

Background: Proteins involved in DNA replication and repair are cleaved by caspases during apoptosis.
Results: Caspase-dependent degradation of human ribonucleotide reductase subunits R2 and p53R2 occurs in apoptotic cells.
Conclusion: Inhibition of deoxyribonucleotide (dNTP) synthesis by ribonucleotide reductase proteolysis favors apoptosis.
Significance: Enzymatic production of dNTPs is a prosurvival metabolic process down-regulated in apoptotic cells.
Keywords: apoptosis, caspase, nucleoside/nucleotide biosynthesis, protein degradation, ribonucleotide reductase
Abstract
Ribonucleotide reductase (RnR) is a key enzyme synthesizing deoxyribonucleotides for DNA replication and repair. In mammals, the R1 catalytic subunit forms an active complex with either one of the two small subunits R2 and p53R2. Expression of R2 is S phase-specific and required for DNA replication. The p53R2 protein is expressed throughout the cell cycle and in quiescent cells where it provides dNTPs for mitochondrial DNA synthesis. Participation of R2 and p53R2 in DNA repair has also been suggested. In this study, we investigated the fate of the RnR subunits during apoptosis. The p53R2 protein was cleaved in a caspase-dependent manner in K-562 cells treated with inhibitors of the Bcr-Abl oncogenic kinase and in HeLa 229 cells incubated with TNF-α and cycloheximide. The cleavage site was mapped between Asp342 and Asn343. Caspase attack released a C-terminal p53R2 peptide of nine residues containing the conserved heptapeptide essential for R1 binding. As a consequence, the cleaved p53R2 protein was inactive. In vitro, purified caspase-3 and -8 could release the C-terminal tail of p53R2. Knocking down these caspases, but not caspase-2, -7, and -10, also inhibited p53R2 cleavage in cells committed to die via the extrinsic death receptor pathway. The R2 subunit was subjected to caspase- and proteasome-dependent proteolysis, which was prevented by siRNA targeting caspase-8. Knocking down caspase-3 was ineffective. Protein R1 was not subjected to degradation. Adding deoxyribonucleosides to restore dNTP pools transiently protected cells from apoptosis. These data identify RnR activity as a prosurvival function inactivated by proteolysis during apoptosis.
Introduction
Production of dNTPs required for DNA replication and repair is a tightly controlled process to ensure faithful duplication of DNA strands, to prevent DNA mutations resulting from dNTP pool imbalance, and to preserve genome integrity. The reduction of ribonucleotides into deoxyribonucleotides (dNs)3 is catalyzed by ribonucleotide reductase (RnR). The activity of the enzyme is rate-limiting for dNTP synthesis and strictly regulated (for reviews, see Refs. 1 and 2). In mammals, RnRs consist of two distinct complexes formed by the association of a unique catalytic dimeric protein called R1 with one of the two homologues R2 and p53R2. These accessory subunits bring a stable free radical centered on a tyrosyl residue and are necessary for catalysis (1). Levels of protein R1 remain stable throughout the cell cycle as a consequence of both a transcriptional induction of the RRM1 gene encoding R1 at the G1/S boundary and a long half-life of the protein. By contrast, R2 is a cell cycle-regulated protein. Transcriptional regulation of the RRM2 gene encoding R2 is similar to that of RRM1, but maximal expression of the protein is limited to the S phase by proteasomal degradation of the subunit in G2 and M/G1 phases mediated, respectively, by Skp1-Cullin-Fbox (cyclin F) and anaphase-promoting complex/cyclosome (Cdh1) ubiquitin ligases (1, 3, 4). Fluctuations in protein R2 levels limit the activity of the R1-R2 complex and coordinate enhanced dNTP production with DNA replication. The p53R2 protein is expressed at a low constant level in cycling and quiescent cells (5). It is essential for mitochondrial DNA replication in some human and mouse tissues, especially skeletal muscle (6). DNA damage induces a transcriptional up-regulation of p53R2 in a p53-dependent manner, and p53R2 participates in repair of DNA lesions requiring a dNTP supply at least in postmitotic cells (7–9). Several lines of evidence also indicate a participation of protein R2 in DNA repair processes (2, 4).
According to the current view, beyond a certain threshold of DNA lesions that overwhelm the repair mechanisms, damaged cells are eliminated by p53-dependent or -independent apoptotic cell death (10). Apoptosis requires a family of aspartate-directed cysteine proteases termed caspases, which are classified as initiator (2, 8, 9, and 10) or effector (3, 6, and 7) caspases according to the timing of their proteolytic activation during the apoptosis signaling cascade (11). Specific cleavage of numerous polypeptide substrates, including structural components of the cytoskeleton, nucleus, and chromatin architecture; key players in signaling transduction pathways; cell cycle regulators; and DNA-binding proteins, ultimately leads to characteristic morphological changes, chromatin condensation, and nucleus fragmentation. Several proteins active during DNA repair or DNA replication are caspase substrates destroyed in the course of apoptosis, including poly(ADP-ribose) polymerase (PARP), the catalytic fragment of the DNA-dependent protein kinase, DNA topoisomerase II, and DNA replication protein RF-C140 (11). RnR supplies dNTPs for DNA synthesis and repair, but the fate of the enzyme subunits during apoptosis is currently unknown. We provide here the first evidence for a caspase-dependent degradation of human R2 and p53R2 small subunits. By contrast, R1, which is a long lived protein present in resting and proliferating cells like p53R2, is not subjected to proteolysis during apoptosis.
Experimental Procedures
Biochemicals and Antibodies
Cycloheximide, propidium iodide, protease-free BSA, isopropyl β-d-thiogalactopyranoside, dNs, and cisplatin (cis-diammineplatinum(II) dichloride) were purchased from Sigma-Aldrich. E-64d ((2S,3S)-trans-epoxysuccinyl-l-leucylamido-3-methylbutane ethyl ester), z-Val-Ala-dl-Asp-fluoromethyl ketone (z-VAD-fmk), Ac-VDVAD-CHO, Ac-IETD-CHO, Ac-DEVD-CHO and Ac-YVAD-CHO were purchased from Enzo Life Science. Imatinib and nilotinib were gifts from Novartis (Basel, Switzerland). Recombinant caspases and Q-VD-OPh were obtained from R&D Systems; TNF-α was purchased from Thermo Fisher Scientific; and lactacystin, Pefabloc, and N-acetyl-Leu-Leu-Nle-CHO were obtained from Merck Millipore. MG-132 was from Selleckchem.
Primary antibodies against α-tubulin (T9026, Sigma-Aldrich), p53R2 (ab8105, Abcam), R1 (AD203, InRo BioMedTek), R2 (I-15 and E-16, Santa Cruz Biotechnology), PARP-1 (C-2-10, Merck Millipore), a V5 epitope (R960-25, Life Technologies), and caspase-3 and cleaved caspase-3 (9665 and 9661, respectively, Cell Signaling Technology) were used for immunoblotting. Fluorescent Alexa Fluor 680-conjugated anti-mouse (A-21057, Life Technologies) and IRDye 800CW-conjugated anti-rabbit (926-32211, Li-Cor Biosciences) IgGs were used for infrared fluorescence detection of primary antibodies in immunoblotting.
Cell Culture and Treatment
The human leukemia and lung carcinoma cell lines K-562 and H1299 were maintained in RPMI 1640 medium supplemented with 5% fetal bovine serum, 2 mm l-glutamine, 25 mm HEPES, and antibiotics. The human adenocarcinoma HeLa 229 cell line and the human mammary MCF-7 cell lines obtained by stable transfection of caspase-3 cDNA (MCF-7c3) or empty vector (MCF-7vc) were cultured in DMEM supplemented with 10% fetal bovine serum and antibiotics. For experiments, K-562, HeLa 229, H1299, and MCF-7 cells were seeded in 6-well culture plates at 0.75, 0.6, 0.3, and 0.7 × 106 cells/well, respectively, in 5 ml of culture medium. The day after, imatinib (0.06–4 μm) or nilotinib (1.9–125 nm) was added to K-562 cells. HeLa 229 and H1299 cells received a combination of TNF-α (10 ng/ml) and cycloheximide (CHX) (10 μg/ml). MCF-7 cells were treated with 25 μg/ml cisplatin. Cells were harvested from 16 to 48 h later for further analysis.
Synchronization of HeLa 229 Cells by a Double Thymidine Block
HeLa 229 cells at 25–30% confluence were maintained for 18 h in culture medium supplemented with 2 mm thymidine. Then thymidine was removed by washing cells once with phosphate-buffered saline (PBS). Fresh medium was added for 9 h before starting a second thymidine treatment for 17 h. Cells arrested at the beginning of S phase and maintained in the presence of 2 mm thymidine were incubated with TNF-α and CHX if required.
Determination of Protein Half-life
Synchronized HeLa 229 cells kept in the presence of 2 mm thymidine were treated with TNF-α and CHX for 19 h. Then cells were incubated in fresh culture medium supplemented with thymidine, CHX (10 μg/ml), and Q-VD-OPh (2 μm). Cells were harvested after 0–24 h, and expression of proteins R2 and R1 was determined by immunoblotting. Kinetics of protein decay was fitted with a first-order exponential equation (A = A0e−kt), and protein half-life was calculated (t½ = ln 2/k).
Immunoblotting Analysis
Cells were washed twice in ice-cold PBS. Crude cell extracts were prepared in a 50 mm Tris·HCl lysis buffer, pH 7.4 supplemented with 150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 1 mm EDTA, 1 mm DTT, and protease inhibitors including 1 mm Pefabloc. SDS-PAGE, protein transfer on a nitrocellulose membrane, and antigen detection were carried out as described previously using α-tubulin as a loading standard (12). The infrared fluorescence signals at 680 and 800 nm were recorded and quantified with an Odyssey scanner (LI-COR Biosciences) and normalized to α-tubulin. The extent of p53R2 protein cleavage was analyzed by measuring the fluorescence signal of the cleaved fragment. It was expressed as the percentage of the total amount of the protein.
Detection of Apoptotic Nuclei
Cells were incubated for 16 h with TNF-α and CHX. In some experiments, nucleosides were added with the proapoptotic agents. Cells were then harvested in PBS, resuspended in 1 ml of ice-cold PBS, and fixed in 3 ml of 75% EtOH kept at −20 °C. An aliquot was transferred on a microscopy slide. Nuclei were stained with DAPI contained in the mounting medium ProLong Gold antifade reagent (Life Technologies). At least 200 nuclei were recorded with a Zeiss Axioplan2 fluorescence microscope, and the percentage of nuclei exhibiting morphological apoptotic features (i.e. chromatin condensation and nuclear fragmentation) was then calculated.
