

Background: Chemokine function is regulated by proteolytic processing.
Results: Cysteine cathepsins activate signaling by ELR CXC chemokines and terminate signaling by non-ELR chemokines.
Conclusion: Cysteine cathepsins process CXC chemokines and promote inflammation by recruitment of CXCR2-expressing cells.
Significance: This is the first comprehensive study on the processing of CXC chemokines by cysteine cathepsins.
Keywords: chemokine, glycosaminoglycan, inflammation, mass spectrometry (MS), proteolysis, CXC chemokine, chemokine processing, cysteine cathepsin
Abstract
Cysteine cathepsins are primarily lysosomal proteases involved in general protein turnover, but they also have specific proteolytic functions in antigen presentation and bone remodeling. Cathepsins are most stable at acidic pH, although growing evidence indicates that they have physiologically relevant activity also at neutral pH. Post-translational proteolytic processing of mature chemokines is a key, yet underappreciated, level of chemokine regulation. Although the role of selected serine proteases and matrix metalloproteases in chemokine processing has long been known, little has been reported about the role of cysteine cathepsins. Here we evaluated cleavage of CXC ELR (CXCL1, -2, -3, -5, and -8) and non-ELR (CXCL9–12) chemokines by cysteine cathepsins B, K, L, and S at neutral pH by high resolution Tris-Tricine SDS-PAGE and matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Whereas cathepsin B cleaved chemokines especially in the C-terminal region, cathepsins K, L, and S cleaved chemokines at the N terminus with glycosaminoglycans modulating cathepsin processing of chemokines. The functional consequences of the cleavages were determined by Ca2+ mobilization and chemotaxis assays. We show that cysteine cathepsins inactivate and in some cases degrade non-ELR CXC chemokines CXCL9–12. In contrast, cathepsins specifically process ELR CXC chemokines CXCL1, -2, -3, -5, and -8 N-terminally to the ELR motif, thereby generating agonist forms. This study suggests that cysteine cathepsins regulate chemokine activity and thereby leukocyte recruitment during protective or pathological inflammation.
Introduction
Chemokines are a group of small structurally related chemotactic cytokines that regulate migration of leukocytes in homeostasis and inflammation but also function in other processes such as embryogenesis, angiogenesis, hematopoiesis, tumor growth, and metastasis (1). Their basic structure is conserved and includes a short unstructured N-terminal region and an extended N-loop followed by three β-strands and a C-terminal α-helix. Two disulfide bonds tether the N-loop and the N-terminal residues to the core of the protein. Based on the presence of conserved cysteine (C) residues in the sequence of the N terminus, chemokines are classified as “CC,” “CXC,” “C,” and “CX3C.” CXC chemokines are further divided into “ELR” and “non-ELR” chemokines based on the presence or absence of the tripeptide glutamate-leucine-arginine motif before the first cysteine.
Upon secretion from cells, chemokines, which are highly basic proteins, bind to negatively charged glycosaminoglycans (GAGs)4 on the surface of cells or in the extracellular matrix, thereby creating a haptotactic gradient. However, there seems to be a considerable topological diversity in the location of the binding sites (2). Binding to GAGs not only prevents the diffusion of chemokines away from the site of release but might also protect them against proteolysis (3), although for the matrix metalloproteinases (MMPs) there is little effect (4, 5).
Chemokine receptors belong to the G protein-coupled receptor superfamily and their activation has been described as a two-site model. The N-loop of the chemokine (site I) is important for binding affinity and receptor selectivity, whereas the N-terminal region (site II) mediates receptor activation (6).
Post-translational modifications such as specific and limited proteolysis, termed proteolytic processing (7) or deamination, have been established as important mechanisms for regulating chemokine activity (8). Several proteases have been demonstrated as contributing to chemokine processing, in particular MMPs, aminopeptidase/CD13, and the serine proteases thrombin, plasmin, membrane-bound peptidylpeptidase IV/CD26/DPPIV, neutrophil cathepsin G, and elastase (9). For example, MMPs convert CC agonists into antagonists (10, 11), switch chemokine receptor specificity (12), inactivate CC (13) and CXC chemokines (5, 14), activate CC (15) and CXC chemokines (16), and shed CX3CL1/fractalkine (17).
N- and C-terminally truncated forms of both ELR and non-ELR CXC chemokines have been described in the supernatant of cultured leukocytes (8, 18–21). With an intact ELR motif, N-terminal truncations of ELR CXC chemokines result in higher activity in calcium mobilization and chemotaxis assays (16, 18, 20, 22, 23). In contrast, N-terminal processing of non-ELR CXC chemokines decreases their chemotactic activity (4, 24, 25). Even so, fragments without the first two N-terminal amino acid residues retain their angiostatic properties, but this is lost upon further truncations (19). Moderate proteolysis at the C terminus generally does not alter chemokine activity, whereas more extensive truncations can affect GAG binding ability (4, 15). Several proteases have been identified that can generate N-terminally truncated forms of CXCL5, -8, -10, and -11 (4, 9, 16). In contrast, the identity of proteases responsible for the N-terminal truncations of CXCL1–3 beyond the proline residue is still unknown except for a set of MMPs that cleave CXCL2 to generate the (5–73)-form (13, 14).
Despite the well established role of the above mentioned proteases in chemokine processing, only limited information is available for cysteine cathepsins. There are 11 human cysteine cathepsins (Cat), CatB, CatC, CatF, CatH, CatK, CatL, CatO, CatS, CatV, CatW, and CatX, which exhibit considerable redundancy and generally a broad substrate specificity (26). Cysteine cathepsins are primarily lysosomal enzymes, and because of limited stability at neutral pH they were initially considered ineffective in the extracellular milieu. However, there are considerable differences in the stability of different cathepsins at neutral pH, ranging from a few minutes in the case of CatL to hours for CatS. Furthermore, the stability and activity of cathepsins can be significantly prolonged by binding to various ligands, including substrates and GAGs (26–28). In particular, CatB, CatK, CatL, CatS, and CatX have been linked to extracellular proteolysis in physiological as well as disease conditions (29–31). Because of a potent but relatively short-lived activity at neutral pH, their potential for protease signaling is high (32), suggesting that cysteine cathepsins are likely candidates for chemokine processing. In support of that, CatL has been identified as a CXCL8-converting enzyme in a model of stimulated fibroblasts (33), and cathepsin B has been found to cleave CCL20 (with no effect on activity) and CXCL9–14, but functional analyses were not performed on these CXCL cleavage products (34). More recently, CatS has been suggested to be involved in CX3CL1/fractalkine shedding in the spinal cord, which contributes to chronic pain in neuronal cells and arthritis models (35–37), and osteoblast-derived CatX in the degradation of CXCL12 (38).
To test the hypothesis that chemokines are extracellular substrates for cysteine cathepsins, we systematically analyzed the processing of human ELR CXC chemokines CXCL1/GROα, CXCL2/GROβ, CXCL3/GROγ, CXCL5/ENA-78, and CXCL8/IL-8 and non-ELR CXC chemokines CXCL9/MIG, CXCL10/IP-10, CXCL11/I-TAC, and CXCL12/SDF by human cysteine cathepsins CatB, CatK, CatL, and CatS at neutral pH and in the absence or presence of the GAG chondroitin sulfate C/chondroitin-6-sulfate (C6S). We found that cysteine cathepsins CatK, CatL, and CatS remove four or five amino acid residues from the N terminus of CXCL1–3. In addition, cysteine cathepsins generate more potent forms of CXCL5 and -8 and inactivate or degrade non-ELR CXC chemokines CXCL9–12. Collectively, our results suggest that cysteine cathepsins complement other proteases in post-translational modulation of CXC chemokine activity.
Experimental Procedures
Chemokines and Proteases
All chemokines were synthesized using t-Boc (tertiary butyloxycarbonyl) solid phase chemistry as described previously (39). Recombinant human cysteine cathepsins were expressed in Escherichia coli (CatB (40)) or Pichia pastoris (CatK (41), CatL, and CatS (42)). CatB and CatS were activated in 100 mm phosphate buffer, pH 6.0, and CatK and CatL in 100 mm acetate buffer, pH 5.5, all in the presence of 5 mm DTT at room temperature for 5 min.
