

Background: MG53, a zinc finger protein, is essential to cell membrane repair. It is not known whether zinc contributes to MG53-mediated membrane repair.
Results: Chelation of Zn2+ or mutation of Zn2+-binding motifs in MG53 affects membrane repair.
Conclusion: Zn2+ binding to MG53 is required for membrane repair.
Significance: This study establishes a base for Zn2+ interaction with MG53 in protection against injury to the cell membrane.
Keywords: E3 ubiquitin ligase, redox regulation, signaling, trafficking, vesicles
Abstract
Zinc is an essential trace element that participates in a wide range of biological functions, including wound healing. Although Zn2+ deficiency has been linked to compromised wound healing and tissue repair in human diseases, the molecular mechanisms underlying Zn2+-mediated tissue repair remain unknown. Our previous studies established that MG53, a TRIM (tripartite motif) family protein, is an essential component of the cell membrane repair machinery. Domain homology analysis revealed that MG53 contains two Zn2+-binding motifs. Here, we show that Zn2+ binding to MG53 is indispensable to assembly of the cell membrane repair machinery. Live cell imaging illustrated that Zn2+ entry from extracellular space is essential for translocation of MG53-containing vesicles to the acute membrane injury sites for formation of a repair patch. The effect of Zn2+ on membrane repair is abolished in mg53−/− muscle fibers, suggesting that MG53 functions as a potential target for Zn2+ during membrane repair. Mutagenesis studies suggested that both RING and B-box motifs of MG53 constitute Zn2+-binding domains that contribute to MG53-mediated membrane repair. Overall, this study establishes a base for Zn2+ interaction with MG53 in protection against injury to the cell membrane.
Introduction
Zinc is an essential element for normal physiology (1), and nutritional Zn2+ deficiency and lack of bioavailability are associated with growth failure, dermatitis, impaired immunity, and delayed wound healing (2–4). One of the examples of impaired wound healing is acrodermatitis enteropathica, caused by reduced Zn2+ uptake in the small intestine and reduced serum Zn2+ levels (2, 3, 5). Surgical patients undergoing total hip replacement procedures have impaired wound healing that correlates with lower levels of serum Zn2+ (6, 7). Zn2+ deficiency also accompanies aging and a number of other pathophysiological conditions, particularly those linked to oxidative stress (8).
Intracellular Zn2+ levels are tightly controlled by transporters and ion channels. Roughly 10% of the structures deposited in the Protein Data Bank have Zn2+ listed in their structure indexes (9, 10). Inside the cell, Zn2+ is bound to numerous structural and regulatory proteins, including transcription factors (11), regulators of hematopoietic stem cells (12) and immune cells (13), and proteins involved in intracellular signaling and neurotransmission (14). As such, metabolically active, labile Zn2+ is present inside cells at pico- to nanomolar concentrations, with extracellular concentrations in the sub-micromolar to micromolar range (15–17). Increased oxidative stress can release Zn2+ from its binding sites, executing the task of the so-called “redox Zn2+ switch” in performing its corresponding biological functions (18).
Dynamic membrane repair is a fundamental process in maintaining cellular integrity. Defective membrane repair is linked to compromised wound healing, muscular dystrophy, and cardiovascular diseases (19–23). We discovered that MG53, a TRIM (tripartite motif) family protein, is an essential component of the membrane repair machinery (24). MG53 acts as a sensor of oxidation to nucleate recruitment of intracellular vesicles to the injury site for membrane patch formation. MG53 ablation results in defective membrane repair, with progressive pathological consequences to skeletal and cardiac muscles (24–27).
Domain homology analysis showed that MG53 contains two Zn2+-binding domains in the RING finger and B-box motifs (24), but whether MG53 binds with Zn2+ to regulate membrane repair is unknown. Here, we present evidence that Zn2+ binding to MG53 is indispensable for repair of cell membrane injury. Chelation of extracellular Zn2+ impacts the movement of intracellular vesicles to the acute membrane injury sites in a MG53-dependent manner. Defective membrane repair was observed in the absence of extracellular Zn2+ or in response to disruption of Zn2+-binding motifs in MG53. Our data suggest that MG53 serves as an acceptor for Zn2+ during cell membrane repair and provide mechanic insight in the biology of Zn2+ in wound healing and regenerative medicine.
Experimental Procedures
Plasmid Construction
Cloning and construction of MG53 expression plasmids were performed as described previously (24, 28). The various MG53 mutants (C29L, H31A, C53A/C55A/C56A, C86A, C105S, and C29L/C105S) were constructed by replacing the appropriate residues in GFP-MG53 using the method described previously (29). The coding sequence of WT MG53 was cloned into the pMAL-p2 vector (New England Biolabs) for expression of the recombinant maltose-binding protein (MBP)4-MG53 fusion protein in Escherichia coli (30). The coding sequences of mutants C29L and C29L/C105S were cloned into the pMAL-p2 vector in the same manner to yield plasmids pMAL-C29L and pMAL-C29L/C105S for production of the MBP-MG53(C29L) and MBP-MG53(C29L/C105S) fusion proteins using E. coli fermentation. All plasmids were confirmed by sequencing.
Flexor Digitorum Brevis (FDB) Muscle Fiber Isolation and Membrane Repair Assay
All animal care and usage followed National Institutes of Health guidelines and was approved by the institutional animal care and use committees of Rutgers University and The Ohio State University. Mice null for MG53 (mg53−/−) or age-matched WT control animals were produced as described previously (24). FDB muscle fiber isolation was performed with a previously published protocol (24, 30, 31). Membrane repair capacity in the FDB muscle was determined using an established technique (32, 33). Briefly, prior to cell wounding, 2.5 μm FM 1-43 (Life Technologies, Inc.) was added to Tyrode's solution containing 140 mm NaCl, 5 mm KCl, 2.5 mm CaCl2, 2 mm MgCl2, and 10 mm HEPES (pH 7.2), and membranes in FDB fibers were damaged using an UV laser on the stage of a Zeiss LSM 510 confocal microscope (Enterprise) to irradiate a 5 × 5 pixel area at a maximum power for 5 s (80 milliwatts, 351/364 nm). Confocal images were acquired (x-y images captured at 6.6-s intervals) to monitor FM 1-43 dye entry at the irradiation site. The mean fluorescence intensity at the irradiation site was calculated based on the following equation: ΔF/F0 = (Ft − F0)/F0, where F0 is the fluorescence intensity at the initial time point, and Ft is the fluorescence intensity at the various time points afterward. In some experiments, the fibers were preincubated with a cell-impermeable cation chelator (Ca-EDTA, Sigma), a Zn2+ ionophore (1-hydroxypyridine-2-thione zinc salt (Zn-HPT), Sigma), or a high affinity Zn2+ chelator (tetrakis(2-pyridylmethyl)enediamine (TPEN), Sigma) for 5 min at room temperature to analyze the Zn2+ effect on muscle membrane repair. Although the free Zn2+ concentration in Tyrode's solution ranges from 1 to 5 μm, the addition of 40 μm Ca-EDTA or 40 μm TPEN essentially chelates all Zn2+ ions.
