

Background: Posttranslational regulation of rictor, a key partner of mTOR complex 2, and its underlying mechanism are largely undefined and thus are the focus of this study.
Results: FBXW7 interacts with rictor and mediates its degradation; this process requires phosphorylation of rictor at threonine 1695 by GSK3.
Conclusion: Rictor undergoes GSK3-dependent, FBXW7-mediated ubiquitination and proteasomal degradation.
Significance: A previously unknown mechanism underlying posttranslational regulation of rictor expression is suggested.
Keywords: Akt PKB, mTOR complex (mTORC), protein degradation, signal transduction, ubiquitylation (ubiquitination), FBXW7, GSK3, rictor
Abstract
Rictor, an essential component of mTOR complex 2 (mTORC2), plays a pivotal role in regulating mTOR signaling and other biological functions. Posttranslational regulation of rictor (e.g. via degradation) and its underlying mechanism are largely undefined and thus are the focus of this study. Chemical inhibition of the proteasome increased rictor ubiquitination and levels. Consistently, inhibition of FBXW7 with various genetic means including knockdown, knock-out, and enforced expression of a dominant-negative mutant inhibited rictor ubiquitination and increased rictor levels, whereas enforced expression of FBXW7 decreased rictor stability and levels. Moreover, we detected an interaction between FBXW7 and rictor. Hence, rictor is degraded through an FBXW7-mediated ubiquitination/proteasome mechanism. We show that this process is dependent on glycogen synthase kinase 3 (GSK3): GSK3 was associated with rictor and directly phosphorylated the Thr-1695 site in a putative CDC4 phospho-degron motif of rictor; mutation of this site impaired the interaction between rictor and FBXW7, decreased rictor ubiquitination, and increased rictor stability. Finally, enforced activation of Akt enhanced rictor levels and increased mTORC2 activity as evidenced by increased formation of mTORC2 and elevated phosphorylation of Akt, SGK1, and PKCα. Hence we suggest that PI3K/Akt signaling may positively regulate mTORC2 signaling, likely through suppressing GSK3-dependent rictor degradation.
Introduction
Rictor is a core component of mammalian target of rapamycin (mTOR)4 complex 2 (mTORC2), which plays an important role in the regulation of cellular homeostasis primarily by phosphorylating several AGC kinases such as Akt, PKC, and serum- and glucocorticoid-induced protein kinase (SGK). Because mTORC2 functions as a critical serine 473 kinase of Akt, a protein that is often hyper-activated in cancer, great attention has recently been focused on the study of the biological roles of this complex in cancer and whether this complex serves as a relevant target for cancer therapy (1–4).
Rictor has also been shown to associate with other proteins independent of mTOR, such as cullin-1 (5), Myo1c (6), integrin-linked kinase (ILK) (7), and F-Box and WD repeat domain-containing 7 (FBXW7; also known as FBW7 and CDC4) (8), implying that it could mediate other biological functions outside mTORC2. One example is that rictor is involved in the regulation of protein degradation. It has been recently demonstrated that rictor contains a novel E3 ubiquitin ligase activity by forming a complex with cullin-1 to promote SGK1 ubiquitination and degradation (5). Moreover, rictor interacts with the E3 ubiquitin ligase FBXW7 to positively regulate ubiquitination and degradation of the oncogenic proteins c-Myc and cyclin E in colon cancer cells (8).
It has been suggested that rictor can be phosphorylated at multiple sites, the majority of which are within the C-terminal half of the protein (9–11). Phosphorylation at a given site of rictor may affect its biological functions. For example, rictor has been shown to be phosphorylated at Thr-1135 by AGC kinases such as p70S6K1 (9–11), leading to disruption of rictor and cullin-1 interaction followed by reduced ability of the rictor/cullin-1 complex to ubiquitinate and degrade SGK1 (5). Rictor is also phosphorylated at Ser-1235 by GSK3β during endoplasmic reticulum stress, resulting in inhibition of mTORC2-Akt binding and mTORC2 downstream signaling (12).
Ubiquitin proteasome-mediated protein degradation regulates many important cellular processes, including differentiation, proliferation, and apoptosis. In general, this degradation process involves the covalent attachment of ubiquitin to a substrate in a chain of four or more (also called polyubiquitination) by an ubiquitin ligase, resulting in it targeting to the 26S proteasome for rapid destruction (13, 14). This is an important process in the maintenance of cellular homeostasis. Accordingly, aberrant protein degradation through this system underlies many diseases, including cancer. The E3 ubiquitin ligase, FBXW7, is a tumor suppressor that is frequently mutated in certain types of human cancer (13). The exact mechanisms underlying its tumor suppressive function remain unclear, although it appears to be associated with regulation of the degradation of multiple oncogenic proteins such as c-Myc, cyclin E, and c-Jun (13). A recent study has shown that mTOR can be degraded through an FBXW7-dependent mechanism, and loss or mutation of FBXW7 increases cell sensitivity to rapamycin due to increased levels of mTOR (15).
While studying the Akt inhibitor perifosine, we reported that perifosine reduced rictor levels, likely by inducing FBXW7-dependent protein degradation (16). However, detailed or systematic studies of rictor degradation are lacking. Here we focused our study on demonstrating rictor degradation and its underlying mechanism. We provide robust evidence to indicate that rictor is degraded through a GSK3-dependent and FBXW7-mediated mechanism.