Chemokine Processing Assays
Chemokine processing assays were performed in 50 mm HEPES, pH 7.4, at an enzyme to substrate ratio of 1:20 (w/w), in the case of CatB, CatK, and CatS, and 1:40 (w/w), in the case of CatL, at 37 °C. The final concentration of chemokine in the reaction mixture was 0.1 mg/ml (43). Additional processing reactions were performed at an enzyme to substrate ratio of 1:500 or 1:600 (w/w) or at acidic pH in 100 mm citrate buffer, pH 4.5. To investigate the effect of GAGs on the processing, C6S was added to the reaction mixture at a final concentration of 0.01, 0.05, 0.25, or 1.0 mg/ml. Samples of the reaction mixture were taken after 15, 30, or 60 min. The reaction was terminated by adding E64d, a broad spectrum cysteine cathepsin inhibitor, at a final concentration of 50 μm. Samples were then analyzed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) on a Voyager-DE STR (Applied Biosystems) in sinapinic acid matrix. To degrade C6S, which interfered with MALDI-TOF MS analysis, hyaluronidase from bovine testis type NS (Sigma) was added to the reaction mixture at a final concentration of 0.1 mg/ml for 1 min after the reaction was terminated with E64d and before the sinapinic acid matrix was added. In addition, spotted samples were washed with 1 μl of dH2O. PeptID software was used to identify the truncated fragments in the MS spectra (43). Reaction mixtures were also analyzed with 15% Tris-Tricine SDS-PAGE, and gels were silver-stained (43).
Transfected and Isolated Cells
Human CXCR3- and CXCR2-transfected B300-19 cells (44), kindly provided by B. Moser (Bern, Switzerland), were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 50 μm β-mercaptoethanol, and 1.0 mg/ml G418. Human neutrophil granulocytes were isolated from peripheral blood of healthy volunteers as approved by the Clinical Ethics Review Board of the Republic of Slovenia. Blood was drawn into sodium citrate-treated Vacutainer tubes and layered on Lympholyte®-poly solution (Cedarlane) according to the manufacturer's instructions.
Calcium Mobilization
CXCR2- and CXCR3-transfected B300-19 cells were stained with 2 mm Fluo-4 AM (Molecular Probes) for 30 min at 37 °C. The assay was performed as described previously (4).
Chemotaxis
Chemotaxis assays were performed in 96-well chemotaxis chambers of MBA96 series (Neuro Probe) across polycarbonate filters with 5-μm pores for CXCR3-transfected B300-19 cells and 3-μm pores for neutrophil granulocytes. Cells and chemokines were diluted in RPMI supplemented with 1% (w/v) BSA, 20 mm HEPES, and 2 mm l-glutamine. Serial dilutions of a chemokine ranging from 100 to 0.1 nm were prepared in lower chambers, and 0.2 × 106 cells were added to upper chambers. After incubation at 37 °C for 60 min with neutrophil granulocytes and 90–120 min with B300-19 cells, the upper chambers were aspirated and washed twice with dH2O. Migrated cells in lower wells were transferred to a black 96-well plate and lysed by freeze-thawing, and the total cell numbers were quantified against a standard curve using a CyQuant assay according to the manufacturer's instructions (Molecular Probes). Chemotactic index was calculated as the ratio of cells migrating in response to chemokines compared with buffer control.
Results
Processing of CXC Chemokines by CatB, CatK, CatL, and CatS at Neutral pH
Human CXCL1–3, -5, and -8–12 were incubated with activated human CatB, CatK, and CatS at an enzyme to substrate ratio of 1:20 (w/w) and with CatL at an enzyme to substrate ratio of 1:40 (w/w) for 15 min at neutral pH. Analysis by Tris-Tricine SDS-PAGE (Fig. 1), with confirmation and identification of cleavage sites by MALDI-TOF MS (Table 1, Fig. 2, and supplemental Fig. 1), showed that in the absence of C6S, the four cathepsins specifically cleaved these chemokines by processing or degradation. The only exception was CXCL1, which was not processed by CatB. Moreover, the presence of bands of slightly reduced molecular weight indicated that CXCL1–3, -5, and -8 were specifically processed by cathepsins, whereas CXCL9–11 were mostly degraded, as judged on the bands of considerably lower molecular weight. CXCL12 was completely degraded, as no smaller processing products were observed with any of the cathepsins.
FIGURE 1.
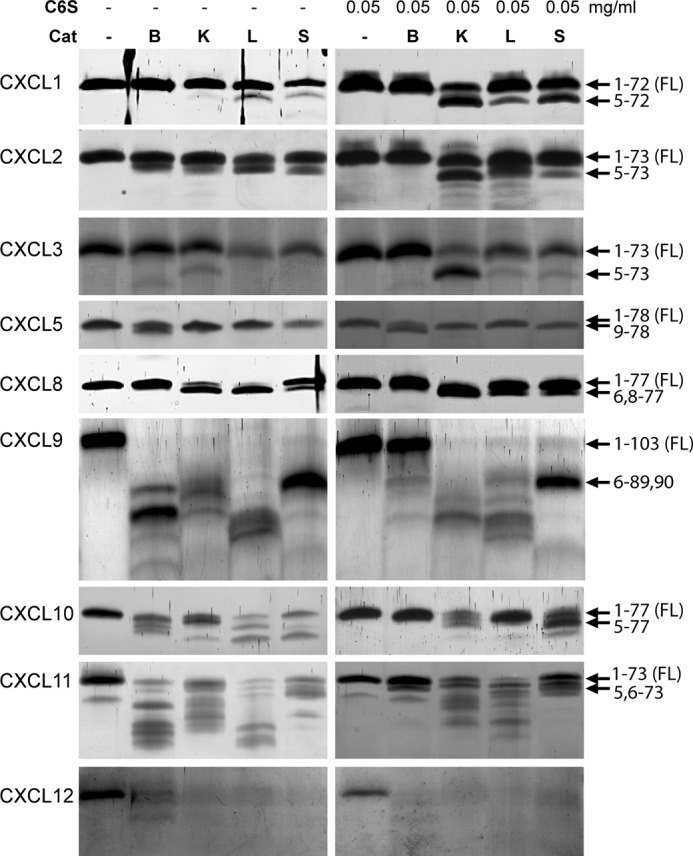
Processing of CXCL1/GROα, CXCL2/GROβ, CXCL3/GROγ, CXCL5/ENA-78, CXCL8/IL-8, CXCL9/MIG, CXCL10/IP-10, CXCL11/I-TAC, and CXCL12/SDF by cysteine cathepsins. Cysteine cathepsins CatB, CatK, CatL, and CatS were incubated with chemokines at an enzyme to substrate ratio of 1:20 (w/w) in 50 mm HEPES buffer, pH 7.4, in the absence or presence of 0.05 mg/ml GAG C6S for 15 min. Samples were separated by 15% Tris-Tricine SDS-PAGE and visualized by silver staining. Arrows indicate cleavage sites identified by MALDI-TOF MS analysis.
TABLE 1.
Fragments produced by cysteine cathepsin processing of CXCL1, CXCL2, CXCL3, CXCL5, CXCL8, CXCL10, and CXCL11
Cysteine cathepsins were incubated with chemokines at an enzyme to substrate ratio of 1:20 (w/w) (for CatB, CatK, and CatS) or 1:40 (w/w) (for CatL) in 50 mm HEPES buffer, pH 7.4, in the absence or presence of 0.05 mg/ml GAG C6S for 15 or 30 min. Samples were analyzed by MALDI-TOF MS on a Voyager-DE STR (Applied Biosystems) in sinapinic acid matrix. PeptID software (43) was used to identify the truncated fragments in the MS spectra. Predicted activity was assigned according to the literature and our cell assays (relevant figures are indicated). Major fragments were determined based on MALDI-TOF MS and Tris-Tricine SDS-PAGE analysis (Fig. 1). ↑, increased activity compared to full-length chemokine; ↓, decreased activity compared to full-length chemokine; ND, activity not determined; *, major fragment; −, fragment generated/stable in the absence of GAGs; +, fragment generated/stable in the presence of GAGs.
