

Background: Pericytes are a major cerebrovasculature component, producing abundant apolipoprotein E (apoE).
Results: Deletion of apoE accelerates pericyte migration and adhesion by increasing RhoA activity. The effects are reversed by exogenous apoE3 but not apoE4.
Conclusion: ApoE suppresses pericyte mobility in a RhoA-mediated manner.
Significance: Pericyte-derived apoE regulates cell mobility and can be explored as a target for neurovascular disease therapy.
Keywords: adhesion; apolipoprotein E (ApoE); migration; Ras homolog gene family, member A (RhoA); vascular biology; LRP1; pericytes
Abstract
Pericytes play a critical role in the cerebrovasculature within the CNS. These small contractile cells produce large quantities of apolipoprotein E (apoE) whose isoforms influence cerebrovascular functions and determine the genetic risk for Alzheimer disease. Despite extensive studies on astrocyte-secreted apoE, which supports synapses by transporting cholesterol to neurons, the biochemical properties and function of apoE secreted by pericytes are not clear. Because pericytes mediate important functions in the CNS, including the initiation of glial scar formation, angiogenesis, and maintenance of the blood-brain barrier, we investigated the potential role of apoE in pericyte mobility. We found that knockdown of apoE expression significantly accelerates pericyte migration, an effect that can be rescued by exogenous apoE3, but not apoE4, a risk factor for Alzheimer disease. ApoE-regulated migration of pericytes also requires the function of the low-density lipoprotein receptor-related protein 1 (LRP1), a major apoE receptor in the brain that is abundantly expressed in pericytes. Because apoE-knockdown also leads to enhanced cell adhesion, we investigated the role of apoE in the regulation of the actin cytoskeleton. Interestingly, we found that the levels of active RhoA are increased significantly in apoE knockdown pericytes and that RhoA inhibitors blocked pericyte migration. Taken together, our results suggest that apoE has an intrinsic role in pericyte mobility, which is vital in maintaining cerebrovascular function. These findings provide novel insights into the role of apoE in the cerebrovascular system.
Introduction
The major apolipoprotein E (apoE) function is to transport cholesterol and lipids through apoE receptors in the periphery and the CNS (1, 2). In addition, apoE also directly regulates cellular functions, including the modulation of inflammatory response (3, 4), cell migration, and proliferation (5, 6). Humans have three polymorphic APOE alleles (ϵ2, ϵ3, and ϵ4). The most common allele found in the general population is ϵ3, followed by ϵ4 and, finally, ϵ2 (2, 4, 7). Growing evidence has shown that APOE ϵ4 carriers are at increased risk for age-related cognitive decline during normal aging as well as Alzheimer disease (2, 4, 7). Furthermore, APOE4 is also shown as an anti-longevity gene by disturbing lipid metabolism (8). Although the roles of apoE have been actively investigated because of its involvements in diverse human diseases, it is still not fully understood how different apoE isoforms in the CNS regulate physiological and pathological pathways.
Several types of cell can produce apoE, with hepatocytes and macrophages in the peripheral tissues and astrocytes in the CNS being the major sources, respectively (9). ApoE derived from astrocytes is a major apolipoprotein present in the brain, exhibiting a primary function in lipid transport and supporting synapses and dendritic spines (2, 4, 7). However, information on the biochemical properties and functions of apoE produced by other cell types in the brain is limited. Cerebral vessels constitutively express apoE (10). In particular, apoE is produced abundantly by cerebrovascular pericytes (11). These are small contractile cells that ensheath endothelial cells and reinforce the structure of the capillaries. Ubiquitously present throughout the body, pericytes support brain capillaries and execute a multitude of functions (12). Through intracellular signaling or direct cell-cell contact via the neurovascular unit, pericytes control cerebral blood flow and angiogenesis and maintain the integrity of the blood-brain barrier (BBB)3 (13, 14). Furthermore, pericytes play a critical role in scar formation, where pericytes accumulate faster than glial cells and seal off the damaged tissue in the injured spinal cord (15). Importantly, recent findings have shown that apoE regulates BBB integrity and cerebral blood flow in an isoform-dependent manner (16, 17). However, apoE-mediated pericyte function remains unclear.
In this study, we investigated the role of endogenous apoE in human brain vascular pericytes by deleting apoE with a specific siRNA. Here we demonstrate that apoE regulates pericyte mobility by controlling RhoA activity.
