give pCY925. This plasmid was then digested with PciI and HindIII and ligated to the expression vector pKK233-2 cut with NcoI and HindIII to give pCY939 (PciI and NcoI make compatible ends), an expression vector encoding wild type E. coli AcpP. To obtain pCY941, an expression vector encoding wild type L. lactis AcpA pNRD73 was digested with NcoI and HindIII, and the acpA sequence was ligated to pKK233-2 cut with the same enzymes.
TABLE 1.
Plasmids used in the work
| Plasmid | Replication origina | Antibiotic selectionb | Promoter: gene | Sources |
|---|---|---|---|---|
| pNRD25 | pUC | Amp | paraBAD: acpP | Ref. 10 |
| pNRD73 | pUC | Amp | paraBAD: acpA | Ref. 4 |
| pKK233–2 | ColE1 | Amp | ptac | Ref. 24 |
| pCY939 | ColE1 | Amp | ptac: acpA | This work |
| pCY941 | ColE1 | Amp | ptac: acpA/acpP II | This work |
| pCY983 | ColE1 | Amp | ptac: acpP/acpA II | This work |
| pCY955 | ColE1 | Amp | ptac: acpP/acpA II VE | This work |
| pCY956 | ColE1 | Amp | ptac: acpP/acpA II VEM | This work |
| pCY957 | ColE1 | Amp | ptac: acpP/acpA II VEMA | This work |
| pCY969 | ColE1 | Amp | ptac: acpP/acpA II F40V | This work |
| pCY970 | ColE1 | Amp | ptac: acpP/acpA II E41Q | This work |
| pCY982 | pUC | Cml | Promoterless acpP | This work |
| pCY983 | ColE1 | Amp | ptac: acpA I + l/acpP II, III, IV | This work |
| pCY985 | ColE1 | Amp | ptac: acpA I/acpP II, III, IV | This work |
| pREP4 | p15A | Kan | Wild type lacI | Qiagen |
| pMS421 | pSC101 | Spc | placIQ lacI | Ref. 2 |
| pCY938 | pUC | Amp | placIQ lacI | This work |
| pNRD136 | RSF1030 | Kan | placIQ lacI:sfp | Ref. 4 |
| pCY947 | RSF1030 | Kan | placIQ lacI:sfp lacIQ | This work |
| pCY948 | RSF1030 | Kan | Δsfp, placIQ lacI | This work |
| pCY765 | p15A | Spc | paraBAD: acpP | This work |
a The pUC origin is that of ColE1 lacking the rop gene.
b The antibiotic abbreviations are: Amp, ampicillin; Kan, kanamycin; Cml, chloramphenicol; and Spc, spectinomycin.
TABLE 2.
Strains used in the work
| E. coli strains | Chromosomea | Plasmids | Antibiotic: replication origin |
|---|---|---|---|
| CY1871 | ΔacpP::cat | pCY765 | Spc: p15A |
| ΔlacIZYA | |||
| CY2155 | ΔacpP::cat | pCY765 | Spc: p15A |
| ΔlacIZYA | pCY947 | Kan: RSF1030 | |
| CY2156 | ΔacpP::cat | pCY765 | Spc: p15A |
| ΔlacIZYA | pCY948 | Kan: RSF1030 | |
| CY2211 | ΔacpP | pCY765 | Spc: p15A |
| ΔlacIZYA | pCY947 | Kan: RSF1030 | |
| ΔpanD::cat | |||
| Derivatives of CY2155 | ΔacpP::cat | pCY765 | Spc: p15A |
| Carrying a pKK233–2 | ΔlacIZYA | pCY947 | Kan: RSF1030 |
| acpP/acpA encoding plasmid | pKK233-2 acpP/acpA | Amp: ColE1 | |
| Derivatives of CY2156 | ΔacpP::cat | pCY765 | Spc: p15A |
| Carrying a pKK233–2 | ΔlacIZYA | pCY947 | Kan: RSF1030 |
| acpP/acpA encoding plasmid | pKK233-2 acpP/acpA | Amp: ColE1 | |
| Derivatives of CY2211 | ΔacpP::cat | pCY765 | Spc: p15A |
| Carrying a pKK233–2 | ΔlacIZYA | pCY947 | Kan: RSF1030 |
| acpP/acpA encoding plasmid | ΔpanD::cat | pKK233-2 acpP/acpA | Amp: ColE1 |
a The Δ symbol denotes deletion, whereas cat indicates the chloramphenicol acetyltransferase gene of transposon Tn9.
TABLE 3.