Microelectrode Cell Wounding and Confocal Imaging
Live cell confocal imaging was used to monitor intracellular trafficking of GFP-MG53 transiently expressed in C2C12 myoblast cells (24). C2C12 cells were plated onto ΔT glass-bottom dishes (Bioptechs Inc.) and visualized using a Radiance 2100 laser scanning confocal microscope (Bio-Rad) with a 40× (1.3 numerical aperture) oil immersion objective at 24 h after transfection with the various GFP-MG53 fusion constructs. For mechanical membrane damage, C2C12 myoblasts were penetrated by the tip of a micropipette attached to a micromanipulator, and the movements of GFP-MG53 were monitored following our published protocols (24, 34).
Lactate Dehydrogenase (LDH) Release Measurements following Glass Microbead-induced Cell Membrane Injury
C2C12 myoblasts were transfected with GFP-MG53, GFP-MG53(C29L), GFP-MG53(C105S), or GFP-MG53(C29L/C105S) and allowed to differentiate into myotubes for 3 days. Cells were subjected to glass microbead-induced mechanical damage as described previously (35, 36). Measurement of LDH enzymatic activity released from C2C12 myotubes following glass microbead damage was used as an index of membrane injury. LDH activity was measured with a LDH cytotoxicity detection kit (Thermo Scientific) under basal conditions (without glass beads), at 7 min after glass bead-induced injury, or after treatment with 1% Triton X-100 to induce total cell lysis.
Purification of Recombinant MG53 and MBP-MG53 Proteins
Plasmid pMAL-MG53, pMAL-C29L, or pMAL-C29L/C105S plasmid was transformed into JM109 bacterial cells. Expression of MBP-MG53 and its mutant proteins was induced with 0.3 mm isopropyl β-d-thiogalactopyranoside (Sigma) for 3.5 h at 30 °C in bacterial cultures at log phase growth. Harvested bacterial pellets were resuspended in buffer containing 20 mm Tris-HCl, 200 mm NaCl, 100 μm ZnSO4, and 0.2 mm PMSF (pH 7.5) supplemented with 0.5% protease inhibitor mixture (Sigma), 0.2 mg/ml lysozyme, and 1 mg/ml MgCl2 and sonicated (Branson Ultrasonics) for 2 min at 30% power output. The lysates were centrifuged twice at 15,000 rpm for 30 min at 4 °C and filtered through a 0.45-μm syringe filter. The filtrates were then diluted 1:4 into the suspension buffer, mixed with amylase resin beads (1.25% final bead volume; New England Biolabs), and incubated overnight at 4 °C on an orbital shaker. The mixture was loaded onto a Poly-Prep chromatography column (Bio-Rad), allowed to precipitate by gravity, and washed with 50 ml of zinc-free column buffer (20 mm Tris-HCl, 200 mm NaCl, and 0.2 mm PMSF (pH 7.5)). MBP-MG53, MBP-MG53(C29L), or MBP-MG53(C29L/C105S) was eluted with 10 mm maltose from the bound amylase resin beads and resuspended in 2 ml of zinc-free column buffer. The final protein concentration was measured with the DC protein assay (Bio-Rad) and verified by SDS-PAGE and colloidal blue staining (Invitrogen). Recombinant human MG53 (rhMG53) was purified from E. coli following our established protocol (30).
Zn2+ Binding Assessment
Zn2+ contents in the recombinant MBP-MG53 proteins were measured by 6-methoxy-(8-p-toluenesulfonamido)quinolone (TSQ) fluorescence assay according to our previous protocol (37). In brief, the Zn2+-specific fluorescent probe TSQ (Life Technologies, Inc.) was added to 2–10 μm MBP-MG53 fusion proteins bound on the amylose resin beads in phosphate-buffered saline to a final concentration of 10 μm. After removal of the unbound TSQ probe, the thiol-bound Zn2+ contents of the MBP-MG53 fusion proteins on the beads were analyzed with the thiol-reactive reagent p-hydroxymercuriphenylsulfonate (Sigma) for 10 min at room temperature to liberate the intramolecular Zn2+ content. Stained beads were excited at 334 nm, and fluorescence emission was recorded at 465 nm with an Axiovert 200M motorized fluorescence microscope (Zeiss) equipped with a mercury lamp. A standard Zn2+ solution (Sigma) was used to calibrate the assay system. The quantification of relative Zn2+ content was performed with MetaMorph imaging analysis software (Molecular Devices).
Western Blotting
C2C12 cells expressing the different GFP-MG53 proteins were harvested and lysed with ice-cold modified radioimmune precipitation assay buffer (150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40, and 20 mm Tris-HCl (pH 7.5)) supplemented with protease inhibitor mixture. The total protein lysates (10 μg each) were separated on 4–12% SDS-polyacrylamide gradient gels (Invitrogen). Proteins were transferred onto PVDF membrane (Millipore) and probed with a custom-made rabbit anti-MG53 polyclonal antibody as described previously (24), and detection was conducted with an ECL Plus kit (Thermo Scientific). To assay the effect of chelating Zn2+ concentration on the redox-dependent oligomerization of MG53, water-dissolved rhMG53 protein (in lyophilization buffer containing 3% mannitol, 1% sucrose, 0.005% Tween 80, and 5 mm phosphate buffer) (30) was treated with varying concentrations of Ca-EDTA or TPEN as indicated. The protein samples were then incubated with sample loading buffer (62.5 mm Tris-Cl (pH 6.8), 2% SDS, 10% glycerol, and 0.002% bromphenol blue with Ca-EDTA or TPEN and with or without 10 mm DTT as indicated), followed by SDS-PAGE separation.
Statistical Analysis
All data are expressed as mean ± S.E. unless indicated otherwise. Statistical analyses were performed using Student's t test (unpaired and two-tailed). Analysis of variance was used for comparisons between more than two groups. A value of p < 0.05 was considered statistically significant.
Results
Chelation of Zn2+ Impacts MG53-mediated Membrane Repair in Skeletal Muscle
To investigate the role of Zn2+ in muscle membrane repair, the FM 1-43 fluorescent dye entry assay following UV laser damage was performed on isolated FDB muscle fibers from WT and mg53−/− mice as described previously (24, 34). Changes in FM 1-43 fluorescent dye entry (ΔF/F0) at the damage sites were assessed by confocal microscope imaging. As shown in Fig. 1A, detectable fluorescent dye entry into WT muscle fiber was observed under control conditions. The addition of 20 μm Zn-HPT increased membrane repair capacity as reflected by the diminished FM 1-43 dye entry following injury compared with the control. Removal of Zn2+ ions by chelation with 40 μm TPEN, a high affinity Zn2+ chelator, led to a significant increase in FM 1-43 dye entry following UV damage, indicating compromised membrane repair capacity in the absence of Zn2+.
FIGURE 1.

