

Background: Age and disease affect cardiac expression and localization of Pim1.
Results: Survival and proliferation are enhanced and senescence is antagonized by intracellular targeting of Pim1.
Conclusion: Cardioprotective properties are emphasized by targeting Pim1 expression in human cardiac progenitor cells.
Significance: Compartmentalizing Pim1 to enhance stem cell characteristics can be used for targeted molecular intervention to improve cell-based regenerative therapy.
Keywords: aging, apoptosis, heart failure, proliferation, senescence, Pim1, human cardiac progenitor cell
Abstract
Human cardiac progenitor cells (hCPC) improve heart function after autologous transfer in heart failure patients. Regenerative potential of hCPCs is severely limited with age, requiring genetic modification to enhance therapeutic potential. A legacy of work from our laboratory with Pim1 kinase reveals effects on proliferation, survival, metabolism, and rejuvenation of hCPCs in vitro and in vivo. We demonstrate that subcellular targeting of Pim1 bolsters the distinct cardioprotective effects of this kinase in hCPCs to increase proliferation and survival, and antagonize cellular senescence. Adult hCPCs isolated from patients undergoing left ventricular assist device implantation were engineered to overexpress Pim1 throughout the cell (PimWT) or targeted to either mitochondrial (Mito-Pim1) or nuclear (Nuc-Pim1) compartments. Nuc-Pim1 enhances stem cell youthfulness associated with decreased senescence-associated β-galactosidase activity, preserved telomere length, reduced expression of p16 and p53, and up-regulation of nucleostemin relative to PimWT hCPCs. Alternately, Mito-Pim1 enhances survival by increasing expression of Bcl-2 and Bcl-XL and decreasing cell death after H2O2 treatment, thereby preserving mitochondrial integrity superior to PimWT. Mito-Pim1 increases the proliferation rate by up-regulation of cell cycle modulators Cyclin D, CDK4, and phospho-Rb. Optimal stem cell traits such as proliferation, survival, and increased youthful properties of aged hCPCs are enhanced after targeted Pim1 localization to mitochondrial or nuclear compartments. Targeted Pim1 overexpression in hCPCs allows for selection of the desired phenotypic properties to overcome patient variability and improve specific stem cell characteristics.
Introduction
The mammalian adult heart harbors niches of clonogenic, self-renewing, multipotent, c-kit+ cardiac progenitor cells (CPCs)3 to initiate myocardial repair in the face of physiological stress and oxidative damage (1, 2). An endogenous survival response and secretion of growth factors recruit the resident population of CPCs following myocardial injury to drive proliferation, self-renewal, and differentiation into new cardiomyocytes (3). The endogenous system of repair in the heart is not independently sufficient to effectively repair and regenerate the failing myocardium, and CPC function further decreases with age (4–6). Use of exogenously expanded CPCs to mitigate myocardial damage has demonstrated promising results in animal models and Phase I clinical trials (7–10). Intracoronary injection of autologous CPCs has been validated as a safe and feasible therapeutic option to reduce scar size and significantly improve left ventricular systolic function in patients with ischemic cardiomyopathy in the Phase I SCIPIO clinical trial (10). However, problems with cell survival and engraftment after myocardial infarction reduce the regenerative capability of adoptively transferred CPCs (7–9), highlighting the need for in vitro genetic manipulation to enhance cellular proliferation, survival, engraftment, and commitment of CPCs (11–14).
Work from our laboratory with Pim1 kinase reveals pleiotropic roles to enhance proliferation, survival, metabolism, rejuvenation, and regeneration of cardiomyocytes and stem cells in the myocardium (12, 15–18). Pim1 is a highly conserved serine-threonine kinase and is a downstream target of cardioprotective nuclear Akt accumulation (12). Pim1 regulates cellular processes such as cell cycle progression, survival, telomere preservation, and senescence by interaction, stabilization, and phosphorylation of many downstream targets (14, 16, 17, 19, 20). Pim1 is the main isoform of the kinase in the heart, and expression level and subcellular localization change over the course of cardiovascular development. Pim1 is highly expressed and predominantly localizes to the nucleus of cardiomyocytes in the neonatal heart, mediating rapid proliferation during cardiac development (12). Additionally, Pim1 promotes proliferation through interactions with cell cycle regulators, cyclins, and cyclin-dependent kinases (CDK) in embryonic, hematopoietic, and cardiovascular stem cells (14, 21). During postnatal development, Pim1 expression decreases and translocates to the cytosol of cardiomyocytes. Pim1 expression is reactivated and shuttled to the mitochondria following injury, coinciding with the role of Pim1 in cell survival (12). Pim1 antagonizes the intrinsic pathway of apoptosis in the heart by elevating anti-apoptotic proteins Bcl-2 and Bcl-XL at the mitochondria (14). Collectively, these findings support a pivotal role for Pim1 in preservation of mitochondrial integrity and structure as well as inhibition of apoptotic signaling during acute heart damage (19).
Regeneration of the heart is significantly improved by the application of Pim1-modified CPCs. Pim1 CPCs have enhanced proliferation, survival, metabolic activity, and cardiac commitment, along with reduction in infarct size and improved cardiac function after injection into an infarcted mouse heart (17). Pim1 antagonizes senescence, elongates telomeres, and rejuvenates phenotypically aged stem cells (11, 17, 18). Patients with end-stage heart failure are a major cohort of the target population that would benefit from Pim1-modified human CPC (hCPC)-based regenerative treatment. The role of Pim1 in cardioprotection is expansive; the kinase has a precocious role in heart development and stem cell-based regeneration, and determining functional effects of Pim1 in hCPCs has provided valuable insight regarding enhancement of stem cell-based myocardial regeneration. Characterization of hCPCs from individual patients delineates the unique properties of each patient isolate and can be used to decipher modifications needed to return the cells to a youthful state. Genetic modification with Pim1 represents a proven strategy to restore CPCs for cardiovascular regenerative therapy despite patient diversity or genetic background. The goal of this study was to demonstrate that employing targeted Pim1 preferentially modifies hCPC characteristics based upon internal localization. Specifically, differential regulation of cellular processes dictated through Pim1 in distinct subcellular organelles could provide for tailored molecular intervention in hCPCs. Targeted Pim1 overexpression independently influences proliferation, survival, and senescence, sidestepping variability in stem cell characteristics, growth rates, and regenerative potential of hCPCs and providing an avenue for increased specificity of genetic intervention to augment patient-specific cell-based cardiac regenerative therapy.
Experimental Procedures
Isolation of Human CPCs
Left ventricular heart tissue samples were collected from patients undergoing left ventricular assist device implantation for the isolation of hCPCs as previously described (17, 18). NIH guidelines for human research declare this study protocol approved by the IRB (120686). In brief, the tissue was minced into small pieces, digested in collagenase (Worthington Bio Corp.), cells were incubated with magnetic beads labeled for c-kit (Miltenyi Biotec) and sorted according to the manufacturer's protocol. The pellet was resuspended in hCPC media and plated at 37 °C overnight in a 5% CO2 incubator. hCPCs from multiple patients were screened for differences in growth kinetics, survival, and response to Pim1 overexpression. H10-001 was chosen for the remainder of the study as the cell line to delineate the effects of Pim1 engineering based on its slow proliferation rate and our previous publications characterizing its cellular phenotype (17). Fetal hCPCs were derived from non-surgically obtained second trimester fetal heart tissue purchased from Novogenix Laboratories. Fetal hCPCs utilized in this study are prototypical youthful stem cells, with enhanced proliferation rates, robust cell protection, and decreased expression level of senescence markers (18) (Fig. 1). Patient backgrounds used for isolation of hCPCs used can be found in Table 2.
