

Background: Proteases cleave protease-activated receptor-2 (PAR2), which activates transient receptor potential (TRP) ion channels to cause inflammation and pain.
Results: Neutrophil elastase cleaves PAR2, resulting in Gαs-mediated cAMP formation, transient receptor potential vanilloid 4 (TRPV4) activation, and sensitization of nociceptive neurons, inflammation, and pain.
Conclusion: Elastase causes PAR2- and TRPV4-mediated inflammation and pain.
Significance: PARs and TRP channels mediate responses to diverse proteases.
Keywords: cell signaling, G protein-coupled receptor (GPCR), inflammation, protease, transient receptor potential channels (TRP channels), biased agonism, nociception, protease-activated receptor
Abstract
Proteases that cleave protease-activated receptor-2 (PAR2) at Arg36↓Ser37 reveal a tethered ligand that binds to the cleaved receptor. PAR2 activates transient receptor potential (TRP) channels of nociceptive neurons to induce neurogenic inflammation and pain. Although proteases that cleave PAR2 at non-canonical sites can trigger distinct signaling cascades, the functional importance of the PAR2-biased agonism is uncertain. We investigated whether neutrophil elastase, a biased agonist of PAR2, causes inflammation and pain by activating PAR2 and TRP vanilloid 4 (TRPV4). Elastase cleaved human PAR2 at Ala66↓Ser67 and Ser67↓Val68. Elastase stimulated PAR2-dependent cAMP accumulation and ERK1/2 activation, but not Ca2+ mobilization, in KNRK cells. Elastase induced PAR2 coupling to Gαs but not Gαq in HEK293 cells. Although elastase did not promote recruitment of G protein-coupled receptor kinase-2 (GRK2) or β-arrestin to PAR2, consistent with its inability to promote receptor endocytosis, elastase did stimulate GRK6 recruitment. Elastase caused PAR2-dependent sensitization of TRPV4 currents in Xenopus laevis oocytes by adenylyl cyclase- and protein kinase A (PKA)-dependent mechanisms. Elastase stimulated PAR2-dependent cAMP formation and ERK1/2 phosphorylation, and a PAR2- and TRPV4-mediated influx of extracellular Ca2+ in mouse nociceptors. Adenylyl cyclase and PKA-mediated elastase-induced activation of TRPV4 and hyperexcitability of nociceptors. Intraplantar injection of elastase to mice caused edema and mechanical hyperalgesia by PAR2- and TRPV4-mediated mechanisms. Thus, the elastase-biased agonism of PAR2 causes Gαs-dependent activation of adenylyl cyclase and PKA, which activates TRPV4 and sensitizes nociceptors to cause inflammation and pain. Our results identify a novel mechanism of elastase-induced activation of TRPV4 and expand the role of PAR2 as a mediator of protease-driven inflammation and pain.
Introduction
Protease-activated receptors (PARs)2 are a unique family of four G protein-coupled receptors that are activated by extracellular and membrane-tethered proteases (1). Proteases such as thrombin and trypsin cleave at specific sites within the extracellular N terminus of PAR1, PAR2, and PAR4, which reveals tethered ligand domains that bind to and activate the cleaved receptors. When activated by this canonical mechanism, PAR1, PAR2, and PAR4 predominantly couple to Gαq and β-arrestins, leading to downstream signaling events that mediate hemostasis, inflammation, pain, and repair mechanisms (2, 3).
PAR2 is an important mediator of protease-driven inflammation and pain. PAR2 is expressed by epithelial, endothelial, and smooth muscle cells in multiple tissues, as well as by cells of the immune system and the central and peripheral nervous systems (4). Proteases that activate PAR2 on nociceptive neurons can stimulate the release of substance P and calcitonin gene-related peptide, which cause neurogenic inflammation in peripheral tissues and mediate pain transmission in the dorsal horn of the spinal cord (5, 6). PAR2 sensitizes transient receptor potential (TRP) ion channels of nociceptors, including TRP vanilloid 1 (TRPV1) (7), TRPV4 (8–10), and TRP ankyrin A1 (TRPA1) (11), which amplifies the proinflammatory and hyperalgesic actions of proteases.
Pancreatic trypsin I and II, the most widely studied and first identified agonists of PAR2, cleave the receptor at Arg36↓Ser37, to reveal the tethered ligand SLIGKV (in human) (4, 12). Trypsin-activated PAR2 couples to Gαq, leading to mobilization of intracellular Ca2+ and activation of second messenger kinases such as protein kinase C (PKC) and D (PKD) (8, 13, 14). Trypsin-activated PAR2 also recruits G protein receptor kinase-2 (GRK2) and β-arrestins, which uncouple PAR2 from G proteins and mediate clathrin- and dynamin-dependent receptor endocytosis (15, 16). By recruiting PAR2 and Src to endosomal signalosomes, β-arrestins can mediate the activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) within the cytosol (15).
Although pancreatic trypsins effectively activate PAR2, the identity of the proteases that activate PAR2 under patho-physiological conditions is uncertain. In particular, given the widespread distribution of PAR2 and the restricted expression of trypsin I/II to the pancreas, where activity is tightly controlled by endogenous inhibitors, the proteases that activate PAR2 in tissues other than the pancreas and intestine remain to be identified. Other proteases that can cleave PAR2 at Arg36↓Ser37 include trypsin IV (mesotrypsin) (17, 18), tryptase (19, 20), coagulation factors VIIa and Xa (21), acrosin (22), granzyme A (23), membrane-type serine protease 1 or matriptase (24), TMPRSS2 (25), and kallikrein 2, 4, 5, 6, and 14 (26–29). These proteases would be expected to trigger the same canonical signaling events as trypsin I/II, leading to similar physiological outcomes. Alternatively, proteases that cleave PAR2 at different sites can either remove the trypsin-revealed tethered ligand domain and thereby prevent activation by the canonical mechanism (i.e. disarm the receptor), or can activate distinct signaling mechanisms (i.e. biased agonism). Such proteases include cathepsin S, which cleaves at Gly41↓Lys42 (30) and Glu56↓Thr57 (31), neutrophil elastase, which cleaves at Ser67↓Val68 (32), cathepsin G, which cleaves at Phe64↓Ser65 (32), and proteinase 3, which cleaves at Val61↓Asp62 (32). However, the precise molecular mechanisms and the physiological consequences of biased protease signaling are poorly defined.
We investigated the mechanisms and patho-physiological outcomes of neutrophil elastase-induced biased agonism of PAR2. Elastase is one of the major proteases released from infiltrating neutrophils in inflamed tissues. Given its high circulating concentration (up to 1 μm) and long half-life (6–8 h) during inflammation, elastase has been proposed as a target for anti-inflammatory therapy (33). Elastase is a biased agonist of both PAR1 and PAR2, but by distinctly different mechanisms. Elastase cleaves PAR1 at Leu45↓Arg46, distal to the thrombin cleavage site, which reveals a tethered ligand domain (RNPNDKYEPF-NH2) that activates Gαi/o-mediated ERK signaling (34). Elastase cleaves PAR2 at Ser67↓Val68, distal to the trypsin cleavage site, which activates PAR2 by a mechanism that does not involve exposure of a tethered ligand domain (32). However, the functional importance of the elastase-biased agonism of PAR2 is uncertain.
Given the important proinflammatory and pro-nociceptive actions of elastase, PAR2, and TRP channels, we investigated whether the elastase-biased agonism of PAR2 activates TRPV4 and causes inflammation and pain. Our results reveal that elastase-activated PAR2 robustly couples to Gαs, leading to a PKA-dependent activation of TRPV4 and hypersensitivity of nociceptive neurons, and PAR2- and TRPV4-mediated inflammatory edema and mechanical hyperalgesia.
Experimental Procedures
Animals
Institutional animal ethics committees approved all experiments. C57BL/6 mice, Par2−/− and Par2+/+ littermates (35) and Trpv4+/+ and Trpv4−/− littermates (36) (8–12 weeks, male) were studied. Mice were maintained under temperature- (22 ± 4 °C) and light- (12-h light/dark cycle) controlled conditions with free access to food and water. Oocytes were collected from Xenopus laevis as described (37).
Materials
Human sputum elastase was from SERVA Electrophoresis GmbH (10 units/mg) for oocyte experiments, and from Elastin Products Company Ltd. (864 units/mg) for other experiments. AlphaScreen SureFire phosphor-ERK and cAMP activity assays were from PerkinElmer Life Sciences Inc. Coelenterazine H was from Nanolight Technology, Prolume Ltd. Adenylyl cyclase inhibitor SQ22536 and PKC inhibitor GF109203X were from Cayman Chemicals. Unless otherwise indicated, other reagents were from Sigma.
Generation of cDNA Constructs, and Cell Culture
Generation of human PAR2 and human TRPV4 constructs for expression in X. laevis oocytes have been described (38). PAR2 constructs for expression in mammalian cells have been described (9, 15). Human embryonic kidney (HEK) 293 cells and sarcoma virus-transformed rat kidney epithelial (KNRK) cells were maintained in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. Generation and maintenance of HEK293 and KNRK cells stably expressing human PAR2 constructs have been described (12, 15, 39, 40).
Elastase Cleavage of N-terminal PAR2 Fragments
Peptides corresponding to N-terminal fragments of human PAR2 (100 μm) were incubated with elastase (10 units/ml (390 nm)) in Hanks' balanced salt solution, pH 7.4, for 1, 15, or 60 min at 37 °C. Reactions were quenched with an equal volume of 50% acetonitrile and 0.1% trifluoroacetic acid in H2O. The reaction products were separated by reverse phase high pressure liquid chromatography using a Phenomenex Luna 3-μm C8 column (100 Å, 100 × 2 mm) with a gradient of 0 to 60% acetonitrile in 0.05% trifluoroacetic acid over 10 min. Products were identified by mass spectrometry using a Shimadzu LCMS 2000.