Protective effect of Zn2+ on the membrane repair capacity of FDB muscle fibers. The effect of Zn2+ on membrane repair capacity was assessed in isolated single FDB muscle fibers derived from WT (A) or mg53−/− (B) mice. FDB muscle fibers were subjected to UV laser wounding under control conditions (dimethyl sulfoxide), after treatment with 20 μm Zn-HPT, or after treatment with 40 μm TPEN, a specific high affinity Zn2+ ion chelator. FM 1-43 dye entry at the irradiation sites (red arrows) upon UV light-induced membrane damage was analyzed. C, summary of data for time-dependent accumulation of FM 1-43 dye. Data are mean ± S.E. for 14 cells from each group derived from three independent experiments. ΔF/F0 = (Ft − F0)/F0, where F0 is the fluorescence intensity at the initial time point, and Ft is the fluorescence intensity at the various time points afterward. *, p < 0.01 versus control (Ctrl) at 50 s following UV laser injury.
Consistent with our previous study (24), the mg53−/− FDB muscle fibers exhibited defective membrane repair function, as shown by the elevated amount of FM 1-43 dye entry following identical treatment (Fig. 1B, left panel). Unlike the WT muscle fibers, the membrane repair capacity observed in mg53−/− muscle fibers did not show Zn2+ dependence: neither Zn-HPT (Fig. 1B, middle panel) nor TPEN (right panel) affected FM 1-43 dye entry following UV damage. Furthermore, the membrane-impermeable Zn2+ chelator Ca-EDTA, which buffers external Zn2+, was also found to cause compromised membrane repair capacity in WT muscle, whereas it had no significant effect on membrane repair capacity in mg53−/− muscle (Fig. 1C). These data suggest that MG53 is a critical player in membrane repair, and Zn2+ ions are part of the regulatory component of the repair machinery.
Extracellular Zn2+ Regulates MG53-mediated Vesicle Translocation to Membrane Injury Sites
Our previous study (24) demonstrated that MG53 facilitates intracellular vesicle trafficking to the membrane disruption site for formation of a repair patch. To examine the role of Zn2+ in the process of MG53-mediated vesicle translocation, we expressed GFP-MG53 fusion protein in C2C12 myoblasts and used live cell imaging to assess microelectrode-generated mechanical membrane damage. As shown in Fig. 2A, under resting conditions, GFP-MG53 localized to intracellular vesicles in proximity to the plasma membrane. Acute injury to the cell membrane caused rapid translocation of MG53-containing vesicles to the injury site. Removal of extracellular Zn2+ by preincubation with 40 μm Ca-EDTA impaired GFP-MG53 translocation to membrane injury sites (Fig. 2B). Moreover, the addition of 20 μm TPEN significantly reduced translocation of GFP-MG53-containing vesicles to the mechanical injury site (Fig. 2C). Data from multiple experiments assessing the role of Ca-EDTA and TPEN in GFP-MG53-mediated membrane repair in C2C12 myoblast cells are summarized in Fig. 2E. Clearly, chelation of extracellular Zn2+ produced significant defects in membrane repair patch formation.
FIGURE 2.
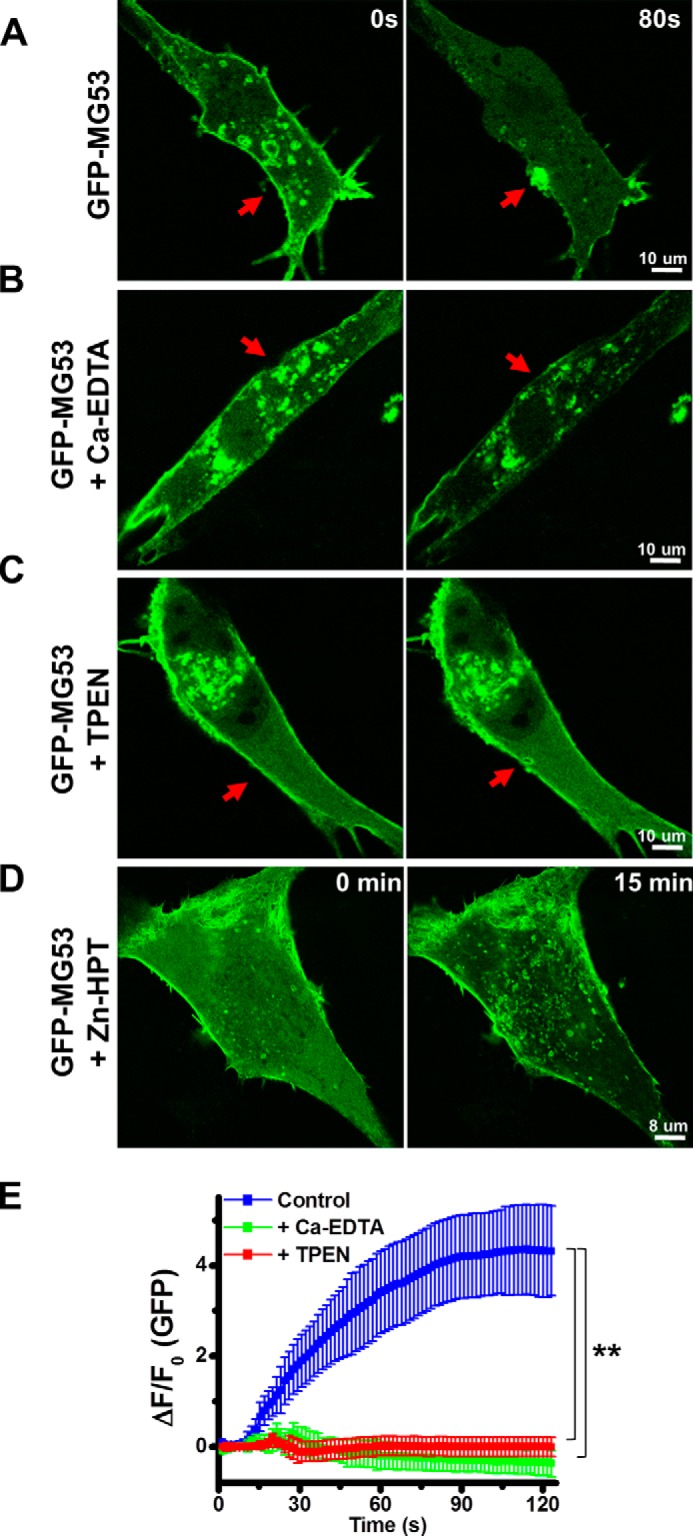
Extracellular Zn2+ regulates MG53-mediated vesicle translocation to acute membrane injury sites. Shown are the results from confocal analysis of GFP-MG53-containing vesicles in C2C12 cells before (0 s; left panels) and after (80 s; right panels) microelectrode-induced membrane damage. A, GFP-MG53 translocation to the acute membrane injury site (arrows). GFP-MG53-containing vesicle translocation was abolished in the presence of 40 μm Ca-EDTA (B) or 20 μm TPEN (C). D, prolonged incubation (15 min) with 20 μm Zn-HPT caused redistribution of intracellular GFP-MG53, which concentrated at plasma membrane and in the intracellular membranes. E, summary of data for the time-dependent accumulation of GFP-MG53 at the injury sites. Data are mean ± S.E. for 14 cells from each group derived from three independent experiments. **, p < 0.001 versus control at 30 s following microelectrode-induced injury.