| Chemokine |
m/z [M+H]+ |
Predicted activity | Cathepsin | |
|---|---|---|---|---|
| Measured | Predicted | |||
| CXCL1-(1–72) | 7751 | 7751 | ||
| CXCL1-(5–72) | 7422 | 7422 | ↑ Ref. 18, Figs. 6A and 7A | *K+, *L, *S |
| CXCL1-(5–69) | 7099 | 7093 | ND | *K+ |
| CXCL2-(1–73) | 7893 | 7893 | ||
| CXCL2-(5–73) | 7541 | 7540 | ↑ Ref. 20, Fig.7, B and C | *K+, *L, *S |
| CXCL2-(1–69) | 7507 | 7506 | ND | B− |
| CXCL2-(1–68) | 7391 | 7392 | ND | K |
| CXCL2-(5–68) | 7040 | 7040 | ND | L− |
| CXCL3-(1–73) | 7865 | 7865 | ||
| CXCL3-(2–73) | 7795 | 7794 | ND | K+, L′ |
| CXCL3-(3–73) | 7708 | 7707 | ND | K+ |
| CXCL3-(1–70) | 7562 | 7563 | ND | B− |
| CXCL3-(5–73) | 7510 | 7509 | ↑ Ref. 18 | *K+, *L+, *S+ |
| CXCL3-(6–73) | 7406 | 7408 | ND | L |
| CXCL5-(1–78) | 8357 | 8357 | ||
| CXCL5-(5–78) | 8065 | 8061 | ↑ Refs. 18 and 22 | *B, *K− |
| CXCL5-(9–78) | 7709 | 7706 | ↑ Ref. 22 and Fig. 6B | *K+, L, *S |
| CXCL5-(10–78) | 7552 | 7550 | ↓ Ref. 22 | L+ (pH 4) |
| CXCL8-(1–77) | 8923 | 8923 | ||
| CXCL8-(1–75) | 8721 | 8721 | ND | B− |
| CXCL8-(1–73) | 8520 | 8521 | ND | B− |
| CXCL8-(6–77) | 8386 | 8386 | ↑ Ref. 70 | *K, *L,*S |
| CXCL8-(8–77) | 8228 | 8228 | ↑ Ref. 23 | *K+, S |
| CXCL9-(1–103) | 11725 | 11725 | ||
| CXCL9-(1–101) | 11525 | 11523 | ND | B |
| CXCL9-(1–90) | 10139 | 10140 | ND | K |
| CXCL9-(1–89) | 10011 | 10012 | ND | K |
| CXCL9-(6–90) | 9457 | 9459 | ↓ Ref. 24 and Fig. 6C | *K, *S |
| CXCL9-(6–89) | 9840 | 9843 | ↓ Ref. 24 and Fig. 6C | *K, *S |
| CXCL10-(1–77) | 8646 | 8646 | ||
| CXCL10-(1–75) | 8461 | 8462 | ND | B− |
| CXCL10-(5–77) | 8248 | 8250 | ↓ Ref. 24 and Figs. 6D and 7, D and E | *K, *L+, *S+ |
| CXCL10-(5–74) | 7906 | 7910 | ↓ | L |
| CXCL10-(5–73) | 7779 | 7781 | ↓ | S |
| CXCL11-(1–73) | 8307 | 8307 | ||
| CXCL11-(5–73) | 7786 | 7784 | ↓ Refs. 4, 24, and 25 | S |
| CXCL11-(6–73) | 7651 | 7649 | ↓ Fig. 7F | *K, *L+, *S |
FIGURE 2.
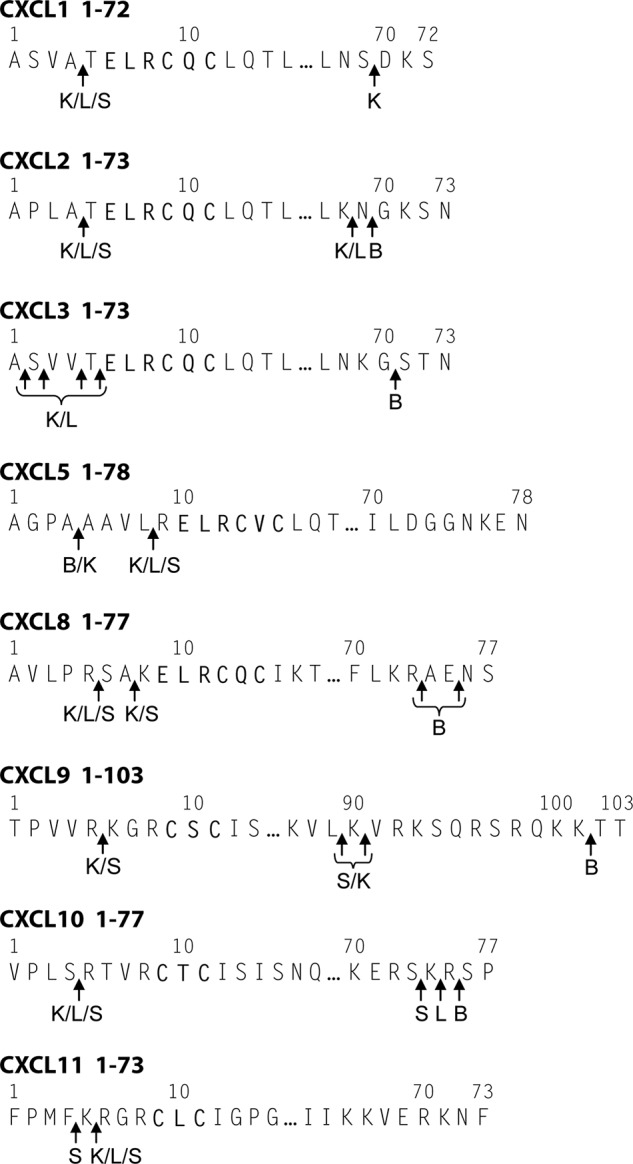
Cysteine cathepsin cleavage sites in human CXCL1, CXCL2, CXCL3, CXCL5, CXCL8, CXCL9, CXCL10, and CXCL11. Cysteine cathepsins CatB, CatK, CatL, and CatS were incubated with chemokines at an enzyme to substrate ratio of 1:20 (w/w) in 50 mm HEPES buffer, pH 7.4, in the absence or presence of 0.05 mg/ml GAG C6S for 15 or 30 min. Cleavage sites were determined on the basis of fragments identified by MALDI-TOF MS analysis (supplemental Fig. 1).
In the presence of C6S, the proteolytic profile changed. Chemokine processing by CatB was largely diminished, whereas processing by CatL and CatS was also reduced but to a smaller extent. In contrast, processing of chemokines by CatK was increased in the presence of C6S, in agreement with the known effect of GAGs on the proteolytic activity of CatK on protein substrates. However, the effect of C6S was also chemokine-specific. In the presence of C6S truncated forms of CXCL1–3, and to a smaller extent CXCL10 and -11, were more readily detected by Tris-Tricine SDS-PAGE and confirmed by MALDI-TOF MS than in the absence of C6S. In contrast, there was little effect of C6S on the cleavage of CXCL5, -8, -9, or -12. Similar effects on chemokine processing were also observed in the presence of C4S (results not shown).
With the exception of CXCL5, CatB cleaved all chemokines at the C terminus (Table 1 and Fig. 2). CatK, CatL, and CatS processed chemokines at sites N-terminal to the conserved CXC sequence. The resultant truncated forms of CXCL1-(5–72), CXCL2-(5–73), CXCL3-(5–73), CXCL5-(9–78), and CXCL8-(6–77, 8–77) were reported previously to have increased chemotactic activity (8, 16, 18, 22, 23), whereas CXCL9-(6–89, 6–90), CXCL10-(5–77), and CXCL11-(5–73) lose their chemotactic activity upon N-terminal truncation (4, 8, 24). In addition to N-terminal truncations of CXCL9, CatK and CatS cleaved in the extended unstructured C-terminal region of CXCL9, whereas CatL rapidly degraded this chemokine. Interestingly, cleavage sites in the N-terminal region of chemokines were often shared by two or more cysteine cathepsins (Fig. 2), in agreement with their generally similar specificity (45).