Detection of Apoptosis by TUNEL Assay
HeLa 229 cells incubated with TNF-α and CHX were processed for terminal deoxynucleotidyltransferase-mediated fluorescent labeling of 3′-OH ends of fragmented DNA using the Click-iT TUNEL Alexa Fluor Imaging assay (Life Technologies) according to the manufacturer's instructions. The kit was adapted to a sample of 1 × 106 cells in suspension. Cells were gently resuspended every 10 min during labeling reactions, and washings were done using centrifugation. Analysis of Alexa Fluor 488-labeled cells was performed with a CyFlow ML flow cytometry system using Summit 6.1 software.
Plasmids and Transfections
All DNA manipulations, mutation, cloning, and transformation experiments in Escherichia coli DH5α were performed according to standard protocols. The pcDNA3.1 vector carrying the cDNA sequence of the human p53R2 gene (pcDNA3-hp53R2) was a generous gift from Prof. Hirofumi Tanaka (Tokyo Medical and Dental University, Japan). The pcDNA3-hp53R2mut plasmid encoding p53R2-D342E was generated by PCR from pcDNA3-hp53R2 using the Phusion Site-Directed Mutagenesis kit (Thermo Scientific) and the following primers: 5′-CCAAGGTGAAGACGTTTTCTGTGGTTTCTGCCATAACTGCA-3′ and 5′-GGCAGAAACCACAGAAAACGTCTTCACCTTGGATGCAGATT-3′ (mutated nucleotide underlined). All steps were performed according to the manufacturer's instructions. The presence of the A to T point mutation in the p53R2 sequence was confirmed by sequencing. The pcDNA3-p53R2-ΔC9 plasmid was created by PCR amplification from the pcDNA3-hp53R2 plasmid using the following primers: 5′-TGGAATTCCAGACCGGCTAGCATGGGCGACCCGGGA-3′ and 5′-CTCGAGTTAATCTGTGGTTTCTGCCATAACTGC-3′. The PCR fragment digested with NheI and XhoI was ligated into the pcDNA3.1 expression vector opened with the same enzymes. Bacterial expression vectors expressing N-terminally His6-tagged wild-type, D342E, and p53R2-ΔC9 proteins were constructed as follows. A DNA fragment containing the sequence of the WT human p53R2 gene was amplified by PCR from the pcDNA3-hp53R2 plasmid using the primers 5′-GTGGTGGAATTCCAGACCGGCTAGCATGGGCGACCCGGAA-3′ and 5′-AAGCCACAGTGGAGGCTGATCA-3′. The fragment was then cleaved by NheI and XhoI and inserted in the pET28b vector opened by the same enzymes. NheI and XhoI were used to release p53R2-D342E and p53R2-ΔC9 fragments from pcDNA3-p53R2mut and pcDNA-p53R2-ΔC9, respectively. These sequences were then ligated into the pET28b vector opened by the same enzymes to create pET28b-p53R2-D342E and pET28b-p53R2-ΔC9. The pET28b-R1His plasmid expressing a N-terminally His6-tagged human R1 protein was built by PCR amplification of the R1 cDNA sequence contained in the pET3a-hR1 plasmid (kindly given by Pr. Lars Thelander, Umeå University, Sweden) using primers 5′-CTTTAAGAAGGAGATGCTAGCATGCATGTGATCAAG-3′ and 5′-GGGCTTTGTCTCGAGCCGGATCCAC, subsequent digestion with NheI and XhoI, and ligation into the pET28b vector cleaved by the same enzymes.
The two complementary primers KV5S (5′-CTAGTCACCATGGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGg) and KV5AS (5′-ctagcCGTAGAATCGAGACCGAGGAGAGGGTTAGGGATAGGCTTACCcatggtga) containing the V5 epitope sequence flanked by NheI and SpeI restriction sites were annealed and then ligated with NheI-digested pcDNA3.1 (Life Technologies) to obtain the KV5-pcDNA3.1 vector. The pcDNA3-hp53R2 and pcDNA3-hp53R2mut plasmids were digested with NheI and XhoI, producing a fragment of 1.1 kb that was purified and ligated into KV5-pcDNA3.1 digested with NheI and XhoI to yield KV5-p53R2 and KV5-p53R2mut vectors encoding an N-terminal V5 epitope followed by wild-type and mutant D342E human p53R2 proteins, respectively. All constructs were sequence-verified.
K-562 cells were transfected by nucleofection with a Nucleofector II device (Lonza) according to the manufacturer's instructions. Briefly, 1 × 106 cells were resuspended in 100 μl of Nucleofector solution V and mixed with 2 μg of plasmid DNA. Electroporation was performed using the program T-016. The next day, cells were harvested for analysis by immunoblotting. Clones 3 and 19 stably expressing the p53R2-ΔC9 mutant were obtained by limiting dilution of transfected K-562 cells selected for 2 weeks in the presence of 1 mg/ml neomycin sulfate. H1299 cells seeded at 0.3 × 106 cells/well were transfected 24 h later with 4 μg of plasmid DNA using 8 μl of Lipofectamine 2000 reagent (Life Technologies). Cells were treated 28 h post-transfection with TNF-α and CHX with or without Q-VD-OPh at 2 μm and then harvested 19 h later.
Production and Purification of Recombinant Proteins
His6-tagged wild-type p53R2, p53R2-D342E, and p53R2-ΔC9 mutants were expressed and purified according to the following protocol. Expression vectors pET28b-p53R2, pET28b-p53R2-D342E, and pET28b-p53R2-ΔC9 were used to transform BL21-CodonPlus (DE3)-RIPL bacteria (Stratagene). Overnight cultures of each strain were diluted 100 times in 1 liter of Luria broth medium (LB) and grown at 37 °C until A600 reached 0.6 and then induced by 400 μm isopropyl β-d-thiogalactopyranoside. Bacteria were harvested 4 h later. Human His6-tagged R1 protein was produced in the same expression vector following transformation with the pET28b-hR1His plasmid. The R1 expression protocol was the same as for p53R2 proteins with the following modifications. Cultures were grown at 30 °C to an A600 of 0.6, then the growth temperature was switched to 20 °C, and induction was done at 50 μm isopropyl β-d-thiogalactopyranoside. Cultures were then grown overnight before harvesting. Bacterial pellets were resuspended in buffer A (20 mm sodium phosphate, 0.5 m NaCl) containing 20 mm imidazole and lysed by sonication. Nucleic acid precipitation and ammonium sulfate fractionation were performed as described previously for the mouse R2 protein (13). Supernatants containing His6-tagged p53R2 proteins were then loaded on a HisTrap HP nickel-Sepharose column (GE Healthcare) equilibrated with buffer A containing 20 mm imidazole and washed with buffer A containing 60 mm imidazole. His-tagged p53R2 proteins were then eluted with buffer A containing 150 mm imidazole and immediately concentrated in 50 mm HEPES, pH 7.4 using Amicon Ultra-4 centrifugal filter devices (Millipore).
Measurement of p53R2 Activity
The iron/radical center in His6-tagged WT p53R2 and p53R2-ΔC9 proteins was reconstituted using an already described anaerobic procedure (14). Activity of p53R2 was assayed using the reduction of [3H]CDP to [3H]dCDP. Active p53R2 or mutant p53R2-ΔC9 was diluted at a final concentration of 4 μm with a 4-fold molar excess of the R1 subunit in 50 mm HEPES, pH 7.6 containing 100 mm KCl, 15 mm MgCl2, 5 mm ATP, 10 mm DTT, 200 μm CDP, and 1 μCi of [5-3H]CDP (American Radiolabeled Chemicals, St. Louis, MO) in a final volume of 90 μl. Each sample was incubated at 30 °C for 10 min. The last steps were performed essentially as described (14). A Nucleosil C18 5 μm HPLC column (Interchrom) connected to a radioactive flow detector (model LB506-C1, Berthold) was used to analyze [5-3H]cytidine and [5-3H]deoxycytidine. Nucleoside separation was done with 10 mm sodium acetate, pH 4.8, 4.25% methanol, 1% low UV PIC-B5 reagent (Waters) as the mobile phase at a flow rate of 0.6 ml/min.
Fluorescent Labeling of Caspases
Evaluation of caspase-positive cells was determined by flow cytometry using FAM fluorochrome-labeled inhibitors of caspases (FLICA) reagent according to the manufacturer's instructions (FLICA Poly Caspases kit, ImmunoChemistry Technologies). Briefly, 1.25 × 106 HeLa 229 cells were treated for 20 h with TNF-α and CHX. Then 1 × 106 cells resuspended in 290 μl of culture medium were incubated with the FAM FLICA pan-caspase reagent for 75 min at 37 °C. The reagent is cell-permeable and binds covalently to activated caspases in apoptotic cells. After two washes with Hanks' balanced salt solution to eliminate the unbound dye, propidium iodide was added at 2.5 μg/ml to distinguish dead cells. Flow cytometry analysis was performed with MoFlo Astrios equipment (Beckman Coulter) using Summit 6.1 software.
RNA Interference
HeLa 229 cells seeded at 0.7 × 106 cells/well were transfected 24 h later with 75 nm siRNA (Silencer Select validated or predesigned siRNA) using the Lipofectamine RNAiMAX reagent (all from Life Technologies). After 48 h, TNF-α and CHX were added, and cells were harvested 24 h later for immunoblotting analysis of p53R2.
Proteolysis of p53R2 by Purified Caspases
In a first set of experiments, lysates from H1299 cells were used as a source of native p53R2 protein. Subconfluent cells grown in two 14-cm culture dishes were harvested by trypsinization. The pellet was resuspended in caspase buffer (50 mm HEPES, pH 7.4, 100 mm KCl, 0.1% (w/v) CHAPS, 5% (w/v) sucrose, 10 mm DTT) supplemented with protease inhibitors including 1 mm Pefabloc and 40 μm N-acetyl-Leu-Leu-Nle-CHO. The lysate prepared with a glass Dounce homogenizer was centrifuged for 20 min at 4 °C in a Beckman TL-100.2 rotor at 100,000 × g. A 100-μl aliquot of the soluble fraction was then applied to a BioSpin 6 column (Bio-Rad) pre-equilibrated with caspase buffer to eliminate protease inhibitors by gel filtration. A volume containing 400 μg of protein was then incubated for 1–4 h at 37 °C in caspase buffer with 400–800 ng of recombinant caspase-8 or -3. Finally, the reaction mixture was analyzed by immunoblotting using 30 μg of protein.
In a second set of experiments, cell lysates were replaced with purified wild-type or mutant p53R2 proteins. Recombinant proteins were diluted in caspase buffer supplemented with protease inhibitors, which were then eliminated by gel filtration as described above. After caspase addition, proteolysis was tested in a reaction mixture containing a p53R2 protein at 20 μm, 10 μg/ml protease-free BSA, and 1 μg of purified caspase. Incubation was carried out at 37 °C, and four aliquots taken every hour were analyzed by immunoblotting.
