

Background: Parkin is proposed to maintain mitochondrial QC through promoting mitophagy.
Results: Under moderate mitochondrial stress conditions, parkin stimulates mitochondrial fusion instead of mitophagy by catalyzing K63-linked ubiquitination and inactivating α-synuclein.
Conclusion: Parkin, PINK1, and α-synuclein form a regulatory circuit to regulate mitochondrial stress response.
Significance: This study provides a physiological context to functionally connect key PARK genes in the pathogenesis of Parkinson disease.
Keywords: α-synuclein (a-synuclein), mitochondria, parkin, Parkinson disease, PTEN-induced putative kinase 1 (PINK1)
Abstract
Mutations in PARKIN (PARK2), an ubiquitin ligase, cause early onset Parkinson disease. Parkin was shown to bind, ubiquitinate, and target depolarized mitochondria for destruction by autophagy. This process, mitophagy, is considered crucial for maintaining mitochondrial integrity and suppressing Parkinsonism. Here, we report that under moderate mitochondrial stress, parkin does not translocate to mitochondria to induce mitophagy; rather, it stimulates mitochondrial connectivity. Mitochondrial stress-induced fusion requires PINK1 (PARK6), mitofusins, and parkin ubiquitin ligase activity. Upon exposure to mitochondrial toxins, parkin binds α-synuclein (PARK1), and in conjunction with the ubiquitin-conjugating enzyme Ubc13, stimulates K63-linked ubiquitination. Importantly, α-synuclein inactivation phenocopies parkin overexpression and suppresses stress-induced mitochondria fission, whereas Ubc13 inactivation abrogates parkin-dependent mitochondrial fusion. The convergence of parkin, PINK1, and α-synuclein on mitochondrial dynamics uncovers a common function of these PARK genes in the mitochondrial stress response and provides a potential physiological basis for the prevalence of α-synuclein pathology in Parkinson disease.
Introduction
Parkinson disease (PD)3 is the most common progressive neurodegenerative movement disorder. Two prominent pathological features are associated with the neurological lesions: the appearance of cytoplasmic inclusion bodies, Lewy bodies, and defective mitochondria. Lewy bodies are mainly composed of fibrillar α-synuclein (PARK1), whose mutations and duplication/triplication have been linked to familial forms of PD (1). Although Lewy bodies have been a focus of PD pathogenesis for decades, the physiological function of α-synuclein and the underlying cause for its prevalent involvement in PD remain poorly understood. On the other hand, the characterization of genes responsible for other familial forms of PD, including PARKIN (PARK2), PINK1 (PARK6), and DJ1 (PARK7), have yielded compelling evidence that mitochondrial dysfunction and oxidative stress are key contributing factors to parkinsonism (2–6). Indeed, brain tissues from PD patients display signs of mitochondrial defects and oxidative stress (7), whereas exposure to mitochondrial toxins, such as rotenone, can cause Parkinsonian-like syndromes in rodent models (8).
The characterization of mitochondrial abnormalities in Drosophila parkin and PINK1 mutants have led to the hypothesis that these two PARK genes work in conjunction to maintain a healthy mitochondrial population. A series of studies have shown that parkin, in a PINK1-dependent manner, undergoes translocation from the cytosol to chemically depolarized mitochondria (9–12). As a RING domain-containing E3 ubiquitin ligase, parkin subsequently catalyzes mitochondrial protein ubiquitination, which results in the recognition and eventual elimination of depolarized mitochondria by autophagy (13–15). As dysfunctional mitochondria are common in PD, the removal of impaired mitochondria by mitophagy has provided an effective model to explain how parkin suppresses Parkinsonism. Indeed, many PD-associated parkin mutants were found defective in mounting mitophagy (11, 12, 14–16). Despite the rapid acceptance of the mitophagy model, whether parkin and PINK1 maintain mitochondrial QC solely through this irreversible process remains uncertain. The mitophagy hypothesis also does not provide an effective mechanism to explain the prevalence of α-synuclein aggregation and Lewy bodies in sporadic PD. The identification of a common biological process regulated by these key PARK genes could provide a framework to more accurately understand the molecular events that lead to PD pathogenesis.
In this report, we present evidence that in response to moderate mitochondrial stress induced by mitochondrial toxins, parkin does not bind or target mitochondria for destruction; instead it promotes mitochondrial fusion. This morphological remodeling requires parkin ubiquitin E3-ligase activity as well as PINK1 (PARK6). The stress-induced mitochondrial connectivity depends on lysine 63 (K63)-linked, ubiquitination mediated by the E2-conjugating enzyme Ubc13. We identified α-synuclein (PARK1) as a regulatory target of parkin in the mitochondrial stress response. We found that mitochondrial toxin treatments stimulate parkin-α-synuclein interaction and ubiquitination, which recruits synphilin 1, a protein that promotes α-synuclein sequestration to inclusion bodies and suppresses α-synuclein toxicity. Indeed, inactivation of α-synuclein phenocopies parkin expression and suppresses mitochondrial fission induced by mitochondrial stresses. The convergence of parkin, PINK1, and α-synuclein on mitochondrial dynamics not only uncovers a common function of these key PARK genes in the mitochondrial stress response independent of mitophagy but also provides a physiological context for the prevalence of α-synuclein pathology in PD.
Experimental Procedures
Cell Culture and Transfection
WT, Mfn1 KO, Mfn2 KO, and Ubc13 KO MEFs were maintained in DMEM supplemented with 10% FCS and penicillin/streptomycin (Gibco). HeLa cells were maintained in DMEM supplemented with 10% FBS and penicillin/streptomycin. The YFP-Parkin permanent HeLa cells and the Flp-In parental HeLa cells were maintained in DMEM supplemented with 10% FBS, penicillin/streptomycin, and a selection of antibiotics, hygromycin, and Zeocin (Invitrogen), respectively. All cell lines were cultured at 37 °C and 5% CO2.
Neuron-enriched cultures were generated from cortex of male and female E16.5 CD1 mouse embryos (Charles River Laboratories). Briefly, the cortex was dissected, cells were dissociated with papain (Worthington Biochemicals), and the cell suspension was plated on poly-d-lysine/laminin-coated glass coverslips in Neurobasal medium with B27 supplements (Invitrogen) and penicillin/streptomycin. Cultures were co-transfected with Parkin-shRNA plasmid and pcDNA3-EGFP using Lipofectamine 2000 (Life Technologies) on day in vitro 3. At day in vitro 6, the cells were treated with rotenone, carbonyl cyanide m-chlorophenylhydrazone (CCCP), or DMSO control for 18 h prior to immunostaining.