Experimental Procedures
Cell Culture
Human brain vascular pericytes were purchased from ScienceCell and have an APOE ϵ3/ϵ3 genotype. Pericytes were cultured in commercial pericyte medium (ScienceCell) under humidified conditions at 37 °C in 5% CO2. Medium was changed every other day until cells reached 90% confluency, at which point they were used for experiments. Immortalized apoE3 astrocytes were a gift from Dr. David M. Holtzman (Washington University). The cell line was generated from primary astrocyte cultures derived from human APOE-targeted replacement (TR) mice in which human APOE3 is knocked into the mouse endogenous Apoe gene locus (9). Immortalized apoE3 astrocytes were cultured in DMEM:F12 (Invitrogen) containing 20% FBS (Gemini Bio), 1 mm sodium pyruvate, 1% penicillin-streptomycin, 0.5% amphotericin B and 0.1% EGF. Medium was changed every other day until cells reached 90% confluency before being harvested for experiments.
FPLC
Conditioned media were generated by culturing cells in serum-free medium and collected after conditioning in T75 flasks for 24 h. Media were concentrated 20 times with a 10-kDa cutoff filter (Millipore) and centrifuged to remove any cellular debris before storage at 4 °C prior to fractionation. Samples were run on an AKTA FPLC system through a single Superose-6 10/300GL column (GE Healthcare) as described previously (18). Fractions were collected and stored at 4 °C until further analysis.
ApoE Immunoprecipitation
Concentrated conditioned media were incubated with Avidin-agarose beads (Pierce) precoupled with biotinylated polyclonal anti-apoE antibody K74180B (Meridian) overnight at 4 °C. Bead-antibody-apoE complexes were washed with TBS three times, and apoE-associated cholesterol was eluted with 0.1% Triton X-100 in TBS. Immunoprecipitated apoE was then eluted with 0.1 m glycine (pH 2.5) and neutralized with 1 m Tris (pH 8.5).
ApoE ELISA
A 96-well plate (Fisher Scientific) was coated overnight with WUE4 antibody (19). After blocking in 1% nonfat milk in PBS, samples were applied at the appropriate dilution and incubated with detection antibody (K74180B), followed by incubation with streptavidin-poly-HRP tertiary antibody (Fitzgerald). Finally, tetramethylbenzidine (Sigma) was applied to each well, and the substrate-peroxidase reaction was stopped with sulfuric acid stop solution. The absorbance was read at 450 nm using a BioTek 600 plate reader. ApoE concentration of each sample was calculated against a standard curve derived from serial dilutions of recombinant human apoE3 protein (Fitzgerald).
Cholesterol Assay
Cholesterol levels were analyzed using the Amplex Red cholesterol assay kit (Life Sciences) according to the instructions of the manufacturer. Briefly, samples were pipetted into an opaque 96-well plate with a transparent bottom and lid. Standards and samples were incubated with Amplex Red reagent (300 μm Amplex Red, 2 units/ml HRP, 2 units/ml cholesterol oxidase, and 0.2 units/ml cholesterol esterase) at 37 °C for 30 min, and fluorescence was measured using excitation in the range of 530–560 nm and emission detection at 590 nm.
Gene Silencing by siRNA
siRNAs were chemically synthesized by Dharmacon (Lafayette, CO). Human apoE siRNA (300 nm) was transfected into pericytes through nucleofection using the NucleofectorTM kit (Lonza) according to the specifications of the manufacturer. The target sequences for siRNAs were 5′-AAG GTG GAG CAA GCG GTG GAG-3′ (apoE-siRNA #1), 5′-GGA GTT GAA GGC CTA CAA A-3′ (apoE-siRNA #2), and 5′-AAG CAG TTT GCC TGC AGA GAT-3′ (LRP1-siRNA). Cells were further cultured for 48 h before use in experiments and analysis.
Quantitative Real-time PCR
Total RNA was extracted from pericytes using RNeasy mini kit (Qiagen) according to the instructions of the manufacturer. After cDNA synthesis using SuperScript® III first-strand synthesis system (Invitrogen), quantitative real-time PCR was carried out using SYBR Green Supermix (Bio-Rad). The relative levels of expression were quantified and analyzed using the iCycler iQ software (Bio-Rad). The real-time value for each sample was averaged and compared using the CT method, where the amount of target RNA (2−ΔΔCT) was normalized to the endogenous actin reference (ΔCT) and related to the amount of target gene in cells. For human apoE, RT2 quantitative PCR Primer (Qiagen) was used. The primer sequence used to amplify β-actin was as follows: forward, 5-CAT GTA CGT TGC TAT CCA GGC-3; reverse, 5-CTC CTT AAT GTC ACG CAC GAT-3.