Results
Induction of p53R2 Proteolysis by Bcr-Abl Inhibitors in K-562 Cells
The Bcr-Abl fusion protein is an oncogenic tyrosine kinase that plays a key role in the pathophysiology of chronic myelogenous leukemia. Inhibitors of Bcr-Abl activity such as imatinib are clinically successful drugs that block the proliferation of Bcr-Abl+ leukemia cells and induce apoptosis. Treatment of the Bcr-Abl+ K-562 leukemia cell line with imatinib induced the appearance of a new band just below the p53R2 full-length polypeptide detected by immunoblotting (Fig. 1A). The effect was dose-dependent. The calculated IC50 of 0.39 ± 0.07 μm and visual examination of the cells indicated that p53R2 proteolysis occurred at cytotoxic concentrations of imatinib (Fig. 1B). The lowest band was revealed using two different primary antibodies recognizing the N-terminal extremity of the p53R2 protein (not shown), suggesting that the additional band was generated from the cleavage of p53R2 at its C-terminal tail. This part of the protein is required for its association with the catalytic subunit R1. Hence, loss of a small C-terminal fragment of p53R2 in K-562 cells undergoing apoptosis should prevent proper binding of p53R2 to R1. Imatinib-induced cleavage of p53R2 increased with time but remained partial even at the latest time points (Fig. 1, C and D). The highest percentage of cleaved p53R2 observed in all our experiments was 46%. The total amount of the protein remained fairly constant (Fig. 1E), suggesting that the cleaved fragment was not subjected to further proteolysis. Imatinib-induced cleavage of p53R2 was also observed using two other chronic leukemia cell lines, AR230 and Lama-84 (not shown). No proteolysis of protein R1 was observed in K-562 cells incubated with imatinib (Fig. 1, A, C, and E). Expression of protein R2, the cell cycle-regulated p53R2 homologue, dramatically decreased during the same time range. Because imatinib also modified K-562 cell cycle distribution (data not shown), we did not further investigate the reasons for R2 breakdown in this experimental model. Very similar results showing p53R2 proteolysis and absence of R1 degradation were obtained using 10–100 nm nilotinib, a second generation Bcr-Abl inhibitor much more potent than imatinib (data not shown). Overall, we observed specific cleavage of a small but essential C-terminal fragment of the p53R2 protein in leukemia cells incubated with proapoptotic anticancer drugs.
FIGURE 1.

Proteolysis of p53R2 in K-562 leukemia cells after treatment with a Bcr-Abl inhibitor. A, cells were incubated for 40 h with the indicated concentrations of imatinib to induce cellular apoptosis. The immunoblot shows the R1 and p53R2 ribonucleotide reductase subunit protein levels. A smaller p53R2 fragment only observed in imatinib-treated cells is indicated by an arrow. α-Tubulin is a loading control. B, quantification of the smaller p53R2 fragment as detected in A by near-infrared immunofluorescence in percentage of total p53R2 polypeptides. Results are mean ± S.E. (error bars) of three determinations. C, immunoblot showing the time course effect of 2 μm imatinib on p53R2 proteolysis as well as R1 and R2 levels. D, quantification of the p53R2 proteolytic fragment as shown in C (n = 3). E, variations in protein R1, R2, and total protein p53R2 levels as depicted in C relative to their expression measured at t = 0.
Cleavage of p53R2 Is Caspase-dependent
Caspases play essential roles in apoptotic cell death. Therefore, we suspected caspases to be involved in the cleavage of p53R2 induced in K-562 by proapoptotic Bcr-Abl inhibitors. A pan-caspase inhibitor, z-VAD-fmk, was tested and compared with lactacystin, a proteasome inhibitor, and with E-64-D, which inhibits calpain and cathepsin activities. As shown in Fig. 2, A and B, z-VAD-fmk completely prevented p53R2 proteolysis induced by imatinib, whereas lactacystin and E-64-D had no effect. Cleavage of the nuclear enzyme PARP-1 by caspases is an early event in the apoptotic process. As expected, PARP-1 cleavage was induced in K-562 cells by imatinib treatment, and the appearance of the 89-kDa proteolytic fragment was completely prevented by z-VAD-fmk (Fig. 2C).
FIGURE 2.

Potent inhibition of p53R2 proteolysis by two pan-caspase inhibitors. A, K-562 cells were cultured for 40 h with or without 2 μm imatinib. Effects of the proteasome inhibitor lactacystin (Lac) at 2.5 μm and the pan-caspase inhibitor z-VAD (z-V) at 50 μm on p53R2 proteolysis (arrow) were evaluated by immunoblotting. B, conditions as described in A except that the calpain and cathepsin inhibitor E-64-D (E64) was also tested at a concentration of 5 μm. Cleavage of p53R2 was measured as described in Fig. 1. Results are mean ± S.E. (error bars) of three independent experiments. *, p < 0.01. C, representative immunoblot in duplicates showing cleavage of the caspase substrate PARP-1 induced by imatinib treatment of K-562 cells and inhibited by 50 μm z-VAD. Native PARP-1 (116 kDa) and its smaller proteolytic fragment (85 kDa) are indicated. D, apoptosis of HeLa 229 cells was induced by TNF-α (10 ng/ml) and CHX (10 μg/ml) treatment for 16 h. Apoptotic nuclei detected by fluorescence microscopy were counted to estimate the extent of apoptosis. The general caspase inhibitor Q-VD-OPh (Q-VD) at 2 μm efficiently inhibited apoptosis. E, immunoblot showing invariant R1 expression and p53R2 proteolysis (arrow) in HeLa 229 cells after apoptosis induction as described in D. F, quantification of the p53R2 cleavage shown in E. G, evaluation of p53R2 proteolysis induced by treatment of K-562 and HeLa 229 cells with 2 μm imatinib (Im) for 33 h or TNF-α and CHX (T/X) for 20 h, respectively. Results are mean ± S.E. (error bars) of at least three independent experiments. *, p < 0.01. Ctrl, control.
To extend the significance of our data, we turned to a different and more classical model of apoptosis. A combination of TNF-α and a protein synthesis inhibitor such as CHX efficiently induces apoptosis in susceptible cell lines via the death receptor extrinsic pathway. When applied for 16 h to HeLa 229 cells, this treatment induced more than 60% apoptosis as measured by nuclear fragmentation and chromatin condensation (Fig. 2D). Q-VD-OPh, a pan-caspase inhibitor more efficient than z-VAD-fmk, strongly inhibited apoptosis in this cellular model. Cleavage of the p53R2 protein was also detected during apoptosis of HeLa 229 cells, whereas no noticeable degradation of protein R1 was evident (Fig. 2E). A time course experiment indicated that p53R2 proteolysis increased gradually from 8 to 20 h after addition of TNF-α and CHX (Fig. 2F). Compared with the very early cleavage of PARP-1 (data not shown), p53R2 proteolysis occurs later in the apoptotic process. In K-562 and HeLa 229 cells undergoing apoptosis, cleavage of p53R2 was totally inhibited by Q-VD-OPh (Fig. 2G), confirming the implication of caspase involvement in p53R2 degradation. To verify activation of caspases in our models, we monitored the proteolytic processing of the main executioner caspase-3 in K-562 cells. Activated caspase-3 fragments of 20 and 17 kDa appeared in K-562 cells incubated with imatinib (Fig. 3A). The kinetics of caspase-3 activation and p53R2 cleavage were similar (compare Fig. 3B with Fig. 1D). In HeLa 229 cells, activation of caspases was evaluated by labeling all activated caspases with fluorochrome-labeled inhibitors and detecting labeled cells by flow cytometry. Results shown in Fig. 3C indicate that 50.6 ± 2.0% of HeLa 229 cells incubated for 20 h with TNF-α and CHX were caspase-positive (n = 3, mean ± S.E.). In conclusion, in two very different experimental settings of apoptosis, p53R2 is subjected to a limited C-terminal proteolysis, and this cleavage is caspase-dependent.
FIGURE 3.
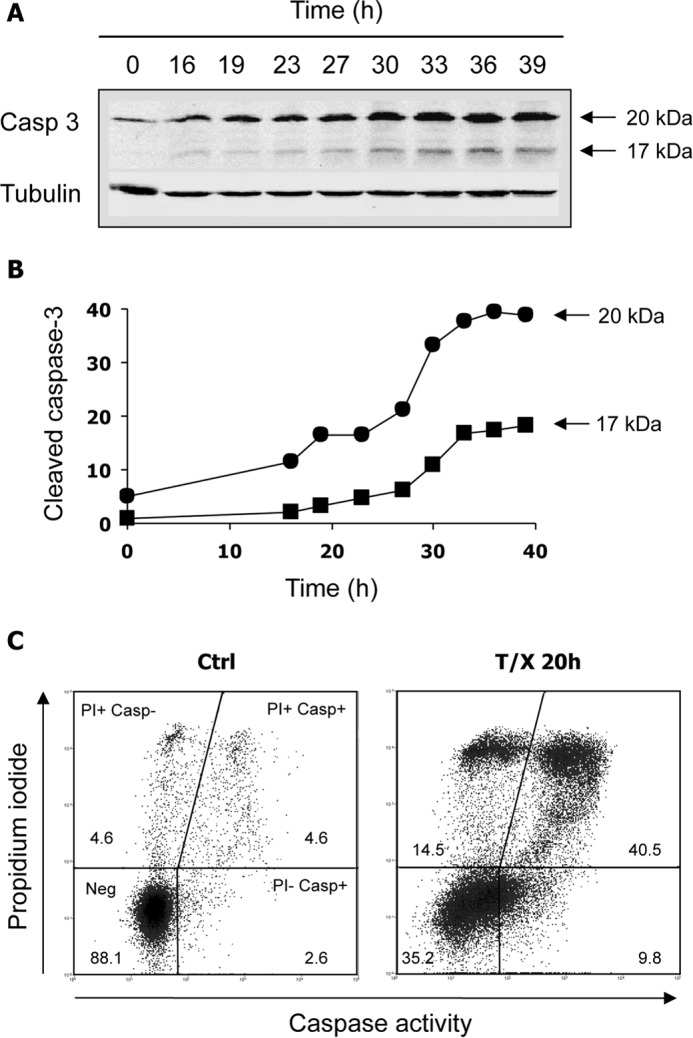
Treatments that lead to p53R2 cleavage also induce caspase activation. A and B, time course of caspase-3 activation in K-562 cells cultured with 2 μm imatinib. An immunoblot of active caspase-3 fragments (A) and corresponding quantification in arbitrary fluorescence units (B) are shown. C, detection of active caspases in HeLa cells incubated for 20 h in the presence of TNF-α (10 ng/ml) and CHX (10 μg/ml). Cells were labeled with propidium iodide (PI) and the caspase-specific FAM-VAD-fmk probe (Casp). Viable unlabeled cells (Neg), necrotic cells (PI+ Casp−), and early (PI− Casp+) or late (PI+ Casp+) apoptotic cells were quantified by flow cytometry. Numbers indicate the percentage of cells in each category. Biparameter diagrams are representative of three experiments. T/X, TNF-α and CHX; Ctrl, control.