Signaling Assays in Cell Lines
For measurement of [Ca2+]i, KNRK-PAR2 or KNRK-empty vector control (VC) cells were seeded into 96-well poly-d-lysine-coated plates at a density of 30,000 cells/well 16–24 h before the assay. Cells were loaded with Fura-2/AM (1 μm) in assay buffer (150 mm NaCl, 2.6 mm KCl, 0.1 mm CaCl2, 1.18 mm MgCl2, 10 mm d-glucose, 10 mm HEPES, pH 7.4) containing 4 mm probenecid and 0.5% BSA for 1 h at 37 °C. Fluorescence was measured at 340 and 380 nm excitation and 530 nm emission using a FlexStation Microplate Reader (Molecular Devices). After a baseline reading for 60 s, cells were exposed to trypsin (1 × 10−4–30 units/ml (1 × 10−12–10−7 m)), elastase (1 × 10−4–30 units/ml (1.2 × 10−11–1.2 × 10−6 m)), or ATP (10 μm, positive control). Peak responses to each agonist were measured. For cAMP accumulation assays, CAMYEL BRET sensors were used (41). KNRK-PAR2 or KNRK-VC cells were plated into 10-cm dishes the day before transfection, and transfected with 4 μg of cDNA encoding CAMYEL sensor (YFP-Epac-RLuc). After 24 h, cells were seeded in poly-d-lysine-coated 96-well plates and cultured overnight. Medium was replaced with Hanks' balanced salt solution 30 min before assays. Cells were loaded with coelenterazine H (5 μm), RLuc8 luminescence (480 nm) and Venus/YFP fluorescence (530 nm) were measured for a 2–3 min baseline and at various times after incubation with trypsin or elastase using LUMIstar Omega (BMG Labtech). Forskolin (10 μm) was used as a positive control.
Bioluminescence Resonance Energy Transfer (BRET) Analysis of PAR2 Association with Heterotrimeric G Proteins
HEK293 cells were transiently transfected with PAR2-RLuc8 (0.18 μg), Gγ2-Venus (0.4 μg), Gβ1 (0.266 μg) and either Gαq or Gαs (0.266 μg) using PEI in 6-well dishes. After 24 h, cells were seeded into poly-d-lysine-coated 96-well plates and the ligand-induced BRET signal was measured as described above (41–43).
BRET Analysis of PAR2 Association with GRKs, β-Arrestins, and Plasma Membrane Proteins
HEK293 cells were transiently transfected with PAR2-RLuc8 (1 μg) and GRK2-Venus, GRK6-Venus, β-arrestin1-YFP, β-arrestin2-YFP, RIT-Venus, or KRas-Venus (4 μg) using PEI in 10-cm dishes. After 24 h, cells were seeded in 96-well plates. After 48 h, cells were equilibrated in Hanks' balanced salt solution for 30 min at 37 °C, and the ligand-induced BRET signal was measured as described above.
Two-electrode Voltage Clamp Studies of X. laevis Oocytes
Oocytes were obtained from adult X. laevis as described (38). Defolliculated stage V–VI oocytes were injected (Nanoject II automatic injector, Drummond) with 0.5 ng of TRPV4 cRNA alone, 10 ng of PAR2 cRNA alone, or both TRPV4 and PAR2 cRNA. Oocytes were studied 2 days after injection using the two-electrode voltage-clamp technique as described (37, 38, 44). A Ca2+-free solution was used to prevent activation of endogenous Ca2+-activated chloride channels by TRPV4-mediated Ca2+ influx and to delay a Ca2+-induced decay of TRPV4 current (31, 45). Oocytes were voltage-clamped at −60 mV. Oocytes were incubated with trypsin (2.48 units/ml (8 nm)), elastase (1 units/ml (3 μm)), or vehicle, and were then challenged with the TRPV4 agonist GSK1016790A (50 nm) and the TRPV4 antagonist HC067047 (100 nm).
Signaling Assays in Neurons
Dorsal root ganglia (DRG) (C1-L5) from C57BL/6 wild-type, PAR2−/− or Trpv4−/− mice were isolated as described (31). Neurons were incubated in serum-free medium overnight before ERK1/2 and cAMP assays. For cAMP accumulation assays, neurons were preincubated with 3-isobutyl-1-methylxanthine (1 mm) for 45 min before expose to agonists. Neurons were challenged with trypsin (10 units/ml (34.7 nm)), elastase (1 units/ml (39 nm) or 10 units/ml (390 nm)), forskolin (10 μm, cAMP positive control), or phorbol 12,13-dibutyrate (200 nm, ERK1/2 positive control) for 45 (cAMP assays) or 30 min (ERK1/2 assays) at 37 °C. cAMP accumulation was measured using AlphaScreen cAMP assay and ERK1/2 activity was measured using AlphaScreen SureFire phosphor-ERK assay (PerkinElmer Life Sciences). For measurement of [Ca2+]i, neurons were loaded with Fura-2/AM (2 μm) for 1.5 h at 37 °C, and the cells were observed using a Leica DMI6000B microscope with a PL APO ×20 NA0.75 objective. Fluorescence was measured at 340 and 380 nm excitation with 530 nm emission using an Andor iXon 887 camera (Andor) and MetaFluor version 7.8.0 software (Molecular Devices). Neurons were challenged sequentially with either trypsin (10 units/ml (34.7 nm)) or elastase (10 units/ml (390 nm)), capsaicin (1 μm), and KCl (50 mm). In some experiments, neurons were assayed in Ca2+-free buffer containing 2 mm EDTA. Neurons were also treated with inhibitors of PKA (PKI, 10 μm), adenylyl cyclase (SQ22536, 20 μm), PKC (GF109203X, 1 μm), or Rho kinase (Y-27632, 10 μm) (60 min preincubation and present through the experiments). Images were analyzed using a custom journal in MetaMorph version 7.8.2 software (Molecular Devices). A maximum intensity image was generated and projected through time to generate an image of all cells. Cells were segmented and binarized from this image using the Multi Wavelength Cell Scoring module on the basis of size and fluorescence intensity. Neurons of interest (<25 μm diameter) were selected.
Hyperexcitability of Nociceptive Neurons
DRG (T9-T13) from C57BL/6 mice were dispersed and cultured overnight (31). Cells were preincubated with elastase (10 units/ml (390 nm)) for 1 h. The PKA inhibitors PKI (10 μm), Rho kinase inhibitor (Y-27632, 10 μm), or the PKC inhibitor GF109203X (1 μm) were applied 30 min before elastase. Perforated patch clamp recordings were made from small-diameter neurons (<30 pF capacitance) in current clamp mode at room temperature as described (31). Changes in excitability were quantified by measuring rheobase and numbers of action potentials discharged at twice rheobase.
Mechanical Hyperalgesia and Edema in Mice
For behavioral assessments, C57BL/6 wild-type, PAR2−/− or Trpv4−/− mice were acclimatized to the experimental room, restraint apparatus, and investigator for 2-h periods on 2 successive days before experiments, and the investigator was blinded to the experimental treatments. von Frey filaments were used to determine mechanical pain response as described (31). An increase in the filament stiffness required to induce paw withdrawal indicates mechanical analgesia, whereas a decrease in the filament stiffness required to induce withdrawal indicates mechanical hyperalgesia. To assess inflammatory edema of the paw, hind paw thickness was measured using digital calipers before and after treatments (46). Baseline von Frey scores were taken the day before the experiments. To examine the effects of elastase, mice were sedated with 5% isoflurane and elastase (100 units/ml (3.9 μm), 10 μl) or vehicle (0.9% NaCl, 10 μl) was injected subcutaneously into the plantar surface of one hind paw. Mechanical hyperalgesia and edema were measured between 30 and 240 min after intraplantar injections.
Statistical Analyses
Results are expressed as mean ± S.E. Differences between two groups were examined using unpaired t-tests. Differences between multiple groups were examined using an analysis of variance and a Bonferroni's or Dunnett's post hoc test. A p value <0.05 was considered to be significant.
Results
Elastase Cleaves PAR2 at Ala66↓Ser67 and Ser67↓Val68
To identify the sites at which elastase cleaves human PAR2, we incubated elastase (10 units/ml (390 nm)) with two synthetic peptides spanning the N terminus of PAR2 including trypsin cleavage site (Arg36↓Ser37), cathepsin S cleavage site (Glu56↓Thr57), and the remaining extracellular domain up to the first transmembrane domain (residues 31–75) (Fig. 1A). Elastase rapidly cleaved PAR2 peptide 2 (residues 61–75) but not PAR2 peptide 1 (residues 31–60) (Fig. 1, B–D). We observed ∼50% degradation of peptide 2 at 15 min and almost complete degradation after 60 min (Fig. 1D). The products that were identified by mass spectrometry are consistent with elastase cleaving human PAR2 at Ala66↓Ser67 and Ser67↓Val68 (Fig. 1B). This finding is consistent with a previous report in which a lower concentration of elastase was used to cleave PAR2 fragments (32).
FIGURE 1.

Elastase cleavage of N-terminal fragments of human PAR2. A, N-terminal domain of human PAR2 showing the synthetic peptides (peptide 1, blue; peptide 2, red) used for cleavage studies. Arrows indicate the established trypsin and cathepsin S (Cat-S) cleavage sites, and the elastase cleavage sites identified in this study. B, elastase degradation of peptide 2 (residues 61–75). The high pressure liquid chromatograms showing the elution of the substrate and products (p) after incubation with elastase for 0 or 60 min. Mass spectrometry analysis identify 4 products, consistent with cleavage at Ala66↓Ser67 and Ser67↓Val68. C, peptide 1 (residues 31–60) was not degraded after incubation with elastase for 60 min. D, peptide 2 (residues 61–75) was rapidly degraded generating products after incubation with elastase for 15 or 60 min. n = 3 experiments.