GFP-MG53-mediated membrane repair patch formation was also examined in the presence of the Zn2+ ionophore Zn-HPT. Interestingly, after a 15-min incubation with Zn-HPT, GFP-MG53 was redistributed toward the cell surface membrane and concentrated in the intracellular membrane compartments (Fig. 2D), suggesting that elevation of intracellular Zn2+ may facilitate the translocation of GFP-MG53 to intracellular vesicles and to the plasma membrane.
Molecular Analysis of Zn2+ Binding to MG53
MG53 is a member of the TRIM family of proteins comprising two to three Zn2+-binding domains (38–40). It has been reported that Zn2+-binding motifs coordinate Zn2+ ions with a cluster of cysteine and histidine residues. The Cys2His2-like structure is by far the best characterized class of zinc fingers and is commonly found in mammalian transcription factors. These domains adopt a simple ββα-fold and have the amino acid sequence motif X2-Cys-X2,4-Cys-X12-His-X3,4,5-His (40), which is present in MG53. As shown in the schematic diagram in Fig. 3A, domain homology analysis revealed that MG53 contains one Zn2+-binding domain in the RING finger motif (amino acids 1–56) and another in the B-box motif (amino acids 86–117).
FIGURE 3.
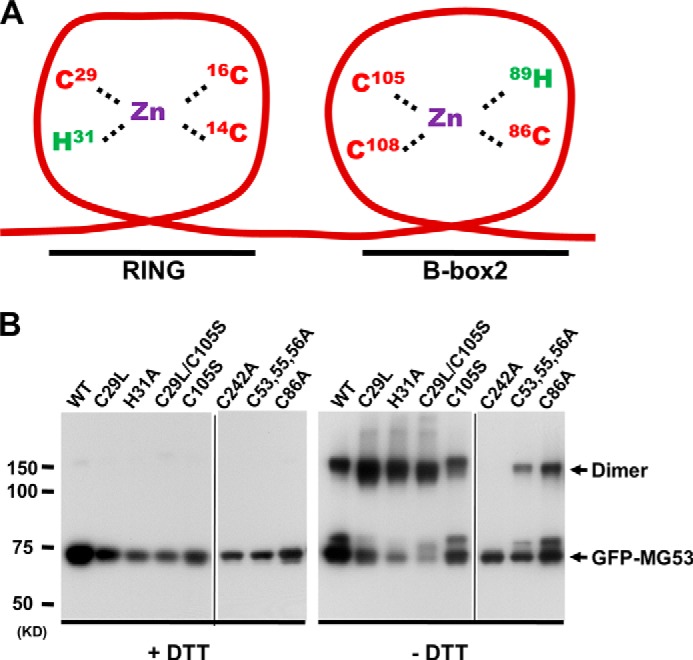
Mutations of Zn2+-binding motifs in MG53 maintain intact intermolecular oligomerization properties. A, schematic diagram of Zn2+-binding motifs in MG53. The specific amino acids that participate in Zn2+ binding are indicated. B, WT and mutant GFP-MG53 proteins expressed in C2C12 myoblasts were analyzed by Western blotting in the presence (left panel) or absence (right panel) of 10 mm DTT. Arrows indicate monomeric or dimeric forms of GFP-MG53. The GFP-MG53(C242A) mutant was defective in dimerization even in the absence of DTT. Both panels are composite images from their original gels.
To understand the molecular mechanisms underlying the role of Zn2+ binding to MG53 in repair of cell membrane injury, we generated several site-specific mutations of the possible Zn2+-binding residues in the RING and B-box motifs of GFP-MG53, including GFP-MG53(C29L), GFP-MG53(H31A), and GFP-MG53(C53A/C55A/C56A) in the RING motif; GFP-MG53(C86A) and GFP-MG53(C105S) in the B-box motif; and GFP-MG53(C29L/C105S) in both motifs. These constructs were transiently transfected into C2C12 myoblasts, and expression of the mutant MG53 proteins was determined by Western blotting. As shown in Fig. 3B, all mutant constructs could be expressed in C2C12 cells, with a predicted molecular mass of ∼75 kDa for GFP-MG53. Although all mutant proteins showed a monomeric form in a reduced environment (with 10 mm DTT) (Fig. 3B, left panel), removal of DTT (right panel) resulted in the appearance of oligomeric forms of GFP-MG53, which were observed for all mutants generated, except for mutant C242A, which was used as a control here because it has been shown to exist only in monomeric form under both reduced and oxidized conditions (24, 33). This finding indicates that all mutants with changes in the Zn2+-binding motifs maintained their intermolecular oligomerization properties, which would be crucial for MG53-mediated membrane repair function.
To examine the Zn2+-binding properties of MG53 in vitro, we purified the recombinant MBP-MG53 fusion proteins following E. coli fermentation. MBP has been proven to enhance the solubility of proteins expressed in E. coli (41). The addition of MBP to the N terminus of MG53 enabled proper folding and purification of the MBP-MG53 fusion proteins. Approximately 100 mg of fusion protein/liter of bacterial culture could be obtained using the amylose affinity column, as shown in Fig. 4A. To characterize the Zn2+-binding capacity, we used the Zn2+-specific fluorescent probe TSQ (42) to quantify the amount of Zn2+ that was bound to the MBP-MG53 proteins. The linearity of TSQ fluorescence as a function of Zn2+ content was established in our previous publication (37). The thiol-bound Zn2+ in MBP-MG53, MBP-MG53(C29L), or MBP-MG53(C29L/C105S) attached to the amylose resin beads was liberated with p-hydroxymercuriphenylsulfonate treatment. After washing away the free TSQ fluorescent dye, the MBP-MG53-bound beads were analyzed by fluorescence microscopy. The images show strong fluorescent signals corresponding to Zn2+ amounts liberated from the beads in striking contrast to the beads under the control conditions (Fig. 4B). Compared with MBP-MG53, TSQ fluorescence was less in the MBP-MG53(C29L) mutant and further decreased in the MBP-MG53(C29L/C105S) double mutant. Quantitative data are presented in Fig. 4C. The Zn2+ content was diminished by approximately half in MBP-MG53(C29L), and Zn2+ binding was further reduced in the MBP-MG53(C29L/C105S) double mutant. Taken together, these data indicate that disruption of zinc finger domains in the RING and B-box motifs of MG53 compromises its Zn2+-binding capacity.
FIGURE 4.