Experimental Procedures
Reagents
MG132, LiCl, SB216763, cycloheximide (CHX), rabbit polyclonal anti-actin antibody, mouse FLAG M2 monoclonal antibody, anti-FLAG M2 affinity gel (A2220), and mouse anti-GST antibody (SAB4200237) were purchased from Sigma. CHIR99021 was purchased from LC laboratories (Woburn, MA). MLN4924 was provided by Millennium Pharmaceuticals, Inc. (Cambridge, MA). λ-Protein phosphatase was purchased from New England BioLabs (Ipswich, MA). Rabbit polyclonal antibody against mTOR, rictor (D16H9) rabbit mAb (Sepharose bead) conjugate, and GSK3α and -β kinases were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Goat polyclonal mTOR (FRAP; N-19), rabbit polyclonal cyclin E (M-20), and mouse monoclonal c-Myc (9E10) antibodies and c-Myc (9E10) AC bead (sc-40AC) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit polyclonal rictor (BL2178) and raptor antibodies were purchased from Bethyl Laboratories, Inc. (Montgomery, TX). Anti-HA antibody was purchased from Abgent (San Diego, CA). HA-ubiquitin (HA-Ub) plasmid and FBXW7 isoform plasmids were kindly provided by Dr. C. Chen (Kunming Institute of Zoology, Chinese Academy of Sciences, Yunnan, China). Dominant negative FBXW7 (dnFBXW7) expression plasmid was obtained from Dr. K. I. Nakayama (Medical Institute of Bioregulation, Kyushu University, Maidashi, Fukuoka, Japan) (17). GSK3β expression vector plasmid was a generous gift from Dr. B. P. Zhou (The University of Kentucky College of Medicine, Lexington, KY). Myc-rictor in pRK-5 (#11367) and GSK3α in pMT2 (#15896) were purchased from Addgene (Cambridge, MA).
Cell Lines and Cell Culture
HCT116-FBXW7+/+ and HCT116-FBXW7−/− cell lines were kindly provided by Dr. B. Vogelstein (Johns Hopkins University School of Medicine, Baltimore, MD). HEK293T cells were provided by Dr. K. Ye (Emory University, Atlanta, GA). Other cancer cell lines were originally purchased from the ATCC. These cell lines were grown in monolayer culture in RPMI 1640 medium, DMEM, or McCoy's medium supplemented with glutamine and 5% fetal bovine serum at 37 °C in a humidified atmosphere consisting of 5% CO2 and 95% air.
Plasmid Construction and Transfection
FLAG-GSK3α was constructed by inserting GSK3α PCR fragment, which was amplified with the primers 5′-ATGAATTCAATGAGCGGCGGCGGGCCTT-3′ and 5′-ATGGATCCTCAGGAGGA GTTAGTGAGGG-3′ using the GSK3 plasmid as a template, into pFLAG-CMV-3 expression vector (Sigma) via EcoRI and HindIII sites. The rictor site mutant construct, T1695G (in which threonine at 1695 was converted to glycine), was created using site-directed mutagenesis with the primers 5′-GAGGCTGTGTTGGCAGGACCACCAAAGCAACCT-3′ and 5′-AGGTGGCTTTGGTGGTCCTGCCAACACAGCCTC-3′. All plasmid transfections were performed with either Lipofectamine 2000 (Invitrogen) (e.g. H460 cells) or FuGENE 6 (Roche Applied Science) (e.g. 293T cells).
Small Interfering RNA (siRNA) and Transfection
GSK3α/β siRNA (#6301) was purchased from Cell Signaling. GSK3α siRNA was described previously (18). FBXW7 siRNA, that targets the sequence of 5′-AACACAAAGCTGGTGTGTGCA-3′ (19), was synthesized by Qiagen (Valencia, CA). siRNA transfection was performed with HiPerFect transfection reagent (Qiagen) following the manufacturer's instructions.
Western Blotting (WB) and Immunoprecipitation (IP)
Preparation of whole-cell protein lysates (WCL) and performance of the WB were the same as described previously (20, 21). For IP, cells were lysed in a CHAPS buffer (40 mm HEPES, pH 7.5, 120 mm NaCl, 1 mm EDTA, pH 8.0, and 0.3% CHAPS) supplemented with protease inhibitors and phosphatase inhibitors. Five hundred μg of lysates were incubated with the appropriate antibody-conjugated beads overnight at 4 °C. Immunocomplexes were washed with the CHAPS buffer twice and HEPES wash buffer (50 mm HEPES, 40 mm NaCl, 2 mm EDTA, pH 8.0) twice and then subjected to WB for detection of the proteins of interest.
Protein Stability Assay
293T cells were transfected with the plasmids of interest. After 24 h, all cells were treated with 10 μg/ml CHX and harvested at different time points for preparation of WCL and subsequent WB as described above for detection of given proteins.
Preparation of GST-rictor Proteins
DNA fragments RF1, encoding amino acids 1586–1708 of wild-type rictor, and RF1-T1695G, encoding amino acids 1586–1708 of mutant rictor T1695, were amplified from plasmids carrying myc-rictor or myc-rictor (T1695G), respectively. They were inserted into pGEX-2TK vector (GE Healthcare) to generate GST fusion protein expression constructs pGST-RF1 and pGST-RF1-T1695G. These plasmids were then transformed into Escherichia coli BL21-DE3 competent cells (Invitrogen). These transformed bacteria were grown and induced with 1 mm isopropyl-β-d-thiogalactopyranoside at 25 °C for 24 h. The proteins of interest were then purified using glutathione-Sepharose 4B (GE Healthcare) following the manufacturer's instructions. Eluted protein was desalted using PD-10 Sephadex G-25 (GE Healthcare), supplemented with 10% glycerol, and flash-frozen at −80 °C.