Elastase Stimulates PAR2-dependent cAMP Accumulation but Not Ca2+ Mobilization
Elastase cleavage of PAR2 at Ser67↓Val68 stimulates Gα12/13- and Rho kinase-dependent phosphorylation of ERK1/2 (32). Whether elastase activates other signaling pathways is uncertain. Cathepsin S, a lysosomal protease that is also a biased agonist of PAR2, stimulates Gαs-mediated cAMP accumulation (31). To investigate whether elastase also stimulates PAR2-dependent cAMP activation, we compared responses of KNRK cells expressing empty vector control (KNRK-VC) or human PAR2 (KNRK-hPAR2). As previously reported, trypsin stimulated a concentration-dependent increase in [Ca2+]i in KNRK-PAR2 cells but not KNRK-VC cells (12, 31) (Fig. 2, A and B). Conversely, elastase had no effect on [Ca2+]i at any concentration in both cell lines (Fig. 2, A and B).
FIGURE 2.
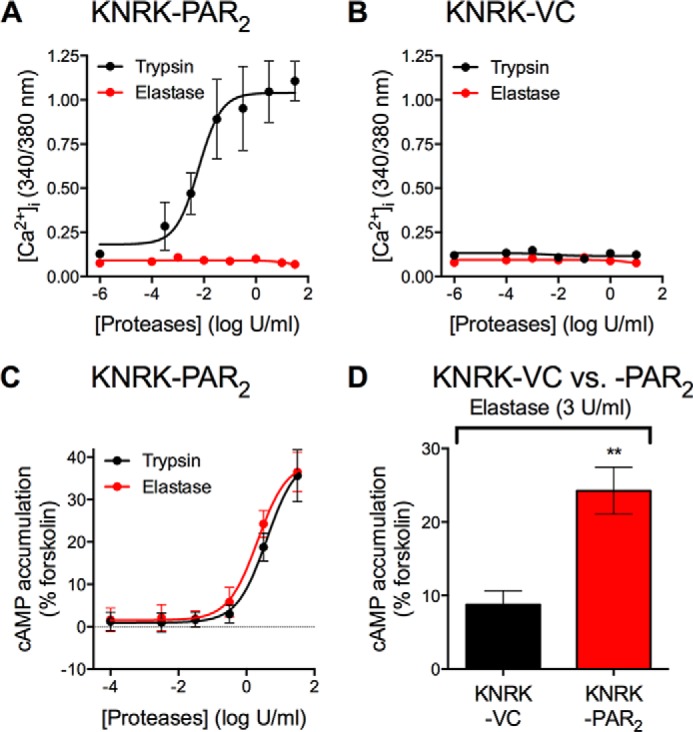
PAR2-dependent [Ca2+]i mobilization and cAMP formation. A and B, effects of graded concentrations of trypsin or elastase on [Ca2+]i mobilization in KNRK-PAR2 (A) and KNRK-VC (B) cells. C and D, effects of graded concentrations of trypsin or elastase on cAMP formation in KNRK-PAR2 cells or KNRK-VC cells. Data were normalized to forskolin control. Peak values from indicated concentrations were plotted. Triplicate measurements of n = 3–4 experiments are shown. **, p < 0.01, unpaired t test.
We studied cAMP activation in response to trypsin or elastase in KNRK-VC and KNRK-PAR2 cells transiently transfected with a CAMYEL YFP-Epac-RFP cAMP sensor (41). Both trypsin and elastase stimulated a concentration-dependent increase in cAMP formation in KNRK-PAR2 cells but not KNRK-VC cells with similar potency (EC50: trypsin, 3.90 ± 0.22 units/ml (12.33 ± 0.22 nm); elastase, 2.13 ± 0.21 units/ml (80.90 ± 0.21 nm)) (Fig. 2, C and D). Thus, elastase stimulates PAR2-dependent cAMP accumulation but not Ca2+ mobilization in KNRK cells.
Elastase Induces PAR2 Coupling to Gαs but Not Gαq
cAMP formation may due to direct activation of Gαs, which activates adenylyl cyclase, or a result of indirect activation of other signaling pathways, such as the Gq pathway via activation of Ca2+-sensitive adenylyl cyclases (47). As an indicator of direct activation of G proteins, we used BRET to examine the change in proximity between PAR2 and heterotrimeric G proteins in HEK293 cells (31). We transiently expressed PAR2-RLuc8 with Gβ1, Gγ2-Venus and different Gα proteins, including Gαs and Gαq. Changes in BRET ratios between PAR2-RLuc8 and Gγ2-Venus were monitored in cells treated with trypsin or elastase. Trypsin (10 units/ml (34.7 nm)) stimulated and increased the BRET signal in cells expressing either Gαq or Gαs (Fig. 3, A and B). In contrast, elastase (10 units/ml (390 nm)) only induced a change in BRET in cells expressing Gαs but not Gαq (Fig. 3, A and B). These findings are consistent with our signaling data showing that trypsin-activated PAR2 mobilizes Ca2+ and generates cAMP, whereas elastase-activated PAR2 generates cAMP but does not mobilize Ca2+. They suggest that elastase activates PAR2-dependent Gαs signaling that leads to cAMP formation. Notably, in Gαs-expressing cells, whereas trypsin increased BRET, elastase decreased BRET. This observation suggests that trypsin-activated PAR2 adopts a different conformation from elastase-activated PAR2 relative to G proteins. The Gγ2-Venus fluorescence was similar in all experiments, suggesting comparable levels of expression (Fig. 3C).
FIGURE 3.
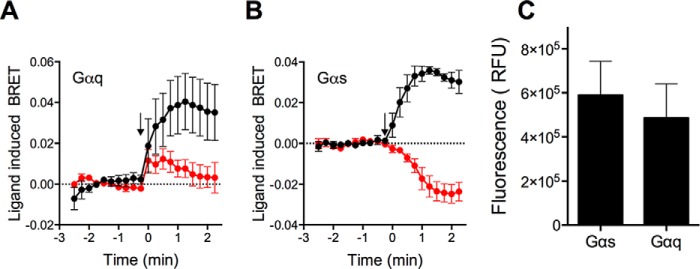
PAR2 and G protein associations. A and B, PAR2-RLuc8 and Gγ-Venus were co-expressed in HEK293 cells with Gβ1 and various Gα subunits. The effects of trypsin or elastase (arrow) on BRET ratios between PAR2-RLuc8 and Gγ-Venus in cells overexpressing Gαq (A) or Gαs (B) were measured. C, Gγ-Venus fluorescence levels were measured. Duplicate measurements of n = 4 experiments are shown.
Elastase Recruits GRK6 but Not GRK2 or β-Arrestin to PAR2
Trypsin-activated PAR2 associates with β-arrestins, which mediate PAR2 desensitization and endocytosis (12, 15, 39). β-Arrestins also recruit Src and PAR2 to endosomes, where it assembles a signalosome that activates ERK1/2 in the cytosol (15). GRKs can phosphorylate activated GPCRs and thereby promote receptor interaction with β-arrestins (48). We examined whether elastase-biased agonism of PAR2 leads to differential GRK and β-arrestin recruitment. We expressed in HEK293 cells PAR2-RLuc8, GRK2-Venus, GRK6-Venus, β-arrestin1-YFP, or β-arrestin2-YFP, and examined BRET after stimulation of cells with trypsin or elastase. Trypsin (10 units/ml (34.7 nm)) but not elastase (10 units/ml (390 nm)) increased BRET between PAR2-RLuc8, GRK2-Venus, β-arrestin1-YFP, and β-arrestin2-YFP (Fig. 4, A, C, and D). These results support the hypothesis that GRK2 phosphorylates trypsin-activated PAR2, which induces β-arrestin recruitment, receptor desensitization, and internalization. Although elastase failed to recruit GRK2 and β-arrestins to PAR2, both elastase and trypsin caused a decrease in BRET between PAR2-RLuc8 and GRK6-Venus (Fig. 4B). Although further studies are required to determine the importance of the potential association of PAR2 with GRK6, it is possible that GRK6 may play an important regulatory role for elastase-activated PAR2.
FIGURE 4.
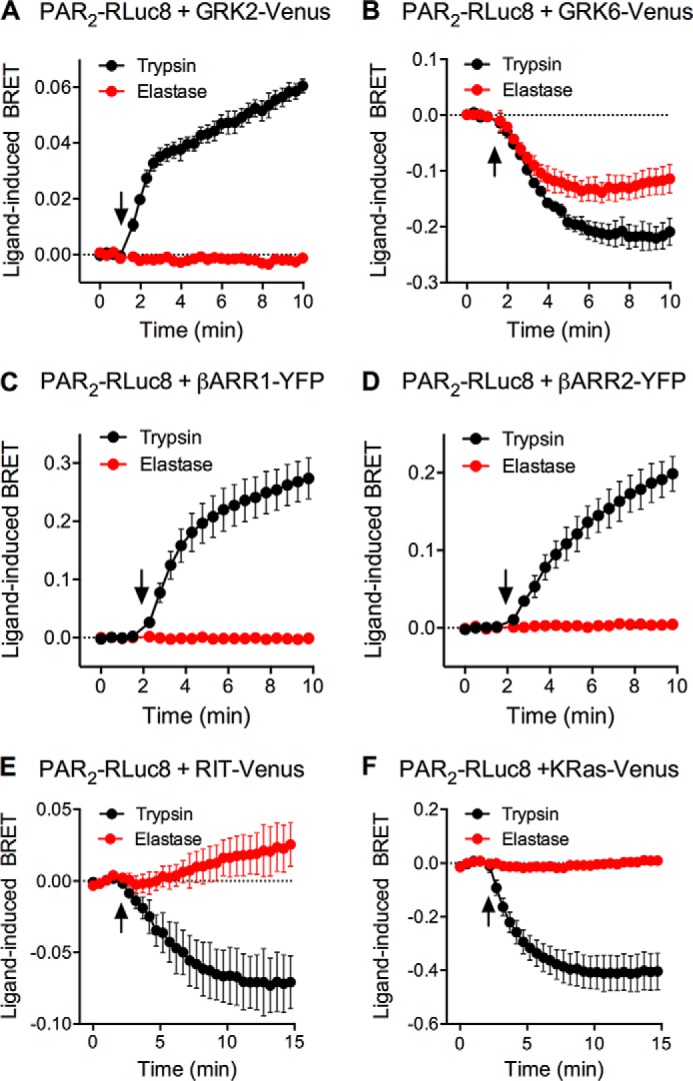
PAR2 association with GRKs, β-arrestins, and plasma membrane proteins. PAR2-RLuc8 was co-expressed in HEK293 cells with GRK2-Venus (A), GRK6-Venus (B), β-arrestin1-YFP (C), β-arrestin2-YFP (D), RIT-Venus (E), or KRas-Venus (F). The effects of trypsin or elastase (arrow) on BRET ratios were measured. Triplicate measurements of n = 3–4 experiments are shown.