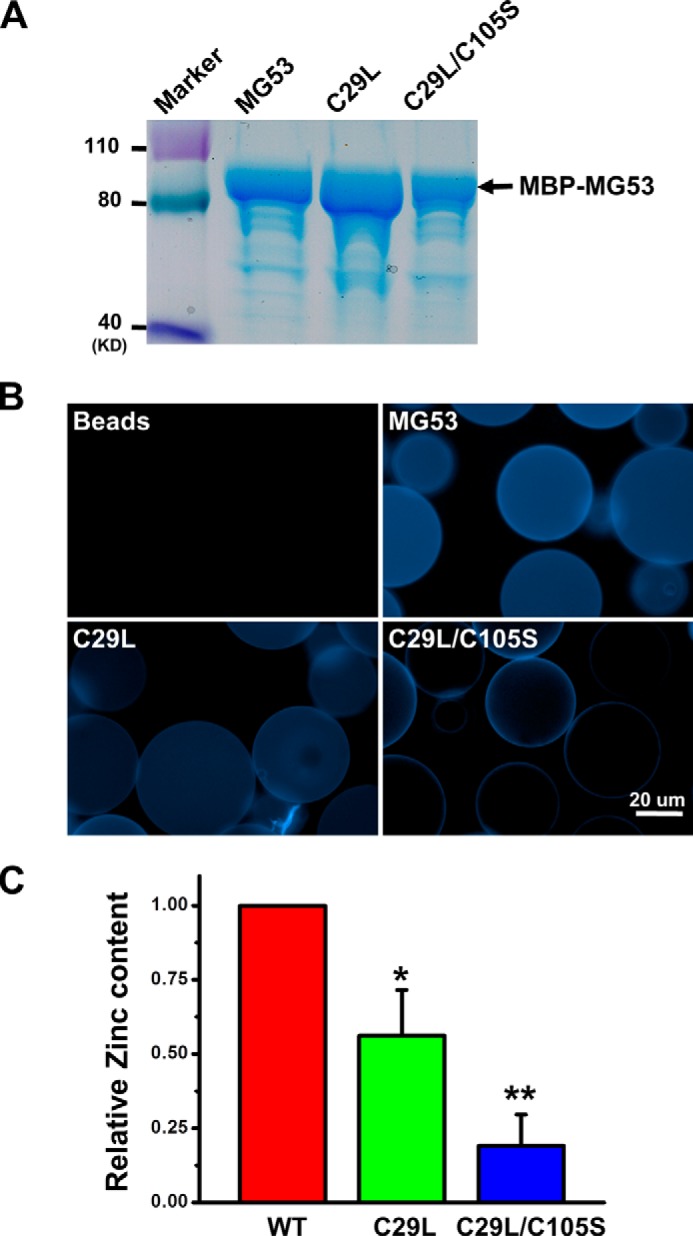
Characterization of Zn2+ binding to mutant MG53 proteins. A, purified MBP-MG53 fusion proteins expressed in E. coli were analyzed by SDS-PAGE. B, the Zn2+-binding capacity of the MBP-MG53 mutant fusion proteins was characterized by TSQ staining of amylase beads. Representative fluorescence images were taken after washing out the unbound TSQ. C, the purified MBP fusion proteins were used to assay for the Zn2+-binding capacity with an established biochemical protocol. The relative Zn2+ concentration in each MBP-MG53 fusion protein was quantified. Data from multiple trials were averaged and normalized to WT MBP-MG53 (mean ± S.E., n = 5). *, p < 0.05; **, p < 0.001.
Due to technical difficulty with expression of the MG53(C105S) mutant in E. coli, its Zn2+-binding capacity could not be determined. In addition, we found that an MG53 mutant with defective E3 ligase activity (C14A), according to our previous work (43), also could not be expressed in sufficient quantity using E. coli fermentation, likely due to protein-misfolding problems.
Disruption of Zn2+ Binding to MG53 Correlates with Defects in MG53-mediated Repair of Membrane Damage
To examine the functional role of Zn2+ binding in MG53-mediated membrane repair, we mechanically damaged membranes of C2C12 cells transfected with the various GFP-MG53 mutants by microelectrode penetration. As shown in Fig. 5A, the GFP-MG53(C29L) mutant displayed compromised movement toward the acute injury site in a nominal zinc-free extracellular solution (middle panel). The addition of Zn-HPT to the extracellular solution partially restored the movement of GFP-MG53(C29L) toward the acute injury site (Fig. 5A, right panel). The GFP-MG53(C105S) mutant with a mutation in the B-box motif also failed to move to the acute injury site following microelectrode penetration in an extracellular nominal zinc-free solution (Fig. 5B, middle panel). Similarly, the addition of Zn-HPT led to partial rescue of the membrane repair capacity of the GFP-MG53(C105S) mutant, as evidenced by its movement toward the injury site (Fig. 5B, right panel).
FIGURE 5.

Zn2+ binding to RING and B-box motifs in MG53 is critical for membrane repair. The effect of Zn2+ binding on MG53-mediated membrane repair was analyzed with different Zn2+-binding motif mutants: GFP-MG53(C29L) (A), GFP-MG53(C105S) (B), and GFP-MG53(C29L/C105S) (C). Confocal images of GFP-MG53 translocation before (0 s; left panels) and after (50 s; middle panels) microelectrode-induced acute membrane damage with extracellular nominal zinc-free solution are shown. In separate experiments, the addition of 20 μm Zn-HPT (right panels) rescued the membrane translocation properties of single mutants, but failed in double mutants. D, summary of mutant GFP-MG53 protein translocation upon acute membrane damage: GFP-MG53(C29L) (left panel), GFP-MG53(C105S) (middle panel), and GFP-MG53(C29L/C105S) (right panel). **, p < 0.001. E, LDH release after glass microbead-induced injury to C2C12 cells transfected with WT GFP-MG53 or the zinc-binding mutants. Data represent the normalized LDH activity relative to cells transfected with WT GFP-MG53 (n = four independent experiments). *, p < 0.05 versus WT.
Further study showed that the GFP-MG53(C29L/C105S) double mutant failed to move to the plasma membrane upon acute membrane damage in nominal zinc-free solution (Fig. 5C, middle panel) and following the addition of extracellular Zn2+ (right panel). Data from multiple experiments are summarized in Fig. 5D. These results demonstrated that the membrane repair function of the MG53(C29L/C105S) mutant was completely abolished and was insensitive to Zn2+ (Fig. 5D, right panel).
We next performed a quantitative LDH release assay to assess the impact of Zn2+-binding motif mutations on MG53-mediated cell membrane repair (35, 36). Populations of C2C12 cells (5 × 105) transfected with GFP-MG53, GFP-MG53(C29L), GFP-MG53(C105S), or GFP-MG53(C29L/C105S) were subjected to glass microbead-induced cell membrane damage. Membrane damage-induced release of LDH into the culture medium was measured using a LDH detection kit. As shown in Fig. 5E, C2C12 cells expressing GFP-MG53(C29L) or GFP-MG53(C105S) did not show a significant change in LDH release compared with cells expressing GFP-MG53. However, C2C12 cells expressing GFP-MG53(C29L/C105S) displayed a significant increase in LDH release, indicating more cell injury. These data are consistent with our live cell imaging of MG53-mediated vesicle translocation to acute membrane injury sites, where mutations in Zn2+ binding to both RING and B-box motifs are required for complete disruption of the membrane repair function of MG53.