In Vitro Phosphorylation Assay
An in vitro GSK3 kinase assay was performed by mixing 0.4 μg of purified RF1, RF1-T1685G, or GST-only (GST control) protein with 30 μl of GSK kinase buffer (containing 4 mm MOPS, pH 7.2, 2.5 mm β-glycerophosphate, 1 mm EGTA, 0.4 mm EDTA, 4 mm MgCl2, 0.05 mm DTT, and 40 μm BSA), 1 μCi of [γ-32P]ATP (PerkinElmer Life Sciences), and GSK3α or -β kinase. After incubation at 37 °C for 30 min, the reaction samples were denatured with 5× SDS-loading buffer at 100 °C for 5 min. Proteins were then fractionated on 10% SDS-PAGE gels and transferred to a PVDF membrane. The phosphorylation of GST fusion rictor protein by GSK3 kinase was detected by exposing to x-ray films for 20 min (GSK3β) or 3 h (GSK3α) at −80 °C deep-freeze. Purified GST fusion protein level was determined using anti-GST antibody (Sigma).
Adenoviral Infection of Cancer Cells
Adenovirus harboring an empty vector (Ad-CMV) or a constitutively activated form of Akt (myristoylated Akt; Ad-myr-Akt) and cell infection were described previously (22).
Results
Rictor Levels Are Modulated by Proteasome-mediated Degradation
Our previous study using perifosine suggested that rictor is degraded through an ubiquitin/proteasome-mediated mechanism (16). To further confirm this finding, we treated two cancer cell lines, H460 and MCF-7, with the proteasome inhibitor MG132 and then determined the levels of rictor accumulation. As shown in Fig. 1A, rictor levels were increased after only 15 min of MG132 treatment and remained high within the tested time ranges (15–120 min). Under the tested conditions, the levels of mTOR and c-Myc, which are known to be degraded by FBXW7-dependent mechanism (15, 17, 23, 24), were also increased in both cell lines. Because Cullin neddylation is an essential step for activation of the SCF-FBXW7 E3 ligase (25), we then further determined whether MLN4924, an inhibitor of NEDD8-activating enzyme that catalyzes attachment of NEDD8 to a cullin (or neddylation) (25), increases rictor levels as well. Indeed, MLN4924 caused time-dependent elevation of rictor as it did to c-Myc in both H460 and MCF-7 cells (Fig. 1B). In these experiments we found that the levels of raptor, an essential component of mTOR complex 1 (mTORC1), were also elevated by both MG132 and MLN4924. Moreover, we further asked whether rictor is ubiquitinated. As presented in Fig. 1C, co-transfection of HA-Ub and rictor expression plasmids increased ubiquitinated levels of rictor, particularly in the presence of MG132, indicating that rictor is indeed a ubiquitinated protein, Taken together, these data clearly demonstrate that rictor is regulated by ubiquitination/proteasome-mediated degradation.
FIGURE 1.

Rictor is regulated by ubiquitination/proteasome-mediated degradation. A and B, both H460 and MCF-7 cells were treated with 10 μm MG132 (A) or 1 μm MLN4924 (B) for the indicated times and then harvested for preparation of WCLs and subsequent WB to detect the given proteins. C, H460 cells were transfected with HA-ubiquitin (HA-Ub) or empty vector for 48 h and then treated with DMSO or 10 μm MG132 for an additional 2 h. The WCLs prepared from these cells were then immunoprecipitated with anti-rictor antibody, and ubiquitinated rictor (Ub-rictor) was detected by WB using anti-HA antibody.
Modulation of FBXW7 Expression Alters Rictor Ubiquitination and Levels
We previously had suggested that perifosine-induced rictor degradation involves FBXW7 (16). Therefore, we conducted detailed experiments to further demonstrate the involvement of FBXW7 in mediating rictor degradation. When comparing the levels of rictor between HCT116/FBXW7−/− and HCT116/FBXW7+/+ cells, we detected higher levels of rictor, mTOR, and cyclin E (known FBXW7 substrates) in HCT116/FBXW7−/− cells than in HCT116/FBXW7+/+ cells (Fig. 2A). Elevated levels of rictor, mTOR, and cyclin E were also detected in both H460 and HCT116 cells transfected with FBXW7 siRNA in comparison with those transfected with control scrambled siRNA (Fig. 2B). We used ectopically expressed FBXW7 to validate the knockdown efficiency as we lacked an anti-FBXW7 antibody able to recognize endogenous FBXW7 as did others (26). Moreover, enforced expression of dnFBXW7 dose-dependently increased the levels of rictor, mTOR, and cyclin E (Fig. 2C). Finally, we compared the impact of FBXW7 inhibition on rictor stability and ubiquitination. We found that rictor was degraded much slower in FBXW−/− HCT116 cells than in FBXWT+/+ HCT116 cells (Fig. 2D). Consistently, the levels of ubiquitinated rictor were substantially reduced in cells transfected with FBXW7 siRNA when compared with cells transfected with control scrambled siRNA (Fig. 2E). These results clearly show that depletion or inhibition of FBXW7 suppresses rictor ubiquitination and degradation.
FIGURE 2.

Inhibition of FBXW7 increases rictor levels (A–C) with increased stability (D) and reduced ubiquitination (E). A, WCLs were prepared from the indicated cell lines. B, twenty-four hours after transfection with control or FBXW7 siRNA, H460 cells were further transfected with FLAG-FBXW7β plasmid. After an additional 48 h, the cells were harvested for preparation of WCLs. For HCT116 cells, WCLs were prepared after transfection of control or FBXW7 siRNA for 48 h. C, 293T cells were transfected with different amounts of dnFBXW7 as indicated and then harvested after 24 h for preparation of WCLs. WB was conducted to detect the indicated proteins in the WCLs. D, the indicated cells line were exposed to CHX (10 μg/ml) and then harvested at the different time points as indicated for preparation of WCLs. The indicated proteins were detected with WB and quantified with NIH ImageJ software (Bethesda, MA). Rictor levels were normalized to tubulin and plotted as the relative rictor levels compared with those at time 0 of CHX treatment (right panel). E, 24 h after transfection with control or FBXW7 siRNA, H460 cells were further transfected with HA-ubiquitin or empty vector for an additional 48 h. The cells were then treated with 10 μm MG132 for 2 h and harvested for preparation of WCLs. These lysates were then immunoprecipitated with an anti-rictor antibody, and ubiquitinated rictor (Ub-rictor) was determined by WB using an anti-HA antibody. The asterisk indicates IgG heavy chain.