Oligonucleotides used in the work (5′ → 3′)
| E-L top | CGACGCTGATTCTCTGGACACCGTTGAGCTGGTTATGGCGCTCGAGGAAG |
| E-L btm | AATTCTTCCTCGAGCGCCATAACCAGCTCAACGGTGTCCAGAGAATCAGCGTCGAGCT |
| l-E top | GGGTGCTGACTCACTTGACCTTTTCCAAATCATCAATGACATCGAAGACGAATTCGACACCGAAAT |
| l-E btm | CCGGGATTTCGGTGTCGAATTCGTCTTCGATGTCATTGATGATTTGGAAAAGGTCAAGTGAGTCAGC |
| Pci-Top | GATCCGCTAGCAGGAGGAAACATGTCTACTATCGAAGAA |
| Pci-Btm | CGCGTTCTTCGATAGTAGACATGTTTCCTCCTGCTAGCG |
| ΔRI-C-top | GTAGAAAACAACAAATAAGGTACCA |
| ΔRI-C-btm | AGCTTGGTACCTTATTTGTTGTTTTCTAC |
| GS53140 | CGCGTTAAAAAAATCATCGGTGAACAGCTGGGTGTTAAACAGGAGGAAGTTACCAACAACGCATCTTTCGTTGAAGATCTGGGTGCTGACTCACTTGACCTTTTCCAAATCATCAATGACATCGAAGACGAATTCGACACCGAAATCCCGGACGAAGAAGCTGAAAAAATCACCACTGTACAGGCTGCTATCGATTACATCAACGGTCACCAGGCTTA |
| VE-top | GATCTGGGTGCTGACTCACTTGACCTTGTTGAAATCATCAATGACATCGAAGACG |
| VE-btm | AATTCGTCTTCGATGTCATTGATGATTTCAACAAGGTCAAGTGAGTCAGCACCCA |
| VEM-top | GATCTGGGTGCTGACTCACTTGACCTTGTTGAAATCATCATGGACATCGAAGACG |
| VEM-btm | AATTCGTCTTCGATGTCCATGATGATTTCAACAAGGTCAAGTGAGTCAGCACCCA |
| VEMA-top | GATCTGGGTGCTGACTCACTTGACCTTGTTGAAATCATCATGGCAATCGAAGACG |
| VEMA-btm | AATTCGTCTTCGATTGCCATGATGATTTCAACAAGGTCAAGTGAGTCAGCACCCA |
| VE-FE top | ATCTGGGTGCTGACTCACTTGACCTTTTTGAAATCATCAATGACATCGAAGACG |
| VE-FE btm | AATTCGTCTTCGATGTCATTGATGATTTCAAAAAGGTCAAGTGAGTCAGCACCCA |
| VE-VQ top | GATCTGGGTGCTGACTCACTTGACCTTGTTCAAATCATCAATGACATCGAAGACG |
| VE-VQ btm | AATTCGTCTTCGATGTCATTGATGATTTGAACAAGGTCAAGTGAGTCAGCACCCA |
| L. lactis αI | CTAGCAACATGTCTACTGTATTTGAAAAAGTACAAGATATTATCGTTGACGAGCTGGGTGTTAAACAGGAGGAAGTTACCAACAACGCATCTTTCGTTGAA |
| WT-top | GATCTGGGTGCTGACTCTCTGGACACCGTTGAGCTGGTTATGGCGCTGGAAGAAG |
| WT-btm | AATTCTTCTTCCAGCGCCATAACCAGCTCAACGGTGTCCAGAGAGTCAGCACCCA |
The chimera constructions of this work were all straightforward substitutions of sequences encoding the wild type protein (e.g. AcpP) with a synthetic sequence encoding a segment of the other ACP encoding gene (e.g. AcpA) by use of naturally occurring restriction sites or sites introduced without altering the sequence of the encoded protein. The synthetic sequences were annealed to produce double-stranded DNAs having protruding overhangs to allow ligation to restriction enzyme-digested plasmids. All constructs were verified by DNA sequence analysis.
To construct a gene encoding a chimera of L. lactis AcpA containing E. coli AcpP helix II, the large NcoI-SnaBI fragment encoding the N-terminal 68 residues of AcpA pNRD73 was ligated to pMTL23P cut with NcoI and HincII. The resulting plasmid was digested with SacI and EcoRI, and the large fragment was purified and ligated to a synthetic DNA made by annealing oligonucleotides E-L top and E-L btm (Table 3). The product of this ligation was confirmed by sequencing and then digested with NcoI and AlwNI and ligated to the large fragment of pNRD73 digested with the same enzymes to incorporate the segment encoding the C terminus of AcpP in plasmid pCY926. Plasmid pCY926 was then digested with NcoI and HindIII and ligated to the expression vector pKK233-2 cut with the same enzymes to give pCY941.
The pCY926 sequence downstream of the AcpA C terminus was subsequently modified to remove unwanted restriction sites by digestion with SnaBI and HindIII and ligation to a synthetic DNA fragment made by annealing oligonucleotides ΔRI-C-top and ΔRI-C-btm (Tables 1 and 2). To construct a gene that encoded a chimera of E. coli AcpP having the helix II of L. lactis AcpA, pCY925 was digested with MluI and HindIII and ligated to a synthetic double-stranded DNA segment called GS53140, the top strand of which is given in Tables 1 and 2, resulting in pCY983.
In the course of these constructions, several new restriction sites were introduced into the widely used synthetic E. coli acpP gene constructed many years ago in this laboratory (11, 12). They were a PciI site overlapping the initiation codon, the BglII and EcoRI sites introduced in insertion of the L. lactis helix II sequence, and a BsrGI site located in the sequence encoding the N terminus of helix IV (all were inserted without changing the coding sequence). To make use of these new sites in future work, we replaced the L. lactis helix II sequence with that of the wild type E. coli helix II by use of the BglII and EcoRI sites plus annealed oligonucleotides WT-top and WT-btm, resulting in plasmid pCY982 (Table 3), which encodes the wild type protein produced from an E. coli acpP gene containing each of the new sites plus a useful NheI site located just upstream of the coding sequence.
The finding that the expression of the AcpP-α2L protein gave weak growth in the presence of Sfp led to mutagenesis of the α2L sequence to conform more closely to the sequence of E. coli helix II. This was done by substitution of the BglII-EcoRI segment of pCY940 with synthetic DNA segments made by annealing oligonucleotides denoted as VE, VEM, VEMA, VE-FE, and VE-VQ in Table 1, although in some cases intermediate plasmids were utilized. The same approach was used to test the function of the highly conserved valine (Val-40) of E. coli acpP by substitution of the GTT valine codon with the TTT phenylalanine codon, the GCT alanine codon, or the ATT isoleucine codon.