To evaluate the efficiency of cleavage, we investigated chemokine processing with reduced amounts of proteases. Figs. 3 and 4, which present MALDI-TOF MS spectra for selected cathepsin/chemokine combinations, illustrate that although the processing efficiency was diminished, characteristic chemokine fragments were generated after as little as 15 min of incubation with cathepsins.
FIGURE 3.

Representative MALDI-TOF spectra of chemokine CXCL5 processed by CatK (A) and CXCL10 processed by CatS (B). Cleavage assays were performed at an enzyme to substrate ratio of 1:20 or 1:500 (w/w) in 50 mm HEPES buffer, pH 7.4, or 100 mm citrate buffer, pH 4.5, in the absence or presence of 0.05 mg/ml GAG C6S for 15, 30, or 60 min. Samples were analyzed by MALDI-TOF MS on a Voyager-DE STR in sinapinic acid matrix. Processing was efficient, and similar chemokine fragments were generated at high or low cathepsin to chemokine ratios. Proteolysis by CatK was increased in the presence of GAGs, most notably at a low cathepsin to chemokine ratio, at acidic pH.
FIGURE 4.

Representative MALDI-TOF spectra of chemokines CXCL1 processed by CatL (A) and CXCL2 processed by CatK (B). Cleavage assays were performed at an enzyme to substrate ratio of 1:20 or 1:500 (w/w) (CatK) and 1:40 or 1:600 (w/w) (CatL) in 50 mm HEPES buffer, pH 7.4, in the absence or presence of 0.05 mg/ml GAG C6S for 15 or 30 min. Samples were analyzed by MALDI-TOF MS on a Voyager-DE STR (Applied Biosystems) in sinapinic acid matrix. Processing was efficient, and similar chemokine fragments were generated at high or low cathepsin to chemokine ratios.
Cathepsins are normally functional within the acidic environment of lysosomes, and thus we also investigated the processing of selected chemokines at pH 4.5. MALDI-TOF MS spectra for the processing of CXCL5 by CatK (Fig. 3A) and CXCL10 by CatS (Fig. 3B) are presented. At acidic pH, CatK was found to be a potent protease both in the presence and absence of C6S, generating even smaller CXCL5 fragments than at neutral pH. In contrast, CatS was a less potent protease at acidic pH, although it generated similar cleavage products as at neutral pH.
The Effect of C6S on Chemokine Processing
To better understand the effect of GAGs on chemokine processing, processing of CXCL1, -2, and -11 was performed at four different C6S concentrations (0.01, 0.05, 0.25, and 1.0 mg/ml, Fig. 5). As visualized by Tris-Tricine SDS-PAGE, the presence of CatL- or CatS-derived proteolytic products of CXCL1 and -2 was reduced with increasing concentration of C6S. In contrast, the processing of these two chemokines by CatK was increased at low C6S concentrations, whereas high C6S concentrations attenuated it. In the case of CXCL11, the effect of C6S was only minor even at the highest concentration used.
FIGURE 5.
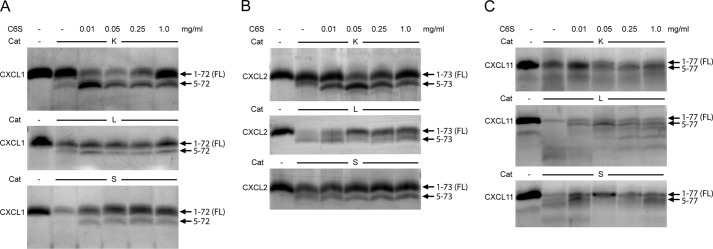
Effect of GAGs on processing of CXCL1, -2, and -11 by CatK, CatL, and CatS. Cysteine cathepsins CatK, CatL, and CatS were incubated with chemokines CXCL1 (A), CXCL2 (B), and CXCL11 (C) at an enzyme to substrate ratio of 1:20, in 50 mm HEPES buffer, pH 7.4, in the absence or presence of 0.01, 0.05, 0.25, or 1.0 mg/ml GAG ChC for 15 min. Samples were analyzed by 15% Tris-Tricine SDS-PAGE, and gels were silver-stained. Arrows indicate fragments identified by MALDI-TOF MS analysis.
Because pH and GAGs considerably modulated chemokine processing by cysteine cathepsins, we evaluated their effects on the catalytic efficiency of cysteine cathepsins using Z-FR-AMC, a small fluorogenic substrate. C6S prolonged the activity of all tested cysteine cathepsins on small fluorogenic substrates at acidic and neutral pH (data not shown), likely through the structural stabilization of the proteases, in agreement with previous studies using different GAGs (46, 47).
Calcium Mobilization of CXCR2- or CXCR3-transfected B300-19 Cells
The effect of cathepsin processing of chemokines on chemokine receptor activation was evaluated by a calcium mobilization assay, performed on human CXCR2- or CXCR3-transfected B300-19 cells with C6S, full-length or C6S/CatS-processed CXCL2, -5, -9, or -10 (Fig. 6). The two receptors were selected based on their binding of ELR (CXCR2) (48) or non-ELR (CXCR3) (44) chemokines. CatS was selected as a model cathepsin because it generated the same major fragments as CatK and CatL. ELR CXC chemokines CXCL2 and -5 induced detectable calcium influx in CXCR2-transfected cells only after cleavage with CatS. Full-length CXCL2-(1–73) exerted no significant activity at chemokine concentrations of up to 100 nm, whereas C6S/CatS-generated CXCL2-(5–73) showed calcium mobilization activity at concentrations as low as 1 nm (Fig. 6A). Similarly, full-length CXCL5-(1–78) had no activity, whereas C6S/CatS-truncated CXCL5-(9–78) induced calcium influx at concentrations above 10 nm (Fig. 6B). In contrast, Ca2+ influx in CXCR3-transfected cells in response to non-ELR chemokine was decreased after cysteine cathepsin processing. Truncated CXCL9-(6–89, 6–90) generated by C6S/CatS had considerably reduced activity (Fig. 6C). Similarly, full-length CXCL10-(1–77) induced a strong calcium influx above 5 nm, whereas truncated CXCL10-(5–77) had no detectable activity (Fig. 6D). The observed low Ca2+ influx with ELR chemokines was a reflection of the low CXCR2 expression on cells relative to the receptor expression in the CXCR3 cells.
FIGURE 6.
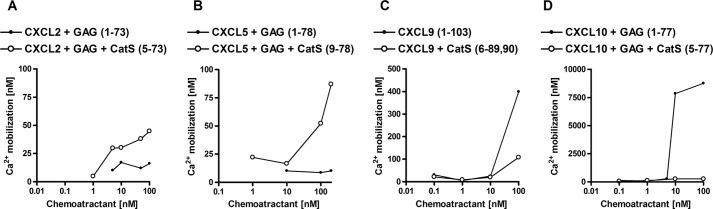
Calcium mobilization is increased in response to cysteine cathepsin-processed ELR CXC chemokines and decreased in response to cysteine cathepsin-processed non-ELR chemokines. Calcium mobilization of CXCR2-transfected B300-19 cells is increased in response to CatS-truncated CXCL2-(5–73) (A) and CatS-truncated CXCL5 (B). In contrast, calcium mobilization of CXCR3-transfected B300-19 cells is decreased in response to CXCL9 processed by CatS (C), which generates (6–89, 6–90)-fragments, or in response to CatS-truncated CXCL10-(5–77) (D). Cysteine cathepsins were incubated with chemokines at the enzyme to substrate ratio of 1:20 (w/w) for CatS in 50 mm HEPES buffer, pH 7.4, in the presence of 0.05 mg/ml GAG C6S for 15 min. Calcium concentrations were calculated from relative fluorescence based on calibration with ionomycin and EGTA.