BrdU Incorporation Assay
Cells were subjected to a BrdU incorporation assay to assess cellular proliferation according to the instructions of the manufacturer (Abcam). Briefly, cells were plated at 1 × 105 cells/ml, 100 μl/well in 96-well plate overnight. BrdU reagent was diluted in culture medium and incubated with cells for 18 h. After incubation with detector antibody followed by HRP-conjugate streptavidin, tetramethylbenzidine-peroxidase substrate was added. The substrate-peroxidase reaction was stopped with sulfuric acid stop solution, and the signal was read at a dual wavelength of 450/550 nm.
MTT Assay
An MTT assay was performed to assess cell viability via mitochondrial activity according to the instructions of the manufacturer (Roche). Briefly, cells were plated at 1 × 105 cells/ml, 250 μl/well in a 24-well plate overnight. MTT reagent was incubated with cells for 4 h at 37 °C before developing solution was added to stop the reaction. The plate was read at 550–600 nm with a reference wavelength of >650 nm.
Transwell Chamber Assay
Cells were plated at 5.0 × 104 cells/well on transwell chamber inserts with 8-μm pore size (Corning). After incubation for 2 h at 37 °C, 0.5% FBS in DMEM was added to the outer chamber and left to incubate for 2 h. Finally, the medium was replenished with pericyte growth medium to initiate migration. Cells were incubated for 18 h before analysis and quantification. After remaining cells on the top surface of the insert were removed by a cotton swab and thorough washing with PBS, migrated cells at the bottom surface of the insert were stained with hematoxylin and eosin (Fisher). Images of cells on the bottom surface were taken with an inverted fluorescence microscope (Olympus), and the number of migrated cells was quantified manually under blind conditions (20, 21).
Wound Healing Assay
Cells were plated at 5.0 × 105 cells/well on 6-well plates or 2.5 × 105 cells/well on 12-well plates. Cells from one half of each well were removed using a cell scraper, and the scratch line (midline) of each well was visualized and imaged using Axio software. Cells were left to heal for 24 h, and the scratch line was visualized and reimaged. Using Photoshop, a fixed crop was applied to the same area on each image, corresponding to the cells migrated past the scratch line. Cell numbers were counted manually and analyzed. In some experiments, apoE3 and apoE4 particles (5 ng/ml), an LRP1 antagonist, RAP (500 nm), and a RhoA inhibitor, Rhodblock 1a (22) (Sigma, 1 μm), were used. Recombinant RAP was produced and purified as described previously (23).
Purification of apoE
Conditioned medium from apoE3 or apoE4 immortalized astrocytes were concentrated and run on an AKTA FPLC system through a heparin column (GE Healthcare). Heparin-bound apoE was eluted with a linear gradient of NaCl from 0–1 m in 20 mm Tris-HCl (pH 7.4). Fractions containing apoE were combined, and the sodium concentration was adjusted to a physiological level through buffer exchange. The final apoE concentrations were measured with an isoform-specific apoE ELISA.
Western Blotting
Samples were lysed in PBS containing 1% Triton X-100 and protease inhibitor mixture from Roche. Protein concentration was determined in each sample using a protein assay kit (Bio-Rad). An equal amount of protein for each sample was used for SDS-PAGE. Immunoreactive bands were detected and quantified using the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE). Mouse monoclonal anti-LRP1 antibody was produced in our laboratory (24). Monoclonal antibodies for adhesion-related proteins were from the zonula adherens sampler kit and integrins sampler kit (BD Transduction Laboratories).
Adhesion Assay
Cells were plated at 0.4 × 106/well in a 6-well plate and left to attach for 30 min. After subsequent washing with PBS, remaining cells were imaged on an Axiovert 40 CFL light microscope (Zeiss) and quantified manually under blind conditions. In some experiments, cells were stained with Alexa Fluor 568 Phalloidin (Invitrogen) and DAPI (Invitrogen). Cells were imaged using confocal laser-scanning fluorescence microscopy (LSM 510 invert, Carl Zeiss).