Finally, the L. lactis helix I sequence was assembled from four annealed oligonucleotides as a NheI-BglII fragment (the sequence of the upper strand given in Tables 1 and 2) and ligated to the large fragment of pCY982 to replace helix I of E. coli AcpP with helix I of L. lactis AcpA. This plasmid in turn was used to replace the EcoRI-HindIII sequence encoding the E. coli acpP helices III and IV together with the intervening loop and C terminus with the EcoRI-HindIII fragment of a derivative of pNRD73 from which a downstream EcoRI site had been removed. The genes encoding these sequences were then moved into the pKK233-2 expression vector as described above to give pCY984.
Construction and Testing of the lacIQ sfp Plasmid pCY947
The wild type lacI gene of pREP4 (Qiagen) was excised with SalI and ligated to SalI-digested pMTL20. The resulting white colonies were the screened for a construct in which lacI was in the orientation opposite that of the lacZ promoter, to give pCY937. To replace the wild type lacI promoter with the 10-fold more powerful lacIQ promoter, pMS421 (13) was digested with PstI and MluI, and the lacIQ lacI′ fragment was ligated to pCY937 cut with the same enzymes, This manipulation restored the LacI coding sequence and introduced the lacIQ promoter. The resulting plasmid (pCY938) was then cut with NheI and SacI and ligated to the large fragment of the Sfp-encoding plasmid pNRD136 (4) cut with the same enzymes to give pCY947. An sfp deletion derivative of this plasmid called pCY948 was constructed by digestion with XbaI and EagI followed by blunting the ends and ligation under dilute conditions. Upon transformation into lacY and ΔlacI strains, both pCY947 and pCY948 showed the very strong lacZ repression characteristic of lacIQ. The strains were also tested for the ability to complement the conditional acpS strain HT253 (14) under nonpermissive conditions. Only pCY947 allowed growth of this strain.
Construction of Strain CY2211 and β-[3H]Alanine Labeling
Strain CY2211 is derived from strain CY1871, which was briefly described previously (15). Strain CY1871 (Table 2) is a derivative of the ΔacpP::cat strain NRD62 (10) in which the lambda prophage has been removed from the host chromosome and the ampicillin resistance of the AcpP encoding plasmid has been replaced by spectinomycin resistance. Efficient labeling with exogenous β-alanine requires inactivation of panD, the gene encoding aspartate 1-decarboxylase, the enzyme responsible for β-alanine synthesis (16). To introduce a ΔpanD mutation into the strain, the chloramphenicol resistance cassette that replaced acpP was removed by use of the site-specific recombinase encoded by plasmid pCP20 (17). The resulting strain was then transduced to chloramphenicol resistance using a phage P1 stock grown on strain NRD1 (ΔpanD::cat) (10) to give a β-alanine requiring derivative. This strain was then transformed with the Sfp encoding plasmid pCY947 to give strain CY2211 (able 1). Strain CY2211 was then transformed with the AcpA, AcpP, and chimera encoding pKK233-2 derivatives. Labeling with β-[2,3-3H]alanine in the presence of isopropyl β-d-1-thiogalactopyranoside (IPTG) was done 37 °C for 6 h essentially as described previously (4, 10). The β-[2,3-3H]alanine was synthesized from l-[2,3-3H]aspartic acid (MP Biomedicals) using purified PanD and purified on a Dowex l-X4 ion exchange column as described previously (18). Synthesis was necessary because the commercial samples available were either inactive or of insufficient specific activity.
Growth Conditions
The strains to be tested were streaked from LB medium plates containing 50 μg/ml each of spectinomycin sulfate and kanamycin sulfate, 100 μg/ml of sodium ampicillin, 10 μg/ml of chloramphenicol and 0.2% l-arabinose to plates of the same medium that lacked arabinose and were supplemented with 50 μm IPTG or left without IPTG supplementation. Thick plates were poured to counter drying and following inoculation were incubated at 37 °C for at least 16 h.
Results and Discussion
Exchanging Helices II of AcpP and AcpA
The chimeric E. coli ACP that contained L. lactis helix II (called AcpP-α2L) and a chimera of L. lactis ACP containing the E. coli helix II (called AcpA-α2E) (Fig. 2) were constructed and tested for the ability to replace E. coli ACP in vivo. The L. lactis acpA gene was chosen over that of E. faecalis because of its higher expression in E. coli (4), the presence of useful restriction sites, and the fact that E. faecalis encodes a putative second ACP of unknown function. The ability of the constructed protein chimeras to support E. coli lipid synthesis was tested in derivatives of strain CY1877 (15) (Table 2). Strain CY1877 contains a complete deletion of the acpP gene that encodes ACP, which was replaced with a chloramphenicol resistance cassette. The strain is viable because of the presence of a spectinomycin-resistant plasmid that expresses acpP under control of an arabinose promoter. This strain grows normally in the presence of arabinose but is unable to form colonies in the absence of arabinose. Two plasmid-carrying derivatives of this strain were made. The kanamycin-resistant plasmid (pCY947) of strain CY2155 encoded LacI from the lacIQ promoter and the promiscuous Sfp 4′-phosphopantetheinyl transferase (4, 19) under control of the lac promoter. The other strain (CY2156) carried pCY948, a plasmid derived from pCY947 by deletion of sfp. The genes encoding the chimeric and wild type ACP genes were placed under control of the LacI-regulated tac promoter of an ampicillin-resistant third plasmid, pKK233-2. The replication origins and antibiotic selections of the three plasmids are mutually compatible.