Antibodies, Constructs, and Reagents
Primary antibodies used in immunofluorescence were as follows: myc (9E10, mouse monoclonal), cytochrome c (mouse monoclonal; BD Biosciences), Tom20 (rabbit polyclonal; Santa Cruz Biotechnology), HA (HA.11, Covance), and FLAG (M2, Sigma). Secondary antibodies used were as follows: Alexa Fluor 594 donkey anti-mouse and anti-rabbit IgG (Molecular Probes), goat anti-rabbit Cy5 (Molecular Probes), and Pacific Blue goat anti-mouse IgG (Molecular Probes). Primary antibodies used in immunoblotting were: GFP (Roche), ubiquitin (total; P4D1, Santa Cruz Biotechnology), ubiquitin (K63-specific; Apu3, Millipore), α-synuclein (C20, Santa Cruz Biotechnology; 2462, Cell Signaling), Tim23 (BD Bioscience), GAPDH (Cell Signaling), and parkin (Santa Cruz Biotechnology). HRP-conjugated secondary antibodies were used for immunoblotting (Promega).
The following plasmids were used: GFP-parkin and mutants, as previously described (14); myc-Ubc13 (human, obtained from Kah-Leong Lim); and HA-tagged Ub and mutant Ub constructs (obtained from Colin S. Duckett). Plasmids encoding shRNA against mouse Parkin (SHCLNV-NM_016694, Sigma) and human PINK1 (TRCN0000199193, Board Institute).
Determination of CCCP Concentrations
As each cell type responds differently to CCCP, we tested a range of doses in each cell line used to determine which dose was appropriate for use in this study. We expressed GFP-parkin (or in the case of the M17 cells, we compared control and PARKIN KD and stained for mitochondria using cytochrome c antibodies. We analyzed the mitochondria in control versus parkin-expressing cells to see at which concentration(s) there was a difference in morphology. We also checked for parkin translocation or mitochondrial aggregation/clearance, to confirm “mitophagy inducing” concentrations of CCCP for each cell line.
Analysis of Mitochondrial Morphology
Mitochondrial morphology was quantified, as previously described (17). For each experiment, cells were divided into three categories: “hyperfused,” where the majority of mitochondria were so interconnected that individual mitochondria were rarely distinguishable, “intermediate,” where mitochondria were interconnected and tubular, but individual mitochondrial units were often distinguishable, and “fragmented,” where mitochondria were mostly short tubules or small, rounded units. 50 cells per condition were quantified in each sample of independent experiments. The average percentage of cells with each type of mitochondrial morphology was plotted ± S.D. or ± S.E. as indicated, and p values were determined using Student's t test.
Fluorescence recovery after photobleaching (FRAP) was performed, as previously described (17). Briefly, cells were transfected with YFP targeted to the mitochondrial matrix (mito-YFP) and either control vector or parkin (FLAG-tagged) with or without treatment. The data from each experiment can be shown in two forms: recovery, as plotted in line graph form, or mobile fraction. In the recovery curves, greater increases in normalized fluorescence intensity (y axis) over time in seconds (x axis) represent more interconnected mitochondria. The mobile fraction of the mito-YFP was plotted in bar graphs, with larger mobile fractions representing more interconnected mitochondria. We utilized the mobile fraction graphing method for all of the FRAP experiments, and only showed the line graph in the first instance, Fig. 1C. In each FRAP experiment, 15–20 cells were analyzed per condition per cell type. The average was plotted ± S.E. FRAP experiments were performed in duplicate.
FIGURE 1.

Mitochondrial morphology in response to CCCP treatment. A, control (top) or 1 μm CCCP-treated (bottom) WT MEFs were transiently transfected with GFP-parkin (green), and mitochondria were immunostained using cytochrome c (red) antibody. Scale bar represents 25 μm. Small squares represent ×4 zoom images of cytochrome c staining (red only). Arrows (green) mark transfected cells and arrowheads (white) are cells not expressing GFP-parkin (untransfected). B, mitochondrial morphology was assessed in WT MEFs, and the percentage of cells (average ± S.D.) with hyperfused (blue bars), intermediate (red bars) or fragmented (green bars) was plotted. Cells were either control (−) or treated (+) with 1 μm CCCP for 18 h, as indicated. C, mitochondrial morphology was analyzed quantitatively using image analysis and Metamorph software to derive the network score, or the ratio of mitochondrial area to number of individual mitochondria (“resolvable units”). For a more complete description, see “Experimental Procedures.” The network score was measured for WT MEFs expressing GFP-parkin (versus control cells) with (red bars) or without (blue bars) treatment with CCCP (1 μm, 18 h). The average score ± S.D. is plotted. D, connectivity of mitochondria in WT MEFs was quantitatively assessed using FRAP. Cells were transfected with YFP targeted to the mitochondrial matrix (mito-YFP) and either control vector or parkin (FLAG-tagged) with or without treatment with 1 μm CCCP. Mito-YFP recovery is plotted, with greater increases in normalized fluorescence intensity (y axis) over time in seconds (x axis) representing more interconnected mitochondria. Data represent mean ± S.E. (n ∼ 20 cells per condition). E, the average mobile fraction (± S.E.) of the mito-YFP from the FRAP experiment in C is plotted, with larger mobile fractions representing more interconnected mitochondria. F, HeLa cells were transiently transfected with GFP-parkin (green) and imaged as in A. Boxes 1 and 4 are transfected cells, boxes 2 and 3 are untransfected cells. G, mitochondrial morphology was assessed in HeLa cells treated with DMSO control or 50 nm rotenone for 2 h, and the data were plotted as in B. H, FRAP was performed in HeLa cells treated with rotenone (100 nm, 2 h) or control (DMSO), and the average mobile fraction was plotted as in D. I, HeLa cells transiently transfected with GFP-parkin were treated with control or H2O2 (100 nm, 2 h), and mitochondrial morphology was assessed as in B. All statistical significance was assessed using two-way ANOVA analysis with Tukey's test.
In addition to FRAP, we performed quantitative analysis of the mitochondrial network using image analysis in Metamorph. Images were acquired using standardized settings on the confocal microscope, with the laser intensity and brightness (gain) maintained at the same level for all images. Mitochondria were immunostained using cytochrome c antibody. Images were acquired at ×63, and 15–20 cells were analyzed per condition. To obtain the network score, Metamorph software was used to measure the mitochondrial area as well as identify the number of resolvable structures (or individual mitochondrial units). The area was divided by the number of resolvable structures, which gave us a network score (or a quantitative measure of how interconnected the mitochondria were). A higher score represented a more fused mitochondrial network. The average network score was plotted for each cell type under the conditions, as listed, ± S.D.