Previous studies using immunofluorescence and confocal microscopy suggest that whereas trypsin-activated PAR2 undergoes endocytosis, elastase-activated PAR2 remains at the plasma membrane (32, 49). To quantitatively assess PAR2 trafficking at the plasma membrane, we measured the BRET signal between PAR2-RLuc8 and two proteins that reside at the plasma membrane: KRas-Venus, which is present in cholesterol-independent microdomains (50), and RIT-Venus, which is uniformly distributed. Trypsin (10 units/ml (34.7 nm)) but not elastase (10 units/ml (390 nm)) induced a rapid decrease in BRET between PAR2-RLuc8 and both RIT-Venus and KRas-Venus (Fig. 4, D and E). These results are consistent with the observation that trypsin- but not elastase-activated PAR2 couples to β-arrestins and internalizes.
Elastase Evokes PAR2-dependent Sensitization of TRPV4 Currents
After activation by canonical agonists such as trypsin, or biased agonists such as cathepsin S, PAR2 activates and sensitizes TRPV4, which contributes to neurogenic inflammation and mechanical hyperalgesia (8, 31). To evaluate whether elastase-activated PAR2 sensitizes TRPV4, we expressed TRPV4 alone or both PAR2 and TRPV4 together in X. laevis oocytes, and measured whole cell currents using the two-electrode voltage-clamp technique. Preincubation of oocytes expressing PAR2 and TRPV4 with elastase (1 units/ml (3 μm)) for 5 min resulted in a 6-fold increase in the response to the TRPV4-selective agonist GSK1016790A (50 nm) compared with vehicle (Fig. 5, A and B, second and fourth columns). In contrast, elastase did not affect the response to GSK1016790A in oocytes expressing TRPV4 but not PAR2 (Fig. 5B, first and third columns). These results suggest that elastase induces a PAR2-dependent sensitization of TRPV4 in X. laevis oocytes, as we have previously described (38). To evaluate the mechanism of this sensitization, we preincubated oocytes with a PKC inhibitor GF109203X (1 μm), adenylyl cyclase inhibitor SQ22536 (20 μm), or PKA inhibitor PKI (10 μm). SQ22536 and PKI prevented elastase-induced and PAR2-mediated sensitization of TRPV4, whereas GF109203X partially inhibited sensitization (Fig. 5, A and B, fifth to seventh columns). All three inhibitors had no effect on GSK1016790A-stimulated TRPV4 currents in oocytes expressing TRPV4 alone (data not shown). In control experiments, elastase activation of the epithelial sodium channel was preserved in the presence of protein kinase inhibitors (data not shown). Thus, the inhibitory effect of the kinase inhibitors on the PAR2-mediated sensitization of TRPV4 by elastase is not due to an inhibition of enzymatic activity. These results suggest that adenylyl cyclase and PKA are critically important for elastase-stimulated and PAR2-dependent sensitization of TRPV4, and are consistent with the capacity of elastase-activated PAR2 to couple to Gαs and cAMP accumulation.
FIGURE 5.

Elastase-induced sensitization of TRPV4 and disarming of PAR2. A and B, X. laevis oocytes co-expressing TRPV4 and PAR2 or expressing TRPV4 alone were preincubated with vehicle or elastase for 5 min. Elastase-treated oocytes were treated with vehicle or inhibitors of adenylyl cyclase (SQ22536), PKA (PKI), or PKC (GF109203X). Oocytes were treated with the TRPV4 agonist GSK1016790A (GSK) and the TRPV4 antagonist HC067047 (HC) to study TRPV4 currents. A, representative traces from oocytes co-expressing TRPV4 and PAR2. B, mean ΔIGSK1016790A values of pooled data from oocytes expressing TRPV4 alone or TRPV4 and PAR2. C, oocytes expressing PAR2 were preincubated with vehicle (veh) or elastase for 5 min, washed (W), and trypsin-evoked whole cell currents were measured. Columns represent mean ΔItrypsin values. n indicates number of individual oocytes measured. N indicates the number of batches of oocytes. ***, p < 0.001, unpaired t test; §§§, p < 0.0001, unpaired t test.
Because elastase cleaves PAR2 distal from the trypsin site, elastase can remove the trypsin-exposed tethered ligand and disarm the receptor to subsequent activation by trypsin (32, 49). To determine whether elastase also disarms PAR2 in oocytes, we expressed PAR2 and examined trypsin-evoked PAR2 activation of whole cell currents. In oocytes preincubated with vehicle, trypsin (2.48 units/ml (8 nm)) stimulated a transient inward current, consistent with the activation of Ca2+-sensitive Cl− channels (Fig. 5C). Non-injected oocytes showed no or minimal response to trypsin (38). Preincubation with elastase (1 units/ml (3 μm), 5 min) inhibited trypsin-evoked currents by ∼80% (Fig. 5C). Thus, elastase disarms PAR2 by removing the tethered ligand.
Elastase Activates Endogenous PAR2 and TRPV4 in DRG Neurons
Proteases can activate PAR2 on nociceptive neurons to stimulate the release of neuropeptides in peripheral tissues, resulting in neurogenic inflammation, and in the dorsal horn of the spinal cord, leading to pain transmission (5, 6). PAR2 activates TRP channels, which amplify the proinflammatory and pro-nociceptive actions of proteases (8, 11, 13). We have previously reported that proteases that activate PAR2 by canonical mechanisms, such as trypsins and tryptase (6, 7), and biased mechanisms, including cathepsin S (31), can signal to nociceptors by activating PAR2 and TRP channels. To determine whether elastase similarly signals to nociceptors, we assessed cAMP accumulation, ERK1/2 activation, and [Ca2+]i in mouse DRG neurons in short term culture.
In DRG from wild-type mice, trypsin (10 units/ml) and elastase (1 or 10 units/ml) stimulated ERK1/2 activation (Fig. 6A). Elastase robustly stimulated ERK1/2 activation to a similar degree at both 1 (39 nm) and 10 units/ml (390 nm). Elastase (10 units/ml) did not stimulate ERK1/2 activation in DRG from Par2−/− mice, but instead inhibited ERK1/2 activation by mechanisms that remain to be elucidated (Fig. 6B). Similarly, trypsin and elastase (both 10 units/ml) stimulated cAMP accumulation in DRG from wild-type mice (Fig. 6C), and the stimulatory effect of elastase was not observed in DRG from Par2−/− mice (Fig. 6D). These results suggest that elastase stimulates ERK1/2 activation and cAMP accumulation in DRG by a PAR2-dependent process, consistent with our current observations and with published studies showing similar stimulator actions of elastase in KNRK-PAR2 but not KNRK-VC cells (32).
FIGURE 6.
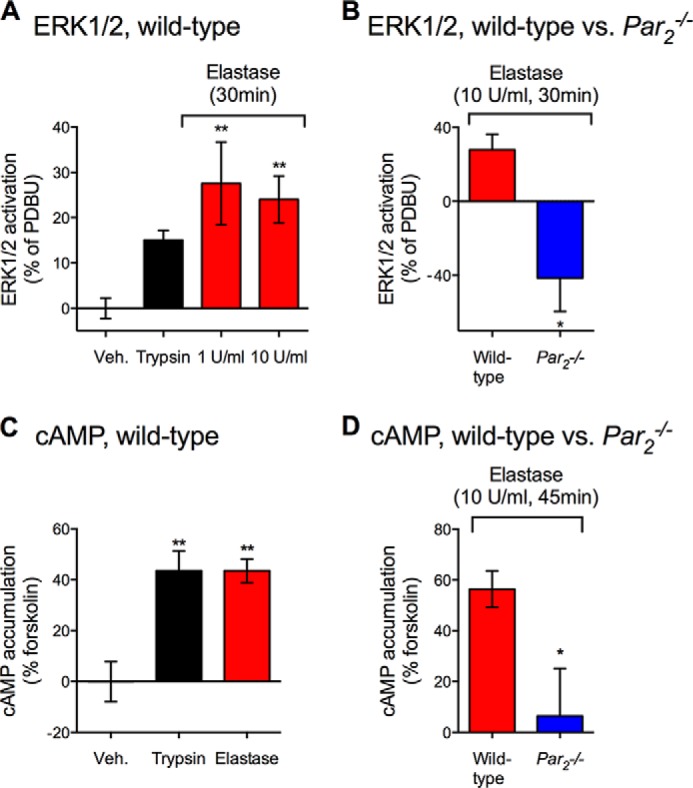
Elastase-evoked ERK1/2 activation and cAMP accumulation in DRG. The effects of trypsin and elastase on ERK1/2 activation (A and B) and cAMP accumulation (C and D) in DRG from wild-type and Par2−/− mice. *, p < 0.05; **, p < 0.01 unpaired t test (B and D) one-way analysis of variance (A and C) compared with control groups.