FIGURE 2.

The AcpP and AcpA proteins and the chimeras constructed. The segments from L. lactis AcpA are given in blue, whereas the E. coli AcpP segments are colored puce. The colors plus the letters A and P denote the origins of the sequences (AcpA and AcpP, respectively). The abilities of the proteins to complement growth an acpP deficient strain of E. coli in either the absence or presence of the promiscuous B. subtilis Sfp 4′-phosphopantetheinyl transferase are given in the right-hand columns. The modified serine is the first residue of helix II. w denotes weak growth (approximately one-tenth of wild type), whereas (w) denotes very weak growth (approximately one-twentieth of wild type). e denotes the unexplained erratic growth of these strains.
Following electroporation of strains CY2155 and CY2156 with expression plasmids encoding each of the chimeric and wild type ACPs, the transformants were spread on LB plates containing arabinose plus the appropriate antibiotics. Single colonies from these plates were then streaked on LB plates that lacked arabinose and contained the appropriate antibiotics with or without IPTG supplementation. The E. coli host strain carries a deletion of the lacY (lactose permease) gene such that IPTG enters by diffusion. This property plus the high levels of LacI repressor allowed well controlled expression of ACP levels by IPTG. Note that growth was scored on solid medium rather than in liquid medium because multiple generations are required to dilute the AcpP produced in the presence of arabinose. This would require numerous successive dilutions if done in liquid medium,. However, dilution proceeds readily on plates because it takes ∼105 generations to form a small colony. Note that maximal IPTG induction of L. lactis AcpA and of the various chimeric proteins failed to inhibit growth of strain CY2156 in the presence of arabinose, indicating that the apo forms of these proteins were not toxic to growth.
As expected from prior work, the strain expressing wild type E. coli AcpP grew well with or without expression of Sfp (the strain requires IPTG for normal growth but grows slowly in its absence; see below). As expected, the strain that expressed wild type L. lactis AcpA showed no detectable growth even upon Sfp expression (Fig. 3A) (4), and the strain that expressed the AcpA-α2E chimera showed the same lack of growth as the AcpA strain. In contrast, the strain that expressed the AcpP-α2L chimera grew, but only in the presence of Sfp (Fig. 3A). However, growth proceeded slowly and largely ceased when the colonies had reached pinpoint size, suggesting that some essential E. coli function had been titrated. Growth required IPTG induction, and upon streaking the pinpoint colonies onto arabinose containing medium, they were viable (as were each of the other constructs described below). All chimeric proteins were sufficiently modified by phosphopantetheinylation in the presence of Sfp (Fig. 3B). Note that even the faintest labeling was sufficient to allow robust growth (e.g. lanes 5 and 8). Labeling was performed only in the presence of Sfp because the supply of tritiated β-alanine available was severely limited (the precursor had to be synthesized from tritiated aspartate). Note that upon a treatment that cleaved acyl-ACP thioester linkages (elevated pH in the presence of a high dithiothreitol concentration), the multiple bands seen in lanes 11–15 collapsed to a single band, suggesting that some bands were acyl-ACP species. ACP disulfide dimers would also be cleaved, but dimers seems less likely because of the presence of 1 mm dithiothreitol in the lysis buffer. It should be noted that small peptides (8–11 residues) have been isolated that are specifically modified by either E. coli AcpS or B. subtilis Sfp (20, 21). Therefore, sequences outside helix II are not expected to influence phosphopantetheinylation. Moreover, consistent with its activity with acceptor proteins from diverse pathways (19), Sfp was found to efficiently modify peptides having markedly diverse sequences downstream of the modified serine residue (20).
FIGURE 3.

Abilities of the parental and helix II chimeras to support growth of the arabinose-dependent ΔacpP strain in the absence of arabinose and to be modified by 4′-phosphopantetheine attachment. A, the strains were grown in the presence of IPTG to induce expression of the ACP genes in the presence or absence of Sfp as shown above the plates. The plates were incubated for 20 h at 37 °C. B, the plasmids tested in A plus all of the other constructed plasmids were transformed into the ΔpanD derivative of the arabinose-dependent ΔacpP Sfp-producing strain CY2211. These strains were then starved for β-alanine and labeled with β-[2,3-3H]alanine synthesized from l-[2,3-3H]aspartic acid as described under “Experimental Procedures.” The gels were of 20% polyacrylamide that lacked a denaturant. The mobility of ACP species in these gels is due to their acidic charge plus their Stokes radii (which can be altered by acylation) (26). In the lower gel, the samples of lanes 11–15 were treated with 10 mm dithiothreitol at pH 9.5 for 1 h prior to loading. This treatment cleaves ACP acyl-thioester and disulfide bonds (27). The same treatment of the samples of lanes 1–10 gave essentially identical results. Lanes 12 and 14 are duplicates from separate colonies, whereas lane 13 was another colony from the same plate that grew poorly. Restriction enzyme mapping of the plasmid in this strain showed that part of the lacI gene had been deleted, which seems likely to account for the high expression of the modified protein. The increased labeling observed indicates that Sfp activity is present in excess.