FIGURE 1.

Characterization of hCPC. A, proliferation rate shows increases in fetal hCPCs versus adult hCPCs on days 2 and 3 as measured by CyQUANT assay. Bars refer to fast-growing fetal hCPCs (black), medium-growth rate adult hCPCs (black stripes), and slow-growing adult hCPCs (gray). B, population doubling time varies in different hCPC lines as measured by CyQUANT assay readings. C, adult hCPC are more susceptible to apoptosis than fetal CPC when treated with 30 μm H2O2 for 3 h as measured by FACS analysis of annexin V and PI staining. D, disparity in endogenous Pim1 expression of adult hCPC represented as fold-change over fetal CPC measured by immunoblot analysis (each sample was normalized to α-tubulin). E, telomere lengths decrease in hCPC as measured by qRT-PCR. F, TERT protein expression measured by immunoblot analysis shows variability in hCPC lines. G, endogenous Pim1 localization, as shown in whole cell versus cytosolic and nuclear subcompartments in fetal versus adult hCPCs, is shown by immunoblot analysis (whole cell and cytosolic fractions were normalized to α-tubulin and nuclear fractions were normalized to Lamin A/C). *, p < 0.05; **, p < 0.01; ***, p < 0.001. Significance is calculated for fetal versus adult hCPC.
TABLE 2.
Clinical profile of hCPC lines isolated from multiple human patients
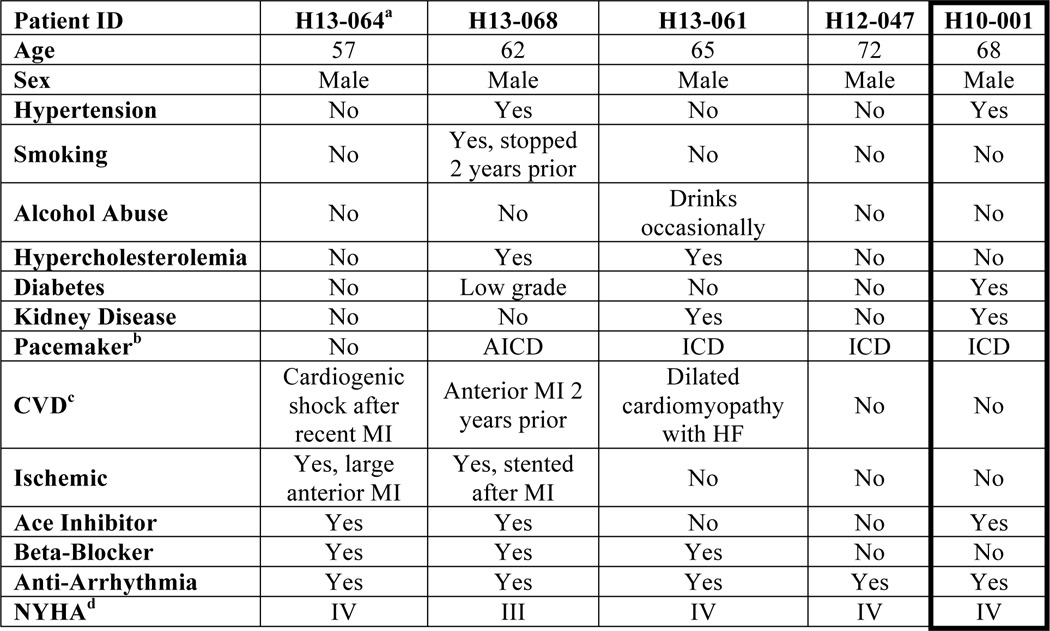
a Patient identification signifies the year the explant was collected (H–) followed by sample number.
b Automatic implantable cardioverter-defibrillator (AICD) and implantable cardioverter-defibrillator (ICD).
c History of cardiovascular disease.
d New York Heart Association Functional Classification (NYHA) describes extent of heart disease.
Lentiviral Vector Generation
Lentiviruses for the control GFP or experimental GFP and Pim1 were cloned with either a mitochondrial targeting sequence or nuclear localization signal for proper intracellular targeting of Pim1 and/or GFP. Sequences were subcloned into a lentiviral backbone by excising from the pShooter vector (Life Technologies, V821-20 and V822-20) with restriction enzymes EcoRI and BsrGI (New England Biolabs, Inc.). After digestion, the pShooter sequences were ligated into a construct (also cut with EcoRI and BsrGI) driven by a myeloproliferative sarcoma virus LTR-negative control region-deleted promoter using the Roche DNA Rapid Ligation Kit and transformed in Bp5α-competent cells (Biopioneer). Proper ligation of backbone and insert was confirmed by PCR and DNA sequencing (Eton Biosciences). Upon translocation, the mitochondrial leader sequence was removed to direct the protein to the mitochondria, and the presence of three nuclear localization sequences direct protein to the nucleus. GFP or GFP and Pim1 were fused to the respective targeting sequences (mitochondrial, MSVLTPLLLRGSTGSARRLPVPRAKIHSL and nuclear, 3× DPKKKRKV) and the c-myc epitope for detection. Lentiviral constructs for expression of GFP and GFP/Pim1 were generated as previously described (18). These constructs were used as controls in this study to compare the effects of targeted versus non-targeted Pim1, in a manner consistent with previous findings (11, 17).
hCPC Transduction
hCPCs were plated in a 6-well plate at a density of 50,000 cells/well and transduced with lentivirus multiplicity of infection of 20. Cells were expanded to generate cell lines expressing GFP (eGFP) or GFP and Pim1 (PimWT), mitochondrial targeted GFP (Mito-GFP) or GFP and Pim1 (Mito-Pim1), nuclear targeted GFP (Nuc-GFP) or GFP and Pim1 (Nuc-Pim1). Efficiency of GFP expression was analyzed by flow cytometry and immunocytochemistry. Up-regulation of Pim1 protein and gene expression was confirmed by immunoblot analysis and real-time quantitative PCR, respectively.
Real Time RT-PCR
Total RNA was isolated from hCPCs with a Quick-RNA MiniPrep kit (Zymo Research) according to the manufacturer's protocol (18). mRNA was reverse transcribed to cDNA with iScript cDNA Synthesis kit (Bio-Rad) and real time PCR was completed in triplicate using iQ SYBR Green (Bio-Rad) on a CFX real time PCR Detection System (Bio-Rad) (18). Samples were normalized to 18S for analysis. Primer sequences are listed in Table 1.
TABLE 1.