Gene Silencing and Analysis
For the α-SYNUCLEIN knockdown, HeLa cells were transfected with a non-targeting, negative siRNA control (Invitrogen) or siRNA targeting human α-SYNUCLEIN (Hs SCNA 1; Qiagen) using RNAiMAX (Invitrogen). After 96 h, the cells were treated with 1 μm CCCP for an additional 18 h. Then, the coverslips of control and α-SYNUCLEIN KD cells were fixed and immunostained with cytochrome c. The whole cell lysates were also collected for analysis of KD efficiency by immunoblotting. To knockdown PARKIN in primary neuronal cells, cortical neurons were co-transfected with plasmids encoding Parkin-shRNA and pcDNA-EGFP (to detect transfected cells). After 48 h, the cells were treated as indicated. PINK1 KD in YFP-parkin cells was performed by co-transfecting the cells with plasmids encoding a validated human PINK1-shRNA and pcDNA-EGFP for 72 h prior to further treatment.
Immunoprecipitation
HeLa cells were transfected with plasmids encoding GFP-tagged parkin (wild type or A240R mutant). 36 h after transfection, cells were treated with CCCP for 18 h. DMSO was used as negative control. Cells were lysed in denaturing buffer (50 mm Tris-HCl, pH 7.4, 0.5 mm EDTA, and 1% SDS). Following a brief centrifugation, the lysates were diluted 10-fold in NETN buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 0.5% Nonidet P-40, 0.5 mm EDTA) supplied with the deubiquitinase inhibitor, N-ethylmaleimide, and a protease inhibitor mixture (Roche). The diluted lysates were subjected to overnight immunoprecipitation using the α-synuclein antibody (C20, Santa Cruz). After extensive washing, the immunoprecipitates were subjected to immunoblot analysis.
Immunofluorescence
Cells were cultured on glass coverslips (VWR) in 6-well dishes for 1 day prior to transfection. Treatment with DMSO control, CCCP, or rotenone was performed the following day. Cells were fixed after treatment with 4% paraformaldehyde for 15 min at room temperature. After one wash in ×1 phosphate-buffered saline (PBS), cells were permeabilized for 15 min (room temperature) in 0.15% Triton X-100 (diluted in 1× PBS). Next, cells were blocked in 10% bovine serum albumin (BSA) for 20 min at room temperature. Cells were incubated in primary antibody solution overnight. Primary antibodies were diluted in 10% BSA. Following primary antibody incubation, cells were washed three times in 1× PBS, then incubated in secondary antibody solution, which was also made with 10% BSA. The incubation in secondary antibody solution was followed by three washes in 1× PBS. Finally, the samples were mounted onto glass slides (VWR) using Fluoromount-G mounting solution (Southern Biotech) for imaging.
Confocal Image Acquisition and Analysis
All images were acquired on a spinning-disk confocal microscope (Leica) using a ×100/1.4–0.70NA oil (Leica Plan Apochromat) objective. Images of fixed cells were acquired at room temperature and processed using the Leica LAS AF program software. FRAP analysis was performed at 37 °C. Image contrast and brightness were adjusted in Photoshop (Adobe). Quantitative recovery data from the FRAP analysis was analyzed in Excel (Microsoft).
Statistical Analysis
Two-way ANOVA with Tukey's test was conducted for statistical analysis of all quantitative data. Standard deviation or standard error was used, as labeled.
Results
Parkin Promotes Mitochondrial Connectivity in Response to Moderate Mitochondrial and Oxidative Stress
In parkin-expressing cells, treatment with high concentrations of the chemical uncoupler, CCCP (10–25 μm), causes parkin translocation to depolarized mitochondria, which are subsequently removed by mitophagy (9, 14, 18). As expected, when lower concentrations of CCCP were applied (100 nm-1 μm, see “Experimental Procedures”), no apparent translocation of parkin to mitochondria or mitophagy was observed (Fig. 1A). Surprisingly, however, mitochondria in MEFs transiently expressing GFP-Parkin underwent marked elongation and networking, resulting in a hyperfused phenotype (Fig. 1A, arrows; Fig. 1B). This is in stark contrast to mitochondria in parkin-negative MEFs, which underwent fission and displayed a fragmented mitochondrial phenotype upon CCCP treatment (Fig. 1, A, arrowheads, and B), as it was previously reported (19). To confirm the morphological phenotype, we devised a mitochondrial “network score,” which was calculated as the mitochondrial area divided by the number of individual mitochondria (Fig. 1C). Indeed, the network score was decreased in parkin-negative MEFs but increased in parkin-positive MEFs upon treatment with low dose CCCP (Fig. 1C).
To further quantify the connectivity of the mitochondrial network, we measured FRAP of a mitochondria-targeted YFP (mito-YFP), where connected mitochondria recover fluorescence more rapidly than isolated and fragmented ones. As shown in Fig. 1, E and F, in cells lacking parkin, CCCP treatment caused a significant decrease in fluorescence recovery of mito-YFP (lower mobile fraction) compared with untreated cells, indicative of more fragmented mitochondria. In contrast, CCCP treatment accelerated the fluorescence recovery and significantly increased the mobile fraction in cells expressing parkin (Fig. 1D, purple line, and E).
We next asked if parkin-dependent mitochondrial fusion could be activated by mitochondrial toxins and oxidative stresses that are linked to PD. As shown in Fig. 1, F–H, treatment with a low concentration of complex I inhibitor, rotenone (50 nm), also induced mitochondrial fusion in parkin-expressing cells but triggered mitochondrial fragmentation in parkin-negative HeLa cells. Similarly, hydrogen peroxide treatment, which produces reactive oxygen species, induced parkin-dependent mitochondrial fusion as well (Fig. 1I). Altogether, these results show that ectopically expressed parkin promotes mitochondrial connectivity in response to moderate mitochondrial stress.
Endogenous Parkin Mediates Mitochondrial Hyperfusion in Neuronal Cells
We next investigated whether endogenous parkin could induce mitochondrial fusion in response to mitochondrial and oxidative stressors in primary cortical neurons. As shown in Fig. 2, A and B, treatment with low-dose CCCP (20 nm) and rotenone (2 nm) also resulted in mitochondrial elongation in primary neurons. In contrast, the same treatment led to fragmented mitochondria when parkin was knocked down (Fig. 2, C and D).
FIGURE 2.