To robustly demonstrate the role of FBXW7 in regulation of rictor degradation, we expressed ectopic FBXW7 and then examined its impact on rictor stability and protein levels. It is known that FBXW7 has three isoforms (i.e. α, β, and γ) that share similar structural features and are, in principle, identical (13). Enforced expression of each isoform of FBXW7 reduced the levels of rictor and mTOR (Fig. 3A). Because FBXW7β is primarily present in the cytoplasm, we selected this isoform to use in our subsequent studies. After transfecting different amounts of FBXW7β into the cells, we observed dose-dependent reduction of rictor and mTOR (Fig. 3B). Moreover, co-transfection of FBXW7β with rictor drastically enhanced the rate of rictor degradation in comparison with transfection of rictor alone (Fig. 3C). These results indicate that enforced expression of FBXW7 enhances rictor degradation. Taking these data together, we conclude that rictor is degraded through an FBXW7-mediated mechanism.
FIGURE 3.

Enforced expression of FBXW7 decreases rictor levels (A and B) and stability (C). A and B, 293T cells were transfected with empty vector, FLAG-FBXW7α, -β, or -γ plasmids (A) or different amounts of FLAG-FBXW7β as indicated (B). After 24 h, the cells were harvested for preparation of WCLs and subsequent WB to detect the indicted proteins. C, 293T cells were co-transfected with myc-rictor plus FLAG-FBXW7β or empty vector. After 24 h, the cells were treated with CHX (10 μg/ml) and then harvested at the different time points as indicated for preparation of WCLs. The indicated proteins were detected with WB. Protein levels were quantified with NIH ImageJ software (Bethesda, MA) and were normalized to actin. The results were plotted as the relative rictor levels compared with those at time 0 of CHX treatment (bottom panel).
Rictor Interacts with FBXW7
Because FBXW7 mediates rictor degradation, we determined whether there is an interaction between rictor and FBXW7. Because of the lack of good antibody against endogenous FBXW7, we transfected FLAG-FBXW7 into 293T cells and then detected its interaction with endogenous rictor through IP of FBXW7 with an anti-FLAG M2 affinity gel or IP of rictor with an anti-rictor-conjugated Sepharose bead. Indeed, we detected both FLAG-FBXW7 and rictor from immunocomplexes pulled down either with an anti-FLAG antibody or with an anti-rictor antibody but not from those pulled down with a control IgG antibody (Fig. 4, A and B), indicating an interaction between FBXW7 and rictor. Moreover, we transfected wild-type (WT) FBXW7 and dnFBXW7 into 293T cells and compared their abilities to interact with rictor. As presented in Fig. 4B, rictor was clearly pulled down with an anti-FLAG antibody from WCLs transfected with either WT FBXW7 or dnFBXW7 (Fig. 4C), indicating that both WT FBXW7 and dnFBXW7 interact with rictor. These results provide additional support for the crucial role of FBXW7 in mediating rictor degradation.
FIGURE 4.

FBXW7 interacts with rictor. 293T cells were transfected with FLAG-FBXW7β plasmid (A, B, and D), empty vector (V), FLAG-dnFBXW7 (dn), or FLAG-FBXW7β (WT) plasmid (C). After 48 h the cells were harvested for preparation of WCLs followed by IP with an anti-FLAG (A and C) or anti-rictor antibody (B) and subsequent WB to detect the indicated proteins. In addition, the WCLs were also treated with λ-phosphatase (1000 units) at 30 °C for 1 h before the indicated IP-WB experiments (D).
Moreover, we determined whether rictor interaction with FBXW7 is phosphorylation-dependent. As presented in Fig. 4D, dephosphorylation of WCLs with λ-phosphatase before IP abolished interaction between rictor and FBXW7 as demonstrated by IP with either FLAG or rictor. Hence, it is clear that this interaction is phosphorylation-dependent.
Identification of a CDC4 Phospho-degron (CPD) Motif in Rictor That Is Critical for FBXW7 and Rictor Interaction
It is known that the interaction between FBXW7 and its substrates is dependent on the presence of a consensus CPD in the substrates in which a central phospho-threonine (pT) is embedded within hydrophobic residues: (I/L)(I/L/P)pTPXX*X (X corresponds to any amino acid, and * indicates phosphorylation (e.g. pS or pT) or an acidic amino acid residue) (13). We identified a putative CPD motif (LATPPKQP) near the C terminus (1693–1700) of rictor (Fig. 5A). To determine whether this is a functional CPD, we mutated the threonine (T) at position 1695 in this motif to glycine (G) to generate a mutant rictor, rictor (T1695G). When co-expressed with FBXW7, the levels of WT rictor were clearly reduced; however, the rictor (T1695G) levels were not decreased at all (Fig. 5B). In a stability assay, we found that rictor (T1695G) was degraded much more slowly than WT rictor (Fig. 5C), indicating that rictor (T1695G) is much more stable than WT rictor. These findings indicate that rictor (T1695G) is resistant to FBXW7-mediated degradation. In agreement, we detected drastically reduced amounts of FBXW7 pulled down by rictor (T1695G) than by WT rictor (Fig. 5D), indicating that the interaction between FBXW7 and rictor (T1695G) is much weaker than that between FBXW7 and WT rictor. Moreover, the ubiquitination of rictor (T1695G) was also substantially reduced in comparison with that of WT rictor (Fig. 5E). Taken together, these data clearly show that this CPD is functional and crucial for FBXW7-mediated degradation of rictor.
FIGURE 5.