Identification of the Caspase Cleavage Site in p53R2
The apparent molecular mass of the C-terminal p53R2 proteolytic fragment was estimated from immunoblotting experiments to be approximately between 1100 and 1300 Da. An in silico analysis of potential caspase cleavage sites using SitePrediction software (15) identified a cleavage site by human caspase-3 between Asp342 and Asn343 in the human p53R2 protein sequence, releasing a 1184-Da polypeptide in good agreement with our experimental estimates. This site ranks second in the cleavage predictions by caspase-3 with a score of 14.1. Scores for the other caspases that could be tested were well below this value (1.89, 1.10, 0.43, and 0.248 for caspase-6, -7, -8, and -1, respectively). Alignment of p53R2 and R2 C-terminal sequences of different mammalian species from rodents to human shows a complete identity among species of the last 20 amino acid residues in the p53R2 proteins, including the four residues ETTD342 defining the important positions P1–P4 upstream of the cleavage site (Fig. 4A). Interestingly, these four residues were not conserved within R2 sequences with the exception of Thr341. In particular, the critical aspartate at position 342 was replaced by a glutamic acid in protein R2. This difference certainly explains why in apoptotic cells the C-terminal tail of R2 is not cleaved similarly to its homologue (Fig. 4A). To confirm p53R2 cleavage at Asp342, a mutant p53R2-ΔC9 protein lacking the last nine residues was ectopically expressed in K-562 cells. As expected, electrophoretic migration of the truncated mutant was similar to migration of the cleaved p53R2 protein generated after imatinib treatment of K-562 cells (Fig. 4B). Next, a mutant p53R2-ΔC9 protein was stably expressed in K-562 cells at levels equal to (clone 3) or twice (clone 19) that of native p53R2. In agreement with our hypothesis, this mutant was resistant to proteolysis induced by imatinib treatment as shown on the immunoblot by the absence of a band of lower molecular weight below the band corresponding to the p53R2-ΔC9 mutant (Fig. 4C). Then we generated a V5-tagged p53R2-D342E mutant protein to determine whether Asp342 was indeed absolutely required for the caspase-dependent cleavage of p53R2. Although a V5-tagged wild-type p53R2 was as the endogenous protein subjected to a proteolytic processing inhibited by Q-VD-OPh, the V5-p53R2-D342E mutant was completely resistant to proteolysis (Fig. 4D). Moreover, the cleaved V5-p53R2WT protein was still recognized by an anti-V5 antibody, confirming that p53R2 proteolysis during apoptosis did not affect the N-terminal end of the protein. All these experimental data indicate that Asp342 is the site of caspase cleavage in p53R2.
FIGURE 4.
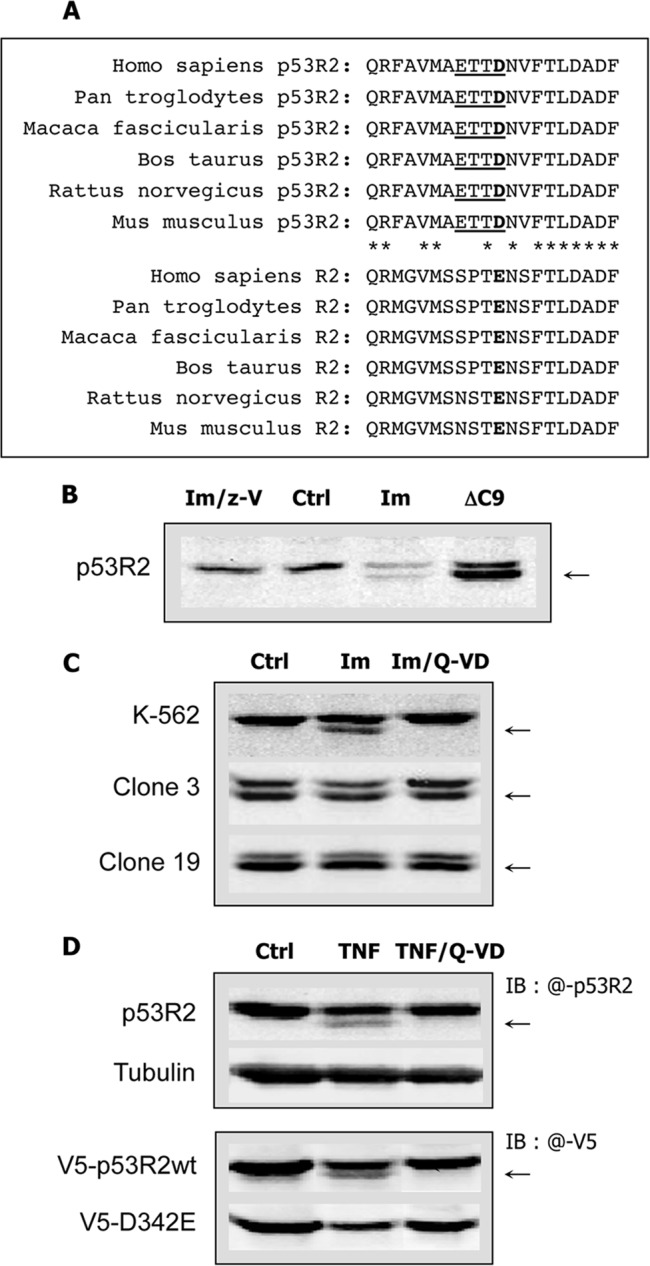
Identification of the caspase cleavage site on p53R2. A, sequence alignments of the last C-terminal 20 amino acid residues of p53R2 and R2 proteins from different species. The putative caspase cleavage site in p53R2 sequences is underlined. The P1 aspartic acid residue in p53R2 and the corresponding glutamic acid at the same position in R2 sequences are in bold. Asterisks indicate p53R2 residues conserved in R2 C-terminal ends. B, identical electrophoretic migration of ectopic p53R2-ΔC9 protein (ΔC9) expressed in untreated K-562 cells and endogenous p53R2 protein cleaved in K-562 cells incubated for 40 h with 2 μm imatinib (Im). Only full-length p53R2 is detected in untreated cells (Ctrl) or after treatment with imatinib and z-VAD (Im/z-V). The arrow indicates the proteolytic fragment of p53R2. C, K-562 cells or clones 3 and 19 derived from the K-562 cell line and stably expressing the p53R2-ΔC9 protein were subjected to a treatment with 5 μm imatinib (Im) or with imatinib and the caspase inhibitor Q-VD-OPh (Im/Q-VD) at 2 μm for 37 h. Immunoblots of p53R2 show that ectopic p53R2-ΔC9 is resistant to caspase-dependent cleavage. D, upper panel, cleavage of p53R2 (arrow) induced in H-1299 cells by treatment with TNF-α (10 ng/ml) and CHX (10 μg/ml) for 19 h (TNF). The caspase inhibitor Q-VD-OPh (Q-VD) at 2 μm prevented p53R2 proteolysis. Ctrl, untreated control. The immunoblot was probed with an anti-p53R2 antibody detected with an IRDye 800CW-conjugated secondary antibody (IB: @-p53R2). Lower panel, H-1299 cells expressing ectopic V5-p53R2WT or V5-p53R2-D342E (V5-D342E) proteins were incubated under the conditions described above. The immunoblot was probed with an anti-V5 antibody labeled with an Alexa Fluor 680-conjugated secondary antibody (IB: @-V5). The endogenous p53R2 protein was not detected under these conditions. Only the V5-p53R2-D342E mutant was resistant to caspase-dependent proteolysis.
Identification of the Caspases Involved in p53R2 Proteolysis during Apoptosis
In a first attempt to determine which caspase might be involved in p53R2 cleavage, we tested a panel of relatively specific caspase inhibitors (Fig. 5A). The caspase-1 and caspase-4 inhibitor Ac-YVAD-CHO had no effects on p53R2 proteolysis induced by imatinib treatment of K-562 cells. Compared with the pan-caspase inhibitor z-VAD-fmk, which completely blocked p53R2 cleavage, the caspase-8 and caspase-6 inhibitor Ac-IETD-CHO, the caspase-3 and caspase-7 inhibitor Ac-DEVD-CHO, and the caspase-2 inhibitor Ac-VDVAD-CHO had intermediate effects at 50 μm. These results suggest that several caspases might cleave p53R2 or alternatively that the low specificity of the different inhibitors precludes a clear-cut identification of the caspase(s) actually involved (16). To gain a better insight into the proteolytic mechanism, we took advantage of the absence of caspase-3 expression in the MCF-7 cell line. A clone (MCF-7c3) genetically complemented to re-express caspase-3 has been derived from MCF-7 cells (17). Treatment with 25 μg/ml cisplatin of MCF-7vc control cells did not induce caspase-dependent proteolytic cleavage of p53R2, whereas the protein was effectively cleaved in MCF-7c3 cells in a z-VAD-fmk-sensitive manner (Fig. 5B). This observation demonstrated the requirement of the effector caspase-3 in p53R2 proteolytic processing during apoptosis. To confirm this result, siRNAs targeting five different caspases were tested in the HeLa-229 model. Knocking down caspase-2, caspase-7, and caspase-10 had no effect on p53R2 processing in cells incubated with TNF-α and CHX (Fig. 5, C and D). A caspase-3-specific siRNA strongly reducing procaspase-3 levels (Fig. 6, A and B) greatly inhibited p53R2 cleavage (Fig. 5, C and E) in good agreement with our data obtained in MCF-7c3 cells. An siRNA directed against caspase-8 messenger nearly completely eliminated caspase-8 (Fig. 6, C and D) and profoundly inhibited p53R2 proteolysis (Fig. 5, C–E). We noticed that knocking down caspase-8 prevented procaspase-3 processing in cells treated with TNF-α and CHX (Fig. 6, A and B). This result is consistent with the caspase activation cascade triggered by death receptor ligands such as TNF-α where the receptor-activated initiator caspase-8 induces procaspase-3 proteolytic activation. Inhibition of p53R2 processing by a caspase-8 siRNA could therefore signify that either caspase-3, caspase-8, or both might cleave p53R2. We conclude from these data that p53R2 is cleaved at least by caspase-3 in apoptotic cells. In addition, when apoptosis is triggered via the extrinsic pathway, caspase-8 may participate directly or indirectly in the processing of p53R2.
FIGURE 5.