Chelation of Zn2+ by Either Ca-EDTA or TPEN Does Not Affect Redox-dependent Oligomerization of MG53
In our previous studies (24, 33), we determined that the redox-dependent oligomerization of MG53 plays an important role in the nucleation process of cell membrane repair. To address whether Zn2+ binding affects the oligomerization process of MG53, we conducted the following experiments. We recently developed a protocol for scale-up production of the rhMG53 protein from CHO cells. Purified rhMG53 showed redox-dependent oligomerization, as removal of DTT from the sample buffer led to the appearance of dimers and oligomers of rhMG53 (Fig. 6). To test whether chelation of Zn2+ affects the oligomerization of MG53, we supplemented the sample buffer with increasing concentrations of Ca-EDTA and TPEN. As shown in Fig. 6, these treatments did not affect the oligomerization of rhMG53. On the basis of these results, we conclude that both Zn2+ binding to MG53 and redox-dependent oligomerization of MG53 contribute to the nucleation process of cell membrane repair.
FIGURE 6.
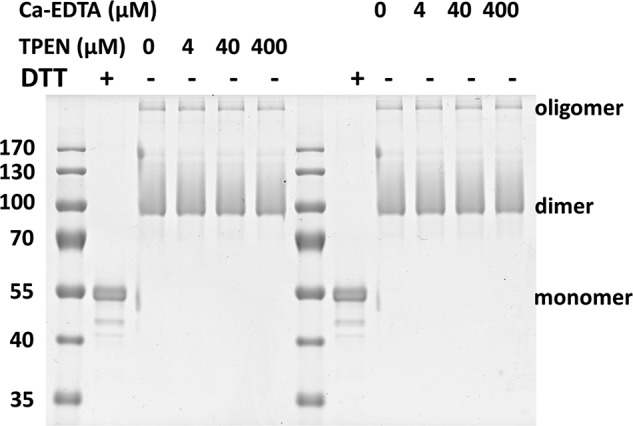
Chelating Zn2+ does not alter the redox-dependent oligomerization of MG53. Purified rhMG53 (1 μg) was run on a SDS-polyacrylamide gel. Colloidal blue staining revealed that DTT (10 mm) treatment disrupted oligomer formation of rhMG53. The redox-dependent oligomerization of rhMG53 was not affected by increasing concentrations of TPEN (0–400 μm) or Ca-EDTA (0–400 μm).
The results from comparative analysis of Zn2+ binding, MG53 oligomerization, and repair function for the various mutant constructs are summarized in Table 1. WT MG53 possesses the ability to form oligomers, bind Zn2+ ions, and function as a membrane repair molecule. MG53(C242A) lost the ability to form oligomers and is defective in membrane repair function (24, 33). The MG53(C29L) and MG53(C105S) single mutants can form dimers, but they have diminished ability to bind Zn2+ and thus cannot repair the injured membrane in the absence of extracellular Zn2+. However, the repair function can be partially recovered in the presence of extracellular Zn2+. The MG53(C29L/C105S) double mutant can form dimers; however, it lost the ability to bind Zn2+ and cannot repair the injured membrane even in the presence of extracellular Zn2+. Altogether, the data suggest that Zn2+ binding to MG53 is indispensable to MG53-mediated function in vesicular trafficking and membrane repair following acute membrane damage in muscle cells.
TABLE 1.
Summary data of MG53 zinc-binding mutants and their corresponding membrane repair capacity
++, normal function; +, ∼50% reduction of function; −, defective function; NA, not assayed.
| MG53 protein | Oligomerization | Zinc binding | Membrane repair |
|
|---|---|---|---|---|
| Without Zn2+ | With Zn2+ | |||
| WT | + | ++ | + | ++ |
| C29L | + | + | − | + |
| H31A | + | NA | − | + |
| C29L/C105S | + | − | − | − |
| C105S | + | NA | − | + |
| C242A | − | NA | − | − |
| C53A/C55A/C56A | + | NA | + | + |
| C86A | + | NA | − | + |
Discussion
Over the past 2 decades, a large body of evidence has determined that an unbalanced homeostasis of two signaling cations, Ca2+ and Zn2+, contributes to the development of various human diseases. It is well known that the extracellular Ca2+ entry is involved in the fusion of intracellular vesicles to reseal the injured plasma membrane (19, 44, 45). Our previous reports have shown that mice null for MG53 display defective muscle membrane repair and progressive myopathy (24, 30). MG53 is a critical component of the membrane repair machinery, and interestingly, the initial movement of MG53 in response to membrane injury is Ca2+-independent (24). Like Ca2+, Zn2+ is involved in a wide array of physiological functions (46), and the deregulation of intracellular Zn2+ has an important role in pathophysiology, including wound healing. Evidence has been obtained that Zn2+ delivered locally provides therapeutic advantages in treatment of both acute and chronic wounds (4, 47–49). However, the definitive role of Zn2+ in the process of plasma membrane repair has not yet been determined. In this study, we established a base for Zn2+ interaction with MG53 in protection against injury to the cell membrane.
Our studies showed that different motifs serve distinct roles in MG53-mediated membrane repair. The C242A mutation in MG53 causes oligomerization loss and has a dominant-negative effect on MG53-mediated membrane repair, indicating that protein oligomerization is important for vesicular nucleation by MG53 at the membrane disruption sites (24, 33). In addition to redox-dependent oligomerization, Zn2+ binding constitutes an important factor for cell membrane repair. We have demonstrated that MG53 is a potential target for Zn2+ during the membrane repair process. We have shown that removing extracellular Zn2+ or disrupting the Zn2+-binding motifs in MG53 alters MG53-mediated vesicular translocation and membrane repair function in muscle cells. We have also shown that the effect of Zn2+ on cell membrane repair was lost in mg53−/− muscle fibers, suggesting that MG53 probably serves as a receptor for Zn2+ during cell membrane repair. Because chelation of free Zn2+ did not appear to affect the redox-dependent oligomerization of MG53, we conclude that both Zn2+ binding to MG53 and redox-dependent oligomerization of MG53 contribute to the nucleation process of cell membrane repair.
Zn2+ deficiency has been linked to many human diseases (50), including cardiovascular diseases (51) and Alzheimer disease (52). Defective membrane repair has been associated with muscular dystrophy and cardiomyopathy (32, 53). Our study highlights a novel potential therapeutic approach in targeting the functional interaction between Zn2+ and MG53 for treatment of human diseases associated with compromised membrane repair capacity.