RhoA Activity Assay
A RhoA pulldown activity assay was performed as directed by the instructions of the manufacturer (Cytoskeleton Inc.). Briefly, cells were lysed using lysis buffer (50 mm Tris (pH 7.5), 10 mm MgCl2, 0.5 m NaCl, and 2% Igepal). Cell lysate (300 μg) was incubated with 50 μg of Rhotekin-RBD beads at 4 °C for 1 h. Beads were pelleted by centrifugation, boiled in Laemmli sample buffer (62.5 mm Tris (pH 6.8), 10% glycerol, 2% SDS, 0.0025% bromphenol blue, 2.5% β-mercaptoethanol) and then loaded into a 12.5% SDS-PAGE gel. Immunoreactive bands were detected using anti-RhoA monoclonal antibody in equal amounts of pulldown samples (GTP-RhoA) and input (total RhoA) and quantified using the Odyssey infrared imaging system. For active RhoA G-LISA® and total RhoA ELISA (Cytoskeleton Inc.), cell lysates were collected using lysis buffer 48 h after knockdown, and equal amounts of lysate were used.
Statistical Analysis
All quantified data were analyzed by Student's t test and one- or two-way analysis of variance with a Tukey's post hoc test. Error bars represent mean ± S.D., and p < 0.05 was considered significant.
Results
Cerebrovascular Pericytes Produce Lipidated apoE
When apoE levels in the conditioned medium from human pericytes with the homozygous APOE3 genotype were analyzed, we found that they produced abundant apoE, ∼25% of that produced by immortalized apoE3 astrocytes when normalized by protein concentrations of cell lysates (Fig. 1A). To assess the molecular size of both pericyte- and astrocyte-produced apoE, conditioned medium from each cell type was concentrated and fractionated using a Superose-6 size exclusion column run on FPLC. A major apoE peak was detected in the fraction, eluting at a molecular size of ∼158 kDa from both samples, as analyzed by ELISA (Fig. 1B). This size is consistent with lipidated apoE particles found in plasma and cerebrospinal fluid (25, 26). In the case of pericyte-derived samples, another peak of apoE close to the 670 kDa molecular size marker was also detected (Fig. 1B). Next, to evaluate the lipidation state of apoE produced by pericytes and astrocytes, apoE in conditioned medium from each cell type was immunoprecipitated using an apoE-specific antibody. ApoE-associated cholesterol levels were analyzed in the immunoprecipitates. We found that significant amounts of cholesterol were detected in apoE-immunoprecipitates from both cell types (Fig. 1C). When the cholesterol level of each fraction was analyzed after fractionation by FPLC, two peaks were detected at around 158 kDa and over 670 kDa in both astrocyte- and pericyte-derived samples (Fig. 1D). The peak at 158 kDa was consistent with that of apoE from both cell types, whereas the peak at around 670 kDa overlapped with that of pericyte-derived apoE (Fig. 1D). In addition, in pericyte-derived samples, a significant amount of cholesterol was detected at smaller molecular sizes (<17 kDa). These results suggest that both pericytes and astrocytes produce lipidated apoE, but their lipidation status and stability may be different.
FIGURE 1.

Human brain vascular pericytes produce lipidated apoE. A, conditioned media and cell lysates were collected from human brain vascular pericytes with the APOE ϵ3/ϵ3 genotype or immortalized astrocytes from apoE3-TR mice. ApoE concentrations in the conditioned media were normalized against the total protein concentrations in cell lysates and compared between pericytes and astrocytes. B, conditioned media from pericytes or astrocytes were concentrated and subjected to fractionation by a Superose-6 size exclusion column run on FPLC. Fractions were subjected to analysis for apoE concentration by ELISA. The amount of apoE in each fraction was plotted as percent of total apoE, calculated by combining those in all fractions. C, the cholesterol levels associating with apoE produced by pericytes or astrocytes were analyzed by Amplex Red cholesterol assay after immunoprecipitation. The amount of cholesterol in each fraction was plotted as percent of total cholesterol, calculated by combining those in all fractions. D, the fractions used in B were further subjected to analysis for cholesterol level by Amplex Red cholesterol assay.