Cleavage of p53R2 is caspase-3- and caspase-8-dependent. A, immunoblot of p53R2 (top) and its quantification (bottom) using K-562 cells after treatment with 2 μm imatinib in the presence or absence of caspase inhibitors with different specificities: z-VAD-fmk (z-VAD), Ac-IETD-CHO (IETD), Ac-DEVD-CHO (DEVD), Ac-VDVAD-CHO (VDVAD), and Ac-YVAD-CHO (YVAD). The cleaved p53R2 fragment is indicated by an arrow. Only the caspase-1 inhibitor Ac-YVAD-CHO was ineffective on p53R2 proteolysis. Ctrl, untreated control cells. B, p53R2 protein expression after treatment of MCF-7vc and MCF-7c3 cells for 16 h with 25 μg/ml cisplatin (Cp) in the presence (Cp/z-V) or absence of z-VAD at 50 μm. The arrow indicates the p53R2 polypeptide cleaved in a caspase-dependent manner. Below this cleavage product, a less abundant, z-VAD-fmk-insensitive proteolytic fragment (*) appears in all cisplatin-treated cell extracts. One representative experiment of four is shown. C and D, immunoblotting of p53R2 after incubation of HeLa 229 cells with TNF-α and CHX (T/X) for 24 h. In one sample, the pan-caspase inhibitor Q-VD-OPh (Q-VD) was added at 2 μm. Forty-eight hours prior to treatment by TNF-α and CHX, cells were transfected with siRNA targeting caspase-2 (siC2), caspase-3 (siC3), caspase-7 (siC7), caspase-8 (siC8), or caspase-10 (siC10). E, quantification of p53R2 proteolysis induced by a treatment of HeLa 229 cells with TNF-α and CHX and showing the protective effect of siRNAs against caspase-3 (siC3) or caspase-8 (siC8) compared with a scrambled siRNA control (siCtrl). Results are mean ± S.E. (error bars) of three independent experiments. *, p < 0.01.
FIGURE 6.
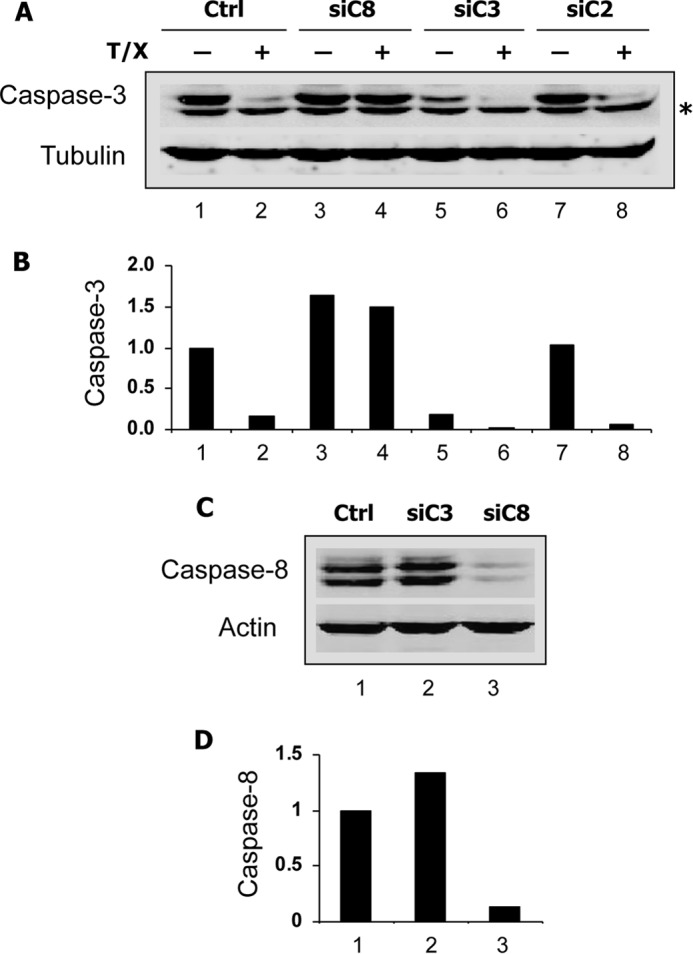
Efficacy of the siRNAs targeting caspase-3 and caspase-8. HeLa 229 cells were transfected with an siRNA directed against caspase-2 (siC2), -3 (siC3), or -8 (siC8) messengers. Forty hours later, TNF-α and CHX (T/X) were added in some cultures as indicated (A and B), and cells were harvested after a further 20-h period. Immunoblots of the 32-kDa caspase-3 zymogen (A) and the caspase-8 doublet (C) and their quantification relative to the respective loading controls α-tubulin (B) and β-actin (D) are presented. * indicates a nonspecific band below procaspase-3. Ctrl, control.
Proteolytic Cleavage of p53R2 by Purified Caspases
Crude soluble extracts prepared from H1299 cells and containing native p53R2 protein were incubated for 1–4 h with recombinant human caspase-3 or -8 (Fig. 7, A and B). In the absence of exogenous caspases, p53R2 protein levels remained stable. However, addition of either caspase-8 or caspase-3 induced a time-dependent cleavage of p53R2. At 2 h, both caspases had also caused PARP-1 cleavage. Because PARP is a preferred caspase-3 substrate, this observation suggests that endogenous procaspase-3 in the cell extract might have been proteolytically activated by purified caspase-8. Hence, it was not possible to conclude from this experiment whether p53R2 was an unexpected genuine substrate for caspase-8 or only cleaved by caspase-3 (i.e. recombinant caspase-3 or caspase-8-activated endogenous caspase-3). To answer this question, wild-type and mutant human recombinant p53R2 proteins were purified and incubated with caspase-3 or -8. As shown in Fig. 7, C and D, wild-type p53R2 was rapidly cleaved by caspase-8 and less efficiently by caspase-3. After 4 h, ∼20% of the protein was processed by caspase-3 compared with 40% in the presence of caspase-8. Even under these cell-free experimental conditions, p53R2 proteolysis never exceeded 50% of the total amount. The mutant p53R2-D342E was completely resistant to proteolysis, confirming that Asp342 is a key residue for the caspase-dependent cleavage of p53R2. Moreover, it was interesting to note that the C-terminal truncated mutant p53R2-ΔC9 protein was also not subjected to proteolysis by caspase-3 and -8. These results are in total agreement with our identification of the caspase-sensitive cleavage site in p53R2 after the Asp342 residue. They also suggest that, at least under cell-free conditions, both caspase-3 and caspase-8 are able to cleave the C-terminal tail of p53R2.
FIGURE 7.
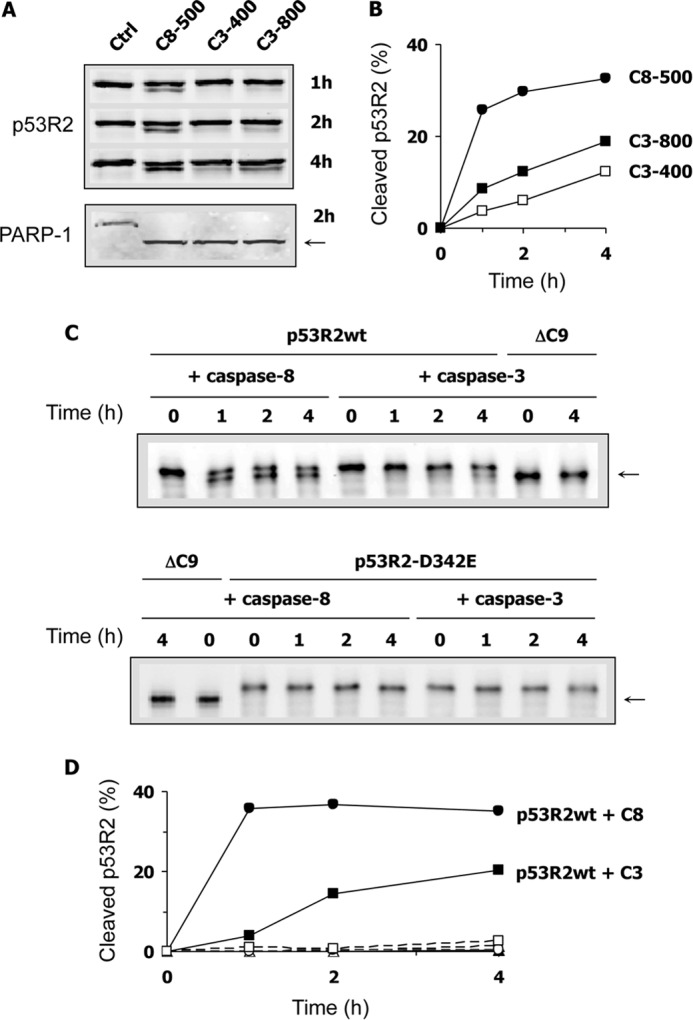
Proteolytic cleavage of the p53R2 protein induced by purified caspase-3 and -8. A, a whole cell extract of H1299 cells was incubated for 1, 2, or 4 h as indicated without (Ctrl) or with 500 ng of recombinant caspase-8 (C8-500) or ng (C3-400) or 800 ng (C3-800) of caspase-3. The immunoblot shows a caspase-dependent cleavage product of p53R2 (upper panel, lower band) and PARP (lower panel, arrow). B, quantification of p53R2 proteolysis as shown in A. C, immunoblots showing purified full-length p53R2 protein (p53R2WT), the C-terminal truncated p53R2-ΔC9 protein (ΔC9), or the caspase-resistant p53R2-D342E mutant after incubation with recombinant caspase-3 or caspase-8 for up to 4 h as indicated. The cleavage product of p53R2 has the same electrophoretic mobility as p53R2-ΔC9 and is indicated by an arrow. D, quantification of p53R2WT (filled symbols) or p53R2-D342E (open symbols) proteolysis in the presence of caspase-3 (squares) or caspase-8 (circles) as a function of time. One experiment repeated twice is shown.
Cleavage of p53R2 at Asp342 Results in a Complete Loss of Activity
To confirm that the truncated p53R2-ΔC9 protein could not form an active holocomplex with R1, we measured RnR activity of wild-type or p53R2-ΔC9 protein in the presence of an excess of protein R1. Mean RnR activity of full-length p53R2 was 40.05 nmol/min/mg, but the p53R2-ΔC9 mutant was inactive (Table 1). This result is in full agreement with previous reports demonstrating that the C-terminal end of β2 RnR subunits is essential for R1 recognition and enzyme activity (18–20).
TABLE 1.