To investigate whether elastase cleavage of PAR2 leads to activation of TRP channels, we measured [Ca2+]i in small to medium diameter neurons. Elastase (10 units/ml) stimulated a rapid and sustained increase in [Ca2+]i in neurons from wild-type mice (Fig. 7A). Responses to elastase were detected in ∼50% of these neurons, similar to the proportion of neurons responding to trypsin (Fig. 7B). The magnitude of response to elastase was markedly diminished in neurons from Par2−/− mice (Fig. 7C) and Trpv4−/− mice (Fig. 7D). Fewer neurons from Par2−/− mice and Trpv4−/− mice responded to elastase (Fig. 7E). Removal of extracellular Ca2+ ions strongly inhibited the proportion of elastase-responsive neurons in wild-type mice (Fig. 7F). Our results suggest that elastase activates PAR2 on nociceptors, which in turn activates TRPV4, allowing the influx of extracellular Ca2+ ions. The residual responses in mice lacking PAR2 may be attributable to activation of other PARs, including PAR1, which is also expressed by nociceptors (51) and which responds to elastase (34), or due to elastase activation of ion channels, such as the epithelial sodium channel (52, 53). Residual responses in TRPV4-deficient mice may be due to PAR2-dependent activation of other TRPs, such as TRPV1 or TRAPA1 (7, 11).
FIGURE 7.

Elastase-evoked Ca2+ influx in DRG neurons. [Ca2+]i was measured in individual small to medium diameter DRG neurons. A, C, and D, representative traces from 15 to 20 individual neurons from wild-type (A), PAR2−/− (C), or Trpv4−/− (D) mice challenged with elastase (arrow). B and E–H, proportion of neurons responding to elastase or trypsin from wild-type mice (B, F–H) or PAR2−/− or Trpv4−/− mice (E). F, proportion of elastase-responsive neurons in 2 mm Ca2+ or Ca2+-free buffer. G, proportion of elastase-responsive neurons treated with vehicle, PKA inhibitor PKI, adenylyl cyclase inhibitor SQ22536, PKC inhibitor GF109203X or Rho kinase inhibitor Y27632. H, proportion of neurons responding to elastase with or without preincubation with elastase inhibitor elafin. *, p < 0.05; **, p < 0.01, unpaired t test (F and H), and one-way analysis of variance (E and G) compared with vehicle or wild-type groups.
To characterize the signaling mechanism underlying elastase-stimulated and PAR2-dependent activation of TRPV4, we pretreated neurons with inhibitors of adenylyl cyclase, PKA, PKC, or Rho kinase. The adenylyl cyclase inhibitor SQ22536 (20 μm), the PKA inhibitor PKI (10 μm), and the Rho kinase inhibitor Y27362 (10 μm) all reduced the percentage of neurons responsive to elastase, whereas the PKC inhibitor GF109203X (1 μm) had no effect (Fig. 7G). These results suggest that elastase-cleaved PAR2 activates TRPV4 in nociceptors by Gαs-, adenylyl cyclase-, and PKA-dependent mechanisms, as well as by a Rho kinase-dependent process. They are consistent with the role of adenylyl cyclase and PKA in mediating elastase-stimulated and PAR2-dependent sensitization of TRPV4 in X. laevis oocytes. Our findings also agree with the reports that elastase-activated PAR2 stimulates Rho kinase (32), and that Rho kinase contributes to PAR2-mediated TRPV4 sensitization in oocytes (38). Preincubation of elastase with its specific inhibitor elafin (10 μm, 30 min) abolished elastase stimulation of [Ca2+]i in nociceptors, indicating a requirement for proteolytic activity (Fig. 7H).
Elastase Evokes PKA-dependent, and PKC- and Rho Kinase-independent Hyperexcitability of Nociceptive Neurons
Hypersensitivity of nociceptors can lead to exacerbated pain transmission. We have previously shown that canonical agonists, such as trypsin and tryptase (54, 55) and biased agonists, such as cathepsin S (31), cause hyperexcitability of DRG neurons from mice. To directly determine whether elastase induces hyperexcitability, we made patch clamp recordings from small diameter DRG neurons in short term culture. We assessed excitability by measuring the rheobase (minimum current required to fire a single action potential) and the action potential discharge frequency at twice rheobase. Preincubation with elastase (10 units/ml (390 nm), 1 h) resulted in a >50% reduction in rheobase, which is indicative of hyperexcitability (Fig. 8, A and B). Elastase-evoked hyperexcitability was abolished by the PKA inhibitor PKI (10 μm), but unaffected by the PKC inhibitor GF109203X (1 μm) or the Rho kinase inhibitor Y-27632 (10 μm) (Fig. 8, A and B). Elastase did not affect the frequency of action potential discharge at the current to twice rheobase (data not shown). Our results suggest that elastase causes hyperexcitability of nociceptive neurons by a PKA-dependent process, consistent with elastase stimulation of PAR2-dependent cAMP accumulation in DRG.
FIGURE 8.
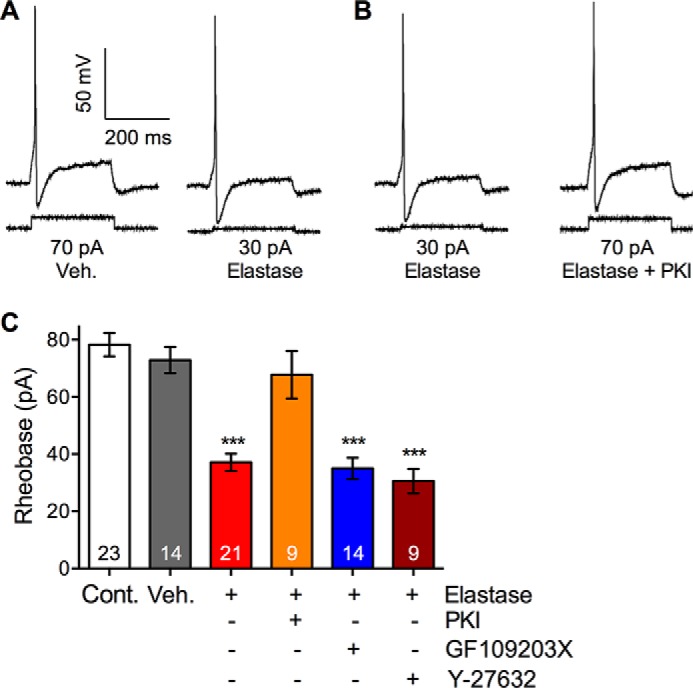
Elastase-evoked and PKA-dependent hyperexcitability of nociceptive neurons. Small diameter DRG neurons were preincubated with vehicle (Veh.) or elastase for 60 min. Rheobase was measured to assess hyperexcitability. In some experiments, neurons were preincubated with PKA inhibitor PKI, or PKC inhibitor GF109203X. or Rho kinase inhibitor Y27632. A and B, representative traces of rheobase. C, pooled results showing rheobase. Numbers of neurons are shown in bars. ***, p < 0.001 elastase compared with vehicle.
Elastase Evokes Inflammation and Mechanical Hyperalgesia by Activating PAR2 and TRPV4
Both canonical agonists, such as trypsin and tryptase, and biased agonists, such as cathepsin S, can activate PAR2 and TRP channels at the periphery terminals of primary nociceptive neurons, which causes neurogenic inflammation and hyperalgesia (5–8, 31). We investigated whether elastase, as a biased agonist for PAR2, induces inflammation and pain by activating PAR2 and TRPV4. We administered elastase (100 units/ml (3.9 μm), 10 μl) to mice by intraplantar injection, and measured mechanical pain using calibrated von Frey filaments applied to the plantar surface of the paw, and assessed inflammatory edema by measuring paw thickness with calipers. In wild-type mice, elastase induced marked mechanical hyperalgesia that was sustained for at least 4 h (Fig. 9A). Elastase also induced a >3-fold increase in paw thickness that declined after 1 h but was sustained for at least 4 h (Fig. 9B). Deletion of Par2 attenuated elastase-evoked mechanical hyperalgesia and inflammatory edema. Deletion of Trpv4 attenuated elastase-induced inflammation to a similar degree as deletion of Par2. However, Trpv4 deletion caused a greater reduction in elastase-induced hyperalgesia than Par2 deletion. Thus, both PAR2 and TRPV4 contribute to elastase-evoked inflammation and pain. The residual pain and inflammation observed in Par2−/− and Trpv4−/− mice may be attributable to elastase activation of other PARs or ion channels.
FIGURE 9.
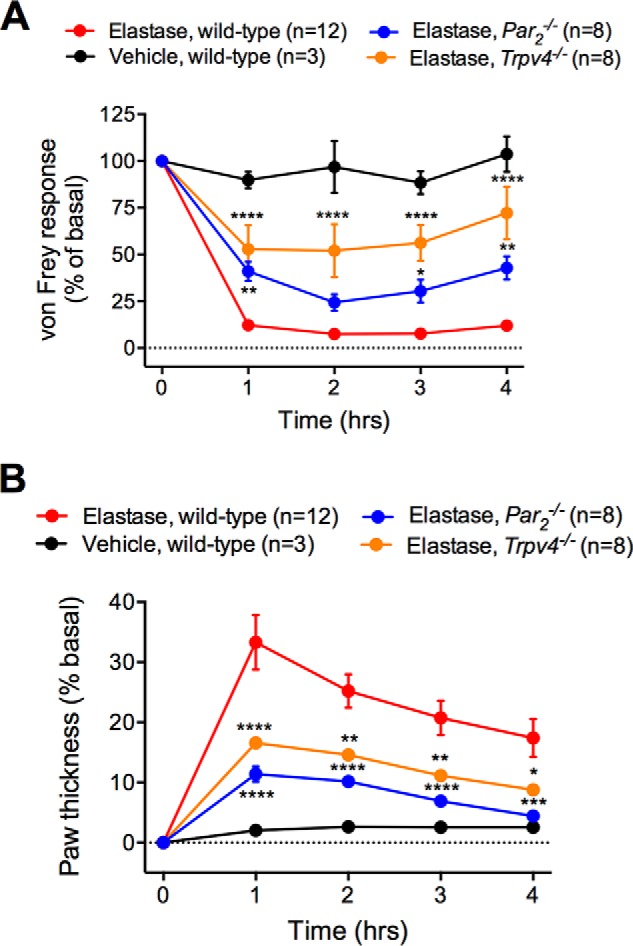
Elastase-evoked and PAR2- and TRPV4-dependent inflammation and pain. Elastase or vehicle was administered by intraplantar injection to wild-type, PAR2−/−, and Trpv4−/− mice. A, mechanical hyperalgesia was measured using von Frey filaments. B, paw edema was assessed by measurement of paw thickness with calipers. *, p < 0.05; ****, p < 0.0001 two-way analysis of variance compared with vehicle. n = 3–12 mice.