ApoE Regulates Pericyte Migration in an Isoform-dependent Manner without Affecting Cell Proliferation and Viability
To test whether apoE regulates pericyte functions, apoE production was suppressed by nucleofection-mediated delivery of specific apoE-siRNAs. When apoE levels in the conditioned medium were determined by ELISA after 48 h of transfection with either apoE-siRNA #1 or #2, it was decreased by ∼90% compared with that in pericytes transfected with control siRNA (Fig. 2A). To assess whether endogenous apoE influences pericyte migration, a critical function of pericytes (13–15), we investigated the effects of apoE silencing on cellular migration through a three-dimensional cell migration assay using transwell chambers (Fig. 2, B and C). When the number of pericytes that migrated from the upper chamber through a micropore membrane to the lower chamber was counted, the number of migrated cells in the apoE knockdown pericytes was increased significantly to 2.5-fold that of control pericytes (Fig. 2B). Another apoE-specific siRNA, #2, also showed similar effects as siRNA #1 on pericyte migration (Fig. 2, B and C), suggesting that such effects are specific rather than a potential off-target effect.
FIGURE 2.
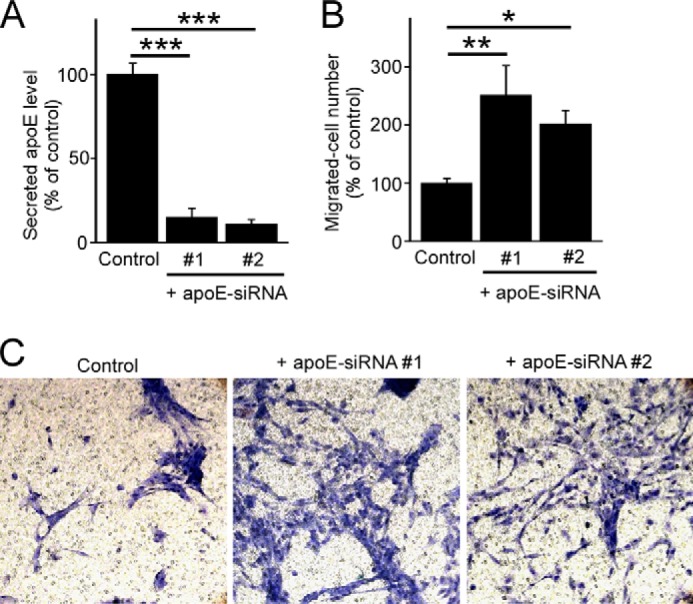
ApoE knockdown leads to increased pericyte migration in three-dimensional assays. A, gene silencing was performed through a transfection of apoE-specific siRNAs in pericytes. Cells were incubated in serum-free medium for 24 h following 48 h of transfection, and apoE levels in the media were analyzed by ELISA. B and C, after 48 h of transfection, control or apoE-knockdown pericytes were plated onto transwell chamber inserts. Following 2 h incubation with 0.5% FBS in DMEM, the medium in the lower chamber was changed back to pericyte growth medium. Cells in the top chamber were removed, and cells migrated through the insert were stained with hematoxylin and eosin after 18 h of incubation. The number of migrated cells under each condition were counted and averaged from multiple experiments. Data are plotted as mean ± S.D. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Consistent with the ELISA results, apoE knockdown by apoE-siRNA #1 resulted in an ∼90% reduction of apoE mRNA in pericytes compared with the control when analyzed by quantitative RT-PCR (Fig. 3A). To test potential apoE-dependent homeostasis of pericytes, both MTT assays and BrdU incorporation assays were performed. When pericyte viability was assessed by MTT assay, no differences were detected between control and apoE knockdown pericytes (Fig. 3B). In addition, the proliferation rate of pericytes was also unchanged after depletion of apoE, as assessed by a BrdU proliferation assay (Fig. 3C). These results indicate that apoE regulates pericyte migration without affecting their proliferation and viability.
FIGURE 3.
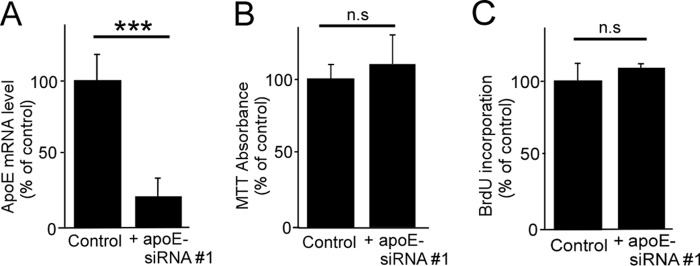
ApoE knockdown in pericytes does not affect cell proliferation and viability. A, gene-silencing was performed through a transfection of a specific apoE siRNA in pericytes. Cells were incubated in serum-free medium for 24 h following 48 h of transfection, and the levels of apoE mRNA were analyzed by quantitative RT-PCR. B, cell viability was assessed by MTT assay. C, cell proliferation was assessed by BrdU incorporation assay. Data are plotted as mean ± S.D. (n = 3). ***, p < 0.001; n.s., not significant.