Primer sequences used for RT-PCR/qPCR assay
| Sequences | Sequences | ||
|---|---|---|---|
| Pim1 (Fwd) | TGCCATTAGGCAGCTCTCCCCA | Pim1 (Rev) | GCGGCTTCGGCTCGGTCTACT |
| P53 (Fwd) | GCAGCGCCTCACAACCTCCG | p53 (Rev) | TGATTCCACACCCCCGCCCG |
| P16 (Fwd) | AGCATGGAGCCTTCGGCTGA | p16 (Rev) | CCATCATCATGACCTGGATCG |
| NS (Fwd) | GGGAAGATAACCAAGCGTGTG | NS (Rev) | CCTCCAAGAAGTTTCCAAAGG |
| 18S (Fwd) | ACGGACCAGAGCGAAAGCAT | 18S (Rev) | TGTCAATCCTGTCCGTGTCC |
| ERRα (Fwd) | CTTCGCTCCTCCTCTCATC | ERRα (Rev) | CTGGAGTCTGCTTGGAGTTAT |
| TFAM (Fwd) | AATCTGTCTGACTCTGAA | TFAM (Rev) | CACATCTCAATCTTCTACTT |
| NRF1 (Fwd) | TTTGTATGCCTTTGAAGAT | NRF1 (Rev) | AACCTGGATAAGTGAGAC |
Immunocytochemistry of Fixed Cells
hCPCs were plated on 2-chamber permanox slides at a density of 1,500 cells/chamber. Cells were fixed with 4% paraformaldehyde overnight at 4 °C, permeabilized with 0.2% Triton X-100 in PBS for 15 min or 100% methanol for 10 min, and blocked with 10% horse serum in PBS for 1 h. Incubation with primary antibodies was carried out overnight at 4 °C. Slides were washed with PBS, followed by secondary antibody incubation for 1 h. TO-PRO-3 iodide was used to stain nuclei and slides were imaged using a Leica TCS SP8 confocal microscope.
Subcellular Fractionation
To isolate nuclear and cytosolic extracts, hCPCs were washed with ice-cold PBS and collected in 200 μl of ice-cold isolation buffer (70 mm sucrose, 190 mm mannitol, 20 mm HEPES solution, 0.2 mm EDTA solution). Samples were repeatedly plunged through a 100-ml insulin needle to homogenize the collected cells. To obtain nuclear fractions, samples were centrifuged at 800 × g for 15 min, washed with isolation buffer, and centrifuged a second time. Supernatant from the preliminary centrifugation was used as the cytosolic fraction and the final nuclear pellet was resuspended in isolation buffer. Total cytosolic and nuclear protein was measured by immunoblot analysis.
Immunoblot Analysis
hCPC protein lysates were collected on ice in sample buffer (pH 6.8), boiled, and sonicated. Protein was separated on a 4–12% NuPage Novex BisTris gel (Life Technologies), transferred onto an Immobilon-P polyvinylidene fluoride (PVDF) membrane (Millipore), and blocked for 1 h in 5% skim milk in TBST (1% Tris-buffered saline, 0.1% Tween). Membranes were probed with primary antibody overnight at 4 °C, washed three times with TBST solution, and incubated with secondary antibody for 1 h at room temperature. Membranes were imaged on a Typhoon scanner. Primary antibodies used were: Pim1 (39–4600) (Life Technologies, discontinued); GFP (A10262) (Life Technologies); myc tag (2278), Bcl-2 (2872), Bcl-XL (2762), Cyclin D1 (2978), αTubulin (2144), phospho-PRAS40 (Thr-246) (2640), PRAS40 (2691), p16 (F12, sc-1661), p53 (F393, sc-6243), Nucleostemin (His-270, sc67012), β-actin (sc-81178) (Santa Cruz); CDK4 (ab-3112) (Abcam); Lamin A (C-terminal) (L1293) (Sigma).
Senescence-associated β-Galactosidase Staining
Senescence-associated β-galactosidase (SA β-gal) assay (Abcam number ab65351) was used according to the manufacturer's protocol to detect senescent cells. In short, hCPCs at late passage (p13) were plated in 2 chamber permanox slides, fixed, and stained overnight at 37 °C with staining supplement, staining solution, X-Gal (lyophilized) and dimethyl sulfoxide. Images were captured on an Olympus IX70 microscope.
Telomere Length Measurements (RT-PCR)
Genomic DNA was prepared from cultured cells using Macherey-Nagel NucleoSpin Tissue Kit (740952.50), according to the manufacturer's protocol. Modified monochrome multiplex quantitative PCR was used to measure telomere lengths by real time as described previously (22).
Cell Morphology Measurements
hCPCs were plated in 2 chamber permanox slides (10,000 cells/well) and imaged on a Leica TCS SP8 Confocal Laser Scanning Microscope after fixation. Cell morphology was measured using ImageJ tracing software.
Cell Death Assay
hCPCs were plated in a 6-well plate (30,000 cells/well) and incubated overnight. Apoptotic challenge was administered by a overnight serum starvation, followed by a 3-h treatment of 30 μm hydrogen peroxide (H2O2) in low serum media, simulating the oxidative stress seen in an infarct region. Adherent and non-adherent cells were collected and stained per the manufacturer's instructions with Annexin V (AnV) (BD Pharmingen) and propidium iodide (PI) (Life Technologies). The simultaneous use of AnV and PI allowed for quantification of early stage apoptosis (AnV+), late stage apoptosis (AnV/PI+), and necrosis (PI+). Apoptotic cells were distinguished on a BD FACS Canto and data were analyzed by FACS Diva software (BD Biosciences).
CyQUANT Direct Assay
hCPCs were plated in triplicate at 2,000 cells/well in growth media in a 96-well black flat bottom plate with a total volume of 100 μl/well. The cellular DNA content was measured by the binding of a fluorescent dye to indirectly determine the cell number using a CyQUANT Direct Cell Proliferation Assay (Life Technologies) on days 0, 1, 2, and 3 as previously described (18). Population doubling time was determined using the values given during the CyQUANT readings with the formula: N(t) = C2t/d, where N(t) = the number of objects at time t, d = doubling period, C = initial number of objects, and t = time.
ATP Assay (Bioluminescent)
hCPCs were plated in full medium at 100 cells/well in a 96-well flat bottom plate. ATP levels were measured using an ADP/ATP Ratio Assay Kit (Abcam number ab65313) according to the manufacturer's protocol.
MitoSOX Red Assay
hCPCs were plated in a 96-well plate in triplicate at 2000 cells/well. Superoxide production in the mitochondria of hCPCs was recorded with and without rotenone treatment using a MitoSOX Red mitochondrial superoxide indicator (Life Technologies, M36008) per the manufacturer's protocol. Production of reactive oxygen species (ROS) was measured by a plate reader quantified as red fluorescence.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 5 software. When comparing two groups, Student's t test was used. One- or two-way analysis of variance with Tukey's and Bonferroni's post-test, respectively, were used to compare multiple groups. p values less than 0.05 were considered statistically significant, and error bars represent mean ± S.E. Each experiment was repeated at least four times to determine statistical significance.
Results
Characterization of hCPC from Various Patients
Adult hCPCs were isolated from cardiac explants of patients undergoing left ventricular assist device implantation. Proliferation rate, survival, endogenous Pim1 expression level, and telomere lengths of adult versus fetal hCPCs (isolated from second trimester fetal heart samples) were consistent with our previous findings (18). Fetal hCPCs exhibit the highest levels of proliferation, survival, telomere preservation, and possess desirable phenotypic characteristics of youthful stem cells and were thus used as a reference standard (18). The proliferation rate was decreased in multiple adult hCPC lines relative to fetal hCPCs (H13-064, −2.47-fold, p < 0.001; H13-068, −2.73-fold, p < 0.001; H13-061, −2.32-fold, p < 0.001; H12-047, −4.34-fold, p < 0.001; and H10-001, −4.59-fold, p < 0.001), as determined by CyQuant Assay 3 days after plating (Fig. 1A). Consistently, population doubling times of adult hCPCs were significantly longer than fetal hCPCs, ranging from 1.74- to 2.95-fold increases (p < 0.001, Fig. 1B). Susceptibility to H2O2-induced apoptosis was increased in adult hCPCs compared with fetal hCPCs, as determined by the percentage of AnV and PI positive cells (10.13 up to 20.48%, p < 0.01; Fig. 1C). Coincident with increased proliferation and survival rate, fetal hCPCs have elevated endogenous Pim1 levels up to 3.79-fold higher relative to adult hCPCs (Fig. 1D). Fetal hCPCs also exhibit elongated telomeres (9.3 kbp) and elevated expression of TERT (the protein responsible for elongation and maintenance of telomeres) compared with adult hCPCs, which exhibit telomere lengths ranging from 4.24 to 7.39 kbp (Fig. 1, E and F). Cell proliferation, susceptibility to apoptosis, and telomere lengths are characteristics used to assess the “biological age” of human stem cells as can be seen collectively in Fig. 1. Furthermore, fetal hCPCs show increased expression of endogenous Pim1 in the whole cell and cytosolic fraction with slightly elevated levels detected in the nucleus relative to adult hCPCs. Variations in loading were corrected by normalization to α-tubulin and Lamin A/C, respectively. Endogenous Pim1 expression is decreased and mainly localized to the cytosol, whereas nuclear expression is undetectable in adult hCPCs.