Specific activity of human p53R2 proteins
The activity of p53R2 was measured at 30 °C by reduction of CDP in the presence of a 4-fold molar excess of protein R1. Results are mean ± S.E. of three determinations.
| p53R2 proteins | nmol CDP/min/mg |
|---|---|
| Wild type | 40.05 ± 1.31 |
| p53R2-ΔC9 | 0.19 ± 0.27 |
Caspase-dependent Proteolytic Degradation of Protein R2
As observed in K-562 cells, R2 expression was also considerably decreased in apoptotic HeLa cells (Fig. 8, A and B, lane 2). To further analyze this phenomenon and to avoid fluctuations in cell cycle distribution that might have changed the quantity of R2, HeLa 229 cells were first synchronized in early S phase by a double thymidine block (Fig. 8C). Thymidine was also conserved during subsequent treatments. Under these conditions, addition of CHX alone did not induce a significant decrease in R2 expression (Fig. 8, A and B, compare lane 8 with lane 3), confirming the enhanced stability of the protein during the S phase (3). Surprisingly, treatment with TNF-α and CHX markedly reduced R2 levels (Fig. 8, A and B, lane 4). This decrease was inhibited by Q-VD-OPh, indicating that R2 degradation was caspase-dependent (Fig. 8D). Thus, siRNAs against caspase-2, -3, and -8 were tested on R2 proteolysis in apoptotic cells. Whereas knocking down caspase-3 or caspase-8 was equally effective on p53R2 cleavage in synchronized cells as observed previously in asynchronous cultures, proteolysis of protein R2 was only prevented by a caspase-8, but not a caspase-3 or a caspase-2, siRNA (Fig. 8, A and B, lanes 5–7). Therefore, activation of the extrinsic apoptotic pathway in HeLa 229 cells induces caspase-dependent proteolysis of R2 and p53R2 via two distinct mechanisms. Degradation of protein R2 is strictly dependent on caspase-8, whereas C-terminal processing of p53R2 is mediated by caspase-3 and possibly by caspase-8.
FIGURE 8.
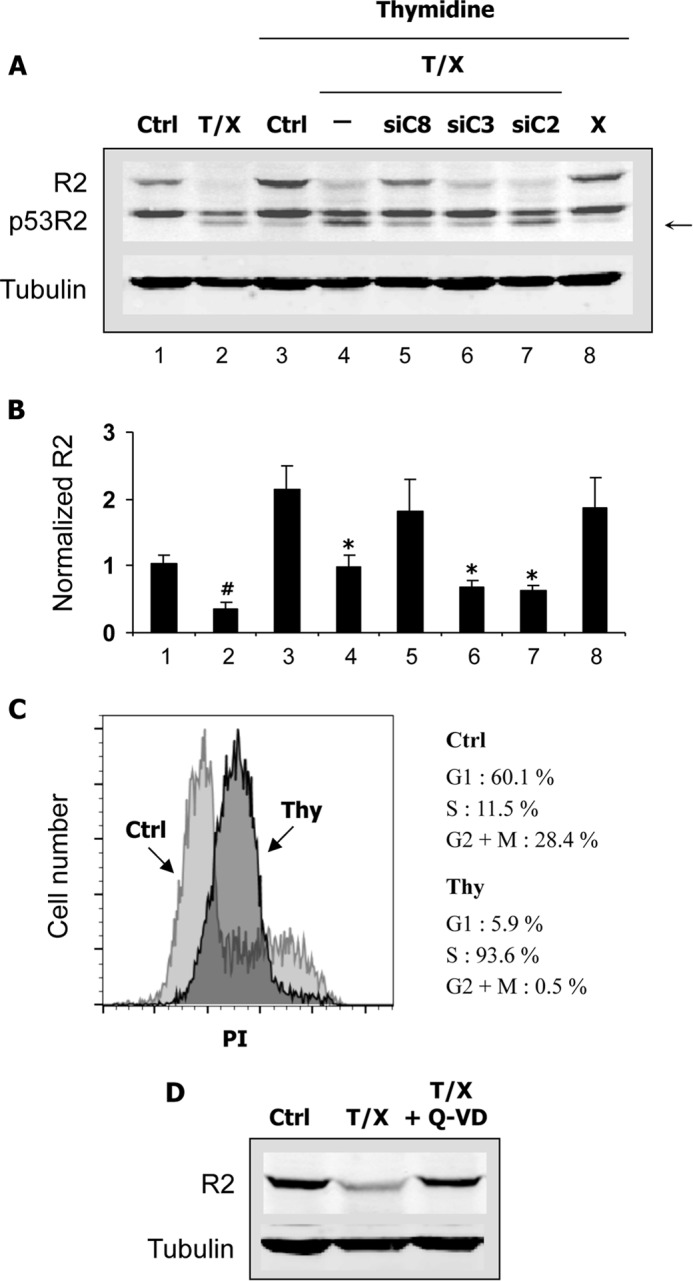
Caspase-dependent proteolysis of the R2 RnR subunit in HeLa 229 cells. A, cells were transfected or not (Ctrl) with siRNA targeting caspase-2 (siC2), -3 (siC3), or -8 (siC8) and synchronized in S phase with a double thymidine block. Asynchronous cultures were also tested for comparison (first two lanes on the left). Apoptosis was induced by TNF-α and CHX (T/X). A sample was incubated with CHX alone (X). Expression of the p53R2 and R2 RnR subunits was analyzed by immunoblotting. Protein R2 was probed with the I-15 antibody. An arrow indicates the p53R2 cleavage product. α-Tubulin is used as a loading control. Note that R2 expression increases more than 2-fold in cells blocked in S phase in agreement with the known regulation of the protein in the cell cycle. B, samples as shown in A. R2 expression was quantified relative to the asynchronous control and to the α-tubulin loading control. Results are mean ± S.E. (error bars) of three experiments. # and *, p < 0.05 versus asynchronous or thymidine-blocked control cells, respectively, as determined by Student's t test. C, cells were synchronized in S phase by a double thymidine block (Thy) or grown as asynchronous cultures (Ctrl). Cells fixed in cold ethanol were labeled with 10 μg/ml propidium iodide (PI), and cell cycle distribution was analyzed by flow cytometry. D, cells were synchronized in S phase before treatment for 22 h with TNF-α and CHX (T/X) with or without the caspase inhibitor Q-VD-OPh (Q-VD) added at 2 μm. Expression of protein R2 was evaluated by immunoblotting.
Decreased Half-life of Protein R2 in S Phase-synchronized Cells Committed to Apoptosis
Using a proteomics approach, a caspase cleavage site releasing the first 29 residues has been previously identified at the N terminus of protein R2 (21). We could not detect any degradation product of protein R2 in the experiments described in Fig. 8 using one antibody recognizing the N-terminal part of the protein (I-15). We therefore tested a second antibody (E-16) that should still recognize a protein R2 truncated from its first 29 amino acid residues according to the supplier. Immunoblots performed with the E-16 antibody revealed the presence of a smaller R2 polypeptide in apoptotic, S phase-synchronized HeLa cells (Fig. 9, A and B). The degradation product was absent from cells incubated with CHX alone or from untreated cells (Fig. 9A). Because truncated proteins are frequently destroyed by a proteasome-dependent pathway, we wonder whether the proteasome machinery was also implicated in R2 degradation during apoptosis. MG-132, a proteasome inhibitor, induced a high increase in R2 levels in apoptotic cells but did not inhibit its cleavage (Fig. 9, A and B). Knocking down caspase-8 increased the levels of full-length protein R2 as shown previously in Fig. 8 and inhibited the production of the smaller R2 polypeptide (Fig. 9B). These data establish that proteolysis of protein R2 during apoptosis was a caspase- and proteasome-dependent phenomenon that leads to a dramatic reduction in R2 levels. To better characterize changes in protein R2 stability during apoptosis, we measured its half-life in S phase-synchronized HeLa 229 cells. After treatment with TNF-α and CHX, R2 decay was monitored as a function of time in a medium containing CHX and Q-VD-OPh to prevent protein neosynthesis and to block caspase-dependent cytotoxic processes, respectively. Under these conditions, a nearly 4-fold reduction in protein R2 half-life was evidenced in apoptotic HeLa 229 cells compared with cells treated with CHX alone or with untreated controls (Fig. 9, C–E). Interestingly, the protein R1 half-life remained unchanged (Fig. 9, C and E), and total p53R2 levels did not decrease (not shown). In conclusion, among the three RnR subunits, only protein R2 expression is strongly down-regulated by proteolysis in apoptotic cells.
FIGURE 9.

Shortened half-life of protein R2 in HeLa 229 cells undergoing apoptosis. Cells were first synchronized in S phase prior to any other treatment. Thymidine at 2 mm was maintained in the culture medium until the end of the experiment to keep cells in S phase. The E-16 antibody was used to detect protein R2 by immunoblotting. A, cells were incubated in medium alone (Ctrl), with CHX (10 μg/ml), or with TNF-α (10 ng/ml) and CHX for 22 h with or without MG-132 at 20 μm. Expression of protein R2 was analyzed by immunoblotting. The smaller R2 cleavage product present in apoptotic cells treated with TNF and CHX is indicated by an arrow. Proteasome inhibition by MG-132 protected R2 from degradation in apoptotic cells. B, cells transfected or not with a caspase-8-specific siRNA were incubated with TNF and CHX (T/X) and with (MG) or without MG-132 as described in A. Knocking down caspase-8 inhibited the formation of the smaller R2 fragment (arrow). C, synchronized cells incubated in culture medium (Ctrl) or treated with CHX or with TNF and CHX for 19 h were then placed in a medium containing CHX (10 μg/ml) and Q-VD-OPh (2 μm). After 0–24 h, cells were harvested, and time-dependent decay of proteins R2 and R1 was monitored by immunoblotting. D, relative amounts of protein R2 as shown in C and normalized to its expression at t = 0. Plots were fitted with a first-order exponential equation to estimate protein half-life. E, calculated half-life of R2 and R1 proteins in cells treated as described in C. A, B, and C, representative immunoblot (n ≥ 2). E, mean ± S.E. (error bars) of three experiments. **, p < 0.01.