Identification of a putative CPD in rictor (A) and demonstration of its function in FBXW7-mediated rictor degradation (B–E). A, a putative CPD motif was identified at positions 1693–1704 of rictor. B and D, 293T cells were co-transfected with FLAG-FBXW7 and myc-rictor or myc-rictor (T1695G) for 24 h (B) or 48 h (D). The cells were then harvested for preparation of WCLs and subsequent WB (B) or IP/WB (D). C, 293T cells were co-transfected with FLAG-FBXW7 and myc-rictor or myc-rictor (T1695G) and 24 h later treated with 10 μg/ml CHX. At the indicated time points, the cells were harvested for preparation of WCLs. The indicated proteins were detected with WB and quantified with NIH Image J. After being normalized to actin, the results were then plotted as the relative protein levels compared with those at time 0 of CHX treatment (bottom panel). E, FBXW7β was co-expressed with myc-rictor or myc-rictor (T1695G) in 293T cells, and after 24 h, cells were transfected with an empty vector (V) or HA-ubiquitin (HA-Ub). After another 24 h, cells were treated with 10 μm MG132 for 2 h and then subjected to preparation of WCLs and subsequent IP/WB to detect ubiquitinated rictor.
Rictor Degradation Requires GSK3
It is also well known that prior substrate phosphorylation is an essential step for FBXW7 binding and subsequent degradation. GSK3 is involved in the degradation of most, it not all, substrates by FBXW7 (13, 27). Thus, we further determined whether GSK3 is involved in FBXW7-dependent degradation of rictor. Treatment of H460 and MCF-7 cells with two GSK3 inhibitors, SB216763 and CHIR99021, increased the levels of rictor and c-Myc (Fig. 6A). Consistently, knockdown of GSK3α/β by specific siRNA resulted in elevated levels of rictor (Fig. 6B). Similarly, the levels of cyclin E, which is known to be regulated by GSK3, were also elevated in GSK3 siRNA-transfected cells. The elevation of rictor levels by LiCl (another GSK3 inhibitor) and SB216763 treatment was abrogated when FBXW7 was silenced (Fig. 6C), suggesting that this elevation occurs through an FBXW7-dependent mechanism. In contrast, enforced expression of GSK3α or GSK3β decreased the levels of rictor (Fig. 6D). These results collectively demonstrate that GSK3 is involved in FBXW7-mediated degradation of rictor. Moreover we determined the effects of GSK3 inhibition on rictor ubiquitination. As presented in Fig. 6E, we detected much lower levels of ubiquitinated rictor in LiCl-treated cells than in DMSO-treated cells, suggesting that inhibition of GSK3 with LiCl inhibits rictor ubiquitination. Consistently, inhibition of GSK3 by silencing GSK3α generated similar results as with LiCl (Fig. 6F). Therefore, it is clear that GSK3 is required for rictor ubiquitination. Finally we asked whether GSK3 interacts with rictor. In an IP assay, we detected rictor when pulling down FLAG-GSK3α or myc-GSK3β (Fig. 7, A and B), indicating that there is indeed an interaction between GSK3 and rictor.
FIGURE 6.

Modulation of GSK3 activity alters rictor levels (A–C) and ubiquitination (E and F). A, H460 and MCF-7 cells were treated with the indicated concentrations of SB216763 or CHIR99021 for 6 h. B, H460 or 293T cells were transfected with control (Ctrl) and GSG3α/β siRNA and harvested after 48 h. C, H460 cells were transfected with control or FBXW7 siRNA and after 48 h treated with 50 mm LiCl or 20 μm SB216763 for an additional 6 h. D, 293T cells were transfected with different amounts of GSK3α or GSK3β as indicated. After 24 h the cells were harvested for preparation of WCLs and subsequent WB. E, H460 cells were transfected with vector or HA-ubiquitin (HA-Ub) and after 24 h were treated with 50 mm LiCl for 4 h followed by exposure to 10 μm MG132 for another 2 h. F, H460 cells were transfected with control (Ctrl) or GSK3α siRNA followed after 24 h with transfection of vector or HA-Ub plasmid. After 24 h the cells were treated with 10 μm MG132 for an additional 2 h. After the aforementioned treatments in E and F, the cells were harvested for preparation of WCLs for subsequent IP/WB to detect the indicated proteins or protein ubiquitination.
FIGURE 7.
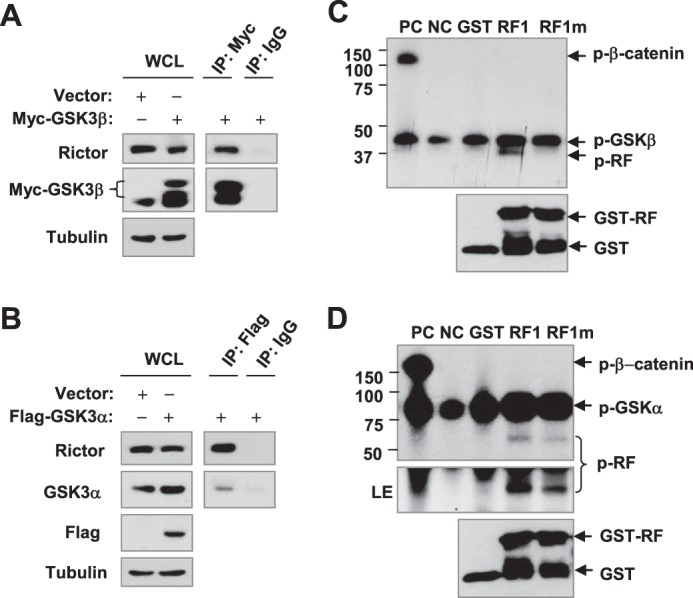
GSK3 interacts with rictor (A and B) and phosphorylates rictor at Thr1965 (C and D). A and B, 293T cells were transfected with Myc-GSK3β or FLAG-GSK3α, and after 48 h IP was performed for GSK3β with an anti-Myc antibody (A) or GSK3α with an anti-FLAG antibody (B) followed by WB to detect the given proteins. C and D, phosphorylation of purified GST-tagged RF1 and RF1m by GSK3 β (C) or GSK3 α (D) was carried out by mixing 1 μCi of [γ-32P]ATP with kinase buffer and incubating at 37 °C for 30 min. β-Catenin (positive control (PC)), non-substrate (negative control (NC)), and GST-only protein (GST) were used as controls. The protein mixtures were separated by 10% SDS-PAGE gels to detect phosphorylation by direct exposure to x-ray film and GST-tagged protein by WB with an anti-GST antibody.