A Single Mutation Allows Full Function of the AcpP-α2L Chimera
The AcpP-α2L protein allowed only weak Sfp-dependent growth (Fig. 3A), which argued that the AcpP-α2L protein was not only defective in E. coli AcpS interaction but also interacted poorly with an essential ACP-requiring enzyme(s). The two defects could be interrelated or independent. Prior work showed that L. lactis AcpA was not a substrate for AcpS (4), and this could be rationalized from the high resolution structure of B. subtilis AcpS with its cognate ACP (9) (PDB accession 1F80). In the alignments of Fig. 1, three residues of L. lactis helix II differ from the residues of the ACPs that function in E. coli (the helix II numbering is that of the E. coli and B. subtilis ACPs, L. lactis numbering is two less). The Val-40 and Glu-41 residues found in all of the complementing ACPs are replaced with a Phe-Gln sequence in both the L. lactis and E. faecalis AcpA proteins. In the B. subtilis AcpS-ACP crystal structure, in silico mutagenesis suggested that substitution of Phe for Val-40 would result in clash of all possible rotamers with neighboring AcpS residues, and hence this residue seemed likely to distort or disrupt enzyme-substrate interactions. Weaker hypotheses could be made concerning the other residues that differ in AcpAs. ACP Glu-41 forms a salt bridge with an AcpS arginine residue proposed to aid in orientation of the ACP substrate (9). Hence loss of this bond could alter enzyme substrate interaction. Finally, third replacement is that of the conserved Met-44 residue by Asn in the two AcpAs. The side chain of ACP Met-44 is sequestered within a hydrophobic pocket formed by three AcpS residues (9), and the polar AcpA helix II Asn residue could weaken or preclude this interaction.
Based on these suppositions, the L. lactis Phe-Gln helix II sequence of the AcpP-α2Ll chimera was replaced with the Val-40–Glu-41 sequence found in the complementing ACPs (Fig. 1). Moreover, Val-40–Glu-41 derivatives were also constructed in which Met was substituted for the L. lactis helix II residue Asn-44 and in which the Ala residue that often follows Met-44 was substituted for the Glu of L. lactis helix II. These mutant genes were designated as the VE, VEM, and VEMA genes, respectively (Fig. 2). Following ligation into the pKK233-2 expression vector, the resulting plasmids were transformed into strains CY2155 and CY2556, and these strains were tested as above (Fig. 4A). Only the VE substitution was required to allow robust growth in the presence or the absence of Sfp (Fig. 4A), and thus the weaker arguments above were not germane. Insertion of the canonical Val-40–Glu-41 sequence into the AcpP-α2L chimera imparted both functional interaction with AcpS and full lipid metabolism to E. coli; the Met-44 and Met-44–Ala-45 substitutions were not required. Indeed, for unknown reasons, the strains expressing the VEMA protein grew poorly and erratically regardless of the presence or absence of Sfp (Fig. 4A).
FIGURE 4.
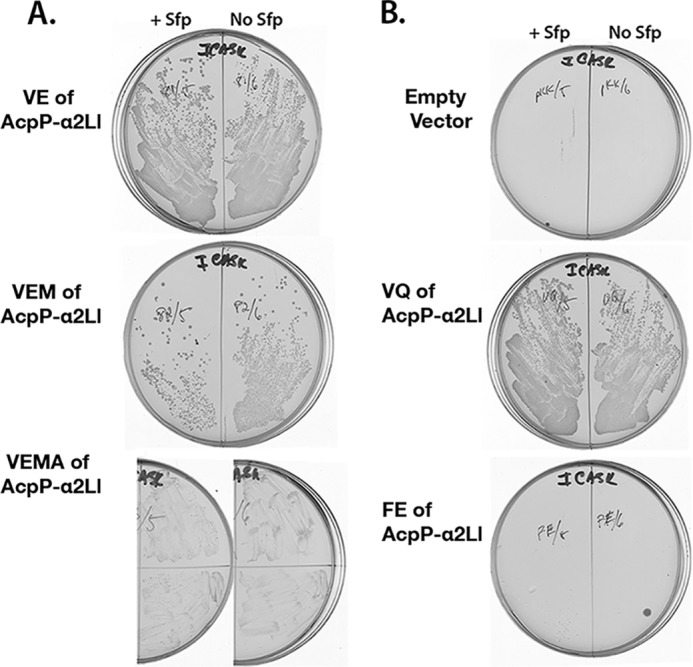
Ability of plasmids with mutations carrying residue substitutions in helix II of the AcpP-α2Ll chimera to support growth of the arabinose-dependent ΔacpP strain in the absence of arabinose. A, growth of strains carrying the VE, VEM, and VEMA derivatives of ACP-α2Ll. B, growth of strains carrying the VQ and FE derivatives of ACP-α2Ll. The strains were grown in the presence of IPTG to induce expression of the ACP genes in the presence or absence of Sfp as shown above the plates. The plates were incubated for 20 h at 37 °C. Further incubation of the FE plate (bottom right corner) for an additional 16 h showed no growth.
These results immediately raised the question of whether Val-40 or Glu-41 (or both residues) were required for activity. Derivatives of the AcpP-α2L chimera were constructed that encoded only the Val-40 or only the Glu-41 substitutions. Strains expressing the Val-40–Gln-41 protein grew robustly in the presence or absence of Sfp (Fig. 4B), and thus the L. lactis Gln residue replaced function of the Glu residue found in the complementing AcpPs (Fig. 1). However, the strains producing the Phe-40–Glu-41 protein showed no detectable growth either plus or minus Sfp expression (Fig. 4B). Therefore, substitution of Val for Phe-40 was the key alteration that allowed the AcpP-α2L chimera to become an AcpS substrate and to function fully in E. coli lipid biosynthesis.