Genetic Modification of hCPC with Pim1
hCPCs from line H10-001 were genetically modified to overexpress GFP or Pim1 and GFP throughout the cell (eGFP, PimWT) or targeted to nuclear (Nuc-GFP, Nuc-Pim1) or mitochondrial (Mito-GFP, Mito-Pim1; Fig. 2A) compartments. Efficiency of lentiviral transduction as measured by GFP positive cells was determined by immunocytochemistry (Fig. 2B). Increased PIM1 gene expression in Pim1-modified cell lines was confirmed by qPCR analysis as compared with relative eGFP controls (Fig. 2C). Genetic modification was confirmed by immunocytochemistry showing co-localization of GFP and Pim1 in hCPCs transduced with WT, Mito-Pim, or Nuc-Pim viruses. Additionally, stainings with Phalloidin, Tom20, and DAPI or TOPRO confirmed localization to the cytosol, mitochondria, or nucleus, respectively (Fig. 2D). Overexpression of Pim1, GFP, and Myc tag protein were also confirmed by immunoblot analysis of lysates from engineered hCPCs (Fig. 2E). Collectively, these results demonstrate the efficacy of lentiviral vectors to overexpress whole cell and targeted Pim1 kinase and GFP in hCPCs.
FIGURE 2.

hCPC engineered with Pim1. A, vectors driving expression of GFP (Lv-eGFP), GFP and Pim1 (Lv-PimWT), mitochondrial targeted GFP (Lv-Mito-GFP), mitochondrial targeted GFP and Pim1 (Lv-Mito-Pim1), nuclear-targeted GFP (Lv-Nuc-GFP), and nuclear-targeted GFP and Pim1 (Lv-Nuc-Pim1) were used in this study. B, percentage of GFP-positive hCPCs after lentiviral modification as measured by confocal microscopy. C, increased PIM1 gene expression in Pim1-modified hCPCs as measured by qPCR analysis. All samples normalized to 18S transcription. D, genetic modification confirmed by localization of GFP and/or Pim1 protein expression in engineered hCPCs by immunocytochemistry. E, expression of Pim1 and GFP confirmed by immunoblot analysis. F, kinase activity of Pim1 lentiviral constructs is intact, as shown by phosphorylation of PraS40 Thr-246 in engineered hCPCs. ***, p < 0.001.
Replicative Senescence Is Antagonized by Nuc-Pim1
hCPCs undergo replicative senescence in culture and exhibit increased SA β-gal activity at late passage (p13) (18). At passage 13, hCPC progression through the cell cycle decreases and cells undergo replicative senescence, as evidenced by a high percentage of SA β-gal+ cells after GFP modification. PimWT and Mito-Pim1 modification resulted in a reduction of SA β-gal+ cells (−16.73%, p < 0.001 and −16.44%, p < 0.05, respectively; Fig. 3, A and B). SA β-gal positive cells were least prevalent in Nuc-Pim1 hCPCs, with a −30.25% decrease (p < 0.001) as compared with Nuc-GFP (Fig. 3, A and B). Cell morphology was assessed to examine cell flattening indicative of a post-mitotic senescent cell withdrawn from undergoing cellular growth or proliferation as previously demonstrated (18). Longer, spindle-shaped hCPCs were evident after Nuc-Pim1 modification as measured by the length to width ratio and cell roundness (Fig. 3, C and D). Concurrently, Nuc-Pim1 modification increased the Hayflick limit of hCPCs as compared with PimWT, prolonging the post-mitotic phenotype as demonstrated by the extended expansion capability of modified cells (Fig. 3E).
FIGURE 3.

Replicative senescence is antagonized by Nuc-Pim1. A, SA β-gal staining of engineered hCPC at passage 13. Scale bar = 100 μm. B, graphical representation of percent of SA β-gal positive cells at passage 13. C, Pim1 modification shows decreased roundness and increased length to width ratio of hCPC at passage 13. Image displays characteristic round, flat cell (right) and a long, spindle shaped cell (left). D, passage at which growth arrest occurs in hCPC, indicative of replicative senescence. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Aged Adult hCPCs Are Rejuvenated with Nuc-Pim1
Maintenance of telomere length was observed in PimWT, Mito-Pim1, and Nuc-Pim1 hCPCs with the addition of 0.8 (ns), 1.37 (p < 0.05), and 1.43 (p < 0.05) kbp, respectively (Fig. 4A). TERT is markedly up-regulated in hCPCs modified with Nuc-Pim1 with a 2.96-fold increase (p < 0.001) (Fig. 4B). Gene and protein expression of the cell cycle arrest/senescence marker p53 decreased after Nuc-Pim1 modification with a 1.15-fold reduction of mRNA and a 1.45-fold reduction of protein, as measured by qPCR and immunoblot analysis, respectively (Fig. 4, C and D). Concurrent down-regulation of the p16 gene and protein expression occurred in Nuc-Pim1 hCPCs as evident by 1.85- and 1.96-fold reductions, respectively (Fig. 4, E and F). Overall, the lowest levels for gene and protein expression of senescent markers were consistently observed in Nuc-Pim1 hCPCs. Nucleostemin (Ns), a marker for youthful stem cells, is a nucleolar protein that plays a role in regulation of stem cell self-renewal, genomic stability, telomere preservation, and regeneration of proliferating cells (23–28). A highly significant increase in NS gene expression resulted from Nuc-Pim1 expression in hCPCs (2.59-fold, p < 0.001) compared with PimWT and Mito-Pim1, as measured by qPCR analysis (Fig. 4G). Concurrent up-regulation of Ns protein expression was also detected in Nuc-Pim1 hCPCs (2.17-fold, p < 0.01) compared with controls (Fig. 4H). Collectively, these results corroborate phenotypic characteristics of rejuvenation for adult hCPCs with Nuc-Pim1 modification with a coincident up-regulation of Ns. Nuc-Pim1 preferentially antagonizes senescence, demonstrated here by reduced SA β-gal positive staining, improved cell morphology, elongated telomeres, increased TERT expression, and decreased senescent markers. Up-regulation of Ns correlates with the attenuation of senescence seen in Nuc-Pim1 hCPCs and indicates a phenotypic shift to a more youthful stem cell.
FIGURE 4.