Parkin-dependent mitochondrial fusion in primary cortical neurons. A, mitochondria were immunostained using Tom20 antibody (red) in primary cortical neurons treated with DMSO, CCCP (20 nm), or rotenone (2 nm) for 18 h. Scale bar represents 10 μm. Right panels show zoomed areas (white squares). B, mitochondrial morphology was assessed in primary neurons and data were plotted as in Fig. 1B. C, primary cortical neurons were co-transfected with plasmids encoding Parkin-shRNA and pcDNA-EGFP. After 3 days, cells were treated with DMSO or rotenone (2 nm) for an additional 18 h. Mitochondria were visualized using Tom20 antibody (red). Transfected cells were tracked with GFP signal (green). Right panels represent zoomed images of the white squares (Tom20 staining only). Cells expressing Parkin-shRNA are marked with green arrows; non-transfected cells are indicated with white arrowheads. D, mitochondrial morphology was analyzed in non-transfected and Parkin-shRNA expressing primary neurons. Cells were treated with DMSO or 2 nm rotenone for 18 h. Knockdown efficiency of the Parkin-shRNA has been confirmed previously (45). E, mitochondrial morphology was analyzed in cKD and PARKIN KD M17 cells treated with DMSO control, CCCP (100 nm), rotenone (200 nm) or H2O2 (100 nm). All statistical significance was assessed using two-way ANOVA analysis with Tukey's test.
Similar parkin-dependent mitochondrial fusion was observed in a neuronal M17 cell line treated with CCCP, rotenone, and hydrogen peroxide (Fig. 2E). Taken together with the data in Fig. 1, these findings show that both ectopic and endogenous parkin promote mitochondrial connectivity in response to moderate mitochondrial stress.
Parkin-mediated Mitochondrial Fusion Requires Mitofusins
Mitochondrial fusion is positively regulated by the mitofusins, Mfn1 and Mfn2 (20–22). To determine whether the parkin-induced hyperfused mitochondrial network requires the mitochondrial fusion machinery, we examined the effect of parkin on mitochondrial connectivity in Mfn2 knock-out (KO) MEFs, which show a fusion deficiency under basal conditions but can undergo fusion when challenged by nutrient starvation (23). As shown in Fig. 3, A and B, expression of GFP-Parkin in Mfn2 KO MEFs did not induce a hyperfused mitochondrial phenotype upon CCCP treatment, a conclusion confirmed by the FRAP-based mitochondrial connectivity assay (Fig. 3C). Similar data were found in Mfn1 KO cells (data not shown). Thus, the mitochondrial fusion machinery is required for the formation of the hyperconnected mitochondrial network induced by parkin and mitochondrial stress.
FIGURE 3.

Mitofusins are required for Parkin-mediated mitochondrial hyperfusion. A, control (top) and 1 μm CCCP-treated (bottom) Mfn2 KO MEFs were transiently transfected with GFP-parkin (green) and imaged as described in the legend to Fig. 1A. Arrows (green) mark transfected cells and arrowheads (white) are untransfected cells. B, mitochondrial morphology was assessed in Mfn2 KO MEFs, and the data were plotted as described in the legend to Fig. 1B. Cells were either control (−) or treated (+) with 1 μm CCCP for 18 h, as indicated. C, connectivity of mitochondria in Mfn2 KO MEFs was quantitatively assessed using FRAP, as in Fig. 1D. All statistical significance was assessed using two-way ANOVA analysis with Tukey's test but p values were not significant in Mfn2 KO experiments (p > 0.05).
Parkin-mediated Mitochondrial Fusion Requires PINK1
Genetic evidence has indicated that PINK1 and parkin work in the same pathway to maintain mitochondrial integrity (4, 5). To determine whether PINK1 is also involved in parkin-mediated mitochondrial morphological remodeling, we inactivated PINK1 by a specific siRNA and assessed CCCP-induced mitochondrial fusion in HeLa cells stably expressing YFP-tagged parkin. As shown in Fig. 4, PINK1 knockdown effectively suppressed parkin-dependent mitochondrial fusion upon treatment with low doses of CCCP. These data indicate that the PINK1-parkin mitochondrial quality control axis also operates to stimulate mitochondrial fusion in response to mitochondrial stress.
FIGURE 4.
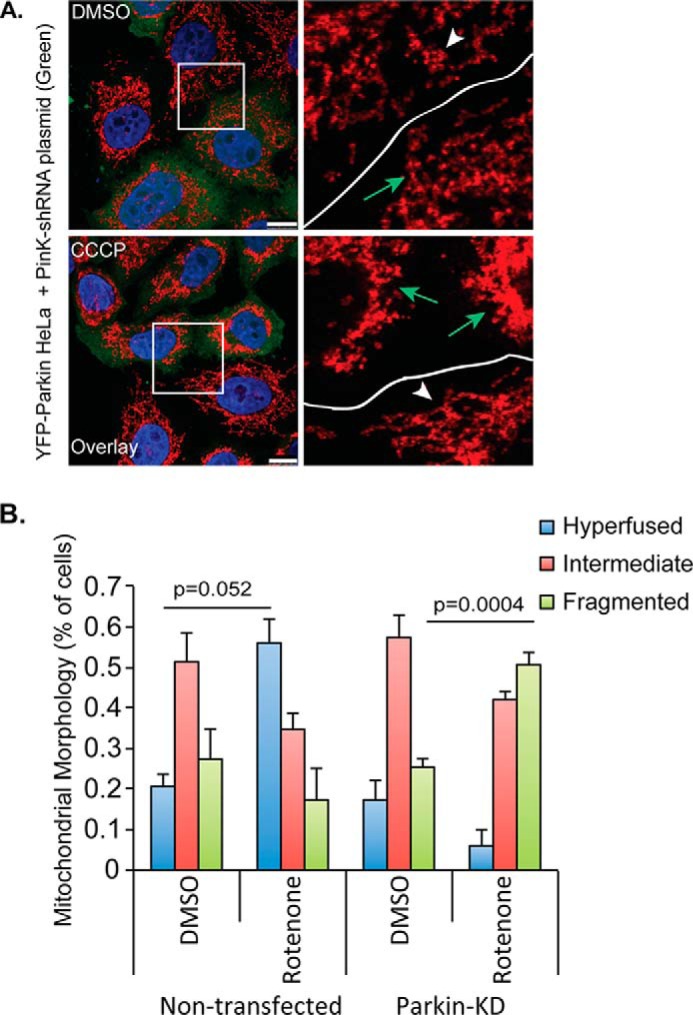
PINK1 is required for parkin-mediated mitochondrial hyperfusion. A, HeLa cells stably expressing YFP-parkin were co-transfected with plasmids encoding PINK1-shRNA and pcDNA-EGFP. Cells were treated with DMSO or CCCP (100 nm, 18 h). Transfected cells were tracked with exogenous EGFP signal (shown in green, cells indicated with green arrows in zoomed in panels) and mitochondria were immunostained using a Tom20 antibody. Scale bar represents 10 μm. B, mitochondrial morphology was assessed in non-transfected and PINK1-shRNA expressing YFP-parkin HeLa cells treated with DMSO or 100 nm CCCP for 18 h. The average ± S.D. was plotted. All statistical significance was assessed using two-way ANOVA analysis with Tukey's test.