As a TRIM family protein, MG53 possesses an intrinsic E3 ligase function, enabling it to execute the ubiquitination of specific target proteins (38–40). Recent studies showed that MG53-mediated IRS-1 ubiquitination negatively regulates insulin signaling in skeletal muscle (43, 54). A single mutation in the Zn2+-binding motif of MG53, C14A, causes complete disruption of E3 ligase function. The data presented in this study show that Zn2+-binding single mutations do not completely abolish the membrane repair function of MG53, unlike the double mutation MG53(C29L/C105S), which results in loss of repair function. Thus, therapeutic approaches for increased expression of MG53 in tissues that bypasses its interaction with IRS-1 may be a novel way to treat human diseases linked to compromised membrane repair capacity.
This work was supported, in whole or in part, by National Institutes of Health Grants AR061385 and HL069000 (to J. M.). This work was also supported by the American Heart Association (to C. C. and H. Z.) and an Ohio State University intramural Lockwood Early Career Development Award (to P. L.). J. M. has an equity interest in TRIM-edicine, which develops MG53 for treatment of human diseases. Patents on the use of MG53 are held by the Rutgers Robert Wood Johnson Medical School.
- MBP
- maltose-binding protein
- FDB
- flexor digitorum brevis
- Zn-HPT
- 1-hydroxypyridine-2-thione zinc salt
- TPEN
- tetrakis(2-pyridylmethyl)enediamine
- LDH
- lactate dehydrogenase
- rhMG53
- recombinant human MG53
- TSQ
- 6-methoxy-(8-p-toluenesulfonamido)quinolone.
References
- 1. Chasapis C. T., Loutsidou A. C., Spiliopoulou C. A., Stefanidou M. E. (2012) Zinc and human health: an update. Arch. Toxicol. 86, 521–534 [DOI] [PubMed] [Google Scholar]
- 2. Moynahan E. J. (1974) Letter: Acrodermatitis enteropathica: a lethal inherited human zinc-deficiency disorder. Lancet 2, 399–400 [DOI] [PubMed] [Google Scholar]
- 3. Braun O. H., Heilmann K., Pauli W., Rossner J. A., Bergmann K. E. (1976) Acrodermatitis enteropathica: recent findings concerning clinical features, pathogenesis, diagnosis and therapy. Eur. J. Pediatr. 121, 247–261 [DOI] [PubMed] [Google Scholar]
- 4. Lansdown A. B., Mirastschijski U., Stubbs N., Scanlon E., Agren M. S. (2007) Zinc in wound healing: theoretical, experimental, and clinical aspects. Wound Repair Regen. 15, 2–16 [DOI] [PubMed] [Google Scholar]
- 5. Gupta M., Mahajan V. K., Mehta K. S., Chauhan P. S. (2014) Zinc therapy in dermatology: a review. Dermatol. Res. Pract. 2014, 709152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zorrilla P., Gómez L. A., Salido J. A., Silva A., López-Alonso A. (2006) Low serum zinc level as a predictive factor of delayed wound healing in total hip replacement. Wound Repair Regen. 14, 119–122 [DOI] [PubMed] [Google Scholar]
- 7. Mozzillo N., Ayala F., Formato A., Forestieri P., Mazzeo F. (1984) First full blown syndrome of acute zinc deficiency in course of long term total parenteral nutrition: a clinical case. Ital. J. Surg. Sci. 14, 229–231 [PubMed] [Google Scholar]
- 8. Herbein G., Varin A., Fulop T. (2006) NF-κB, AP-1, zinc-deficiency and aging. Biogerontology 7, 409–419 [DOI] [PubMed] [Google Scholar]
- 9. Wang C., Vernon R., Lange O., Tyka M., Baker D. (2010) Prediction of structures of zinc-binding proteins through explicit modeling of metal coordination geometry. Protein Sci. 19, 494–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berg J. M., Shi Y. (1996) The galvanization of biology: a growing appreciation for the roles of zinc. Science 271, 1081–1085 [DOI] [PubMed] [Google Scholar]
- 12. van der Meer L. T., Jansen J. H., van der Reijden B. A. (2010) Gfi1 and Gfi1b: key regulators of hematopoiesis. Leukemia 24, 1834–1843 [DOI] [PubMed] [Google Scholar]
- 13. Haase H., Rink L. (2014) Multiple impacts of zinc on immune function. Metallomics 6, 1175–1180 [DOI] [PubMed] [Google Scholar]
- 14. Marger L., Schubert C. R., Bertrand D. (2014) Zinc: An underappreciated modulatory factor of brain function. Biochem. Pharmacol. 91, 426–435 [DOI] [PubMed] [Google Scholar]
- 15. Vallee B. L., Falchuk K. H. (1993) The biochemical basis of zinc physiology. Physiol. Rev. 73, 79–118 [DOI] [PubMed] [Google Scholar]
- 16. Frederickson C. J., Giblin L. J., Krezel A., McAdoo D. J., Mueller R. N., Zeng Y., Balaji R. V., Masalha R., Thompson R. B., Fierke C. A., Sarvey J. M., de Valdenebro M., Prough D. S., Zornow M. H. (2006) Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp. Neurol. 198, 285–293 [DOI] [PubMed] [Google Scholar]
- 17. Bozym R. A., Chimienti F., Giblin L. J., Gross G. W., Korichneva I., Li Y., Libert S., Maret W., Parviz M., Frederickson C. J., Thompson R. B. (2010) Free zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Exp. Biol. Med. 235, 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Korichneva I. (2005) Redox regulation of cardiac protein kinase C. Exp. Clin. Cardiol. 10, 256–261 [PMC free article] [PubMed] [Google Scholar]
- 19. McNeil P. L., Kirchhausen T. (2005) An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 6, 499–505 [DOI] [PubMed] [Google Scholar]
- 20. Towler M. C., Kaufman S. J., Brodsky F. M. (2004) Membrane traffic in skeletal muscle. Traffic 5, 129–139 [DOI] [PubMed] [Google Scholar]
- 21. Glover L., Brown R. H., Jr. (2007) Dysferlin in membrane trafficking and patch repair. Traffic 8, 785–794 [DOI] [PubMed] [Google Scholar]
- 22. Shen S. S., Tucker W. C., Chapman E. R., Steinhardt R. A. (2005) Molecular regulation of membrane resealing in 3T3 fibroblasts. J. Biol. Chem. 280, 1652–1660 [DOI] [PubMed] [Google Scholar]
- 23. McNeil P. L., Vogel S. S., Miyake K., Terasaki M. (2000) Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci. 113, 1891–1902 [DOI] [PubMed] [Google Scholar]
- 24. Cai C., Masumiya H., Weisleder N., Matsuda N., Nishi M., Hwang M., Ko J. K., Lin P., Thornton A., Zhao X., Pan Z., Komazaki S., Brotto M., Takeshima H., Ma J. (2009) MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 11, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cao C. M., Zhang Y., Weisleder N., Ferrante C., Wang X., Lv F., Zhang Y., Song R., Hwang M., Jin L., Guo J., Peng W., Li G., Nishi M., Takeshima H., Ma J., Xiao R. P. (2010) MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation 121, 2565–2574 [DOI] [PubMed] [Google Scholar]