Aged adult hCPC are reverted to youthful phenotypes with nuclear targeted Pim1. A, telomere length measured by RT-qPCR indicates telomere elongation with Pim1 modification. B, increased protein expression of TERT is observed by immunoblot analysis in Nuc-Pim1 hCPC (each sample is normalized to α-Tubulin). C, E, and G, Nuc-Pim1 modification decreases mRNA levels of senescence-associated markers P53 and P16 and increases NS as measured by real time PCR (samples normalized to 18S). D, F, and H, immunoblot analysis for p53, p16, and Ns with graphical representation of fold-change as compared with eGFP, Mito-GFP, and Nuc-GFP (each sample is normalized to β-actin). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Increased Resistance to Apoptosis with Mito-Pim1
Pim1 modification in hCPCs results in increased survival during apoptotic challenge, consistent with previously published findings (17, 18). Survival was compared between hCPCs modified with Pim1-targeted or PimWT constructs utilizing flow cytometric analysis in conjunction with AnV and PI staining. PimWT modification in hCPCs blunted progression from early to late stage apoptosis, evidenced by a decrease of 18.3 to 12.17% (p < 0.05) in AnV/PI+ cells (Fig. 5A). Inhibition of apoptotic shift was more pronounced with Mito-Pim1 modification, with a decrease of 17.58 to 7.58% (p < 0.01) AnV/PI+ cells, and Nuc-Pim1 did not significantly affect survival of hCPCs (Fig. 5, A and B).
FIGURE 5.

Mitochondrial targeted Pim1 enhances survival. A and B, percent annexin V/PI double positive cells after apoptotic stimuli of 30 μm H2O2 for 3 h as measured by FACS analysis. C, Bcl-2 protein expression in untreated control, serum starved and H2O2-treated hCPC by immunoblot analysis. D, protein expression of untreated control, serum starved, and H2O2-treated hCPC by immunoblot analysis. Graph analyses are of fold-change over eGFP, Mito-GFP, or Nuc-GFP untreated control for Bcl-2 and Bcl-XL expression (each sample normalized to β-actin). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Pim1 interacts with pro- and anti-apoptotic signaling molecules at the mitochondria (19). Mito-Pim1 mediated up-regulation of anti-apoptotic Bcl-2 protein as confirmed by immunoblot analysis of hCPCs treated with serum starvation or H2O2. Bcl-2 protein expression was similar in all groups of untreated control and serum-starved hCPCs. Upon treatment with 30 μm H2O2, however, Mito-Pim1 induced a 2.05-fold (p < 0.001) increase in protein expression with a more modest increase in PimWT hCPCs, yielding a 1.38-fold (p < 0.05) increase (Fig. 5C). Nuc-Pim1 did not up-regulate Bcl-2 expression. Coincidently, Bcl-XL expression was induced by serum starvation with a greater fold increase of Bcl-XL in Mito-Pim1 compared with PimWT and Nuc-Pim1 hCPCs, with 1.78- (p < 0.001), 1.53- (p < 0.001), and 1.22-fold (ns) increases, respectively (Fig. 5D). Similar results were evident upon treatment with H2O2, wherein Bcl-XL expression increased 1.73- (p < 0.01) and 1.35-fold (p < 0.01) in Mito-Pim1- and PimWT-modified hCPCs, respectively. Nuc-Pim1 did not cause an increase in Bcl-XL protein expression after H2O2 treatment. Collectively, these findings support mitochondrial localization-enhanced protective effects of Pim1, and induces a greater anti-apoptotic response consistent with up-regulation of Bcl-2 signaling proteins.
Enhanced Proliferation Prompted by Mito-Pim1
The effect of targeted Pim1 upon hCPC proliferation was determined using CyQuant assay. The proliferation rate was increased in all Pim1-modified hCPC lines relative to controls, with Mito-Pim1 showing the most profound effect with a 2.78-fold (p < 0.001) increase on day 3. PimWT and Nuc-Pim1 increased the proliferation rate on day 3 by 1.46- (ns) and 2.21-fold (p < 0.05), respectively (Fig. 6A). Population doubling time as measured by CyQUANT assay readings showed significantly decreased population doubling times by 1.47-fold in PimWT (p < 0.05), 1.94-fold in Mito-Pim1 (p < 0.001), and 1.45-fold (p < 0.05) in Nuc-Pim1 hCPCs relative to GFP modified controls (Fig. 6B).
FIGURE 6.

Growth kinetics are increased by Mito-Pim1. A, increased proliferation rate as measured by CyQUANT analysis after Pim1 modification. B, doubling time decreases, as calculated by CyQUANT measurements, with Pim1 overexpression. C, protein expression of cell cycle regulator Cyclin D as measured by immunoblot analysis. Fold-change as relative to eGFP, Mito-GFP, or Nuc-GFP. D, immunoblot analysis depicts an increase in protein expression of Cdk4 in Mito-Pim1 hCPC relative to controls. E, graphical representation of immunoblot analysis shows fold-change of phospho-Rb relative to total Rb protein expression. Each sample was normalized to β-actin. F, increased ATP levels in PimWT and Mito-Pim1 hCPCs as meansured by a ATP bioluminescence assay. G, difference in ROS levels not apparent with Pim1 modification. H, increase in mitochondrial biogenesis genes is dependent upon cMyc. *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Enhanced proliferation due to mitochondrial localization of Pim1 was confirmed by analysis of cell cycle regulating proteins in modified hCPCs. Transition from G1 to S phase of the cell cycle is driven in part by the Cyclin D/CDK4 complex and inhibited by the tumor suppressor retinoblastoma (Rb) (15, 29). Protein expression of Cyclin D was up-regulated by 1.87- (p < 0.05) and 2.44-fold (p < 0.001) in PimWT and Mito-Pim1 hCPCs, respectively, as revealed by immunoblot analysis (Fig. 6C). Concurrent up-regulation of Cdk4 was observed in Mito-Pim1 hCPCs (2.83-fold, p < 0.001) as compared with PimWT and Nuc-Pim1 (Fig. 6D). PimWT and Mito-Pim1 demonstrated elevated Phospho-Rb, indicating inactivation downstream of Cyclin D (Fig. 6E).
A decrease in energy reserve is reminiscent of loss of mitochondrial function, which can be measured by ATP production. ATP levels were increased by 1.66- and 1.9-fold in PimWT and Mito-Pim1 hCPCs, respectively (Fig. 6F), consistent with previous publications (16, 19) and the premise that Mito-Pim1 maintains mitochondrial function. Reactive oxygen species (ROS) levels did not vary between engineered hCPC samples with and without rotenone treatment as measured by superoxide production via MitoSOX Red assay (Fig. 6G). Concurrent with mitochondrial function, Mito-Pim1 enhances mitochondrial biogenesis as seen by increased gene expression of TFAM (mitochondrial transcription factor A) and NRF1 by qPCR analysis (Fig. 6H). Our recent study demonstrates that the Pim1-mediated increase of regulators of mitochondrial biogenesis is dependent upon c-Myc (16), a downstream target of Pim1 kinase (15, 35). Consistently, silencing c-Myc by shRNA decreases mRNA levels of mitochondrial biogenesis regulators TFAM, NRF1, and ERRα, regardless of Pim1 overexpression (Fig. 6H), suggesting a role for c-Myc in the beneficial effects of Mito-Pim1 overexpression in hCPCs. Collectively, increased proliferation is associated with Pim1 targeting to the mitochondria of hCPCs, consistent with enhanced mitochondrial biogenesis and function and up-regulation of drivers of cell cycle progression.