Because Phe-40 blocked AcpS function and severely limited function in lipid biosynthesis of the AcpP-α2L chimera, a similar result should occur if Phe replaced Val-40 in E. coli AcpP. Indeed, expression of an otherwise wild type E. coli AcpP in which Phe was substituted for Val-40 resulted in a protein that supported only very weak growth and required Sfp expression for activity (Fig. 5).
FIGURE 5.
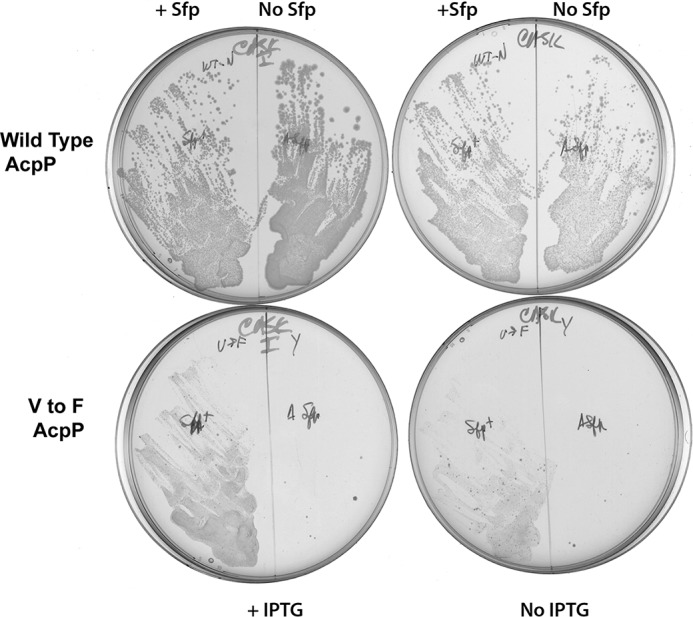
Ability of a mutant AcpP with Phe in place of Val-40 to support growth of the arabinose-dependent ΔacpP strain in the absence of arabinose. The strains were grown in the presence or absence of IPTG to induce expression of the ACP genes in the presence or absence of Sfp as shown below and above the plates, respectively. The plates were incubated for 24 h at 37 °C.
The Converse Question: Why Does E. coli Helix II Fail to Functionally Replace L. lactis Helix II?
Replacement of L. lactis AcpA-helix II residue Phe-40 with Val in the AcpP-α2L chimera allowed both AcpS function and function in E. coli lipid synthesis (Fig. 4B). This result argued that the modified L. lactis helix II was compatible with E. coli AcpP helices I, III, and IV and the connecting loops. Why then did the AcpA-α2E protein fail to function? This failure argued that a segment(s) of AcpA was incompatible with E. coli lipid metabolism. The alignments of Fig. 1 argue that the L. lactis and E. faecalis AcpAs either lack helix III or have a very minimal helix III. In E. coli AcpP, helix III appears of lower stability than the other helices and is found in various positions in the available crystal and NMR structures with the extremes being almost parallel and almost perpendicular to the bundle formed by the other three helices (3). Moreover, AcpP helix III seems the most dynamic segment of the protein as judged from NMR ensembles and crystal structure B-factors. Indeed, the recent crystal structure of E. coli FabA complexed with an analogue of its cognate acyl-ACP implicates helix III reorientation as providing access of the enzyme to the acyl chain (22).
As a first test of the hypothesized role of helix III in the failure of AcpA-α2E protein to support growth, we replaced AcpA helices III and IV (plus the loops that connect the helices) with those of wild type AcpP (Fig. 2). Although we expected this protein to be functional, it was completely inactive in complementation both in the presence and in the absence of Sfp (Fig. 6). Hence, neither helix III or helix IV was responsible for the failure of the AcpA-α2E chimera to support growth. This result argued that the failure was due to defective interaction of AcpA helix I (and/or the helix I-II loop) with the downstream AcpP sequences. Given that loop sequences tend to be permissive and that the long loops connecting helices I and II show little sequence conservation in the ACPs that function in E. coli (Fig. 1), the loop composition seemed unlikely to play a role in the observed incompatibility. This was directly tested by construction of a chimera that fused AcpA helix I to the loop that connects AcpP helices I-II, resulting in an AcpP protein altered only in that the helix I was that of AcpA (Fig. 2). This protein was also completely nonfunctional both in the presence or in the absence of Sfp expression (data not shown). Therefore, the incompatible elements lie within AcpA helix I rather than within the connecting loop.
FIGURE 6.