Deoxyribonucleosides Protect Cells from Apoptosis
Considering that the two small RnR subunits R2 and p53R2 were submitted to caspase-dependent proteolytic degradation, we hypothesized that RnR products might have a negative effect on the apoptotic process. To test this possibility, dNs were added to HeLa 229 cells together with the apoptotic agents. Phosphorylation of dNs by intracellular kinases of the salvage pathway produces dNTPs that can efficiently compensate a deficiency in de novo dNTP synthesis caused by RnR inhibition (22). Addition of dNs to cells treated with TNF and CHX induced a dose-dependent protective effect on the formation of apoptotic nuclei (Fig. 10A). Identical concentrations of ribonucleosides did not similarly decrease the number of apoptotic nuclei (not shown). As RnR-dependent dNTP production has been shown to participate in the DNA repair processes, proteolysis of small RnR subunits might increase the efficiency of apoptotic DNA fragmentation mediated by caspase-activated endonucleases. Therefore, a TUNEL assay was performed to measure DNA cleavage in apoptotic cells. DNA fragmentation was measured by the rightward shift in the Alexa Fluor 488 fluorescence profile of HeLa 229 cells after treatment with TNF and CHX (Fig. 10B, thick line). Supplementation of the culture medium with dNs decreased fluorescence labeling of the cells (Fig. 10B, gray shaded area). Inhibition of DNA fragmentation by dNs was highly significant by comparison of median fluorescence intensities (12.09 ± 0.36 versus 17.77 ± 0.30 with or without dNs, respectively; p < 0.01, n = 3). Importantly, dN-mediated protection against DNA fragmentation was no longer observed at later time points when the percentage of apoptotic nuclei reached 80–90% of the cellular population (data not shown). These data demonstrate a significant but transient protection by dNs of DNA cleavage in apoptotic cells, supporting the notion that restricting dNTP synthesis by proteolytic inactivation of RnR should improve the efficacy of DNA cleavage in apoptotic cells.
FIGURE 10.
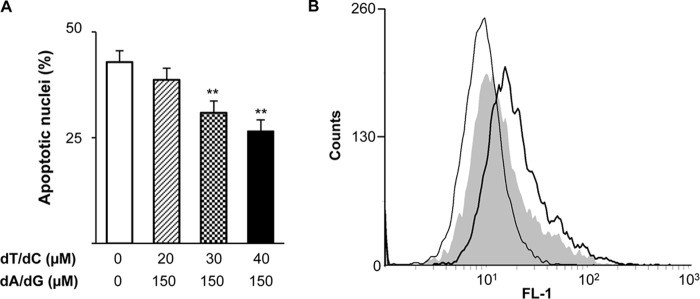
Deoxynucleosides protect cells from apoptosis. A, HeLa 229 cells were cultured with TNF-α (10 ng/ml) and CHX (10 μg/ml) in the presence of the indicated concentrations of thymidine (dT), deoxycytidine (dC), deoxyadenosine (dA), and deoxyguanosine (dG). Apoptotic nuclei were counted after cell fixation and fluorescent labeling of DNA. Results are mean ± S.E. (error bars) of three experiments. **, p < 0.01. B, apoptosis was determined by a fluorometric TUNEL assay. Cells were either left untreated (thin line) or treated with TNF and CHX in the absence (thick line) or presence of dT/dC, each at 40 μm, and dA/dG at 150 μm (gray shaded area). Fixed cells were submitted to the terminal deoxynucleotidyltransferase reaction, and fluorescent nuclear labeling was analyzed by flow cytometry.
Discussion
Class I RnR present in all mammals has been initially described as an α2β2 complex, but higher oligomeric states, (α2)x(β2)y, are also formed in the presence of ATP/dATP effectors (23). Proper binding of β2 (R2 or p53R2) to α2 (R1) is essential for activity of the multisubunit protein complex. The C-terminal tail of purified protein R2 alone is highly mobile and unstructured, but this flexibility is lost when protein R1 is bound to protein R2, suggesting recognition of the R2 tail by a particular surface on R1 (24). Other studies using mouse, viral, or bacterial class I RnR have contributed to establish the essential role of the C-terminal end of protein R2 in subunit interaction and enzyme activity (18, 25). Moreover, heptapeptides similar to β C termini of various species prevented formation of the α2β2 complex in the same species and competitively inhibited enzyme activity (19, 20, 26). All these data demonstrated an essential function of the β C-terminal end in R1 recognition and binding as well as the lack of activity of mammalian β2 proteins truncated from the last C-terminal seven residues. The present work reports for the first time the existence of a caspase-mediated proteolytic regulation of p53R2, releasing the C-terminal extremity involved in R1 recognition. Using in silico prediction tools and mutagenesis experiments, we were able to map the caspase cleavage site (↓) between Asp342 and Asn343 in the highly conserved p53R2 polypeptide ETTD↓NV. As expected, the cleaved p53R2-ΔC9 protein is mostly inactive in vitro. In our study, caspase-dependent cleavage of p53R2 was limited to 30–40% of the total amount, but the overall effect on RnR activity might be amplified if heterodimers consisting of full-length and truncated p53R2 polypeptides are produced (27).
It is noteworthy that, even using a purified p53R2 protein, proteolysis never exceeded 50% of the total amount of the protein as observed in a cellular context. Partial proteolysis of p53R2 might arise as a consequence of the cleavage of only one protomer in β2. In the reported crystal structure of human p53R2, the disordered C-terminal tail is absent (28). It is therefore not possible to predict whether loss of the C-terminal tail in one protomer might influence the structural environment of the second C-terminal tail in the other protomer. There are indeed some indications of a limited structural cross-talk between the two protomers, but it seems to involve the N-terminal extremity of the protein rather than the C-terminal one. Another explanation for limited p53R2 cleavage might be a steric inhibition by protein R1. In the E. coli class I RnR, a prototype for mammalian enzymes, a 20-mer peptide similar to the C terminus of R2 is inserted in a hydrophobic cleft between two α helices in R1 (29). Extrapolation of this binding mode to the C terminus of human β2 proteins can reasonably be expected and should prevent recognition of the C-terminal tail of p53R2 by caspases.
To identify the caspase(s) that might process p53R2, an in silico analysis was undertaken using two non-redundant software programs, SitePrediction and GraBCas. SitePrediction uses known proteolytic sites available in the literature (15), whereas GraBCas is based on the work of Thornberry et al. (31) and uses a combinatorial peptide library to define caspase selectivity. Caspase-3 and caspase-8 emerged as the most probable candidates able to process the C-terminal tail of p53R2 during apoptosis. Pharmacological and RNAi approaches excluded caspase-1, -2, -4, -7, and -10 from the possible candidates, but we could implicate caspase-3 and -8 in the cleavage of p53R2. Because caspase-8 contributes to the proteolytic activation of procaspase-3 in the death receptor signaling cascade, its action on p53R2 cleavage might be indirect and mediated by caspase-3. The fact that caspase-3 is able to cleave p53R2 is supported (i) by the inhibition of p53R2 cleavage after knocking down caspase-3 in HeLa 229 cells; (ii) by the absence of p53R2 proteolysis in caspase-3-deficient MCF-7 cells committed to apoptosis by treatment with cisplatin, which activates the apoptotic intrinsic pathway; and (iii) by the efficient cleavage of purified p53R2 by caspase-3 in a cell-free system. Caspase-8 is also probably able to attack p53R2 in apoptotic cells because it is more efficient than caspase-3 to cleave p53R2 in our cell-free experiments. However, others have noted substantial differences in the caspase activation cascade between dATP-activated cell-free systems and intact cells, suggesting that caspase specificity and efficiency may significantly differ in the two situations (30). This discrepancy led us to carefully consider our results obtained in a cell-free system. In fact, we do not have definitive evidence that p53R2 is a substrate of caspase-8 in cells committed to apoptotic cell death.
On the N-terminal side of the caspase cleavage site, residue P1 (Asp) is essential, residues P2 and P3 have a limited influence on substrate recognition, and residue P4 contributes effectively to caspase specificity (16, 31). Whereas a long sequence including P1–P4 residues is well conserved among mammalian p53R2 proteins, substantial divergence exists between C-terminal tail sequences of p53R2 and its homologue R2 with only six conserved residues among the last 13 residues. In particular, the crucial P1 residue (Asp) and the P4 residue (Glu) are not conserved in the R2 protein sequences analyzed. This discrepancy could explain why we did not observe a similar C-terminal cleavage of the R2 subunit in our experiments. However, in contrast to p53R2 whose total amounts (i.e. the native protein and the cleaved fragment) remained almost constant during apoptosis, we noticed a profound decrease in protein R2 levels in K-562 and HeLa 229 cells undergoing apoptosis. Because protein R2 expression and stability are cell cycle-dependent, this decrease might result from changes in cell cycle distribution eventually induced by the apoptotic stimulus as observed with imatinib treatment. To address this question, we took advantage of enhanced R2 stability during the S phase of the cell cycle (3) to demonstrate unambiguously a caspase-8- but not caspase-3-dependent degradation of protein R2 in apoptotic cells. This is at odds with proteolysis of the p53R2 protein, which implicated caspase-3. We could also detect a protein R2 of lower molecular weight in apoptotic cells. The cleavage product was not recognized by one antibody (I-15) recognizing the N terminus of R2, suggesting that caspase attack occurs at this extremity of the protein. A previous proteomics analysis of caspase substrates in apoptotic Jurkat cells already identified protein R2 (21). The proposed proteolytic cleavage site SLVD↓KE is located in the R2 N-terminal segment missing in p53R2 and just upstream of two motifs (i.e. a KEN box and a phosphorylated threonine residue) already shown to be involved in proteasome-dependent degradation of the R2 protein, respectively, during M/G1 and G2 phases (3, 4). Unmasking a lysine residue at the N-terminal extremity of R2 should have a destabilizing effect according to the N-end rule for proteasome-dependent degradation of truncated proteins (32). Although we did not investigate in our experiments whether the caspase-mediated cleavage of protein R2 occurred at the site identified previously in the Mahrus et al. (21) study, we observed that R2 proteolysis was prevented by a proteasome inhibitor. Moreover, protein R2 half-life was considerably reduced in cells undergoing apoptosis. Degradation of protein R2 in apoptotic cells was therefore mediated by caspase- and proteasome-dependent mechanisms. Clearly, further studies are needed to explore in depth this proteolytic mechanism. The large RnR subunit R1 did not exhibit any obvious changes in half-life, expression levels, or apparent molecular weight. Therefore, proteolytic processes taking place in apoptotic cells selectively target the two small RnR subunits, which are less abundant than protein R1.
Bioinformatics analysis of quantitative protein changes in the ApoptoProteomics database has classified DNA repair within the 10 biological processes most affected during apoptosis (33). Indeed, in addition to PARP-1 and the catalytic fragment of the DNA-dependent protein kinase identified early as caspase substrates, proteomics methods have allowed the identification of more than 20 other proteins participating in various DNA repair processes and cleaved by caspases during apoptosis (11, 21). Several DNA repair mechanisms such as base excision repair, nucleotide excision repair, and homologous recombination require a dNTP supply for de novo synthesis of DNA fragments. Besides recycling and phosphorylation of dNs to produce dNTPs via the salvage pathway, the RnR-mediated reduction of ribonucleosides represents a major metabolic route for dNTP synthesis. Although RnR activity in DNA repair processes was implicated earlier, a direct demonstration of this involvement in mammals was only recently reported for the R1 and R2 subunits (2, 4). A role for p53R2 in the resolution of DNA lesions has also emerged from recent studies (7, 8, 34). Thus, our findings add RNR β2 subunits to the list of caspase substrates involved in DNA repair and shed light on dNTP-generating enzymes as additional new targets submitted to proteolysis in apoptotic cells. The importance of RnR inactivation for apoptosis is suggested by the protective effect of a combination of dNs, but not ribonucleosides, on DNA cleavage and nucleus fragmentation. The salvage pathway can phosphorylate dNs and replenish dNTP pools depleted as a consequence of RnR inhibition. Because protection offered by dN supplementation is transient, proteolytic inactivation or destruction of the β2 RnR subunits promotes apoptosis but is not essential to completion of the cell death program.