To further assess the roles of apoE in pericyte migration, a two-dimensional cell migration assay was conducted in control and apoE knockdown pericytes with or without apoE particles derived from immortalized astrocytes. Pericytes efficiently migrated from the scratch line during a wound healing assay for 18 h. Consistent with the results from transwell chamber assays, we found that apoE knockdown in pericytes significantly accelerated cell migration in these wound healing assays. After application of exogenous apoE particles, the accelerated migratory phenotype in apoE knockdown pericytes was also reversed in an apoE isoform-dependent manner (E3 > E4) (Fig. 4, A and B). These results indicate that the effect of apoE knockdown on pericyte migration is due to reduced apoE levels in the extracellular space and that apoE isoforms have different functional roles in pericyte mobility.
FIGURE 4.
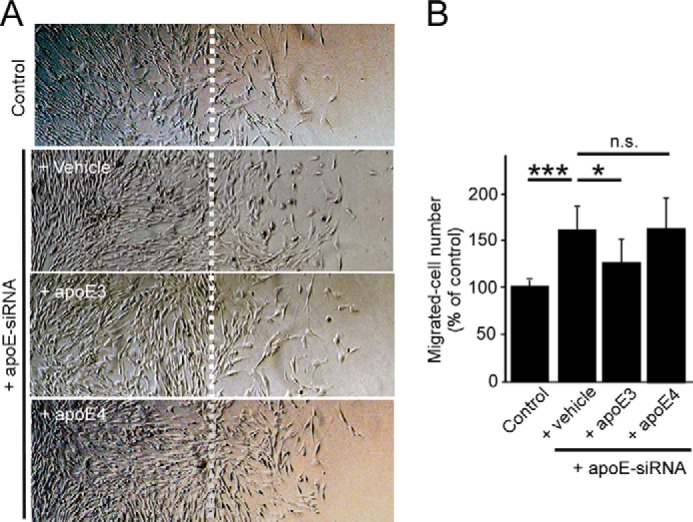
ApoE suppresses two-dimensional pericyte migration in an isoform-dependent manner. A, a wound healing assay was performed in control and apoE knockdown pericytes in the presence or absence of purified apoE3 or apoE4 particles (5 ng/ml). Cells from half of each well from the midline (dashed line) were removed and left to heal for 24 h before reimaging. B, migrated cell numbers were calculated as the number of cells beyond the scratch line. Data are plotted as mean ± S.D. (n = 7). *, p < 0.05; ***, p < 0.001; n.s., not significant.
Low-density Lipoprotein Receptor-related Protein 1 (LRP1) Is Required for apoE-mediated Pericyte Migration
LRP1 is a major apoE receptor in the brain, expressed abundantly in pericytes (17). To investigate the potential involvement of LRP1 in an apoE-dependent pathway for pericyte migration, we performed a knockdown of LRP1. Although LRP1 levels were suppressed by its specific siRNA in pericytes (Fig. 5A), we found significantly increased levels of apoE in the conditioned medium from these cells when analyzed by ELISA (Fig. 5B), suggesting a critical role of LRP1 as an apoE metabolic receptor in pericytes. When a two-dimensional wound healing assay was conducted with these cells, the accelerated migratory phenotype because of apoE-depletion was completely eliminated in LRP1 knockdown pericytes (Fig. 5, C and D). Similar results were obtained when LRP1 function was blocked by an LRP1 antagonist, RAP (23, 27) (Fig. 5, E and F). These results suggest that apoE suppresses pericyte mobility, likely in a manner that depends on the function of the cell surface apoE receptor LRP1.
FIGURE 5.