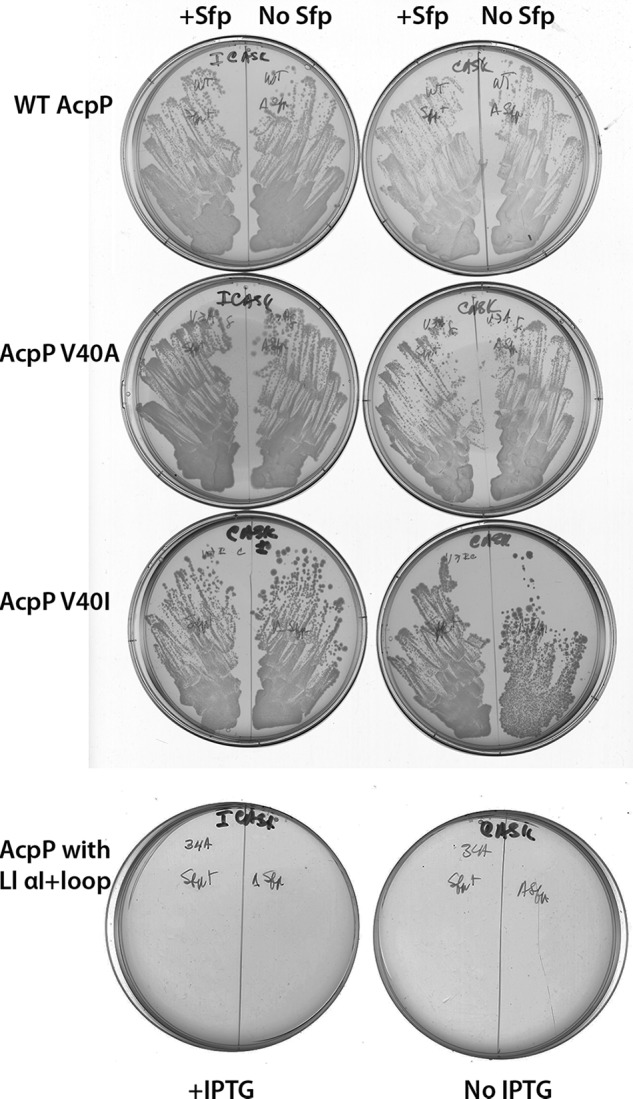
Abilities of mutant AcpP plasmids containing Ala or Ile in place of Val-40 and of a plasmid encoding a chimera containing L. lactis AcpA (plus the downstream loop) and AcpP sequences from helix II to the C terminus to support growth of the arabinose-dependent ΔacpP strain in the absence of arabinose. The strains were grown in the presence of IPTG to induce expression of the ACP genes in the presence or absence of Sfp as shown above the plates. The plates were incubated for 24 h at 37 °C.
The Highly Conserved Val Residue That Imparts Function to the AcpP-α2L Protein Can Be Fully Replaced by Other Aliphatic Residues
The conservation of Val at residue 40 of ACPs seems remarkable. In a recent survey of the AcpP and AcpA sequences, Val was found to be the fourth residue downstream of the modified serine in ∼500 proteins, whereas only ∼50 proteins had Phe and Ala was present in only a handful of proteins. More remarkable was that no instances of Ile or Leu at residue 40 were found, although the three branched chain residues often replace one another elsewhere in ACPs (Fig. 1) (particularly so for Val and Ile). Moreover, changing the Val codon to the codon of another of these residues would require only a single base change. However, despite the observed Val conservation, substitution of Ile or Ala for Val-40 of AcpP gave proteins that allowed robust growth of the E. coli mutant strain (Fig. 6).
Conclusions
Although alignment of the protein sequences and the essentially superimposable nature of the available ACP structures (∼28 x-ray structures and 21 NMR structures) makes it tempting to consider ACP structural components as interchangeable modules, this is not the case. The lack of module interchangeability was most dramatically seen upon replacement of L. lactis AcpA helix II with E. coli helix II, which resulted in a protein that was completely unable to support E. coli growth, although Sfp efficiently modified the protein. This lack of function was not overcome by substitution of E. coli sequences for all except AcpA helix I. In contrast, the opposite helix II substitution, replacement of E. coli helix II with that of AcpA, gave weak Sfp-dependent growth. Robust growth was subsequently obtained by making a single residue change in helix II: substitution of Val for the Phe residue located four residues downstream of the modified serine. Moreover, this substitution allowed growth in the absence of Sfp, indicating that this single substitution allowed E. coli AcpS to modify this chimeric protein. Thus, Phe in this position blocked AcpS activity and greatly compromised function of the protein in E. coli lipid synthesis. This was also the case when Phe was substituted for Val-40 in the otherwise wild type AcpP protein. That protein supported growth only very weakly, and growth required Sfp, indicating that the Phe residue blocked modification by AcpS.
Four recent crystal structures indicate that enzymes cope with the shielding of the acyl chain and thioester by the ACP four-helix bundle by flipping the entire acyl chain plus much of the prosthetic group from the ACP bundle into the hydrophobic lumen of the enzyme (see Ref. 3 for review). This process is called chain flipping, the dynamics of which have been recently explored by indirect means (23). Following enzyme catalysis, the chain must be “reverse flipped” back into the ACP four-helix sheath for presentation to the next enzyme in the pathway. Hence, ACP and its partner enzymes must choreograph a subtle and reversible exchange of acyl chains. An inability to properly perform this choreography might be the cause of the incompatibility of L. lactis helix I with the downstream E. coli AcpP sequences and the poor compatibility of L. lactis helix II with the lipid synthetic pathway of E. coli.
Acknowledgment
We thank Dr. Peter Yau of the Carver Biotechnology Center for help in protein characterization.
This work was supported, in whole or in part, by National Institutes of Health Grant AI15650.
- ACP
- acyl carrier protein
- IPTG
- isopropyl β-d-1-thiogalactopyranoside.