GSK3 Directly Phosphorylates Rictor
Because Thr-1695 in the CPD of rictor is crucial for FBXW7-mediated rictor degradation, we asked whether this site is phosphorylated by GSK3. Thus, we conducted an in vitro kinase assay to determine whether GSK3 directly phosphorylates rictor at Thr-1695. To this end, we cloned and expressed a fragmented form of rictor (RF1) that contains the Thr-1695 residue in the CPD motif and a mutant form (RF1m) in which Thr-1695 was converted to Gly-1695 (T1695G). As presented in Fig. 7, C and D, both GSK3α and GSK3β could phosphorylate RF1, but the mutant RF1m was phosphorylated only weakly or not at all. As a positive control, β-catenin was strongly phosphorylated. Thus, we believe that GSK3 can directly phosphorylate GSK3 at Thr-1695.
Activated Akt Increases Rictor Stability and mTORC2 Activity
Akt is known to phosphorylate GSK3, leading to inactivation of GSK3 (28). Because GSK3 directly phosphorylates rictor, facilitating its degradation as demonstrated above, we asked whether activated Akt positively regulates the mTORC2 activity and signaling through enhancing rictor stability. Hence, we infected two NSCLC cell lines, A549 and H460, with Ad-myr-Akt (to expression activated Akt) and Ad-CMV (as a control) for 48 h and then examined rictor levels and phosphorylation of several proteins known to be phosphorylated by mTORC2. As presented in Fig. 8A, we detected high levels of myr-Akt and p-GSK3α/β in cells infected with Ad-myr-Akt, indicating that expression of activated Akt does increase GSK3 phosphorylation. In addition, we detected increased levels of rictor, p-Akt (Ser-473), p-Akt (Thr-450), p-SGK1 (Ser-422), and p-PKCα (Ser-659) in cells infected with Ad-myr-Akt as compared with cells infected with control Ad-CMV, suggesting that enforced Akt activation may stabilize rictor and increase mTORC2 activity. Moreover, we compared the effects of enforced Akt activation on mTORC2 formation with IP of mTOR followed by WB for detection of mTOR and rictor. Indeed, we detected increased levels of rictor in immunocomplexes from both H460 and A549 cells infected with Ad-myr-Akt in comparison with those from their corresponding Ad-CMV-infected cells, indicating that Akt activation increases mTORC2 formation. Collectively, we suggest that Akt activation increases mTORC2 activity and signaling via inactivation of GSK3 activity and subsequent stabilization of rictor.
FIGURE 8.

Enforced activation of Akt increases the levels of rictor and phosphorylation of Akt, SGK1, and PKCα (A) and enhances the assembly of the mTORC2 (B); these data suggest a model for Akt positive regulation of the mTORC2 activity and signaling (C). A and B, The given cell lines were infected with the indicated adenoviruses for 48 h and then harvested for preparation of WCLs and subsequent WB to detect proteins as indicated (A), The WCLs were also used for IP with mTOR antibody followed with WB to detect the indicated proteins (B). C, a working model for Akt positive regulation of the mTORC2. Akt may enhance the assembly and activity of the mTORC2 by inhibiting GSK3-dependent rictor degradation.
Discussion
As a core component of the mTORC2, how rictor is regulated at the degradation level is largely unknown. The present study provides robust evidence indicating that rictor is degraded through a GSK3-dependent and FBXW7-mediated ubiquitination/proteasome pathway based on the following findings; 1) rictor is regulated by ubiquitination/proteasome-mediated degradation; 2) rictor degradation is mediated by FBXW7; 3) a functional CPD motif is present in rictor; 4) rictor degradation is GSK3-dependent; 5) GSK3 directly phosphorylates the rictor CPD motif at Thr-1695. It has been previously shown that FBXW7 is involved in mediating mTOR degradation (15). The current findings provide another piece of evidence supporting the involvement of protein degradation in the regulation of mTOR signaling. In this study we noted that raptor levels were also elevated upon treatment with MG132 or MLN4924 (Fig. 1). Whether these observations suggest that raptor is regulated through a similar mechanism needs further evaluation.
FBXW7 is the substrate recognition component of an evolutionarily conserved complex of SKP1, cullin-1, and F-box protein (SCF)-type ubiquitin ligase. The binding of FBXW7 to its substrates is an essential step for ubiquitination and subsequent proteasome-mediated degradation of the substrates (13). In our IP/WB assay, we could use anti-FLAG (FBXW7) antibody to pull down rictor or anti-rictor antibody to pull down FLAG-FBXW7 (Fig. 4), suggesting that FBXW7 is associated with rictor. In a previous study on rictor regulation of FBXW7-dependent c-Myc and cyclin E degradation, FBXW7 and rictor interaction was also detected in human colon cancer cells (8). Although this study did not explore FBXW7-mediated rictor degradation, the results support our finding on interaction of rictor with FBXW7 in cells. Interestingly, another recent study has demonstrated that rictor specifically interacts with cullin-1 to function as an ubiquitin E3 ligase (5), indirectly supporting the association of rictor with the SCF (SKP1, cullin-1, and F-box protein). Collectively it is very likely that rictor is also degraded through an FBXW7-mediated mechanism, whereas it functions as a ubiquitin E3 ligase to promote degradation of other proteins.