ApoE-mediated pericyte migration depends on LRP1. A, gene-silencing was performed through a transfection of specific apoE-siRNA#1 and/or an LRP1 siRNA in pericytes. LRP1 expression levels were analyzed by Western blotting 48 h after transfection. B, cells were incubated in serum-free medium for 24 h following 48 h of transfection, and apoE levels in the media were analyzed by ELISA. C and D, a wound healing assay was performed in these apoE- and/or LRP1-depeleted pericytes (C), and cells migrated past the scratch line were imaged and quantified (D). Data are plotted as mean ± S.D. (n = 5). E, similarly, a wound healing assay was performed in control and apoE-knockdown pericytes in the absence or presence of RAP (500 nm). F, cells migrated past the scratch line were imaged and quantified. Data are plotted as mean ± S.D. (n = 3). **, p < 0.01; ***, p < 0.001; n.s., not significant.
ApoE Regulates Cellular Adhesion in Pericytes
To further assess the roles of apoE in pericytes, we performed cell adhesion assays. Control and apoE knockdown pericytes were seeded on uncoated dishes for 30 min, and cellular adhesion was analyzed by microscopy (Fig. 6A). When the number of adhered cells was counted, apoE knockdown had increased pericyte adhesion to 1.9-fold of control conditions (Fig. 6B). To further examine the effects of apoE depletion on pericyte adhesion, F-actin was stained by phalloidin. Morphological differences were evident after phalloidin staining, with apoE knockdown pericytes presenting with a greater spread morphology compared with the compact control cells. Next we investigated the expression levels of major adhesion-related proteins in pericytes. Although N-cadherin, α-catenin, β-catenin, γ-catenin, fibronectin, integrin α2, integrin α5, integrin αV, and integrin β1 were detectable in pericytes (Fig. 6C), apoE knockdown significantly increased the fibronectin level but slightly reduced integrin α2 compared with control cells (Fig. 6, C and D). These results indicate that apoE knockdown is likely to enhance cellular adhesion by inducing rapid spreading and modifying the components of adhesion molecules in pericytes.
FIGURE 6.

ApoE knockdown leads to increased pericyte adhesion. Control or apoE knockdown pericytes were plated on a non-coated plate and allowed to adhere for 30 min. A, after vigorous washing with PBS, the attached cells to the plate were imaged by differential interference contrast (DIC) imaging mode. In addition, F-actin was stained by phalloidin. B, the number of adhered pericytes was quantified. Data are plotted as mean ± S.D. (n = 3). C and D, expression levels of adhesion-related proteins were analyzed by Western blotting in control and apoE knockdown pericytes and quantified. Data are plotted as mean ± S.D. (n = 4). *, p < 0.05; ***, p < 0.001.
ApoE Regulates Pericyte Migration by Modulating RhoA Activity
Because pericyte migration, adhesion, and spreading are increased by apoE knockdown, we next investigated the effects of apoE knockdown on RhoA activity. RhoA GTPase activity regulates the dynamics of the actin cytoskeleton, which then modulates cell adhesion and migration (28). RhoA GTPase serves as an activated form by binding to GTP, whereas it interacts with GDP in inactive states (29). Our results show that, in comparison with control cells, apoE depletion results in an increase in GTP-loaded RhoA within the cell (Fig. 7A). When the amount of GTP-loaded RhoA was normalized by total RhoA levels, apoE knockdown significantly increased the RhoA activity to 2.8-fold that of the control in pericytes (Fig. 7A). Significant increases of RhoA activity were also confirmed in apoE knockdown pericytes by G-LISA® RhoA activation assays (Fig. 7B). Because enhanced RhoA activity was detected in apoE knockdown cells, we compared the migration of control and apoE-depleted pericytes in the presence or absence of a RhoA inhibitor, Rhodblock 1a (22). Wound healing assays showed that the migration was reduced to baseline levels in apoE-depleted pericytes in the presence of Rhodblock 1a, eliminating the difference with control pericytes (Fig. 7C). These results indicate that apoE controls pericyte migration by regulating RhoA activity.
FIGURE 7.