Parkin-mediated Mitochondrial Fusion Requires Its Ubiquitin E3 Ligase Activity
PARKIN encodes for an ubiquitin E3-ligase. To investigate whether stress-induced mitochondrial fusion requires parkin-mediated ubiquitination, we assessed the PD-associated parkin mutants that are deficient in E3-ligase activity, A240R and T415N. We found that these mutants cannot promote a hyperfused mitochondrial phenotype upon rotenone (Fig. 5, A and B) or CCCP treatment (data not shown). These results indicate that parkin-mediated ubiquitination is required for stimulating mitochondrial fusion in response to mitochondrial stress.
FIGURE 5.

Ubiquitination-deficient parkin mutants fail to induce mitochondrial fusion. A, confocal microscope images of HeLa cells transiently transfected with GFP-tagged parkin mutants (A240R, top two rows; T415N, bottom two rows) and either left untreated or treated with 100 nm rotenone for 2 h are shown. Mitochondria were visualized as in Fig. 1A. Scale bar represents 25 μm. Far right images are ×5 zoom magnification of the white boxed region of the overlay (cytochrome c staining only, red). B, mitochondrial morphology was assessed as described in the legend to Fig. 1B. HeLa cells expressing empty vector (control), GFP-parkin WT, A240R, or T415N were treated with DMSO (−) or rotenone (+; 100 nm, 2 h). Average values ± S.D. are shown. All p values were assessed using two-way ANOVA analysis with Tukey's test. C, expression of GFP-tagged parkin was tested by immunoblotting with the indicated antibody. Tubulin served as internal control.
Parkin-dependent Mitochondrial Fusion Requires K63-linked Ubiquitination
Our results with the A240R and T415N parkin mutants indicate that the ubiquitin E3-ligase activity of parkin is required for stress-induced mitochondrial fusion. Parkin has been shown to catalyze both K48-linked and K63-linked ubiquitin chains during mitophagy (13, 24). To determine which type of ubiquitin chain is responsible for mitochondrial fusion, we overexpressed mutant ubiquitin, K48R or K63R, to suppress K48- and K63-linked ubiquitin chain production, respectively. As shown in Fig. 6, A and B, the expression of a K63R, but not a K48R, ubiquitin mutant significantly inhibited CCCP-induced, parkin-dependent mitochondrial fusion. Supporting this observation, inactivation of Ubc13, an ubiquitin-conjugating enzyme that assembles K63 ubiquitin chains with parkin (25), prevented parkin-dependent mitochondrial fusion in response to CCCP (Fig. 6, C and D) or rotenone treatment (data not shown). Mitochondrial fusion in Ubc13-deficient cells was restored by re-introduction of an Ubc13 expression plasmid (Fig. 6, C and D). Taken together, these data indicate that parkin-mediated mitochondrial fusion requires Ubc13-mediated K63-linked ubiquitination.
FIGURE 6.

Parkin-mediated hyperfusion is K63 ubiquitin-dependent. A, HeLa cells treated with CCCP (100 nm, 18 h) were transiently transfected with GFP-parkin (green) and HA-tagged wild type, K63R or K48R mutant Ub (blue), as labeled. Mitochondria (mito) were immunostained using a Tom20 (red) antibody. The scale bar represents 25 μm. Small squares represent ×3 zoom images (cytochrome c staining only, red). Transfected cells are marked with green arrows; untransfected cells are indicated with white arrowheads. B, mitochondrial morphology was assessed in HeLa cells and plotted as in Fig. 1B. Cells were treated with DMSO (−) or 100 nm CCCP (+) for 18 h. The constructs used for transient transfection are marked. C, top two panels, Ubc13 KO MEFs were transiently transfected with GFP-parkin (green) and mitochondria were immunostained as described in the legend to Fig. 1A. The scale bar represents 25 μm. Small squares represent ×4 zoom images (cytochrome c staining only, red). Control cells were treated with DMSO, whereas treated cells were incubated with 1 μm CCCP for 18 h, as indicated. Bottom two panels, Ubc13 KO MEFs were transfected, stained, and treated as described in the legend to Fig. 5A, except Ubc13 expression was reconstituted using transient transfection of a myc-tagged Ubc13 construct, shown by anti-myc immunofluorescence (blue). Transfected cells are marked with green arrows; untransfected cells are indicated with white arrowheads. D, mitochondrial morphology was assessed in Ubc13 KO MEFs, and data were plotted as described in the legend to Fig. 1B. Cells were either control (−) or treated (+) with 1 μm CCCP, as indicated. All statistical significance was assessed using two-way ANOVA analysis with Tukey's test.
Parkin Promotes Ubiquitination-dependent α-Synuclein and Synphilin 1 Complex Formation in Response to Mitochondrial Stress
Overexpression of α-synuclein (PARK1) has been shown to induce mitochondrial fission (26, 27). We therefore asked if α-synuclein is a regulatory target of parkin in response to mitochondrial stress. Co-immunoprecipitation assays showed that α-synuclein and parkin did not form stable complexes under basal conditions (Fig. 7A (i), lane 1), as it was previously reported (28). However, upon CCCP treatment, endogenous α-synuclein became markedly associated with parkin (Fig. 7A (i), lane 2). Remarkably, the formation of the parkin·α-synuclein complex is accompanied by the prominent appearance of ubiquitinated species (Fig. 7A (ii), lane 2, bracket). In contrast, the ubiquitin-ligase-deficient A240R parkin mutant, which cannot support mitochondrial fusion (Fig. 5), did not stably associate with α-synuclein (Fig. 7A (i), lane 4) or induce ubiquitinated species (Fig. 7A (ii), lane 4) in response to CCCP. Low-dose rotenone treatment similarly induced parkin-α-synuclein association and ubiquitination (Fig. 7B, lane 3). Consistent with the finding that parkin stimulates mitochondrial fusion by catalyzing Ubc13-dependent K63-linked ubiquitin chain formation (Fig. 6), the parkin-mediated ubiquitinated species were recognized by a K63-linked ubiquitin chain-specific antibody (Fig. 7A (iii), lane 2, bracket). Furthermore, knockdown of Ubc13 markedly reduced α-synuclein-associated ubiquitination upon treatment with CCCP or rotenone (Fig. 7B, lanes 5 and 6).
FIGURE 7.