The presence of a CPD in a substrate of FBXW7 is absolutely required for FBXW7 to bind to, ubiquitinate, and degrade the substrate. In addition to a central phosphothreonine (Thr(P)), a negative charge at the +4 position (relative to the central threonine residue) of CPD is preserved in most known FBXW7 substrates either by phosphorylation (e.g. Ser(P) or Thr(P)) or by acidic amino acid residues. This negative charge makes direct contact with WD40 repeats of FBXW7 (13). The +4 position in rictor is the acidic amino acid residue glutamine. Characterization of the putative CPD in rictor has shown that this is a functional CPD because mutation of the central threonine, Thr-1695, drastically inhibited its association with FBXW7, ubiquitination, and degradation (Fig. 5).
It is also known that the interaction of FBXW7 and substrate requires prior phosphorylation of substrates within their CPDs; this phosphorylation is often mediated by GSK3 (13). Indeed, rictor and FBXW7 interaction is also phosphorylation-dependent because dephosphorylation abolishes this interaction (Fig. 4D). In general, GSK3 phosphorylates the central threonine of the CPD in each substrate with a priming site (e.g. cyclin E, c-Myc, and Jun) because GSK3 often uses a priming phosphorylation as a binding site to target an acceptor site in the +4 position (13). In the instance of mouse c-Myb degradation by FBXW7, GSK3 is also involved in mediating phosphorylation of T572 within the CPD without a priming phosphorylation site (29). In the CPD of rictor, there is no priming phosphorylation site at the +4 position. However, our in vitro kinase assay clearly showed that GSK3 specifically phosphorylated Thr-1695 as mutation of this site abolished or attenuated the phosphorylation (Fig. 7), thus providing experimental evidence that GSK3 phosphorylates rictor at Thr-1695. In agreement, rictor and GSK3 were co-immunoprecipitated, suggesting that rictor is associated with GSK3. In addition, modulation (inhibition or activation) of GSK3 activity by chemical inhibitors or genetic approaches (e.g. knockdown or enforced expression) altered the levels of rictor accompanied with corresponding changes in rictor ubiquitination (Fig. 6). Together, these results clearly show that rictor degradation is GSK3-dependent.
The PI3K/Akt signaling is known to be hyper-activated in many types of cancer primarily due to frequent mutation of PTEN and PIK3CA and hence plays a critical role in supporting cancer cells survival and growth (28). This signaling positively regulates the mTORC1 signaling through inhibition of TSC2 (30). In this study, we found that enforced activation of Akt through expression of constitutively activated Akt increased GSK3 phosphorylation and rictor levels. Moreover we detected increased levels of p-Akt (Ser-473), p-Akt (Thr-450), p-SGK1 (Ser-422) and p-PKCα (Ser-659) (Fig. 8A), which are known to be the substrates of mTORC2 (31). Moreover increased Akt activation enhanced the formation or assembly of mTORC2 (Fig. 8B). These results suggest that the PI3K/Akt signaling may also positively regulate the mTORC2 activity and signaling through inactivation of GSK3 and subsequent stabilization of rictor followed by enhancement of the assembly of mTOR and rictor complex. Hence, the PI3K/Akt signaling positively regulates not only the mTORC1 but also the mTORC2 signaling (Fig. 8C).
FBXW7 is a tumor suppresser that is frequently mutated in certain types of human cancer (13). The exact mechanisms underlying its tumor suppressive function remain unclear, although it appears to be associated with regulation of the degradation of multiple oncogenic proteins such as c-Myc, cyclin E, and c-Jun (13). Rictor is known to be an essential component of mTORC2, which is a critical Akt Ser-473 kinase (32) and is implicated in cancer development (1, 3, 4). Our findings presented in this study may suggest a putative tumor-suppressive mechanism of FBXW7 involving negative regulation of rictor stability. Hence further study in this direction is warranted.
Acknowledgments
We are grateful to Dr. B. Vogelstein, C. Chen, and K. I. Nakayama for providing cell lines or plasmids. We also thank Dr. A. Hammond in our department for editing the manuscript.
This work was supported by National Institutes of Health Grant R01 CA118450 (to S.-Y. S.) and R01 CA160522 (to S.-Y. S.).
- mTOR
- mammalian target of rapamycin
- mTORC2
- mTOR complex 2
- GSK3
- glycogen synthase kinase 3
- SGK
- serum- and glucocorticoid-induced protein kinase
- FBXW7 (CDC4)
- F-Box and WD repeat domain-containing 7
- CHX
- cycloheximide
- WB
- Western blot
- IP
- immunoprecipitation
- WCL
- whole-cell lysate
- Ub
- ubiquitin
- CPD
- CDC4 phospho-degron.