Chemotactic Migration of CXCR3-transfected B300-19 Cells or Human Neutrophils
To further confirm that the processing of chemokines by cysteine cathepsins has functional consequences, we tested full-length and processed chemokines in Transwell migration assays (Fig. 7). Because of the low expression of CXCR2 in transfected cells, we used primary neutrophil granulocytes to test the activity of ELR CXC chemokines CXCL1 and CXCL2. Compared with full-length CXCL1-(1–72), which did not induce chemotaxis at any concentration in the cells, an increased chemotactic response upon C6S/CatK treatment of the chemokine was observed but only at the highest concentration tested (100 nm) (Fig. 7A), and so this is unlikely to be physiologically relevant. In contrast, full-length CXCL2-(1–73) was processed by CatL (Fig. 7B) and CatS (Fig. 7C) in the presence of C6S to generate mostly CXCL2-(5–73), which induced a dose-dependent migration of neutrophil granulocytes with a typical bell-shaped chemotactic response, with the maximum activity at 1 nm. Thus, cathepsins L and S activate CXCL2. In contrast, non-ELR chemokine CXCL10 lost activity upon processing by cysteine cathepsins. CXCR3-transfected cells migrated in response to full-length CXCL10-(1–77) with maximum migration activity at 10 nm but showed no chemotactic response to truncated CXCL10-(5–77) generated by CatS (Fig. 7D) or CatL (Fig. 7E) in the presence of C6S. Similarly, CXCL11 processed by CatL in the presence of C6S lost chemotactic activity (Fig. 7F), which was presumably due to the generation of an inactive fragment CXCL11-(6–73) and further chemokine degradation.
FIGURE 7.

Chemotactic migration is increased in response to cysteine cathepsin-processed ELR CXC chemokines and abolished in response to cysteine cathepsin-processed non-ELR chemokines. Chemotaxis of neutrophil granulocytes is increased in response to CXCL1 processed by CatK, which generates the (5–72)-fragment (A), and in response to CXCL2 processed by CatL (B) or CatS (C), which generates the (5–73)-fragment. In contrast, chemotaxis of CXCR3-transfected B300-19 cells is abolished in response to CXCL10 processed by CatS (D) or CatL (E), which generates the (5–77)-fragment, or in response to CXCL11 processed by CatL (F), which generates the (6–73)-fragment and also degrades CXCL11. Cysteine cathepsins were incubated with chemokines at an enzyme to substrate ratio of 1:20 (w/w) for CatS and 1:40 (w/w) for CatL in 50 mm HEPES buffer, pH 7.4, in the presence of 0.05 mg/ml GAG C6S for 15 min. Chemotaxis was measured across 3-μm pore filters for 60 min with neutrophil granulocytes and across 5-μm pore filters for 90–120 min with CXCR3-transfected B300-19 cells. Migrated cells were quantified by CyQUANT assay, and a chemotactic index was calculated, defined as the ratio of cells migrating in response to stimulus compared with the buffer control.
Discussion
Although the involvement of extracellular cysteine cathepsins in inflammation-associated diseases is well established (49), a comprehensive analysis of their potential as chemokine-processing enzymes has not been published previously. Herein, we show that cysteine cathepsins CatB, CatL, CatS, and CatK rapidly processed CXC ELR (CXCL1–3, -5, and -8) and non-ELR (CXCL9–12) chemokines, with several cathepsins generating the same truncated forms, and we also show that the processing was functionally relevant. The differential processing of ELR and non-ELR chemokines by cysteine cathepsins, generating more active forms of the former and inactivating the latter, suggests that cathepsins are potential regulators of chemokine activities in vivo. Several other proteases have already been found to process CXC chemokines, including plasmin, thrombin, several MMPs, and DPPIV. We found that CatS, CatL, and CatK process several of the chemokines in a unique way, with the most interesting being N-terminal processing of CXCL1, which generates the (5–72)-form, and of CXCL2 and CXCL3, which generates the (5–73)-forms. Notably, MMP1, -9, -12, and -25 also generate the CXCL2-(5–73) form (13, 14). However, other proteases that generate fragments of CXCL1–3 with increased chemotactic activity in the supernatant of cultured monocytes (8, 18, 20) have not been identified.
Although cysteine cathepsins have broad substrate specificity, consistent with their function in lysosomal degradation (28), they specifically generated CXCL1-(5–72), CXCL2-(5–73), and CXCL3-(5–73, 6–73) forms with valine or leucine in the P2 position, in agreement with their canonical specificity for aromatic or aliphatic residues in this position (45, 50). Similarly, processing of the N-terminal region in CXCL9, CXCL10, and CXCL11 chemokines by CatK, CatL, and CatS matches the canonical specificity of cysteine cathepsins. Although the arginine in the ELR motif of ELR chemokines would be a good candidate for the P1 position, no cleavages were observed within or downstream of the ELR sequence, which would inactivate the chemokines, as demonstrated for MMP12-mediated proteolysis (14). However, chemokine processing by CatB was quite different from the other three cathepsins, as it occurred predominantly in the C-terminal region. An exception was the cleavage in the N-terminal region of CXCL5, perhaps because its N-terminal region is a few amino acid residues longer than in other chemokines. CatB is different from other cysteine cathepsins in that it acts as a dipeptidyl carboxypeptidase at acidic pH, due to the presence of occluding loop in its structure, and as an endopeptidase at neutral pH (51). The removal of two amino acid residues from the C terminus, which we observed in CXCL8–10, suggests that CatB may function as a carboxydipeptidase also at neutral pH. Consistent with this observation, CatB processing of chemokines was almost completely abolished in the presence of GAGs, which bind chemokines via basic residues. In vivo chemokines bind to GAGs to form haptotactic gradients and oligomers (52). Similarly, GAGs are shown to facilitate autocatalytic activation of cathepsins (46, 53, 54), stabilize the structure of mature proteases (55), and potentiate their activity (47). We propose that GAGs sterically hinder the C-terminal interaction between CatB and chemokines. In addition, GAG binding was found to delay or even prevent further processing of the intermediates, suggesting that this binding either stabilized the structure of the intermediates and/or prevented secondary cleavages in the C-terminal region. In contrast to the effects of GAGs on Cat B activity, our results show that processing by CatL and CatS in the N-terminal part of chemokines was preserved in the presence of GAGs, suggesting that GAGs are less likely to interact with this region of chemokines. The collagenase activity of CatK depends on its association with GAGs, including C4S (56, 57). Herein, we show a profound effect of C4S and C6S on the activity of CatK, CatL, and CatS on small substrates at neutral and slightly acidic pH, presumably because of the increased cathepsin stability. Although cathepsin activity in the presence of GAGs was increased, the truncated forms of chemokines were generally more abundant, suggesting that GAGs restricted processing. This is different from processing by MMPs, which does not appear to be affected by the presence of GAGs (4, 10, 15).
CatK differed from CatL and CatS, as it was the only cathepsin with increased processing of chemokines CXCL1, -2, -3, -5, and -8 in the presence of GAGs. This seems to be a direct effect of GAGs on CatK proteolytic activity and not just on its structural stability. Similar to the acquisition of the collagenolytic activity by CatK, the increased processing of chemokines may be explained by the formation of the oligomeric CatK-GAG complex. This effect is mediated by both C4S and C6S, unlike the collagenolytic activity of the CatK-GAG oligomers, which is mediated primarily by C4S and to a lesser extent by some other GAGs including C6S (56, 57). However, at higher GAG concentrations, chemokine processing by CatL, CatS, and even CatK was further decreased, suggesting that cathepsin and chemokine molecules become spatially separated by association to different GAG chains. Because both chemokines and cathepsins associate with GAGs in vivo, these observations suggest that the availability of binding sites on GAG molecules can determine the extent of proteolysis.
Another factor that may influence chemokine processing by cathepsins is the pH. Although cysteine cathepsins as lysosomal enzymes have adapted to acidic pH, they remain active also at neutral pH but only for a limited amount of time because of structural instability and oxidative stress. CatS is an exception and, as the most stable cathepsin, remains active under these conditions for several hours (58). The pH in the pericellular and extracellular region of inflamed tissues and also in the tumor microenvironment can be slightly acidic (∼pH 6.5), which would improve the structural stability of cysteine cathepsins and thereby prolong their activity (29). The combination of GAGs and decreased pH is therefore of physiological importance for the prolonged activity of cysteine cathepsins.