Parkin binds α-synuclein under stress and results in α-synuclein-associated protein ubiquitination. A, HeLa cells transiently expressing GFP-parkin WT or A240R were treated with DMSO control (−) or 1 μm CCCP (+) for 18 h. Endogenous α-synuclein was immunoprecipitated (IP) and the samples were immunoblotted (IB), as indicated. Ubiquitinated species are marked by brackets. B, HeLa cells stably expressing YPF-parkin were transfected with either control or ubc13 siRNA. 48 h after transfection, cells were treated with DMSO control, CCCP (0.5 μm), or rotenone (50 nm) for 18 h. Endogenous immunoprecipitation and whole cell lysate input were analyzed by Western blotting with anti-α-synuclein and other antibodies, as indicated.
We found that CCCP and rotenone treatments also recruited synphilin 1, a negative regulator of α-synuclein toxicity (28–30), to the α-synuclein·parkin complex (Fig. 7B, lanes 2 and 3). Importantly, knockdown of Ubc13 not only reduced α-synuclein-associated ubiquitination but also synphilin 1 recruitment, indicating that parkin-mediated K63-linked ubiquitination promotes an interaction of α-synuclein and synphilin 1 (Fig. 7B). These findings show that in response to mitochondrial stress, parkin becomes physically associated with α-synuclein, catalyzes ubiquitination, and recruits synphilin 1.
α-Synuclein Is Required for Stress-induced Mitochondrial Fission
The parkin-dependent recruitment of synphilin 1 suggests that parkin negatively regulates α-synuclein in response to mitochondrial stress. To directly assess the role of α-synuclein in stress-induced mitochondrial remodeling, we knocked down α-SYNUCLEIN by siRNA in parkin-negative HeLa cells followed by modest levels of CCCP treatment. As shown in Fig. 8, A–C, inactivation of α-synuclein, similar to parkin expression, effectively suppressed mitochondrial fission normally induced by CCCP. These results support the notion that parkin negatively regulates α-synuclein in response to mitochondrial stress.
FIGURE 8.

α-Synuclein is required for stress-induced mitochondrial fission. A, confocal micrographs of control (cKD) and α-SYNUCLEIN (syn) KD HeLa cells treated with DMSO or 1 μm CCCP for 18 h, as labeled, are shown. Mitochondria were imaged using cytochrome c antibodies (shown in green). Scale bar represents 25 μm. Small squares represent ×5 zoom images. B, mitochondrial morphology was assessed in control (cKD) and syn KD HeLa cells treated with DMSO (−) or 1 μm CCCP for 18 h (+). The average ± S.D. was plotted. The statistical significance was assessed using two-way ANOVA analysis with Tukey's test. C, Western blot of whole cell lysates from control (cKD) and syn KD samples, treated with DMSO control (−) or 1 μm CCCP and immunoblotted with α-synuclein and tubulin, as labeled, are shown.
Discussion
The loss of mitochondrial integrity and appearance of Lewy bodies are two prevailing pathological features of PD. Parkin has been proposed to play a key role in maintaining mitochondrial QC by disposing of damaged mitochondria through mitophagy. We have discovered that under moderate mitochondrial stress conditions, parkin promotes mitochondrial connectivity instead of parkin translocation and mitophagy. Several lines of evidence support the physiological relevance of parkin-mediated mitochondrial fusion. First, parkin activates mitochondrial fusion in response to mitochondrial stresses known to associate with PD pathogenesis. Second, several PD-causing parkin mutants are defective in stimulating mitochondrial fusion. Last, parkin-dependent fusion of stressed mitochondria requires PINK1 and involves parkin-α-synuclein interaction and ubiquitination. These findings connect three key PARK genes to mitochondrial morphological remodeling in response to mitochondrial stresses.
Although low levels of CCCP do not fully collapse the mitochondrial membrane potential and activate parkin-mediated mitophagy, this treatment likely elevates mitochondrial stress by stimulating oxidative phosphorylation to maintain the mitochondrial proton gradient. Modest levels of rotenone and H2O2 could similarly increase mitochondrial oxidative stress. Under these modest mitochondrial stress conditions, parkin-negative cells did not show acute mitochondrial failure (<24 h treatment), judging by ATP production, or increased cell death (data not shown). These findings are in line with a rat PD model where low levels of rotenone did not induce acute bioenergetic defects but chronic treatment resulted in PD-like phenotypes, including the appearance of Lewy body-like inclusions (8). Our evidence indicates that parkin-mediated mitochondrial fusion is an adaptive response to mitochondrial stress. Consistent with our findings, mitochondria in parkin-mutant fibroblasts from AR-JP patients are also much more prone to fragmentation than those in control cells when challenged by low-dose rotenone (31). We propose that adaptive mitochondrial fusion mediated by parkin confers protection to mitochondria against low but chronic mitochondrial stress, which is likely more physiologically relevant to PD pathogenesis than high dose mitochondrial toxins commonly used to induce acute neuron dysfunction and death.
In principle, active fusion could protect mitochondrial integrity by complementing damaged mitochondria with their healthy counterparts (32) or by limiting the production of reactive oxygen species (33). Supporting this view, prominent accumulation of damaged mitochondria and neurodegeneration have been observed in fusion-deficient Mfn1/Mfn2 knock-out cells and mice (34–36). We suggest that PINK1 and parkin form a sensor-effector pair that monitors internal mitochondrial status and enforces mitochondrial quality control accordingly. In response to mitochondrial stresses, cytosolic parkin, in conjunction with PINK1, initially promotes the formation of a connected mitochondrial network to protect mitochondria from further damage. Irreparably damaged mitochondria are subsequently targeted by parkin and PINK1 to undergo destruction by mitophagy. The stepwise ubiquitin-dependent triage system could offer more efficient and better management of mitochondrial QC.
Our study has identified α-synuclein (PARK1) as a regulatory target of parkin in the mitochondrial stress response. Despite the extensive characterization of α-synuclein in PD, its physiological function remains elusive. We found that inactivation of α-synuclein by siRNA essentially phenocopies the effect of parkin and suppresses stress-induced mitochondrial fission (Fig. 8). This finding indicates that α-SYNUCLEIN normally promotes mitochondrial fragmentation in response to mitochondrial stress. We suspect that α-synuclein-mediated mitochondrial fission is part of an adaptive response to mitochondrial stress. Indeed, although α-synuclein has been predominantly considered a PD-causing gene, genetic ablation of α-synuclein in mice actually leads to aberrant mitochondrial lipid composition and function (37). Dopaminergic neurons from α-SYNUCLEIN knock-out mice are more susceptible to oxidative stress than their wild type counterparts (38, 39). These findings suggest a physiological and protective role of α-synuclein in the mitochondrial stress response. However, prolonged or unchecked activation of α-synuclein caused by chronic mitochondrial stress or mutations in PARKIN or PINK1 would lead to excessive mitochondrial fission, mitochondrial failure, and eventually neuron cell death and PD. Indeed, overexpression of α-synuclein induces mitochondrial fragmentation and dysfunction along with neurodegeneration (26, 27).