- 26. Cai C., Weisleder N., Ko J. K., Komazaki S., Sunada Y., Nishi M., Takeshima H., Ma J. (2009) Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 284, 15894–15902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang X., Xie W., Zhang Y., Lin P., Han L., Han P., Wang Y., Chen Z., Ji G., Zheng M., Weisleder N., Xiao R. P., Takeshima H., Ma J., Cheng H. (2010) Cardioprotection of ischemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ. Res. 107, 76–83 [DOI] [PubMed] [Google Scholar]
- 28. Cai C., Masumiya H., Weisleder N., Pan Z., Nishi M., Komazaki S., Takeshima H., Ma J. (2009) MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 284, 3314–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ko J. K., Ma J. (2005) A rapid and efficient PCR-based mutagenesis method applicable to cell physiology study. Am. J. Physiol. Cell Physiol 288, C1273–C1278 [DOI] [PubMed] [Google Scholar]
- 30. Weisleder N., Takizawa N., Lin P., Wang X., Cao C., Zhang Y., Tan T., Ferrante C., Zhu H., Chen P. J., Yan R., Sterling M., Zhao X., Hwang M., Takeshima M., Cai C., Cheng H., Takeshima H., Xiao R. P., Ma J. (2012) Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 4, 139ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park K. H., Weisleder N., Zhou J., Gumpper K., Zhou X., Duann P., Ma J., Lin P. H. (2014) Assessment of calcium sparks in intact skeletal muscle fibers. J. Vis. Exp. 84, e50898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bansal D., Miyake K., Vogel S. S., Groh S., Chen C. C., Williamson R., McNeil P. L., Campbell K. P. (2003) Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423, 168–172 [DOI] [PubMed] [Google Scholar]
- 33. Hwang M., Ko J. K., Weisleder N., Takeshima H., Ma J. (2011) Redox-dependent oligomerization through a leucine zipper motif is essential for MG53-mediated cell membrane repair. Am. J. Physiol. Cell Physiol. 301, C106–C114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weisleder N., Lin P., Zhao X., Orange M., Zhu H., Ma J. (2011) Visualization of MG53-mediated cell membrane repair using in vivo and in vitro systems. J. Vis. Exp. 52, 2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu H., Lin P., De G., Choi K. H., Takeshima H., Weisleder N., Ma J. (2011) Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J. Biol. Chem. 286, 12820–12824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin P., Zhu H., Cai C., Wang X., Cao C., Xiao R., Pan Z., Weisleder N., Takeshima H., Ma J. (2012) Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 26, 1875–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Korichneva I., Hoyos B., Chua R., Levi E., Hammerling U. (2002) Zinc release from protein kinase C as the common event during activation by lipid second messenger or reactive oxygen. J. Biol. Chem. 277, 44327–44331 [DOI] [PubMed] [Google Scholar]
- 38. Ikeda K., Inoue S. (2012) TRIM proteins as RING finger E3 ubiquitin ligases. Adv. Exp. Med. Biol. 770, 27–37 [DOI] [PubMed] [Google Scholar]
- 39. Meroni G. (2012) Genomics and evolution of the TRIM gene family. Adv. Exp. Med. Biol. 770, 1–9 [DOI] [PubMed] [Google Scholar]
- 40. Micale L., Chaignat E., Fusco C., Reymond A., Merla G. (2012) The tripartite motif: structure and function. Adv. Exp. Med. Biol. 770, 11–25 [PubMed] [Google Scholar]
- 41. Kapust R. B., Waugh D. S. (1999) Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8, 1668–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frederickson C. J., Burdette S. C., Frederickson C. J., Sensi S. L., Weiss J. H., Yin H. Z., Balaji R. V., Truong-Tran A. Q., Bedell E., Prough D. S., Lippard S. J. (2004) Method for identifying neuronal cells suffering zinc toxicity by use of a novel fluorescent sensor. J. Neurosci. Methods 139, 79–89 [DOI] [PubMed] [Google Scholar]
- 43. Yi J. S., Park J. S., Ham Y. M., Nguyen N., Lee N. R., Hong J., Kim B. W., Lee H., Lee C. S., Jeong B. C., Song H. K., Cho H., Kim Y. K., Lee J. S., Park K. S., Shin H., Choi I., Lee S. H., Park W. J., Park S. Y., Choi C. S., Lin P., Karunasiri M., Tan T., Duann P., Zhu H., Ma J., Ko Y. G. (2013) MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat. Commun. 4, 2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steinhardt R. A., Bi G., Alderton J. M. (1994) Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 263, 390–393 [DOI] [PubMed] [Google Scholar]
- 45. Terasaki M., Miyake K., McNeil P. L. (1997) Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J. Cell Biol. 139, 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sensi S. L., Paoletti P., Bush A. I., Sekler I. (2009) Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 10, 780–791 [DOI] [PubMed] [Google Scholar]
- 47. Agren M. S., Ostenfeld U., Kallehave F., Gong Y., Raffn K., Crawford M. E., Kiss K., Friis-Møller A., Gluud C., Jorgensen L. N. (2006) A randomized, double-blind, placebo-controlled multicenter trial evaluating topical zinc oxide for acute open wounds following pilonidal disease excision. Wound Repair Regen. 14, 526–535 [DOI] [PubMed] [Google Scholar]
- 48. Lansdown A. B. (1993) Influence of zinc oxide in the closure of open skin wounds. Int. J. Cosmet. Sci. 15, 83–85 [DOI] [PubMed] [Google Scholar]
- 49. Grommes J., Binnebösel M., Klink C. D., von Trotha K. T., Rosch R., Oettinger A. P., Lindlar I., Krones C. J. (2011) Balancing zinc deficiency leads to an improved healing of colon anastomosis in rats. Int. J. Colorectal Dis. 26, 295–301 [DOI] [PubMed] [Google Scholar]
- 50. Prasad A. S. (2013) Discovery of human zinc deficiency: its impact on human health and disease. Adv. Nutr. 4, 176–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Frustaci A., Sabbioni E., Fortaner S., Farina M., del Torchio R., Tafani M., Morgante E., Ciriolo M. R., Russo M. A., Chimenti C. (2012) Selenium- and zinc-deficient cardiomyopathy in human intestinal malabsorption: preliminary results of selenium/zinc infusion. Eur. J. Heart Fail. 14, 202–210 [DOI] [PubMed] [Google Scholar]
- 52. Brewer G. J., Kaur S. (2013) Zinc deficiency and zinc therapy efficacy with reduction of serum free copper in Alzheimer's disease. Int. J. Alzheimers Dis. 2013, 586365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Han R., Bansal D., Miyake K., Muniz V. P., Weiss R. M., McNeil P. L., Campbell K. P. (2007) Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J. Clin. Invest. 117, 1805–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Song R., Peng W., Zhang Y., Lv F., Wu H. K., Guo J., Cao Y., Pi Y., Zhang X., Jin L., Zhang M., Jiang P., Liu F., Meng S., Zhang X., Jiang P., Cao C. M., Xiao R. P. (2013) Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 494, 375–379 [DOI] [PubMed] [Google Scholar]