There is increasing evidence that cathepsins are linked with the physiological processing of chemokines. One such example are the N-terminally truncated forms of CXCL1–3 with increased chemotactic activity for neutrophil granulocytes, which have been isolated from cultured peripheral blood monocytes; however, the enzymes generating them have been only partially identified. DPPIV/CD26 can generate only (3–73)-fragments (8, 59), whereas MMP1, -9, -12, and -25 can generate CXCL2-(5–73) (13, 14). In contrast, MMP12 inactivates CXCL1–3 by cleaving these chemokines at the E↓LR and inactivates all other CXCL chemokines by cleavage C-terminal to the ELR motif (14). The functional consequence of the absence of inactivating cleavages by MMP12 in the Mmp12−/− mouse is readily apparent in vivo as a highly exaggerated neutrophil influx in collagen-induced arthritis (60). Because CatK, CatL, and CatS can generate truncated CXCL1-(5–72), CXCL2-(5–73), and CXCL3-(5–73, 6–73) forms with increased chemotactic activity, we hypothesized that cathepsins are involved in in vivo processing of these chemokines. Although CatL was previously identified as a protease that processes CXCL8 at the N terminus to generate forms with elevated activity (33), we show here that CatK and CatS can also generate more active CXCL8-(6–77, 8–77) forms cleaving N-terminal to the ELR motif. Similarly, CatK, CatL, CatS, and CatB truncated CXCL5 to generate more active CXCL5-(5–78, 9–78) forms, as confirmed by calcium mobilization assay. All of these chemokines contain the ELR motif and can attract neutrophil granulocytes and promote angiogenesis (8, 61).
The four non-ELR CXC chemokines tested were all inactivated and/or degraded by cathepsins. In the presence of GAGs, CXCL10 was processed to the CXC10-(5–77) form. We have demonstrated with calcium mobilization and chemotaxis assays that this cleavage inactivates the chemokine; this response is similar to that observed following N-terminal truncation of CXCL11 (4, 19). CD26/DPPIV-truncated CXCL9, CXCL10, and CXCL11, which lack the first two amino acids at the N terminus, are impaired in receptor signaling and lymphocyte chemotaxis but retain angiostatic activity (24). Further processing of the N terminus of CXCL11 was shown to abolish the angiostatic activity (19), which would also apply to CXCL10-(5–77) and CXCL11-(5–73, 6–73) generated by cysteine cathepsins. All ELR CXC chemokines are angiogenic, whereas non-ELR CXCL9, CXCL10, and CXCL11 inhibit angiogenesis. Differential processing of ELR and non-ELR CXC chemokines by cysteine cathepsins suggests different functional consequences (61). Moreover, ELR CXC chemokines and angiostatic non-ELR chemokines are classified as inflammatory chemokines, in which expression is inducible and up-regulated by inflammatory stimuli. In contrast, CXCL12 is a homeostatic chemokine and is expressed constitutively. Although a non-ELR chemokine, CXCL12 is unique here in that it has angiogenic activity; it acts through the CXCR4 receptor, whereas angiostatic CXCL9, CXCL10, and CXCL11 act through CXCR3 (1). Unlike inflammatory chemokines, homeostatic CXCL12 was degraded by all four cathepsins, which extends the relevance of cysteine cathepsins during inflammation.
It is now well established that extracellular cysteine cathepsins contribute to tissue destruction under arthritic conditions, to tumor cell invasion, and to angiogenesis during tumor progression (29, 30, 62, 63). Inflammatory mediators including TNFα, IL-1β, IL-6, IFNγ, and IL-4 rapidly up-regulate cathepsin expression and induce their secretion in many cell types (64–69). Therefore, it is likely that under inflammatory conditions both chemokines and cathepsins will co-localize in the extracellular space, providing an opportunity for processing, as described herein, to occur.
Differential processing of ELR and non-ELR chemokines by cysteine cathepsins suggests another role for these proteases during inflammation. The complexity of an inflammatory response, including the abundance of active cysteine cathepsins and chemokines, the nature and the abundance of GAGs, the abundance of cathepsin inhibitors, and the pH in the microenvironment, makes it difficult to fully integrate the potential roles of cathepsins in inflammation. However, their potent but short-lived proteolytic activity at neutral or slightly acidic pH is suitable for the regulation of rapidly occurring processes such as infiltration of inflammatory cells. Herein we have shown roles for cathepsins in generating chemokine forms that attract neutrophil granulocytes and promote angiogenesis and also in inactivating and degrading chemokines that would attract T-cells and are angiostatic (Fig. 8). Cysteine cathepsins thereby complement the chemokine modulatory activity of other proteases such as MMPs, DPPIV/CD26, neutrophil cathepsin G, and elastase (21).
FIGURE 8.

Cysteine cathepsins promote signaling by ELR CXC chemokines and terminate signaling by non-ELR CXC chemokines. Under inflammatory conditions, cysteine cathepsins are secreted extracellularly, where they encounter and process chemokines. The source of cysteine cathepsins can be tissue-resident cells, such as fibroblasts or smooth muscle cells, or/and tissue-infiltrating cells, such as macrophages and neutrophil granulocytes. Our results show that cysteine cathepsins N-terminally truncate ELR CXC chemokines to produce fragments with increased activity, whereas they inactivate or degrade non-ELR chemokines. By chemokine processing, cysteine cathepsins could promote recruitment of neutrophil granulocytes and angiogenesis.
Supplementary Material
Acknowledgments
We thank Gregor Kosec, Marko Mihelič, Matej Vizovišek, Mojca Prebanda, and Urska Požgan for preparing recombinant cysteine cathepsins.
Note Added in Proof
The legend to Fig. 6 was missing in the version of this article that was published as a Paper in Press on April 1, 2015. The correct version including the legend to Fig. 6 is now shown.
This work was supported by Grants P1-0140, J1-6739, and N1-0022 from the Slovene Research Agency (to B. T.). This research was also supported by a Canadian Institutes of Health Research (CIHR) grant and an Infrastructure Grant from the Michael Smith Research Foundation (University of British Columbia Centre for Blood Research) and the British Columbia Proteomics Network.

This article contains supplemental Fig. 1.
- GAG
- glycosaminoglycan
- MMP
- matrix metalloproteinase
- DPPIV
- dipeptidylpeptidase IV
- Cat
- cathepsin
- C6S
- chondroitin-6-sulfate
- t-Boc
- tertiary butyloxycarbonyl
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Vandercappellen J., Van Damme J., Struyf S. (2008) The role of CXC chemokines and their receptors in cancer. Cancer 267, 226–244 [DOI] [PubMed] [Google Scholar]
- 2. Proudfoot A. E. (2006) The biological relevance of chemokine-proteoglycan interactions. Biochem. Soc. Trans. 34, 422–426 [DOI] [PubMed] [Google Scholar]
- 3. Salanga C. L., Handel T. M. (2011) Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp. Cell Res. 317, 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cox J. H., Dean R. A., Roberts C. R., Overall C. M. (2008) Matrix metalloproteinase processing of CXCL11/I-TAC results in loss of chemoattractant activity and altered glycosaminoglycan binding. J. Biol. Chem. 283, 19389–19399 [DOI] [PubMed] [Google Scholar]
- 5. McQuibban G. A., Butler G. S., Gong J. H., Bendall L., Power C., Clark-Lewis I., Overall C. M. (2001) Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J. Biol. Chem. 276, 43503–43508 [DOI] [PubMed] [Google Scholar]