References
- 1. Byers D. M., Gong H. (2007) Acyl carrier protein: structure-function relationships in a conserved multifunctional protein family. Biochem. Cell Biol. 85, 649–662 [DOI] [PubMed] [Google Scholar]
- 2. Chan D. I., Vogel H. J. (2010) Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem. J. 430, 1–19 [DOI] [PubMed] [Google Scholar]
- 3. Cronan J. E. (2014) The chain-flipping mechanism of ACP (acyl carrier protein)-dependent enzymes appears universal. Biochem. J. 460, 157–163 [DOI] [PubMed] [Google Scholar]
- 4. De Lay N. R., Cronan J. E. (2007) In vivo functional analyses of the type II acyl carrier proteins of fatty acid biosynthesis. J. Biol. Chem. 282, 20319–20328 [DOI] [PubMed] [Google Scholar]
- 5. Butland G., Peregrín-Alvarez J. M., Li J., Yang W., Yang X., Canadien V., Starostine A., Richards D., Beattie B., Krogan N., Davey M., Parkinson J., Greenblatt J., Emili A. (2005) Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433, 531–537 [DOI] [PubMed] [Google Scholar]
- 6. Wang H., Cronan J. E. (2004) Only one of the two annotated Lactococcus lactis fabG genes encodes a functional β-ketoacyl-acyl carrier protein reductase. Biochemistry 43, 11782–11789 [DOI] [PubMed] [Google Scholar]
- 7. Wang H., Cronan J. E. (2004) Functional replacement of the FabA and FabB proteins of Escherichia coli fatty acid synthesis by Enterococcus faecalis FabZ and FabF homologues. J. Biol. Chem. 279, 34489–34495 [DOI] [PubMed] [Google Scholar]
- 8. Zhu L., Bi H., Ma J., Hu Z., Zhang W., et al. (2013) The two functional enoyl-acyl carrier protein reductases of Enterococcus faecalis do not mediate triclosan resistance. MBio 4, e00613-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parris K. D., Lin L., Tam A., Mathew R., Hixon J., Stahl M., Fritz C. C., Seehra J., Somers W. S. (2000) Crystal structures of substrate binding to Bacillus subtilis holo-(acyl carrier protein) synthase reveal a novel trimeric arrangement of molecules resulting in three active sites. Structure 8, 883–895 [DOI] [PubMed] [Google Scholar]
- 10. De Lay N. R., Cronan J. E. (2006) Gene-specific random mutagenesis of Escherichia coli in vivo: isolation of temperature-sensitive mutations in the acyl carrier protein of fatty acid synthesis. J. Bacteriol. 188, 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cronan J. E., Jr., Narasimhan M. L., Rawlings M. (1988) Insertional restoration of β-galactosidase α-complementation (white-to-blue colony screening) facilitates assembly of synthetic genes. Gene 70, 161–170 [DOI] [PubMed] [Google Scholar]
- 12. Rawlings M., Cronan J. E., Jr. (1992) The gene encoding Escherichia coli acyl carrier protein lies within a cluster of fatty acid biosynthetic genes. J. Biol. Chem. 267, 5751–5754 [PubMed] [Google Scholar]
- 13. Graña D., Gardella T., Susskind M. M. (1988) The effects of mutations in the ant promoter of phage P22 depend on context. Genetics 120, 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takiff H. E., Baker T., Copeland T., Chen S. M., Court D. L. (1992) Locating essential Escherichia coli genes by using mini-Tn10 transposons: the pdxJ operon. J. Bacteriol. 174, 1544–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Volkmann G., Murphy P. W., Rowland E. E., Cronan J. E., Jr., Liu X. Q., Blouin C., Byers D. M. (2010) Intein-mediated cyclization of bacterial acyl carrier protein stabilizes its folded conformation but does not abolish function. J. Biol. Chem. 285, 8605–8614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cronan J. E., Jr. (1980) β-Alanine synthesis in Escherichia coli. J. Bacteriol. 141, 1291–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cronan J. E., Jr. (1980) Enzymatic synthesis of β-[U-14C]alanine and d-[1,2,3-14C]pantothenate of high specific radioactivity. Anal. Biochem. 103, 377–380 [DOI] [PubMed] [Google Scholar]
- 19. Beld J., Sonnenschein E. C., Vickery C. R., Noel J. P., Burkart M. D. (2014) The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life. Nat. Prod. Rep. 31, 61–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou Z., Cironi P., Lin A. J., Xu Y., Hrvatin S., Golan D. E., Silver P. A., Walsh C. T., Yin J. (2007) Genetically encoded short peptide tags for orthogonal protein labeling by Sfp and AcpS phosphopantetheinyl transferases. ACS Chem. Biol. 2, 337–346 [DOI] [PubMed] [Google Scholar]
- 21. Zhou Z., Koglin A., Wang Y., McMahon A. P., Walsh C. T. (2008) An eight residue fragment of an acyl carrier protein suffices for post-translational introduction of fluorescent pantetheinyl arms in protein modification in vitro and in vivo. J. Am. Chem. Soc. 130, 9925–9930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nguyen C., Haushalter R. W., Lee D. J., Markwick P. R., Bruegger J., Caldara-Festin G., Finzel K., Jackson D. R., Ishikawa F., O'Dowd B., McCammon J. A., Opella S. J., Tsai S. C., Burkart M. D. (2014) Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature 505, 427–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beld J., Cang H., Burkart M. D. (2014) Visualizing the chain-flipping mechanism in fatty-acid biosynthesis. Angew. Chem. Int. Ed. Engl. 53, 14456–14461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Amann E., Brosius J. (1985) “ATG vectors” for regulated high-level expression of cloned genes in Escherichia coli. Gene 40, 183–190 [DOI] [PubMed] [Google Scholar]
- 25. Yang Z., Lasker K., Schneidman-Duhovny D., Webb B., Huang C. C., Pettersen EF, Goddard T. D., Meng E. C., Sali A., Ferrin T. E. (2012) UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J. Struct. Biol. 179, 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cronan J. E., Thomas J. (2009) Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. Methods Enzymol. 459, 395–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cronan J. E., Jr., Klages A. L. (1981) Chemical synthesis of acyl thioesters of acyl carrier protein with native structure. Proc. Natl. Acad. Sci. U.S.A. 78, 5440–5444 [DOI] [PMC free article] [PubMed] [Google Scholar]