References
- 1. Guertin D. A., Stevens D. M., Saitoh M., Kinkel S., Crosby K., Sheen J. H., Mullholland D. J., Magnuson M. A., Wu H., Sabatini D. M. (2009) mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15, 148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sparks C. A., Guertin D. A. (2010) Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene 29, 3733–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roulin D., Cerantola Y., Dormond-Meuwly A., Demartines N., Dormond O. (2010) Targeting mTORC2 inhibits colon cancer cell proliferation in vitro and tumor formation in vivo. Mol Cancer 9, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee K., Nam K. T., Cho S. H., Gudapati P., Hwang Y., Park D. S., Potter R., Chen J., Volanakis E., Boothby M. (2012) Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J. Exp. Med. 209, 713–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gao D., Wan L., Inuzuka H., Berg A. H., Tseng A., Zhai B., Shaik S., Bennett E., Tron A. E., Gasser J. A., Lau A., Gygi S. P., Harper J. W., DeCaprio J. A., Toker A., Wei W. (2010) Rictor forms a complex with Cullin-1 to promote SGK1 ubiquitination and destruction. Mol. Cell 39, 797–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hagan G. N., Lin Y., Magnuson M. A., Avruch J., Czech M. P. (2008) A Rictor-Myo1c complex participates in dynamic cortical actin events in 3T3-L1 adipocytes. Mol. Cell. Biol. 28, 4215–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McDonald P. C., Oloumi A., Mills J., Dobreva I., Maidan M., Gray V., Wederell E. D., Bally M. B., Foster L. J., Dedhar S. (2008) Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res. 68, 1618–1624 [DOI] [PubMed] [Google Scholar]
- 8. Guo Z., Zhou Y., Evers B. M., Wang Q. (2012) Rictor regulates FBXW7-dependent c-Myc and cyclin E degradation in colorectal cancer cells. Biochem. Biophys. Res. Commun. 418, 426–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dibble C. C., Asara J. M., Manning B. D. (2009) Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 29, 5657–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Treins C., Warne P. H., Magnuson M. A., Pende M., Downward J. (2010) Rictor is a novel target of p70 S6 kinase-1. Oncogene 29, 1003–1016 [DOI] [PubMed] [Google Scholar]
- 11. Julien L. A., Carriere A., Moreau J., Roux P. P. (2010) mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell. Biol. 30, 908–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen C. H., Shaikenov T., Peterson T. R., Aimbetov R., Bissenbaev A. K., Lee S. W., Wu J., Lin H. K., Sarbassov dos D. (2011) ER stress inhibits mTORC2 and Akt signaling through GSK-3β-mediated phosphorylation of rictor. Sci. Signal. 4, ra10. [DOI] [PubMed] [Google Scholar]
- 13. Welcker M., Clurman B. E. (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 83–93 [DOI] [PubMed] [Google Scholar]
- 14. Hoeller D., Dikic I. (2009) Targeting the ubiquitin system in cancer therapy. Nature 458, 438–444 [DOI] [PubMed] [Google Scholar]
- 15. Mao J. H., Kim I. J., Wu D., Climent J., Kang H. C., DelRosario R., Balmain A. (2008) FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321, 1499–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fu L., Kim Y. A., Wang X., Wu X., Yue P., Lonial S., Khuri F. R., Sun S. Y. (2009) Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components in the mTOR axis and induces autophagy. Cancer Res. 69, 8967–8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yada M., Hatakeyama S., Kamura T., Nishiyama M., Tsunematsu R., Imaki H., Ishida N., Okumura F., Nakayama K., Nakayama K. I. (2004) Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23, 2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen S., Cao W., Yue P., Hao C., Khuri F. R., Sun S. Y. (2011) Celecoxib promotes c-FLIP degradation through Akt-independent inhibition of GSK3. Cancer Res. 71, 6270–6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mao J. H., Perez-Losada J., Wu D., Delrosario R., Tsunematsu R., Nakayama K. I., Brown K., Bryson S., Balmain A. (2004) Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 432, 775–779 [DOI] [PubMed] [Google Scholar]
- 20. Sun S. Y., Yue P., Wu G. S., El-Deiry W. S., Shroot B., Hong W. K., Lotan R. (1999) Mechanisms of apoptosis induced by the synthetic retinoid CD437 in human non-small cell lung carcinoma cells. Oncogene 18, 2357–2365 [DOI] [PubMed] [Google Scholar]
- 21. Liu X., Yue P., Zhou Z., Khuri F. R., Sun S. Y. (2004) Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J. Natl. Cancer Inst. 96, 1769–1780 [DOI] [PubMed] [Google Scholar]
- 22. Elrod H. A., Lin Y. D., Yue P., Wang X., Lonial S., Khuri F. R., Sun S. Y. (2007) The alkylphospholipid perifosine induces apoptosis of human lung cancer cells requiring inhibition of Akt and activation of the extrinsic apoptotic pathway. Mol. Cancer Ther. 6, 2029–2038 [DOI] [PubMed] [Google Scholar]
- 23. Kubbutat M. H., Jones S. N., Vousden K. H. (1997) Regulation of p53 stability by Mdm2. Nature 387, 299–303 [DOI] [PubMed] [Google Scholar]
- 24. Welcker M., Orian A., Jin J., Grim J. E., Grim J. A., Harper J. W., Eisenman R. N., Clurman B. E. (2004) The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. U.S.A. 101, 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soucy T. A., Dick L. R., Smith P. G., Milhollen M. A., Brownell J. E. (2010) The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer 1, 708–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanei-Ishii C., Nomura T., Takagi T., Watanabe N., Nakayama K. I., Ishii S. (2008) Fbxw7 acts as an E3 ubiquitin ligase that targets c-Myb for nemo-like kinase (NLK)-induced degradation. J. Biol. Chem. 283, 30540–30548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Z., Inuzuka H., Zhong J., Wan L., Fukushima H., Sarkar F. H., Wei W. (2012) Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett. 586, 1409–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hennessy B. T., Smith D. L., Ram P. T., Lu Y., Mills G. B. (2005) Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 4, 988–1004 [DOI] [PubMed] [Google Scholar]
- 29. Kitagawa K., Hiramatsu Y., Uchida C., Isobe T., Hattori T., Oda T., Shibata K., Nakamura S., Kikuchi A., Kitagawa M. (2009) Fbw7 promotes ubiquitin-dependent degradation of c-Myb: involvement of GSK3-mediated phosphorylation of Thr-572 in mouse c-Myb. Oncogene 28, 2393–2405 [DOI] [PubMed] [Google Scholar]
- 30. Guertin D. A., Sabatini D. M. (2007) Defining the role of mTOR in cancer. Cancer Cell 12, 9–22 [DOI] [PubMed] [Google Scholar]
- 31. Oh W. J., Jacinto E. (2011) mTOR complex 2 signaling and functions. Cell Cycle 10, 2305–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]