- 6. Rajagopalan L., Rajarathnam K. (2006) Structural basis of chemokine receptor function: a model for binding affinity and ligand selectivity. Biosci. Rep. 26, 325–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Overall C. M., Blobel C. P. (2007) In search of partners: linking extracellular proteases to substrates. Nat. Rev. Mol. Cell Biol. 8, 245–257 [DOI] [PubMed] [Google Scholar]
- 8. Mortier A., Van Damme J., Proost P. (2008) Regulation of chemokine activity by posttranslational modification. Pharmacol. Ther. 120, 197–217 [DOI] [PubMed] [Google Scholar]
- 9. Mortier A., Gouwy M., Van Damme J., Proost P. (2011) Effect of posttranslational processing on the in vitro and in vivo activity of chemokines. Exp. Cell Res. 317, 642–654 [DOI] [PubMed] [Google Scholar]
- 10. McQuibban G. A., Gong J. H., Wong J. P., Wallace J. L., Clark-Lewis I., Overall C. M. (2002) Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 100, 1160–1167 [PubMed] [Google Scholar]
- 11. McQuibban G. A., Gong J. H., Tam E. M., McCulloch C. A., Clark-Lewis I., Overall C. M. (2000) Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 289, 1202–1206 [DOI] [PubMed] [Google Scholar]
- 12. Zhang K., McQuibban G. A., Silva C., Butler G. S., Johnston J. B., Holden J., Clark-Lewis I., Overall C. M., Power C. (2003) HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci 6, 1064–1071 [DOI] [PubMed] [Google Scholar]
- 13. Starr A. E., Bellac C. L., Dufour A., Goebeler V., Overall C. M. (2012) Biochemical characterization and N-terminomics analysis of leukolysin, the membrane-type 6 matrix metalloprotease (MMP25): chemokine and vimentin cleavages enhance cell migration and macrophage phagocytic activities. J. Biol. Chem. 287, 13382–13395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dean R. A., Cox J. H., Bellac C. L., Doucet A., Starr A. E., Overall C. M. (2008) Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 112, 3455–3464 [DOI] [PubMed] [Google Scholar]
- 15. Starr A. E., Dufour A., Maier J., Overall C. M. (2012) Biochemical analysis of matrix metalloproteinase activation of chemokines CCL15 and CCL23 and increased glycosaminoglycan binding of CCL16. J. Biol. Chem. 287, 5848–5860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tester A. M., Cox J. H., Connor A. R., Starr A. E., Dean R. A., Puente X. S., López-Otín C., Overall C. M. (2007) LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS One 2, e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dean R. A., Overall C. M. (2007) Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell. Proteomics 6, 611–623 [DOI] [PubMed] [Google Scholar]
- 18. Wuyts A., Govaerts C., Struyf S., Lenaerts J. P., Put W., Conings R., Proost P., Van Damme J. (1999) Isolation of the CXC chemokines ENA-78, GROα and GROγ from tumor cells and leukocytes reveals NH2-terminal heterogeneity: functional comparison of different natural isoforms. Eur. J. Biochem. 260, 421–429 [DOI] [PubMed] [Google Scholar]
- 19. Proost P., Mortier A., Loos T., Vandercappellen J., Gouwy M., Ronsse I., Schutyser E., Put W., Parmentier M., Struyf S., Van Damme J. (2007) Proteolytic processing of CXCL11 by CD13/aminopeptidase N impairs CXCR3 and CXCR7 binding and signaling and reduces lymphocyte and endothelial cell migration. Blood 110, 37–44 [DOI] [PubMed] [Google Scholar]
- 20. King A. G., Johanson K., Frey C. L., DeMarsh P. L., White J. R., McDevitt P., McNulty D., Balcarek J., Jonak Z. L., Bhatnagar P. K., Pelus L. M. (2000) Identification of unique truncated KC/GROβ chemokines with potent hematopoietic and anti-infective activities. J. Immunol. 164, 3774–3782 [DOI] [PubMed] [Google Scholar]
- 21. Fortelny N., Cox J. H., Kappelhoff R., Starr A. E., Lange P. F., Pavlidis P., Overall C. M. (2014) Network analyses reveal pervasive functional regulation between proteases in the human protease web. PLoS Biol. 12, e1001869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nufer O., Corbett M., Walz A. (1999) Amino-terminal processing of chemokine ENA-78 regulates biological activity. Biochemistry 38, 636–642 [DOI] [PubMed] [Google Scholar]
- 23. Van den Steen P. E., Proost P., Wuyts A., Van Damme J., Opdenakker G. (2000) Neutrophil gelatinase B potentiates interleukin-8 tenfold by amino-terminal processing, whereas it degrades CTAP-III, PF-4, and GRO-α and leaves RANTES and MCP-2 intact. Blood 96, 2673–2681 [PubMed] [Google Scholar]
- 24. Proost P., Schutyser E., Menten P., Struyf S., Wuyts A., Opdenakker G., Detheux M., Parmentier M., Durinx C., Lambeir A. M., Neyts J., Liekens S., Maudgal P. C., Billiau A., Van Damme J. (2001) Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood 98, 3554–3561 [DOI] [PubMed] [Google Scholar]
- 25. Ludwig A., Schiemann F., Mentlein R., Lindner B., Brandt E. (2002) Dipeptidyl peptidase IV (CD26) on T cells cleaves the CXC chemokine CXCL11 (I-TAC) and abolishes the stimulating but not the desensitizing potential of the chemokine. J. Leukoc. Biol. 72, 183–191 [PubMed] [Google Scholar]
- 26. Turk V., Stoka V., Vasiljeva O., Renko M., Sun T., Turk B., Turk D. (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta 1824, 68–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Turk B., Dolenc I., Lenarčič B., Križaj I., Turk V., Bieth J. G., Björk I. (1999) Acidic pH as a physiological regulator of human cathepsin L activity. Eur. J. Biochem. 259, 926–932 [DOI] [PubMed] [Google Scholar]
- 28. Turk B., Turk V. (2009) Lysosomes as “suicide bags” in cell death: myth or reality? J. Biol. Chem. 284, 21783–21787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bromme D., Wilson S. (2011) Role of cysteine cathepsins in extracellular proteolysis, in Extracellular Matrix Degradation (Parks W. C., Mecham R. P., eds.) pp. 23–51, Springer-Verlag, Berlin Heidelberg [Google Scholar]
- 30. Fonović M., Turk B. (2014) Cysteine cathepsins and extracellular matrix degradation. Biochim. Biophys. Acta 1840, 2560–2570 [DOI] [PubMed] [Google Scholar]
- 31. Mohamed M. M., Sloane B. F. (2006) Cysteine cathepsins: multifunctional enzymes in cancer. Nat. Rev. Cancer 6, 764–775 [DOI] [PubMed] [Google Scholar]
- 32. Turk B., Turk D., Turk V. (2012) Protease signalling: the cutting edge. EMBO J. 31, 1630–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohashi K., Naruto M., Nakaki T., Sano E. (2003) Identification of interleukin-8 converting enzyme as cathepsin L. Biochim. Biophys. Acta 1649, 30–39 [DOI] [PubMed] [Google Scholar]
- 34. Hasan L., Mazzucchelli L., Liebi M., Lis M., Hunger R. E., Tester A., Overall C. M., Wolf M. (2006) Function of liver activation-regulated chemokine/CC chemokine ligand 20 is differently affected by cathepsin B and cathepsin D processing. J. Immunol. 176, 6512–6522 [DOI] [PubMed] [Google Scholar]
- 35. Clark A. K., Yip P. K., Malcangio M. (2009) The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J. Neurosci. 29, 6945–6954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clark A. K., Grist J., Al-Kashi A., Perretti M., Malcangio M. (2012) Spinal cathepsin S and fractalkine contribute to chronic pain in the collagen-induced arthritis model. Arthritis Rheum. 64, 2038–2047 [DOI] [PubMed] [Google Scholar]
- 37. Jones B. A., Riegsecker S., Rahman A., Beamer M., Aboualaiwi W., Khuder S. A., Ahmed S. (2013) Role of ADAM-17, p38 MAPK, cathepsins, and the proteasome pathway in the synthesis and shedding of fractalkine/CX(3) CL1 in rheumatoid arthritis. Arthritis Rheum. 65, 2814–2825 [DOI] [PubMed] [Google Scholar]
- 38. Staudt N. D., Maurer A., Spring B., Kalbacher H., Aicher W. K., Klein G. (2012) Processing of CXCL12 by different osteoblast-secreted cathepsins. Stem Cells Dev. 21, 1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clark-Lewis I., Vo L., Owen P., Anderson J. (1997) Chemical synthesis, purification, and folding of C-X-C and C-C chemokines. Methods Enzymol. 287, 233–250 [DOI] [PubMed] [Google Scholar]