Our data suggest that parkin terminates the α-synuclein-mediated mitochondrial stress response by catalyzing Ubc13-dependent, K63-linked ubiquitination. We further found that this ubiquitination induces formation of a protein complex of parkin, α-synuclein, and synphilin 1 (Fig. 7). These findings are of potential significance for several reasons. First, synphilin 1 overexpression can suppress neurotoxicity caused by a PD-associated α-synuclein (A53T) mutant transgene (30). Thus, recruitment of synphilin 1 by parkin would inhibit α-synuclein. Second, in both cells and mice, synphilin 1 stimulates α-synuclein to form inclusion bodies that resemble Lewy bodies (28–30, 40). Last, we have previously shown that K63-linked ubiquitin chains are critical for both biogenesis and clearance of the aggresome, a cellular model for Lewy bodies (41, 42). These findings lead us to propose that parkin terminates stress-activated α-synuclein by catalyzing K63-linked ubiquitination and recruiting synphilin 1, which could activate transport and autophagic degradation of α-synuclein·synphilin 1 complex at the perinuclear region. Thus, the prevalence of Lewy bodies in PD patients may reflect rampant production of α-synuclein·synphilin 1 complexes induced by parkin in an attempt to resolve chronic mitochondrial stress. Accordingly, for AR-JP patients deficient in parkin, the inability to terminate α-synuclein activity through K63-linked ubiquitination results in premature mitochondrial failure, and early onset neurodegeneration in the absence of Lewy bodies (43).
The convergence of α-synuclein, parkin, and PINK1 on mitochondrial dynamics, along with the contribution of DJ-1 (26, 31, 44), underscores the significance of adaptive mitochondrial fusion in maintaining mitochondrial integrity when challenged by stress conditions relevant to PD. The interplay of parkin, α-synuclein, and synphilin 1 in response to mitochondrial toxins further provides a physiological basis for the prevalence of Lewy bodies in PD. It is of interest to note that no ortholog of α-synuclein has been found in Drosophila. Therefore, the observed relationship between parkin and α-synuclein must have evolved later in evolution, possibly reflecting the need to better manage mitochondrial stresses and protect neurons in animals with much longer life spans than fruit flies. In the future, elucidating the specific functions of α-synuclein would be crucial to understand how mitochondria adapt to endogenous and exogenous stresses and the physiological basis of neurodegenerative diseases characterized by α-synuclein pathology.
Acknowledgments
We thank Dr. J. Chen for Ubc13 KO MEFs, and Dr. D. C. Chan for the Mfn1 and Mfn2 KO MEFs. The Flp and YFP-parkin HeLa cell lines were a generous gift from Dr. R. Youle. We also thank Dr. A. McClure for her thoughtful comments on the manuscript.
Note Added in Proof
The images of untreated mouse embryonic fibroblasts and mitochondria in Fig. 1A were not correct in the version of this article that was published on April 10, 2015 as a Paper in Press. The correct images are now shown.
This work was supported, in whole or in part, by National Institutes of Health Grants T32-CA-059365 (to K. L. N.), 1R01DA033610 (to A. E. W.), and 2R01-NS054022 (to T. P. Y.) and Leukemia and Lymphoma Society Grant 5396-11 (to K. L .N.).
- PD
- Parkinson disease
- CCCP
- carbonyl cyanide m-chlorophenylhydrazone
- MEF
- mouse embryonic fibroblast
- FRAP
- fluorescence recovery after photobleaching
- Mfn
- mitofusin
- DMSO
- dimethyl sulfoxide
- ANOVA
- analysis of variance
- EGFP
- enhanced green fluorescent protein.
References
- 1. Abou-Sleiman P. M., Muqit M. M., Wood N. W. (2006) Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat. Rev. Neurosci. 7, 207–219 [DOI] [PubMed] [Google Scholar]
- 2. Greene J. C., Whitworth A. J., Kuo I., Andrews L. A., Feany M. B., Pallanck L. J. (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U. S. A. 100, 4078–4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whitworth A. J., Theodore D. A., Greene J. C., Benes H., Wes P. D., Pallanck L. J. (2005) Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 102, 8024–8029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 5. Park M., Salgado J. M., Ostroff L., Helton T. D., Robinson C. G., Harris K. M., Ehlers M. D. (2006) Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron 52, 817–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang B., Zhao D., Verkman A. S. (2006) Evidence against functionally significant aquaporin expression in mitochondria. J. Biol. Chem. 281, 16202–16206 [DOI] [PubMed] [Google Scholar]
- 7. Dexter D. T., Sian J., Rose S., Hindmarsh J. G., Mann V. M., Cooper J. M., Wells F. R., Daniel S. E., Lees A. J., Schapira A. H. (1994) Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann. Neurol. 35, 38–44 [DOI] [PubMed] [Google Scholar]
- 8. Betarbet R., Sherer T. B., MacKenzie G., Garcia-Osuna M., Panov A. V., Greenamyre J. T. (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 3, 1301–1306 [DOI] [PubMed] [Google Scholar]
- 9. Narendra D., Tanaka A., Suen D. F., Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geisler S., Holmström K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., Springer W. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 [DOI] [PubMed] [Google Scholar]
- 11. Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C. A., Sou Y. S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., Tanaka K. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R. L., Kim J., May J., Tocilescu M. A., Liu W., Ko H. S., Magrané J., Moore D. J., Dawson V. L., Grailhe R., Dawson T. M., Li C., Tieu K., Przedborski S. (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U. S. A. 107, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chan N. C., Salazar A. M., Pham A. H., Sweredoski M. J., Kolawa N. J., Graham R. L., Hess S., Chan D. C. (2011) Human Mol. Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee J. Y., Nagano Y., Taylor J. P., Lim K. L., Yao T. P. (2010) Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 189, 671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshii S. R., Kishi C., Ishihara N., Mizushima N. (2011) Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Narendra D., Kane L. A., Hauser D. N., Fearnley I. M., Youle R. J. (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy: VDAC1 is dispensable for both. Autophagy 6, 1090–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karbowski M., Norris K. L., Cleland M. M., Jeong S. Y., Youle R. J. (2006) Role of Bax and Bak in mitochondrial morphogenesis. Nature 443, 658–662 [DOI] [PubMed] [Google Scholar]