Discussion
Current heart failure treatment options are increasingly expensive and at best modestly effective. Aged CPCs have a reduced capacity to repair the myocardium due to a decline in replication and differentiation potential (5–7). The endogenous CPC response to injury in an adult heart is insufficient to produce the amount of regeneration needed to recover lost cardiac function (30). Isolation, expansion, and injection of adult CPCs after myocardial infarction augment myocardial regeneration and mediate functional cardiac repair in the rodent model (4). The SCIPIO clinical trial provides evidence to support autologous transfer of c-kit+ hCPCs as a safe and efficacious treatment for patients with chronic ischemic cardiomyopathy as seen with improvements in cardiac function (10). Although the outcome of the trial was promising, there is ample room for further improvement. Autologous transplantation of hCPCs requires isolation and expansion of cells originating from an already aged and injured environment, potentially contributing to modest functional improvement observed in patients (10). Isolated cells benefit from reinforcement through ex vivo modification to improve their regenerative potential before reintroduction into a failing heart. Therefore, our group has focused upon modifying aged hCPCs to bolster these cells with the help of Pim1. Pim1 modification “turns back” the biological clock of senescent hCPCs, increasing proliferative potential, decreasing susceptibility to apoptosis, elongating telomeres, antagonizing senescence, and ultimately extending myocardial youth (17, 18). Pim1 increases the cardioprotective potential of aged and diseased adult hCPCs derived from heart failure patients and augments the benefits of autologous transplantation (17).
Genetic and environmental (Table 2) influences contribute to biological aging of hCPCs irrespective of chronological age. DNA damage and environmental stressors contribute to a senescent phenotype that decreases stem cell function in the diseased heart (31). Factors such as disease etiology, alcohol and cigarette consumption, medication, and diabetes contribute to the variability in hCPC populations isolated from multiple patients, as evident by differences in proliferation rate, cell death, and telomere lengths (Fig. 1). hCPCs isolated from various patients respond to Pim1 modification differently, and targeted Pim1 provides the option to individualize hCPC modification. A patient with slow growing hCPCs that are highly susceptible to cell death (H13-061) may benefit more from Mito-Pim1 modification, whereas Nuc-Pim1 would ideally be used to modify hCPCs that display a more pronounced senescent phenotype (H13-064), but are resistant to cell death (Fig. 1).
This study demonstrates for the first time that subcellular targeting of Pim1 preferentially regulates cardioprotective properties, enhancing proliferation or survival, and supporting the antagonism of senescence in hCPCs. Pim1 expression and localization in the heart changes with age; nuclear accumulation is associated with neonatal development and postnatal proliferation, whereas Pim1 in the adult heart acts in the mitochondrial context with apoptotic-signaling molecules (11, 12, 19, 21). Pim1 also rejuvenates aged and diseased hCPCs to a youthful phenotype as demonstrated by overexpression of PimWT in our recent study (18). However, mechanisms of nuclear or mitochondrial targeted Pim1 sharpen and clearly illuminate the varying roles of this kinase depending upon the subcellular location in senescent hCPCs.
eGFP and PimWT constructs used in this study to demonstrate the effects of Pim1 engineering in hCPCs have been previously published (11, 17, 18). The four targeting vectors used in this study contain a small 29-base pair myc epitope. The myc epitope has been extensively used to tag and detect proteins as an alternative to commercially available antibodies that fail to detect endogenous protein expression by immunoblot or confocal microscopy (41). In anticipation of this issue with Pim1 antibodies, we deliberately left the myc epitope in the four targeting vectors. Mito- and Nuc-Pim1 perform analogous roles when compared with PimWT in hCPCs (Figs. 3–6), suggesting that the presence of the myc epitope does not adversely affect the function of these constructs. In addition, the Mito-Pim1 and Nuc-Pim1 constructs exhibit comparable kinase activity to PimWT, as seen by an increase in phosphorylation of Pras40 (Fig. 2F), a negative regulator of mammalian target of rapamycin activity that is directly phosphorylated at Thr-246 by Pim1 kinase (40). The Mito- construct, which uses the cytochrome c oxidase VIIIa MLS sequence to localize Pim1 to the mitochondria, specifically targets to the mitochondrial matrix. Whether or not the PimWT construct also localizes to the mitochondrial matrix or to other compartments in the mitochondria is an important and interesting question to address and remains a topic of future investigations. This finding will help shed light on identifying mitochondrial specific targets and functions of Pim1.
Expression of SA β-gal activity, enlarged and flattened cellular morphology, induction of senescence regulators and shortened telomeres comprise the phenotypic characteristics of senescence (18, 32–34). Nuc-Pim1 successfully alters senescence-associated phenotypic characteristics relative to PimWT and Mito-Pim1 overexpression; significant blunting of hCPC senescence was shown by a 30% decrease in SA β-gal positivity, restoration to a youthful morphology, suppression of senescent markers p16 and p53, and the ability to undergo several successive passages indicative of a more youthful cell (Fig. 3). Nuc-Pim1 hCPCs also displayed elongated telomeres with a coincident up-regulation of TERT and Ns compared with whole cell Pim1 overexpression. Nuc-Pim1-mediated up-regulation of Ns could mechanistically contribute to maintenance of youth, consistent with our recent findings demonstrating that Ns is required to maintain properties of stemness and antagonize senescence in a p53-dependent mechanism (35). In conclusion, nuclear overexpression of Pim1 supports both phenotypic and biological changes in senescent hCPCs to enhance stem cell youthfulness associated with increased growth potential, telomere maintenance, and reduced markers of senescence (Fig. 4).
The role of Pim1 expression at the mitochondria has been previously addressed in our laboratory, as Pim1 preferentially localizes to the mitochondria in heart lysates subjected to ischemia/reperfusion injury (19). Indeed, Mito-Pim1 hCPCs had increased resistance to apoptosis as compared with PimWT, with an almost 2-fold reduction in cell death (Fig. 5). Pim1 up-regulates anti-apoptotic protein Bcl-XL during serum starvation, which is further increased after H2O2 treatment. Similarly, Bcl-2 was up-regulated in Mito-Pim1 hCPCs after oxidative stress. Increased resistance to apoptosis is an indispensable trait for stem cells that are to be used for injections into a region of the myocardium bombarded by oxidative stress. These data indicate that mitochondrial targeted Pim1 works synergistically with Bcl-2 and Bcl-XL proteins to inhibit apoptosis and preserve mitochondrial integrity far more effectively than PimWT alone.
Heightened proliferative potential, desirable for regenerative cell therapy, is improved by modification of hCPCs with Mito-Pim1 (Fig. 6). Pim-1, -2, and -3 kinases exhibit a vital role in maintaining functional cellular metabolic pathways (16), which are directly related to regulation of proliferation of stem cells (36, 37). Pim1 stabilizes mitochondrial membrane permeability and organelle morphology (19), both of which impact mitochondrial function and energy metabolism (38). Cardiac metabolism utilizes oxidative phosphorylation to generate ATP, unfortunately creating ROS as a byproduct (36). Efficient cellular metabolism produces lower levels of ROS, which correlates with greater proliferative rates in stem cells (39). Based on previous and current data, we speculate that Mito-Pim1 preserves metabolic function and stimulates fatty acid oxidation, thereby boosting the pro-proliferative and anti-apoptotic effects of targeting Pim1 to the mitochondria. Therefore, mitochondrial targeted Pim1 likely supports maintenance of energy metabolism and mitochondrial biogenesis evidenced by increased ATP levels and up-regulation of gene regulators, respectively. These effects collectively prompt an upsurge of proliferative capacity of Mito-Pim1 hCPCs (Fig. 6).