In conclusion, we have established in this work that the small RnR subunits R2 and p53R2 are caspase substrates undergoing proteolytic cleavage during apoptosis. The two proteins are processed by distinct mechanisms. Whereas a short C-terminal fragment essential for binding to R1 is released from p53R2 in a caspase-3- and possibly caspase-8-dependent manner, protein R2 caspase cleavage occurs at the N-terminal side and is accompanied by a proteasome-dependent destruction of the protein. The C-terminal tail in p53R2 and the N-terminal extremity in R2 are key regions that determine the stability of these two homologues during apoptosis.
Acknowledgments
We thank Jacqueline Bréard (INSERM U1004, Hôpital Paul Brousse, Villejuif, France) for providing MCF-7c3 and MCF-7vc cell lines. We are grateful to Hanaa Safya for assistance in FLICA labeling and flow cytometry analysis. We acknowledge the contribution of Delphine Patin to RnR activity measurements. This work has benefited from the facilities and expertise of the cytometry platform of IMAGIF (Centre de Recherche de Gif), which is supported by the Conseil Général de l'Essonne.
This work was supported in part by a grant from the Groupement des Entreprises Françaises dans la Lutte contre le Cancer Paris Ile-de-France (to M. L.).
- dN
- deoxyribonucleoside
- CHX
- cycloheximide
- RnR
- ribonucleotide reductase
- PARP
- poly(ADP-ribose) polymerase
- z-VAD-fmk
- carbobenzoxy-valyl-alanyl-aspartyl-[o-methyl]-fluoromethyl ketone
- Q-VD-OPh
- quinolyl-valyl-O-methylaspartyl-(2,6-difluorophenoxy)methyl ketone
- Nle
- norleucine
- FLICA
- fluorochrome-labeled inhibitors of caspases.
References
- 1. Nordlund P., Reichard P. (2006) Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706 [DOI] [PubMed] [Google Scholar]
- 2. Niida H., Shimada M., Murakami H., Nakanishi M. (2010) Mechanisms of dNTP supply that play an essential role in maintaining genome integrity in eukaryotic cells. Cancer Sci. 101, 2505–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chabes A. L., Pfleger C. M., Kirschner M. W., Thelander L. (2003) Mouse ribonucleotide reductase R2 protein: a new target for anaphase-promoting complex-Cdh1-mediated proteolysis. Proc. Natl. Acad. Sci. U.S.A. 100, 3925–3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. D'Angiolella V., Donato V., Forrester F. M., Jeong Y. T., Pellacani C., Kudo Y., Saraf A., Florens L., Washburn M. P., Pagano M. (2012) Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 149, 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pontarin G., Ferraro P., Håkansson P., Thelander L., Reichard P., Bianchi V. (2007) p53R2-dependent ribonucleotide reduction provides deoxyribonucleotides in quiescent human fibroblasts in the absence of induced DNA damage. J. Biol. Chem. 282, 16820–16828 [DOI] [PubMed] [Google Scholar]
- 6. Bourdon A., Minai L., Serre V., Jais J. P., Sarzi E., Aubert S., Chrétien D., de Lonlay P., Paquis-Flucklinger V., Arakawa H., Nakamura Y., Munnich A., Rötig A. (2007) Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 39, 776–780 [DOI] [PubMed] [Google Scholar]
- 7. Pontarin G., Ferraro P., Bee L., Reichard P., Bianchi V. (2012) Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. U.S.A. 109, 13302–13307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guittet O., Tebbi A., Cottet M. H., Vésin F., Lepoivre M. (2008) Upregulation of the p53R2 ribonucleotide reductase subunit by nitric oxide. Nitric Oxide 19, 84–94 [DOI] [PubMed] [Google Scholar]
- 9. Tanaka H., Arakawa H., Yamaguchi T., Shiraishi K., Fukuda S., Matsui K., Takei Y., Nakamura Y. (2000) A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 404, 42–49 [DOI] [PubMed] [Google Scholar]
- 10. Nowsheen S., Yang E. S. (2012) The intersection between DNA damage response and cell death pathways. Exp. Oncol. 34, 243–254 [PMC free article] [PubMed] [Google Scholar]
- 11. Earnshaw W. C., Martins L. M., Kaufmann S. H. (1999) Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68, 383–424 [DOI] [PubMed] [Google Scholar]
- 12. Tebbi A., Guittet O., Cottet M. H., Vesin M. F., Lepoivre M. (2011) TAp73 induction by nitric oxide: regulation by checkpoint kinase 1 (CHK1) and protection against apoptosis. J. Biol. Chem. 286, 7873–7884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mann G. J., Gräslund A., Ochiai E., Ingemarson R., Thelander L. (1991) Purification and characterization of recombinant mouse and herpes simplex virus ribonucleotide reductase R2 subunit. Biochemistry 30, 1939–1947 [DOI] [PubMed] [Google Scholar]
- 14. Guittet O., Ducastel B., Salem J. S., Henry Y., Rubin H., Lemaire G., Lepoivre M. (1998) Differential sensitivity of the tyrosyl radical of mouse ribonucleotide reductase to nitric oxide and peroxynitrite. J. Biol. Chem. 273, 22136–22144 [DOI] [PubMed] [Google Scholar]
- 15. Verspurten J., Gevaert K., Declercq W., Vandenabeele P. (2009) SitePredicting the cleavage of proteinase substrates. Trends Biochem. Sci. 34, 319–323 [DOI] [PubMed] [Google Scholar]
- 16. McStay G. P., Salvesen G. S., Green D. R. (2008) Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 15, 322–331 [DOI] [PubMed] [Google Scholar]
- 17. Prunet C., Lemaire-Ewing S., Ménétrier F., Néel D., Lizard G. (2005) Activation of caspase-3-dependent and -independent pathways during 7-ketocholesterol- and 7β-hydroxycholesterol-induced cell death: a morphological and biochemical study. J. Biochem. Mol. Toxicol. 19, 311–326 [DOI] [PubMed] [Google Scholar]
- 18. Filatov D., Ingemarson R., Gräslund A., Thelander L. (1992) The role of herpes simplex virus ribonucleotide reductase small subunit carboxyl terminus in subunit interaction and formation of iron-tyrosyl center structure. J. Biol. Chem. 267, 15816–15822 [PubMed] [Google Scholar]
- 19. Cooperman B. S. (2003) Oligopeptide inhibition of class I ribonucleotide reductases. Biopolymers 71, 117–131 [DOI] [PubMed] [Google Scholar]
- 20. Qiu W., Zhou B., Darwish D., Shao J., Yen Y. (2006) Characterization of enzymatic properties of human ribonucleotide reductase holoenzyme reconstituted in vitro from hRRM1, hRRM2, and p53R2 subunits. Biochem. Biophys. Res. Commun. 340, 428–434 [DOI] [PubMed] [Google Scholar]
- 21. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lepoivre M., Flaman J. M., Bobé P., Lemaire G., Henry Y. (1994) Quenching of the tyrosyl free radical of ribonucleotide reductase by nitric oxide. Relationship to cytostasis induced in tumor cells by cytotoxic macrophages. J. Biol. Chem. 269, 21891–21897 [PubMed] [Google Scholar]
- 23. Fairman J. W., Wijerathna S. R., Ahmad M. F., Xu H., Nakano R., Jha S., Prendergast J., Welin R. M., Flodin S., Roos A., Nordlund P., Li Z., Walz T., Dealwis C. G. (2011) Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 18, 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lycksell P. O., Ingemarson R., Davis R., Gräslund A., Thelander L. (1994) 1H NMR studies of mouse ribonucleotide reductase: the R2 protein carboxyl-terminal tail, essential for subunit interaction, is highly flexible but becomes rigid in the presence of protein R1. Biochemistry 33, 2838–2842 [DOI] [PubMed] [Google Scholar]
- 25. Thelander L., Gräslund A. (1994) in Metal Ions in Biological Systems (Sigel H., ed) pp. 109–129, Marcel Dekker, Inc., New York [Google Scholar]
- 26. Yang F. D., Spanevello R. A., Celiker I., Hirschmann R., Rubin H., Cooperman B. S. (1990) The carboxyl terminus heptapeptide of the R2 subunit of mammalian ribonucleotide reductase inhibits enzyme activity and can be used to purify the R1 subunit. FEBS Lett. 272, 61–64 [DOI] [PubMed] [Google Scholar]
- 27. Sjöberg B. M., Karlsson M., Jörnvall H. (1987) Half-site reactivity of the tyrosyl radical of ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 262, 9736–9743 [PubMed] [Google Scholar]
- 28. Smith P., Zhou B., Ho N., Yuan Y. C., Su L., Tsai S. C., Yen Y. (2009) 2.6 Å x-ray crystal structure of human p53R2, a p53-inducible ribonucleotide reductase. Biochemistry 48, 11134–11141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uhlin U., Eklund H. (1994) Structure of ribonucleotide reductase protein R1. Nature 370, 533–539 [DOI] [PubMed] [Google Scholar]
- 30. Inoue S., Browne G., Melino G., Cohen G. M. (2009) Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ. 16, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 31. Thornberry N. A., Rano T. A., Peterson E. P., Rasper D. M., Timkey T., Garcia-Calvo M., Houtzager V. M., Nordstrom P. A., Roy S., Vaillancourt J. P., Chapman K. T., Nicholson D. W. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272, 17907–17911 [DOI] [PubMed] [Google Scholar]
- 32. Varshavsky A. (1997) The N-end rule pathway of protein degradation. Genes Cells 2, 13–28 [DOI] [PubMed] [Google Scholar]
- 33. Arntzen M. O., Thiede B. (2012) ApoptoProteomics, an integrated database for analysis of proteomics data obtained from apoptotic cells. Mol. Cell. Proteomics 11, M111.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Devlin H. L., Mack P. C., Burich R. A., Gumerlock P. H., Kung H. J., Mudryj M., deVere White R. W. (2008) Impairment of the DNA repair and growth arrest pathways by p53R2 silencing enhances DNA damage-induced apoptosis in a p53-dependent manner in prostate cancer cells. Mol. Cancer Res. 6, 808–818 [DOI] [PubMed] [Google Scholar]