- 18. Tanaka A., Cleland M. M., Xu S., Narendra D. P., Suen D. F., Karbowski M., Youle R. J. (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Legros F., Lombès A., Frachon P., Rojo M. (2002) Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 13, 4343–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen H., Detmer S. A., Ewald A. J., Griffin E. E., Fraser S. E., Chan D. C. (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rapaport D., Brunner M., Neupert W., Westermann B. (1998) Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J. Biol. Chem. 273, 20150–20155 [DOI] [PubMed] [Google Scholar]
- 22. Hermann G. J., Thatcher J. W., Mills J. P., Hales K. G., Fuller M. T., Nunnari J., Shaw J. M. (1998) Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J. Cell Biol. 143, 359–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rambold A. S., Kostelecky B., Elia N., Lippincott-Schwartz J. (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. U. S. A. 108, 10190–10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doss-Pepe E. W., Chen L., Madura K. (2005) α-Synuclein and parkin contribute to the assembly of ubiquitin lysine 63-linked multiubiquitin chains. J. Biol. Chem. 280, 16619–16624 [DOI] [PubMed] [Google Scholar]
- 26. Kamp F., Exner N., Lutz A. K., Wender N., Hegermann J., Brunner B., Nuscher B., Bartels T., Giese A., Beyer K., Eimer S., Winklhofer K. F., Haass C. (2010) Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakamura K., Nemani V. M., Azarbal F., Skibinski G., Levy J. M., Egami K., Munishkina L., Zhang J., Gardner B., Wakabayashi J., Sesaki H., Cheng Y., Finkbeiner S., Nussbaum R. L., Masliah E., Edwards R. H. (2011) Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J. Biol. Chem. 286, 20710–20726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chung K. K., Zhang Y., Lim K. L., Tanaka Y., Huang H., Gao J., Ross C. A., Dawson V. L., Dawson T. M. (2001) Parkin ubiquitinates the α-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat. Med. 7, 1144–1150 [DOI] [PubMed] [Google Scholar]
- 29. Engelender S., Kaminsky Z., Guo X., Sharp A. H., Amaravi R. K., Kleiderlein J. J., Margolis R. L., Troncoso J. C., Lanahan A. A., Worley P. F., Dawson V. L., Dawson T. M., Ross C. A. (1999) Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat. Genet. 22, 110–114 [DOI] [PubMed] [Google Scholar]
- 30. Smith W. W., Liu Z., Liang Y., Masuda N., Swing D. A., Jenkins N. A., Copeland N. G., Troncoso J. C., Pletnikov M., Dawson T. M., Martin L. J., Moran T. H., Lee M. K., Borchelt D. R., Ross C. A. (2010) Synphilin-1 attenuates neuronal degeneration in the A53T α-synuclein transgenic mouse model. Hum. Mol. Genet. 19, 2087–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mortiboys H., Thomas K. J., Koopman W. J., Klaffke S., Abou-Sleiman P., Olpin S., Wood N. W., Willems P. H., Smeitink J. A., Cookson M. R., Bandmann O. (2008) Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 64, 555–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakada K., Sato A., Hayashi J. (2009) Mitochondrial functional complementation in mitochondrial DNA-based diseases. Int. J. Biochem. Cell Biol. 41, 1907–1913 [DOI] [PubMed] [Google Scholar]
- 33. Lee J. Y., Kapur M., Li M., Choi M. C., Choi S., Kim H. J., Kim I., Lee E., Taylor J. P., Yao T. P. (2014) MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J. Cell Sci. 127, 4954–4963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen S., Owens G. C., Makarenkova H., Edelman D. B. (2010) HDAC6 regulates mitochondrial transport in hippocampal neurons. PloS One 5, e10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen H., Vermulst M., Wang Y. E., Chomyn A., Prolla T. A., McCaffery J. M., Chan D. C. (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pham A. H., Meng S., Chu Q. N., Chan D. C. (2012) Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 21, 4817–4826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ellis C. E., Murphy E. J., Mitchell D. C., Golovko M. Y., Scaglia F., Barceló-Coblijn G. C., Nussbaum R. L. (2005) Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking α-synuclein. Mol. Cell. Biol. 25, 10190–10201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dauer W., Kholodilov N., Vila M., Trillat A. C., Goodchild R., Larsen K. E., Staal R., Tieu K., Schmitz Y., Yuan C. A., Rocha M., Jackson-Lewis V., Hersch S., Sulzer D., Przedborski S., Burke R., Hen R. (2002) Resistance of α-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U. S. A. 99, 14524–14529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Musgrove R. E., King A. E., Dickson T. C. (2013) α-Synuclein protects neurons from apoptosis downstream of free-radical production through modulation of the MAPK signalling pathway. Neurotox. Res. 23, 358–369 [DOI] [PubMed] [Google Scholar]
- 40. Lim K. L., Chew K. C., Tan J. M., Wang C., Chung K. K., Zhang Y., Tanaka Y., Smith W., Engelender S., Ross C. A., Dawson V. L., Dawson T. M. (2005) Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25, 2002–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tan J. M., Wong E. S., Kirkpatrick D. S., Pletnikova O., Ko H. S., Tay S. P., Ho M. W., Troncoso J., Gygi S. P., Lee M. K., Dawson V. L., Dawson T. M., Lim K. L. (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 17, 431–439 [DOI] [PubMed] [Google Scholar]
- 42. Hao R., Nanduri P., Rao Y., Panichelli R. S., Ito A., Yoshida M., Yao T. P. (2013) Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 51, 819–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miyakawa S., Ogino M., Funabe S., Uchino A., Shimo Y., Hattori N., Ichinoe M., Mikami T., Saegusa M., Nishiyama K., Mori H., Mizuno Y., Murayama S., Mochizuki H. (2013) Lewy body pathology in a patient with a homozygous parkin deletion. Movement Disorders 28, 388–391 [DOI] [PubMed] [Google Scholar]
- 44. Irrcher I., Aleyasin H., Seifert E. L., Hewitt S. J., Chhabra S., Phillips M., Lutz A. K., Rousseaux M. W., Bevilacqua L., Jahani-Asl A., Callaghan S., MacLaurin J. G., Winklhofer K. F., Rizzu P., Rippstein P., Kim R. H., Chen C. X., Fon E. A., Slack R. S., Harper M. E., McBride H. M., Mak T. W., Park D. S. (2010) Loss of the Parkinson's disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 19, 3734–3746 [DOI] [PubMed] [Google Scholar]
- 45. Kim K. Y., Stevens M. V., Akter M. H., Rusk S. E., Huang R. J., Cohen A., Noguchi A., Springer D., Bocharov A. V., Eggerman T. L., Suen D. F., Youle R. J., Amar M., Remaley A. T., Sack M. N. (2011) Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Investig. 121, 3701–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]