Cell cycle progression is regulated by cyclins and CDKs, and Pim1 has previously been shown to stabilize the interaction of Cyclin D with CDKs (20, 29). The Cyclin D/CDK4 complex promotes G1-S transition of the cell cycle, which is inhibited by Rb protein. The tumor suppressor Rb controls gene expression of cell cycle regulators based on its phosphorylation status. Rb phosphorylation results in inactivation and release of an inhibitory hold on the Cyclin D/CDK4 complex (29). This study shows that mitochondrial localization of Pim1 increases protein expression of Cyclin D and Cdk4 more effectively than PimWT. A concurrent increase of phosphorylated Rb relative to total Rb protein was measured in Mito-Pim1 hCPCs (Fig. 6). Collectively, mitochondrial localization of Pim1 results in increased proliferation and subsequent up-regulation of modulators of cell cycle progression, two processes that could correlate with the stabilization of hCPC energy metabolism.
This study demonstrates that Pim1 localization preferentially enhances proliferation, survival, and youthful properties of aged hCPCs dependent upon intracellular localization. cMyc regulates a large portion of human genes and is a downstream target of Pim1 kinase with known roles in a wide variety of biological functions (42, 43). Research from our laboratory has shown that Pim1-mediated stimulation of TERT activity and telomeric lengthening is dependent upon cMyc activation (20), metabolic dysfunction associated with loss of Pim kinase is rescued by cMyc overexpression (16), and cMyc silencing blunts the Pim1-mediated up-regulation of nucleostemin contributing to cellular senescence (35). Silencing cMyc results in substantial alterations in mitochondrial biogenesis regulators in the presence of both targeted and whole cell Pim1 overexpression (Fig. 6H), providing evidence that cMyc is a likely target of Pim1 at the mitochondria and nucleus of hCPCs. The functional effect of Pim1 is dependent upon intracellular localization, suggesting that the location of the kinase within the cell determines its interaction with cMyc to mediate multifarious downstream effects. The differential outcome of targeted Pim1 overexpression provides an attractive and clinically applicable solution to address patient-to-patient variability. Heart failure patients possess variability in disease etiology, contributing to individually distinct biological age and characteristics of hCPCs (Fig. 1). Controlled localization of Pim1 allows for preferential enhancement of specific stem cell properties, customizing the benefits of modification. This study brings us one step closer to a more personalized method of approaching cardiac regenerative therapy.
Acknowledgments
We acknowledge Sharp Memorial Hospital in San Diego and Dr. Walter P. Dembitsky for their time and contribution of human left ventricular assist device explant tissue. We also thank the SDSU FACS core facility and all members of Dr. Sussman's laboratory for their intellectual collaboration and support.
This work was supported, in whole or in part, by National Institutes of Health Grants R01HL067245, R37HL091102, R01HL105759, R01 HL113656, R01 HL117163, and R01 HL113647, and an award from Fondation Leducq (to M. A. S.).
- CPC
- cardiac progenitor cell
- CDK
- cyclin-dependent kinase
- hCPC
- human cardiac progenitor cell
- eGFP
- green fluorescent protein
- PimWT
- whole cell Pim1
- Mito-GFP
- mitochondrial-targeted GFP
- Mito-Pim1
- mitochondrial-targeted Pim1
- Nuc-GFP
- nuclear-targeted GFP
- Nuc-Pim1
- nuclear-targeted Pim1
- SA β-gal
- senescence-associated β-galactosidase
- AnV
- annexin V
- PI
- propidium iodide
- TERT
- telomerase reverse transcriptase
- NS
- nucleostemin
- Rb
- retinoblastoma
- ROS
- reactive oxygen species
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- qPCR
- quantitative PCR.
References
- 1. Reinecke H., Minami E., Zhu W. Z., Laflamme M. A. (2008) Cardiogenic differentiation and transdifferentiation of progenitor cells. Circ. Res. 103, 1058–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fransioli J., Bailey B., Gude N. A., Cottage C. T., Muraski J. A., Emmanuel G., Wu W., Alvarez R., Rubio M., Ottolenghi S., Schaefer E., Sussman M. A. (2008) Evolution of the c-kit-positive cell response to pathological challenge in the myocardium. Stem Cells 26, 1315–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ellison G. M., Torella D., Dellegrottaglie S., Perez-Martinez C., Perez de Prado A., Vicinanza C., Purushothaman S., Galuppo V., Iaconetti C., Waring C. D., Smith A., Torella M., Cuellas Ramon C., Gonzalo-Orden J. M., Agosti V., Indolfi C., Galiñanes M., Fernandez-Vazquez F., Nadal-Ginard B. (2011) Endogenous cardiac stem cell activation by insulin-like growth factor-1/hepatocyte growth factor intracoronary injection fosters survival and regeneration of the infarcted pig heart. J. Am. Coll. Cardiol. 58, 977–986 [DOI] [PubMed] [Google Scholar]
- 4. Beltrami A. P., Barlucchi L., Torella D., Baker M., Limana F., Chimenti S., Kasahara H., Rota M., Musso E., Urbanek K., Leri A., Kajstura J., Nadal-Ginard B., Anversa P. (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114, 763–776 [DOI] [PubMed] [Google Scholar]
- 5. Urbanek K., Torella D., Sheikh F., De Angelis A., Nurzynska D., Silvestri F., Beltrami C. A., Bussani R., Beltrami A. P., Quaini F., Bolli R., Leri A., Kajstura J., Anversa P. (2005) Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc. Natl. Acad. Sci. U.S.A. 102, 8692–8697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Berlo J. H., Kanisicak O., Maillet M., Vagnozzi R. J., Karch J., Lin S. C., Middleton R. C., Marbán E., Molkentin J. D. (2014) c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509, 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Segers V. F., Lee R. T. (2008) Stem-cell therapy for cardiac disease. Nature 451, 937–942 [DOI] [PubMed] [Google Scholar]
- 8. Rota M., Padin-Iruegas M. E., Misao Y., De Angelis A., Maestroni S., Ferreira-Martins J., Fiumana E., Rastaldo R., Arcarese M. L., Mitchell T. S., Boni A., Bolli R., Urbanek K., Hosoda T., Anversa P., Leri A., Kajstura J. (2008) Local activation or implantation of cardiac progenitor cells rescues scarred infarcted myocardium improving cardiac function. Circ. Res. 103, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gonzalez A., Rota M., Nurzynska D., Misao Y., Tillmanns J., Ojaimi C., Padin-Iruegas M. E., Müller P., Esposito G., Bearzi C., Vitale S., Dawn B., Sanganalmath S. K., Baker M., Hintze T. H., Bolli R., Urbanek K., Hosoda T., Anversa P., Kajstura J., Leri A. (2008) Activation of cardiac progenitor cells reverses the failing heart senescent phenotype and prolongs lifespan. Circ. Res. 102, 597–606 [DOI] [PubMed] [Google Scholar]
- 10. Bolli R., Chugh A. R., D'Amario D., Loughran J. H., Stoddard M. F., Ikram S., Beache G. M., Wagner S. G., Leri A., Hosoda T., Sanada F., Elmore J. B., Goichberg P., Cappetta D., Solankhi N. K., Fahsah I., Rokosh D. G., Slaughter M. S., Kajstura J., Anversa P. (2011) Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378, 1847–1857 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11. Fischer K. M., Cottage C. T., Wu W., Din S., Gude N. A., Avitabile D., Quijada P., Collins B. L., Fransioli J., Sussman M. A. (2009) Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation 120, 2077–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muraski J. A., Rota M., Misao Y., Fransioli J., Cottage C., Gude N., Esposito G., Delucchi F., Arcarese M., Alvarez R., Siddiqi S., Emmanuel G. N., Wu W., Fischer K., Martindale J. J., Glembotski C. C., Leri A., Kajstura J., Magnuson N., Berns A., Beretta R. M., Houser S. R., Schaefer E. M., Anversa P., Sussman M. A. (2007) Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat. Med. 13, 1467–1475 [DOI] [PubMed] [Google Scholar]