Discussion
We report that neutrophil elastase is a biased agonist of PAR2 that stimulates Gαs-mediated cAMP accumulation and ERK1/2 activation in cell lines expressing human PAR2 and in DRG neurons from mice. In X. laevis oocytes and mouse nociceptive neurons, elastase activation of PAR2 sensitizes and activates TRPV4 by adenylyl cyclase- and PKA-dependent mechanisms. Elastase causes PKA-dependent hyperexcitability of nociceptors, and evokes PAR2- and TRPV4-mediated mechanical hyperalgesia and inflammation.
Elastase Is a Biased Agonist of PAR2 That Stimulates a Distinct Signaling Profile
We observed that elastase-activated PAR2 generates a signaling profile that is distinctly different from that evoked by trypsin. By using BRET to examine the proximity of PAR2 to Gγ in HEK293 cells, we found that elastase stimulated Gαs- but not Gαq-dependent BRET, whereas trypsin stimulated a Gαs- and Gαq-dependent BRET. These results suggest that whereas elastase-activated PAR2 couples to Gαs alone, trypsin-activated PAR2 couples to Gαs and Gαq. Our BRET results are consistent with the observations that elastase stimulates accumulation of cAMP but not mobilization of intracellular Ca2+ in KNRK-PAR2 cells, whereas trypsin stimulates both cAMP accumulation and Ca2+ mobilization. These responses are PAR2-dependent, because elastase and trypsin did not affect cAMP or Ca2+ levels in KNRK-VC cells. Our results confirm the inability of elastase to mobilize intracellular Ca2+ in KNRK-PAR2 cells (32). Elastase also stimulates a PAR2-dependent activation of ERK1/2 in KNRK-PAR2 cells that is blocked by a Rho kinase inhibitor, suggesting a Gα12/13-mediated mechanism (32). Considered together, our findings are consistent with the proposal that elastase is a biased agonist of PAR2 that stimulates PAR2-coupling to Gαs, accumulation of cAMP, and activation of ERK1/2, but which is unable to evoke Gαq-mediated Ca2+ signaling. In this regard, elastase resembles cathepsin S, another PAR2 biased agonist that selectively stimulates coupling to Gαs, cAMP accumulation, but not Ca2+ mobilization (31).
The structural basis that underlies the different signaling properties of trypsin-, elastase-, and cathepsin S-cleaved PAR2 remains to be determined. However, because these proteases cleave PAR2 at distinct sites, and promote activation by different mechanisms, differences in receptor conformation are likely to underlie these divergent signaling mechanisms. Whereas trypsin cleaves human PAR2 at Arg36↓Ser37 to reveal the tethered ligand SLIGKV (4), and cathepsin S cleaves at Glu56↓Thr57 to expose the tethered ligand TVFSVDEFSA, elastase acts distal to both the trypsin and cathepsin S sites at Ser67↓Val68 (32). Given the close proximity of the elastase site to the first transmembrane domain, it is not surprising that elastase activates PAR2 by a mechanism that does not involve exposure of a tethered ligand (32). The observation in Gαs-expressing cells that elastase and trypsin induced opposite changes in BRET between PAR2-RLuc8 and Gγ-Venus suggests that elastase-cleaved PAR2 adopts a different conformation from trypsin-cleaved PAR2 relative to Gγ. BRET analysis also suggest that cathepsin S-cleaved PAR2 adopts a different conformation from the trypsin-cleaved receptor (31). Whether these differences underlie the biased agonism of elastase and cathepsin S remains to be determined.
Elastase cleavage of PAR2 was first reported in vitro in Escherichia coli expressing the N-terminal domain of the receptor (56). Because elastase cleaves PAR2 distal to the trypsin cleavage site, elastase pretreatment can prevent subsequent activation by trypsin, and thereby “disarm” the receptor (49, 56). We found that pretreatment with elastase prevented trypsin signals in oocytes, which confirms this mechanism. Cathepsin S can similarly disarm PAR2 in both mammalian cells and oocytes (31).
Regulation and Trafficking of Elastase-activated PAR2
After activation by trypsin, PAR2 is phosphorylated by second messenger kinases and GRKs, and associates with β-arrestins, which mediate receptor desensitization and endocytosis (15, 39). We observed that trypsin stimulated a large and sustained increase in BRET between PAR2 and GRK2 and β-arrestins, consistent with GRK2 and β-arrestin recruitment. This recruitment coincided with a decreased BRET between PAR2 and the resident plasma membrane proteins RIT and K-Ras, suggesting trypsin-induced endocytosis of PAR2. Conversely, we observed that elastase-activated PAR2 was unable to recruit GRK2, interact with β-arrestins, or undergo endocytosis. The lack of GRK2 recruitment by elastase-activated PAR2 is consistent with the lack of β-arrestin recruitment, which is known to rely on GRK-mediated phosphorylation of agonist-occupied receptors (57). Our findings support a previous report that elastase-activated PAR2 is unable to recruit β-arrestins (32). Cathepsin S-activated PAR2 also fails to recruit β-arrestins and does not internalize (31). Further studies are required to define the mechanisms that regulate signaling of elastase- and cathepsin S-activated PAR2.
The inability of elastase to promote the recruitment of GRK2 and β-arrestins to PAR2 and to stimulate receptor endocytosis is likely to affect signaling of elastase-activated PAR2. Trypsin-activated PAR2 undergoes β-arrestin-dependent endocytosis in KNRK-PAR2 cells. β-Arrestins assemble an endosomal signalosome comprising PAR2, Raf, and MEKK that is necessary for activation and cytosolic retention of ERK1/2 (15). A mutant PAR2 that is unable to associate with β-arrestins and internalize activates nuclear ERK1/2 by a mechanism that involves transactivation of the epidermal growth factor receptor. Because elastase-cleaved PAR2 neither recruits β-arrestins nor internalizes, it may only activate nuclear ERK1/2 but not cytosolic ERK1/2. Further experiments will be required to investigate the mechanisms by which elastase-activated PAR2 stimulates ERK1/2, and to determine the importance of PAR2 trafficking for protease-evoked inflammation and pain.
Because elastase was unable to stimulate GRK2 translocation to PAR2 at the plasma membrane, we investigated the possibility that elastase alters the potential association of PAR2 with plasma membrane-localized GRKs. Of 7 known GRKs, GRK4, GRK5, and GRK6 are primarily localized to plasma membrane (58, 59), and GRK6 is expressed in spinal cord and enteric neurons that also express PAR2 (60–62). Deletion of GRK6 leads to an increase in capsaicin-induced post-colitis hyperalgesia, suggesting that GRK6, like PAR2, contributes to post-inflammatory pain (62). Here we show that both trypsin and elastase lead to a decrease in BRET between PAR2 and GRK6. These data suggests that GRK6 contributes to PAR2 regulation or signaling in response to proteases that activate the receptor by canonical and biased mechanisms. Because GRK6 is primarily located at the plasma membrane, a decrease in the BRET signal may reflect PAR2 moving away from the membrane. However, this possibility is unlikely because elastase does not cause PAR2 internalization, demonstrated by both immunofluorescence (32) and our BRET analysis of the association of PAR2 with plasma membrane-resident proteins.
Elastase Activates Nociceptive DRG Neurons, and Leads to PAR2- and TRPV4-dependent Inflammation and Pain
Our results show that elastase stimulates cAMP accumulation and ERK1/2 activation in DRG from wild-type but not Par2−/− mice, which is consistent with our findings in KNRK-PAR2 and KNRK-VC cells. However, in marked contrast to observations in KNRK-PAR2 cells, in which elastase failed to increase [Ca2+]i, elastase stimulated a robust increase in [Ca2+]i in DRG neurons from wild-type mice. Elastase-evoked Ca2+ signals in neurons from wild-type mice were strongly inhibited by removal of extracellular Ca2+ ions, and also suppressed by deletion of Par2 or Trpv4. These results are consistent with the proposal that elastase-activated PAR2 triggers the activation of TRPV4, which allows influx of extracellular Ca2+ ions. Residual responses to elastase in Par2−/− and Trpv4−/− mice may be due to activation of other PARs and TRP channels.
We used pharmacological inhibitors to characterize the mechanism by which elastase-activated PAR2 may stimulate TRPV4 in DRG neurons. Adenylyl cyclase, PKA, and PKC are the major effectors of Gαq and Gαs signaling. To investigate their involvement in elastase-mediated Ca2+ signaling, we treated neurons with inhibitors of adenylyl cyclase, PKA, and PKC. Both adenylyl cyclase and PKA inhibitors attenuated elastase-evoked Ca2+ influx. However, inhibition of PKC did not affect Ca2+ influx. Considered together, these results suggest that in DRG neurons elastase-activated PAR2 causes an adenylyl cyclase- and PKA-dependent activation of TRPV4. They are consistent with the known involvement of PKA in regulating TRPV4 (63), and our observation that cathepsin S also causes a PKA-mediated activation of TRPV4 in DRG neurons (31). PAR2 coupling to TRPV4 has been observed in multiple systems with a variety of PAR2 agonists. However, the mechanism of coupling is agonist-dependent. Trypsin-activated PAR2 stimulates TRPV4 in sensory neurons by PKC- and tyrosine kinase-dependent processes, and by the generation of arachidonic acid metabolites (8, 9). In contrast, diesel exhaust particles can activates the PAR2-TRPV4 axis in airway epithelial cells by a Gi/o and phosphatidylinositide 3-kinase-dependent mechanism (64).