- 40. Rozman J., Stojan J., Kuhelj R., Turk V., Turk B. (1999) Autocatalytic processing of recombinant human procathepsin B is a bimolecular process. FEBS Lett. 459, 358–362 [DOI] [PubMed] [Google Scholar]
- 41. Brömme D., Nallaseth F. S., Turk B. (2004) Production and activation of recombinant papain-like cysteine proteases. Methods 32, 199–206 [DOI] [PubMed] [Google Scholar]
- 42. Mihelič M., Doberšek A., Gunčar G., Turk D. (2008) Inhibitory fragment from the p41 form of invariant chain can regulate activity of cysteine cathepsins in antigen presentation. J. Biol. Chem. 283, 14453–14460 [DOI] [PubMed] [Google Scholar]
- 43. Starr A. E., Overall C. M. (2009) Chapter 13. Characterizing proteolytic processing of chemokines by mass spectrometry, biochemistry, neo-epitope antibodies and functional assays. Methods Enzymol. 461, 281–307 [DOI] [PubMed] [Google Scholar]
- 44. Loetscher M., Gerber B., Loetscher P., Jones S. A., Piali L., Clark-Lewis I., Baggiolini M., Moser B. (1996) Chemokine receptor specific for IP10 and Mig: structure, function, and expression in activated T-lymphocytes. J. Exp. Med. 184, 963–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Turk D., Gunčar G., Podobnik M., Turk B. (1998) Revised definition of substrate binding sites of papain-like cysteine proteases. Biol. Chem. 379, 137–147 [DOI] [PubMed] [Google Scholar]
- 46. Caglič D., Rozman Pungerčar J., Pejler G., Turk V., Turk B. (2007) Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J. Biol. Chem. 282, 33076–33085 [DOI] [PubMed] [Google Scholar]
- 47. Almeida P. C., Nantes I. L., Chagas J. R., Rizzi C. C., Faljoni-Alario A., Carmona E., Juliano L., Nader H. B., Tersariol I. L. (2001) Cathepsin B activity regulation: heparin-like glycosaminogylcans protect human cathepsin B from alkaline pH-induced inactivation. J. Biol. Chem. 276, 944–951 [DOI] [PubMed] [Google Scholar]
- 48. Ahuja S. K., Murphy P. M. (1996) The CXC chemokines growth-regulated oncogene (GRO) α, GROβ, GROγ, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J. Biol. Chem. 271, 20545–20550 [DOI] [PubMed] [Google Scholar]
- 49. Vasiljeva O., Reinheckel T., Peters C., Turk D., Turk V., Turk B. (2007) Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr. Pharm. Des. 13, 387–403 [DOI] [PubMed] [Google Scholar]
- 50. Biniossek M. L., Nägler D. K., Becker-Pauly C., Schilling O. (2011) Proteomic identification of protease cleavage sites characterizes prime and non-prime specificity of cysteine cathepsins B, L, and S. J. Proteome Res. 10, 5363–5373 [DOI] [PubMed] [Google Scholar]
- 51. Illy C., Quraishi O., Wang J., Purisima E., Vernet T., Mort J. S. (1997) Role of the occluding loop in cathepsin B activity. J. Biol. Chem. 272, 1197–1202 [DOI] [PubMed] [Google Scholar]
- 52. Proudfoot A. E., Handel T. M., Johnson Z., Lau E. K., LiWang P., Clark-Lewis I., Borlat F., Wells T. N., Kosco-Vilbois M. H. (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U.S.A. 100, 1885–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ishidoh K., Kominami E. (1995) Procathepsin L degrades extracellular matrix proteins in the presence of glycosaminoglycans in vitro. Biochem. Biophys. Res. Commun. 217, 624–631 [DOI] [PubMed] [Google Scholar]
- 54. Vasiljeva O., Dolinar M., Rozman Pungerčar J., Turk V., Turk B. (2005) Recombinant human procathepsin S is capable of autocatalytic processing at neutral pH in the presence of glycosaminoglycans. FEBS Lett. 579, 1285–1290 [DOI] [PubMed] [Google Scholar]
- 55. Almeida P. C., Nantes I. L., Rizzi C. C., Júdice W. A., Chagas J. R., Juliano L., Nader H. B., Tersariol I. L. (1999) Cysteine proteinase activity regulation: a possible role of heparin and heparin-like glycosaminoglycans. J. Biol. Chem. 274, 30433–30438 [DOI] [PubMed] [Google Scholar]
- 56. Li Z., Hou W. S., Escalante-Torres C. R., Gelb B. D., Bromme D. (2002) Collagenase activity of cathepsin K depends on complex formation with chondroitin sulfate. J. Biol. Chem. 277, 28669–28676 [DOI] [PubMed] [Google Scholar]
- 57. Li Z., Yasuda Y., Li W., Bogyo M., Katz N., Gordon R. E., Fields G. B., Brömme D. (2004) Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J. Biol. Chem. 279, 5470–5479 [DOI] [PubMed] [Google Scholar]
- 58. Kirschke H., Wiederanders B., Brömme D., Rinne A. (1989) Cathepsin S from bovine spleen: purification, distribution, intracellular localization and action on proteins. Biochem. J. 264, 467–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ajami K., Pitman M. R., Wilson C. H., Park J., Menz R. I., Starr A. E., Cox J. H., Abbott C. A., Overall C. M., Gorrell M. D. (2008) Stromal cell-derived factors 1α and 1β, inflammatory protein-10 and interferon-inducible T cell chemo-attractant are novel substrates of dipeptidyl peptidase 8. FEBS Lett. 582, 819–825 [DOI] [PubMed] [Google Scholar]
- 60. Bellac C. L., Dufour A., Krisinger M. J., Loonchanta A., Starr A. E., Auf dem Keller U., Lange P. F., Goebeler V., Kappelhoff R., Butler G. S., Burtnick L. D., Conway E. M., Roberts C. R., Overall C. M. (2014) Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep. 9, 618–632 [DOI] [PubMed] [Google Scholar]
- 61. Belperio J. A., Keane M. P., Arenberg D. A., Addison C. L., Ehlert J. E., Burdick M. D., Strieter R. M. (2000) CXC chemokines in angiogenesis. J Leukoc. Biol. 68, 1–8 [PubMed] [Google Scholar]
- 62. Gocheva V., Joyce J. A. (2007) Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 6, 60–64 [DOI] [PubMed] [Google Scholar]
- 63. Vasiljeva O., Turk B. (2008) Dual contrasting roles of cysteine cathepsins in cancer progression: apoptosis versus tumour invasion. Biochimie 90, 380–386 [DOI] [PubMed] [Google Scholar]
- 64. Mohamed M. M., Cavallo-Medved D., Rudy D., Anbalagan A., Moin K., Sloane B. F. (2010) Interleukin-6 increases expression and secretion of cathepsin B by breast tumor-associated monocytes. Cell. Physiol. Biochem. 25, 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gocheva V., Wang H. W., Gadea B. B., Shree T., Hunter K. E., Garfall A. L., Berman T., Joyce J. A. (2010) IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 24, 241–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lemaire R., Huet G., Zerimech F., Grard G., Fontaine C., Duquesnoy B., Flipo R. M. (1997) Selective induction of the secretion of cathepsins B and L by cytokines in synovial fibroblast-like cells. Br. J. Rheumatol. 36, 735–743 [DOI] [PubMed] [Google Scholar]
- 67. Hou W. S., Li W., Keyszer G., Weber E., Levy R., Klein M. J., Gravallese E. M., Goldring S. R., Brömme D. (2002) Comparison of cathepsins K and S expression within the rheumatoid and osteoarthritic synovium. Arthritis Rheum. 46, 663–674 [DOI] [PubMed] [Google Scholar]
- 68. Caglič D., Globisch A., Kindermann M., Lim N. H., Jeske V., Juretschke H. P., Bartnik E., Weithmann K. U., Nagase H., Turk B., Wendt K. U. (2011) Functional in vivo imaging of cysteine cathepsin activity in murine model of inflammation. Bioorg. Med. Chem. 19, 1055–1061 [DOI] [PubMed] [Google Scholar]
- 69. Caglič D., Repnik U., Jedeszko C., Kosec G., Miniejew C., Kindermann M., Vasiljeva O., Turk V., Wendt K. U., Sloane B. F., Goldring M. B., Turk B. (2013) The proinflammatory cytokines interleukin-1alpha and tumor necrosis factor α promote the expression and secretion of proteolytically active cathepsin S from human chondrocytes. Biol. Chem. 394, 307–316 [DOI] [PubMed] [Google Scholar]
- 70. Walz A., Dewald B., von Tscharner V., Baggiolini M. (1989) Effects of the neutrophil-activating peptide NAP-2, platelet basic protein, connective tissue-activating peptide III, and platelet factor 4 on human neutrophils. J. Exp. Med. 170, 1745–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.