- 13. Bailey B., Izarra A., Alvarez R., Fischer K. M., Cottage C. T., Quijada P., Díez-Juan A., Sussman M. A. (2009) Cardiac stem cell genetic engineering using the αMHC promoter. Regen. Med. 4, 823–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Del Re D. P., Sadoshima J. (2012) Enhancing the potential of cardiac progenitor cells: pushing forward with Pim-1. Circ. Res. 110, 1154–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cottage C. T., Bailey B., Fischer K. M., Avitable D., Collins B., Tuck S., Quijada P., Gude N., Alvarez R., Muraski J., Sussman M. A. (2010) Cardiac progenitor cell cycling stimulated by pim-1 kinase. Circ. Res. 106, 891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Din S., Konstandin M. H., Johnson B., Emathinger J., Völkers M., Toko H., Collins B., Ormachea L., Samse K., Kubli D. A., De La Torre A., Kraft A. S., Gustafsson A. B., Kelly D. P., Sussman M. A. (2014) Metabolic dysfunction consistent with premature aging results from deletion of Pim kinases. Circ. Res. 115, 376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mohsin S., Khan M., Toko H., Bailey B., Cottage C. T., Wallach K., Nag D., Lee A., Siddiqi S., Lan F., Fischer K. M., Gude N., Quijada P., Avitabile D., Truffa S., Collins B., Dembitsky W., Wu J. C., Sussman M. A. (2012) Human cardiac progenitor cells engineered with Pim-I kinase enhance myocardial repair. J. Am. Coll. Cardiol. 60, 1278–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mohsin S., Khan M., Nguyen J., Alkatib M., Siddiqi S., Hariharan N., Wallach K., Monsanto M., Gude N., Dembitsky W., Sussman M. A. (2013) Rejuvenation of human cardiac progenitor cells with Pim-1 kinase. Circ. Res. 113, 1169–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borillo G. A., Mason M., Quijada P., Völkers M., Cottage C., McGregor M., Din S., Fischer K., Gude N., Avitabile D., Barlow S., Alvarez R., Truffa S., Whittaker R., Glassy M. S., Gustafsson A. B., Miyamoto S., Glembotski C. C., Gottlieb R. A., Brown J. H., Sussman M. A. (2010) Pim-1 kinase protects mitochondrial integrity in cardiomyocytes. Circ. Res. 106, 1265–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cottage C. T., Neidig L., Sundararaman B., Din S., Joyo A. Y., Bailey B., Gude N., Hariharan N., Sussman M. A. (2012) Increased mitotic rate coincident with transient telomere lengthening resulting from pim-1 overexpression in cardiac progenitor cells. Stem Cells 30, 2512–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bachmann M., Möröy T. (2005) The serine/threonine kinase Pim-1. Int. J. Biochem. Cell Biol. 37, 726–730 [DOI] [PubMed] [Google Scholar]
- 22. Cawthon R. M. (2009) Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 37, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsai R. Y., McKay R. D. (2002) A nucleolar mechanism controlling cell proliferation in stem cells and cancer cells. Genes Dev. 16, 2991–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qu J., Bishop J. M. (2012) Nucleostemin maintains self-renewal of embryonic stem cells and promotes reprogramming of somatic cells to pluripotency. J. Cell Biol. 197, 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meng L., Lin T., Peng G., Hsu J. K., Lee S., Lin S. Y., Tsai R. Y. (2013) Nucleostemin deletion reveals an essential mechanism that maintains the genomic stability of stem and progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 110, 11415–11420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu Q., Yasumoto H., Tsai R. Y. (2006) Nucleostemin delays cellular senescence and negatively regulates TRF1 protein stability. Mol. Cell. Biol. 26, 9279–9290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsu J. K., Lin T., Tsai R. Y. (2012) Nucleostemin prevents telomere damage by promoting PML-IV recruitment to SUMOylated TRF1. J. Cell Biol. 197, 613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shugo H., Ooshio T., Naito M., Naka K., Hoshii T., Tadokoro Y., Muraguchi T., Tamase A., Uema N., Yamashita T., Nakamoto Y., Suda T., Kaneko S., Hirao A. (2012) Nucleostemin in injury-induced liver regeneration. Stem Cells Dev. 21, 3044–3054 [DOI] [PubMed] [Google Scholar]
- 29. Liu Z., Rader J., He S., Phung T., Thiele C. J. (2013) CASZ1 inhibits cell cycle progression in neuroblastoma by restoring pRb activity. Cell Cycle 12, 2210–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jesty S. A., Steffey M. A., Lee F. K., Breitbach M., Hesse M., Reining S., Lee J. C., Doran R. M., Nikitin A. Y., Fleischmann B. K., Kotlikoff M. I. (2012) c-kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc. Natl. Acad. Sci. U.S.A. 109, 13380–13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leri A., Kajstura J. (2003) Myocardial damage and repair. J. Mol. Cell. Cardiol. 35, 595–597 [DOI] [PubMed] [Google Scholar]
- 32. Kim J. H., Kim J. H., Lee G. E., Kim S. W., Chung I. K. (2003) Identification of a quinoxaline derivative that is a potent telomerase inhibitor leading to cellular senescence of human cancer cells. Biochem. J. 373, 523–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chang B. D., Xuan Y., Broude E. V., Zhu H., Schott B., Fang J., Roninson I. B. (1999) Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 18, 4808–4818 [DOI] [PubMed] [Google Scholar]
- 34. Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hariharan N., Quijada P., Mohsin S., Joyo A., Samse K., Monsanto M., De La Torre A., Avitabile D., Ormachea L., McGregor M. J., Tsai E. J., Sussman M. A. (2014) Nucleostemin rejuvenates cardiac progenitor cells and antagonizes myocardial aging. J. Am. Coll. Cardiol. 65, 133–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Varum S., Rodrigues A. S., Moura M. B., Momcilovic O., Easley C. A., 4th, Ramalho-Santos J., Van Houten B., Schatten G. (2011) Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PloS One 6, e20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pereira S. L., Rodrigues A. S., Sousa M. I., Correia M., Perestrelo T., Ramalho-Santos J. (2014) From gametogenesis and stem cells to cancer: common metabolic themes. Hum. Reprod. Update 20, 924–943 [DOI] [PubMed] [Google Scholar]
- 38. López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G. (2013) The hallmarks of aging. Cell 153, 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kimura W., Muralidhar S., Canseco D., Puente B., Zhang C. C., Xiao F., Abderrahman Y. H., Sadek H. A. (2014) Redox signaling in cardiac renewal. Antioxid. Redox Signal. 21, 1660–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang F., Beharry Z. M., Harris T. E., Lilly M. B., Smith C. D., Mahajan S., Kraft A. S. (2009) PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells. Cancer Biol. Ther. 8, 846–853 [DOI] [PubMed] [Google Scholar]
- 41. Evan G. I., Lewis G. K., Ramsay G., Bishop J. M. (1985) Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 5, 3610–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ahuja P., Zhao P., Angelis E., Ruan H., Korge P., Olson A., Wang Y., Jin E. S., Jeffrey F. M., Portman M., Maclellan W. R. (2010) Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J. Clin. Investig. 120, 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Y., Wang Z., Li X., Magnuson N. S. (2008) Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 27, 4809–4819 [DOI] [PubMed] [Google Scholar]