Elastase also increased the excitability of DRG nociceptors, as revealed by a decrease in the input current required to fire action potentials. Although further studies are required to identify the mechanism of elastase-evoked neuronal sensitization, this sensitization was also suppressed by an inhibitor of PKA but unaffected by a PKC inhibitor, in line with our previous observations of cathepsin S-evoked neuronal hyperexcitability (31).
In X. laevis oocytes co-expressing PAR2 and TRPV4, we observed that pretreatment with elastase caused a 6-fold increase in GSK1016790A-stimulated current, indicative of TRPV4 sensitization. This effect was not observed in oocytes expressing TRPV4 alone, and is thus PAR2-dependent. Inhibitors of adenylyl cyclase and PKA abolished elastase-evoked sensitization of TRPV4, which is consistent with observations in neurons. However, in oocytes and PKC the inhibitor also partially inhibited sensitization, whereas a PKC inhibitor had no effect on elastase-induced sensitization of TRPV4 in DRG neurons. Species differences may account for this discrepancy.
We found that intraplantar injection of elastase leads to sustained mechanical hyperalgesia and inflammatory edema. Deletion of Par2 partly blocked elastase-mediated inflammation and pain, whereas deletion of Trpv4 strongly inhibited both pain and inflammation. The residual effects of elastase in Par2−/− and Trpv4−/− mice may be attributable to activation of other PARs and TRP channels that are expressed by nociceptors.
Infiltration of neutrophils is a essential component of the host defense mechanism against invading pathogens during acute inflammation. Proteases such as elastase, proteinase 3, and cathepsin G, which are released from neutrophils during inflammation, mediate killing of invading microorganisms. Our results suggest that proteases such as elastase can also cleave PAR2 and TRPV4 on sensory nerves to induce the acute neurogenic inflammation and pain, which may also serve as an acute protective mechanism.
Acknowledgment
We thank Cameron Nowell for technical assistance.
This work was supported by NHMRC Grants 63303, 1049682, and 1031886, grants from the ARC Centre of Excellence in Convergent Bio-Nano Science and Technology and Monash University (to N. W. B.), a Ph.D. fellowship from the Bayerische Forschungsstiftung (to S. S.), and the Else Kröner-Fresenius-Stiftung (to S. H.).
- PAR2
- protease-activated receptor 2
- TRP
- transient receptor potential
- GRK
- G protein-coupled receptor kinases
- PKA/C
- protein kinase A/C
- DRG
- dorsal root ganglia
- BRET
- bioluminescence resonance energy transfer
- VC
- vector control.
References
- 1. Ossovskaya V. S., Bunnett N. W. (2004) Protease-activated receptors: contribution to physiology and disease. Physiol. Rev. 84, 579–621 [DOI] [PubMed] [Google Scholar]
- 2. Vergnolle N. (2009) Protease-activated receptors as drug targets in inflammation and pain. Pharmacol. Ther. 123, 292–309 [DOI] [PubMed] [Google Scholar]
- 3. Macfarlane S. R., Seatter M. J., Kanke T., Hunter G. D., Plevin R. (2001) Proteinase-activated receptors. Pharmacol. Rev. 53, 245–282 [PubMed] [Google Scholar]
- 4. Nystedt S., Emilsson K., Larsson A. K., Strömbeck B., Sundelin J. (1995) Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur. J. Biochem. 232, 84–89 [DOI] [PubMed] [Google Scholar]
- 5. Vergnolle N., Bunnett N. W., Sharkey K. A., Brussee V., Compton S. J., Grady E. F., Cirino G., Gerard N., Basbaum A. I., Andrade-Gordon P., Hollenberg M. D., Wallace J. L. (2001) Proteinase-activated receptor-2 and hyperalgesia: a novel pain pathway. Nat. Med. 7, 821–826 [DOI] [PubMed] [Google Scholar]
- 6. Steinhoff M., Vergnolle N., Young S. H., Tognetto M., Amadesi S., Ennes H. S., Trevisani M., Hollenberg M. D., Wallace J. L., Caughey G. H., Mitchell S. E., Williams L. M., Geppetti P., Mayer E. A., Bunnett N. W. (2000) Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat. Med. 6, 151–158 [DOI] [PubMed] [Google Scholar]
- 7. Amadesi S., Nie J., Vergnolle N., Cottrell G. S., Grady E. F., Trevisani M., Manni C., Geppetti P., McRoberts J. A., Ennes H., Davis J. B., Mayer E. A., Bunnett N. W. (2004) Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J. Neurosci. 24, 4300–4312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grant A. D., Cottrell G. S., Amadesi S., Trevisani M., Nicoletti P., Materazzi S., Altier C., Cenac N., Zamponi G. W., Bautista-Cruz F., Lopez C. B., Joseph E. K., Levine J. D., Liedtke W., Vanner S., Vergnolle N., Geppetti P., Bunnett N. W. (2007) Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J. Physiol. 578, 715–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poole D. P., Amadesi S., Veldhuis N. A., Abogadie F. C., Lieu T., Darby W., Liedtke W., Lew M. J., McIntyre P., Bunnett N. W. (2013) Protease-activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J. Biol. Chem. 288, 5790–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sipe W. E., Brierley S. M., Martin C. M., Phillis B. D., Cruz F. B., Grady E. F., Liedtke W., Cohen D. M., Vanner S., Blackshaw L. A., Bunnett N. W. (2008) Transient receptor potential vanilloid 4 mediates protease activated receptor 2-induced sensitization of colonic afferent nerves and visceral hyperalgesia. Am. J. Physiol. Gastrointest Liver Physiol. 294, G1288–G1298 [DOI] [PubMed] [Google Scholar]
- 11. Dai Y., Wang S., Tominaga M., Yamamoto S., Fukuoka T., Higashi T., Kobayashi K., Obata K., Yamanaka H., Noguchi K. (2007) Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J. Clin. Invest. 117, 1979–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Böhm S. K., Khitin L. M., Grady E. F., Aponte G., Payan D. G., Bunnett N. W. (1996) Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J. Biol. Chem. 271, 22003–22016 [DOI] [PubMed] [Google Scholar]
- 13. Amadesi S., Bunnett N. (2004) Protease-activated receptors: protease signaling in the gastrointestinal tract. Curr. Opin. Pharmacol. 4, 551–556 [DOI] [PubMed] [Google Scholar]
- 14. Amadesi S., Grant A. D., Cottrell G. S., Vaksman N., Poole D. P., Rozengurt E., Bunnett N. W. (2009) Protein kinase D isoforms are expressed in rat and mouse primary sensory neurons and are activated by agonists of protease-activated receptor 2. J. Comp. Neurol. 516, 141–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DeFea K. A., Zalevsky J., Thoma M. S., Déry O., Mullins R. D., Bunnett N. W. (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ayoub M. A., Pin J. P. (2013) Interaction of protease-activated receptor 2 with G proteins and β-arrestin 1 studied by bioluminescence resonance energy transfer. Front. Endocrinol. (Lausanne) 4, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cottrell G. S., Amadesi S., Grady E. F., Bunnett N. W. (2004) Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J. Biol. Chem. 279, 13532–13539 [DOI] [PubMed] [Google Scholar]
- 18. Knecht W., Cottrell G. S., Amadesi S., Mohlin J., Skåregärde A., Gedda K., Peterson A., Chapman K., Hollenberg M. D., Vergnolle N., Bunnett N. W. (2007) Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J. Biol. Chem. 282, 26089–26100 [DOI] [PubMed] [Google Scholar]
- 19. Corvera C. U., Déry O., McConalogue K., Böhm S. K., Khitin L. M., Caughey G. H., Payan D. G., Bunnett N. W. (1997) Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J. Clin. Invest. 100, 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Molino M., Barnathan E. S., Numerof R., Clark J., Dreyer M., Cumashi A., Hoxie J. A., Schechter N., Woolkalis M., Brass L. F. (1997) Interactions of mast cell tryptase with thrombin receptors and PAR-2. J. Biol. Chem. 272, 4043–4049 [DOI] [PubMed] [Google Scholar]
- 21. Camerer E., Huang W., Coughlin S. R. (2000) Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc. Natl. Acad. Sci. U.S.A. 97, 5255–5260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith R., Jenkins A., Lourbakos A., Thompson P., Ramakrishnan V., Tomlinson J., Deshpande U., Johnson D. A., Jones R., Mackie E. J., Pike R. N. (2000) Evidence for the activation of PAR-2 by the sperm protease, acrosin: expression of the receptor on oocytes. FEBS Lett. 484, 285–290 [DOI] [PubMed] [Google Scholar]
- 23. Hansen K. K., Sherman P. M., Cellars L., Andrade-Gordon P., Pan Z., Baruch A., Wallace J. L., Hollenberg M. D., Vergnolle N. (2005) A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc. Natl. Acad. Sci. U.S.A. 102, 8363–8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takeuchi T., Harris J. L., Huang W., Yan K. W., Coughlin S. R., Craik C. S. (2000) Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J. Biol. Chem. 275, 26333–26342 [DOI] [PubMed] [Google Scholar]
- 25. Wilson S., Greer B., Hooper J., Zijlstra A., Walker B., Quigley J., Hawthorne S. (2005) The membrane-anchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem. J. 388, 967–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bledsoe G., Shen B., Yao Y. Y., Hagiwara M., Mizell B., Teuton M., Grass D., Chao L., Chao J. (2008) Role of tissue kallikrein in prevention and recovery of gentamicin-induced renal injury. Toxicol. Sci. 102, 433–443 [DOI] [PubMed] [Google Scholar]
- 27. Oikonomopoulou K., Hansen K. K., Saifeddine M., Vergnolle N., Tea I., Blaber M., Blaber S. I., Scarisbrick I., Diamandis E. P., Hollenberg M. D. (2006) Kallikrein-mediated cell signalling: targeting proteinase-activated receptors (PARs). Biol. Chem. 387, 817–824 [DOI] [PubMed] [Google Scholar]
- 28. Ramsay A. J., Dong Y., Hunt M. L., Linn M., Samaratunga H., Clements J. A., Hooper J. D. (2008) Kallikrein-related peptidase 4 (KLK4) initiates intracellular signaling via protease-activated receptors (PARs). KLK4 and PAR-2 are co-expressed during prostate cancer progression. J. Biol. Chem. 283, 12293–12304 [DOI] [PubMed] [Google Scholar]
- 29. Ramsay A. J., Reid J. C., Adams M. N., Samaratunga H., Dong Y., Clements J. A., Hooper J. D. (2008) Prostatic trypsin-like kallikrein-related peptidases (KLKs) and other prostate-expressed tryptic proteinases as regulators of signalling via proteinase-activated receptors (PARs). Biol. Chem. 389, 653–668 [DOI] [PubMed] [Google Scholar]
- 30. Elmariah S. B., Reddy V. B., Lerner E. A. (2014) Cathepsin S signals via PAR2 and generates a novel tethered ligand receptor agonist. PLoS One 9, e99702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao P., Lieu T., Barlow N., Metcalf M., Veldhuis N. A., Jensen D. D., Kocan M., Sostegni S., Haerteis S., Baraznenok V., Henderson I., Lindström E., Guerrero-Alba R., Valdez-Morales E. E., Liedtke W., McIntyre P., Vanner S. J., Korbmacher C., Bunnett N. W. (2014) Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J. Biol. Chem. 289, 27215–27234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramachandran R., Mihara K., Chung H., Renaux B., Lau C. S., Muruve D. A., DeFea K. A., Bouvier M., Hollenberg M. D. (2011) Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J. Biol. Chem. 286, 24638–24648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Henriksen P. A. (2014) The potential of neutrophil elastase inhibitors as anti-inflammatory therapies. Curr. Opin. Hematol. 21, 23–28 [DOI] [PubMed] [Google Scholar]
- 34. Mihara K., Ramachandran R., Renaux B., Saifeddine M., Hollenberg M. D. (2013) Neutrophil elastase and proteinase-3 trigger G protein-biased signaling through proteinase-activated receptor-1 (PAR1). J. Biol. Chem. 288, 32979–32990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lindner J. R., Kahn M. L., Coughlin S. R., Sambrano G. R., Schauble E., Bernstein D., Foy D., Hafezi-Moghadam A., Ley K. (2000) Delayed onset of inflammation in protease-activated receptor-2-deficient mice. J. Immunol. 165, 6504–6510 [DOI] [PubMed] [Google Scholar]
- 36. Liedtke W., Friedman J. M. (2003) Abnormal osmotic regulation in trpv4−/− mice. Proc. Natl. Acad. Sci. U.S.A. 100, 13698–13703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haerteis S., Krappitz M., Bertog M., Krappitz A., Baraznenok V., Henderson I., Lindström E., Murphy J. E., Bunnett N. W., Korbmacher C. (2012) Proteolytic activation of the epithelial sodium channel (ENaC) by the cysteine protease cathepsin-S. Pflugers Arch. 464, 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sostegni S., Diakov A., McIntyre P., Bunnett N., Korbmacher C., Haerteis S. (2015) Sensitisation of TRPV4 by PAR is independent of intracellular calcium signalling and can be mediated by the biased agonist neutrophil elastase. Pflugers Arch., 467, 687–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Déry O., Thoma M. S., Wong H., Grady E. F., Bunnett N. W. (1999) Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein: β-arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 274, 18524–18535 [DOI] [PubMed] [Google Scholar]
- 40. Dulon S., Leduc D., Cottrell G. S., D'Alayer J., Hansen K. K., Bunnett N. W., Hollenberg M. D., Pidard D., Chignard M. (2005) Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am. J. Respir. Cell Mol. Biol. 32, 411–419 [DOI] [PubMed] [Google Scholar]
- 41. Jensen D. D., Godfrey C. B., Niklas C., Canals M., Kocan M., Poole D. P., Murphy J. E., Alemi F., Cottrell G. S., Korbmacher C., Lambert N. A., Bunnett N. W., Corvera C. U. (2013) The bile acid receptor TGR5 does not interact with β-arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J. Biol. Chem. 288, 22942–22960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Galés C., Rebois R. V., Hogue M., Trieu P., Breit A., Hébert T. E., Bouvier M. (2005) Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2, 177–184 [DOI] [PubMed] [Google Scholar]
- 43. Kocan M., Dalrymple M. B., Seeber R. M., Feldman B. J., Pfleger K. D. (2011) Enhanced BRET technology for the monitoring of agonist-induced and agonist-independent interactions between GPCRs and β-arrestins. Front. Endocrinol. (Lausanne) 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rauh R., Diakov A., Tzschoppe A., Korbmacher J., Azad A. K., Cuppens H., Cassiman J. J., Dötsch J., Sticht H., Korbmacher C. (2010) A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J. Physiol. 588, 1211–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Watanabe H., Vriens J., Janssens A., Wondergem R., Droogmans G., Nilius B. (2003) Modulation of TRPV4 gating by intra- and extracellular Ca2+. Cell Calcium 33, 489–495 [DOI] [PubMed] [Google Scholar]
- 46. Vergnolle N., Cenac N., Altier C., Cellars L., Chapman K., Zamponi G. W., Materazzi S., Nassini R., Liedtke W., Cattaruzza F., Grady E. F., Geppetti P., Bunnett N. W. (2010) A role for transient receptor potential vanilloid 4 in tonicity-induced neurogenic inflammation. Br. J. Pharmacol. 159, 1161–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Willoughby D. (2012) Organization of cAMP signalling microdomains for optimal regulation by Ca2+ entry. Biochem. Soc. Trans. 40, 246–250 [DOI] [PubMed] [Google Scholar]
- 48. Moore C. A., Milano S. K., Benovic J. L. (2007) Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 69, 451–482 [DOI] [PubMed] [Google Scholar]
- 49. Dulon S., Candé C., Bunnett N. W., Hollenberg M. D., Chignard M., Pidard D. (2003) Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am. J. Respir. Cell Mol. Biol. 28, 339–346 [DOI] [PubMed] [Google Scholar]
- 50. Prior I. A., Muncke C., Parton R. G., Hancock J. F. (2003) Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 160, 165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. de Garavilla L., Vergnolle N., Young S. H., Ennes H., Steinhoff M., Ossovskaya V. S., D'Andrea M. R., Mayer E. A., Wallace J. L., Hollenberg M. D., Andrade-Gordon P., Bunnett N. W. (2001) Agonists of proteinase-activated receptor 1 induce plasma extravasation by a neurogenic mechanism. Br. J. Pharmacol. 133, 975–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Caldwell R. A., Boucher R. C., Stutts M. J. (2005) Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am. J. Physiol. Lung. Cell Mol. Physiol. 288, L813–L819 [DOI] [PubMed] [Google Scholar]
- 53. Diakov A., Bera K., Mokrushina M., Krueger B., Korbmacher C. (2008) Cleavage in the γ-subunit of the epithelial sodium channel (ENaC) plays an important role in the proteolytic activation of near-silent channels. J. Physiol. 586, 4587–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kayssi A., Amadesi S., Bautista F., Bunnett N. W., Vanner S. (2007) Mechanisms of protease-activated receptor 2-evoked hyperexcitability of nociceptive neurons innervating the mouse colon. J. Physiol. 580, 977–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reed D. E., Barajas-Lopez C., Cottrell G., Velazquez-Rocha S., Dery O., Grady E. F., Bunnett N. W., Vanner S. J. (2003) Mast cell tryptase and proteinase-activated receptor 2 induce hyperexcitability of guinea-pig submucosal neurons. J. Physiol. 547, 531–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Loew D., Perrault C., Morales M., Moog S., Ravanat C., Schuhler S., Arcone R., Pietropaolo C., Cazenave J. P., van Dorsselaer A., Lanza F. (2000) Proteolysis of the exodomain of recombinant protease-activated receptors: prediction of receptor activation or inactivation by MALDI mass spectrometry. Biochemistry 39, 10812–10822 [DOI] [PubMed] [Google Scholar]
- 57. Gurevich E. V., Tesmer J. J., Mushegian A., Gurevich V. V. (2012) G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Ther. 133, 40–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pitcher J. A., Freedman N. J., Lefkowitz R. J. (1998) G protein-coupled receptor kinases. Annu. Rev. Biochem. 67, 653–692 [DOI] [PubMed] [Google Scholar]
- 59. Stoffel R. H., Randall R. R., Premont R. T., Lefkowitz R. J., Inglese J. (1994) Palmitoylation of G protein-coupled receptor kinase, GRK6. Lipid modification diversity in the GRK family. J. Biol. Chem. 269, 27791–27794 [PubMed] [Google Scholar]
- 60. Zhou Y., Huang X., Wu H., Xu Y., Tao T., Xu G., Cheng C., Cao S. (2013) Decreased expression and role of GRK6 in spinal cord of rats after chronic constriction injury. Neurochem. Res. 38, 2168–2179 [DOI] [PubMed] [Google Scholar]
- 61. Sun B., Gao Y., Lou D., Wu X., Wei H., Wen H., Deng X., Zhang F. (2013) Expression of G-protein-coupled receptor kinase 6 (GRK6) after acute spinal cord injury in adult rat. J. Mol. Histol. 44, 259–270 [DOI] [PubMed] [Google Scholar]
- 62. Eijkelkamp N., Heijnen C. J., Elsenbruch S., Holtmann G., Schedlowski M., Kavelaars A. (2009) G protein-coupled receptor kinase 6 controls post-inflammatory visceral hyperalgesia. Brain Behav. Immun. 23, 18–26 [DOI] [PubMed] [Google Scholar]
- 63. Fan H. C., Zhang X., McNaughton P. A. (2009) Activation of the TRPV4 ion channel is enhanced by phosphorylation. J. Biol. Chem. 284, 27884–27891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li J., Kanju P., Patterson M., Chew W. L., Cho S. H., Gilmour I., Oliver T., Yasuda R., Ghio A., Simon S. A., Liedtke W. (2011) TRPV4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ. Health Perspect. 119, 784–793 [DOI] [PMC free article] [PubMed] [Google Scholar]