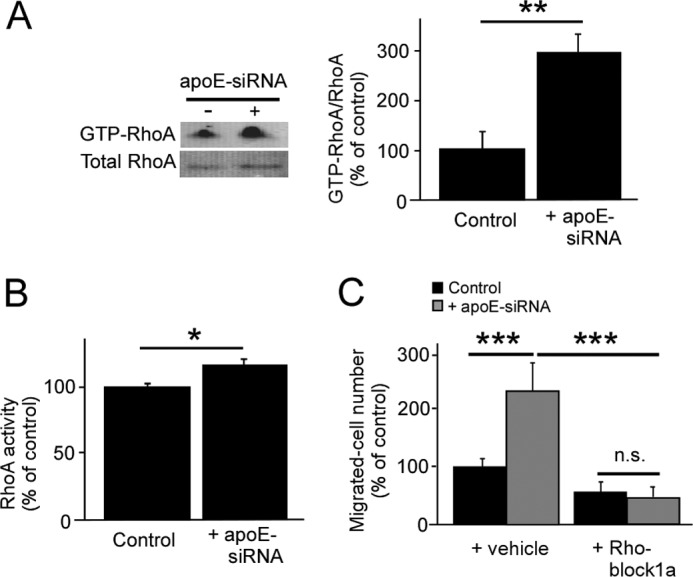
ApoE depletion accelerates pericyte migration by activating RhoA. A, RhoA activation assays were performed in control and apoE knockdown pericytes. Active RhoA (GTP-RhoA) was pulled down with rhotekin-Rho binding domain beads and analyzed by Western blot. The amount of GTP-RhoA was normalized against total RhoA and quantified. B, G-LISA® RhoA activation assay was performed in control and apoE knockdown pericytes. The amount of GTP-RhoA detected by G-LISA was normalized against total RhoA detected by ELISA. C, a wound healing assay was performed in control and apoE knockdown pericytes with or without Rhodblock 1a (1 μm). Cells migrated past the scratch line were imaged and quantified. Data are plotted as mean ± S.D. (n = 3). **, p < 0.01; ***, p < 0.001; n.s., not significant.
Discussion
Pericytes have multiple functions in the CNS, including the initiation of glial scar formation, angiogenesis, and the maintenance of BBB integrity (13). To carry out these functions, pericytes have an intrinsic ability to proliferate and migrate in response to extracellular signals such as inflammatory cytokines or stress (30). In this study, we demonstrated that pericytes produce significant amounts of lipidated apoE, which is comparable with those produced by astrocytes. Importantly, we found that pericyte-derived apoE critically regulates pericyte functions. Knockdown of apoE expression in pericytes leads to an enhanced migratory phenotype without significant effects on proliferation or viability. Furthermore, this phenotype could be rescued to below baseline migratory levels with the addition of exogenous apoE3 but not apoE4, suggesting an isoform-specific effect of apoE on pericyte migratory function.
During the event of traumatic spinal cord injury in mice, pericytes detach from the basal lamina of the cerebrovasculature, migrate through the extracellular matrix, and form the core component of scar tissue surrounding the site of injury (15). Although glial scar formation is critical to prevent an overwhelming inflammatory response and limit cellular degeneration (31), the excess responses disturb axonal regeneration (32). Interestingly, apoE is also up-regulated in astrocytes, neurons, and capillaries after complete spinal cord transection in a mouse model (33). Furthermore, APOE4 was associated with worse neurological recovery and a longer length of stay in rehabilitation in individuals with traumatic cervical spinal cord injury (34). Therefore, pericyte-mediated scar formation may be regulated in an apoE isoform-dependent manner during CNS injury.
In addition to glial scar formation, the migration of pericytes is critically involved in the angiogenesis process (35), where pericytes are recruited to endothelial cell-lined tubes along the ablumenal surface to facilitate vessel maturation by inducing vascular basement membrane matrix deposition (36). Because angiogenesis is essential during development and organ repair, the imbalance in the angiogenesis process contributes to numerous human disorders, including cancer, diabetes, infection, and ischemic diseases (37). In fact, apoE has been shown to be an antiangiogenic and metastasis-suppressive factor in melanoma (38). Therefore, it is possible that apoE-regulated pericyte migration contributes to the pathogenesis of these diseases. Furthermore, pericytes are a vital component to maintaining the integrity of the BBB by forming tight junctions with endothelial cells along the capillary wall (13, 39). In several neurodegenerative diseases, such as stroke and vascular dementia, there is evidence of impaired cerebrovascular integrity, primarily through BBB breakdown (40, 41). Therefore, when pericytes are depleted at capillaries because of external stress or toxic conditions, the BBB likely becomes vulnerable to invasion by peripheral immune cells and substances which can exacerbate any pathological conditions within the CNS (39). It has also been shown that traumatic brain injury results in a thinning of the basal lamina caused by pericyte migration into the parenchyma (42). The BBB would become more vulnerable because of decreased pericyte volume when the pericytes are migrating away from the BBB toward sites of injury. Therefore, the migratory phenotype of pericytes depending on apoE may influence BBB integrity. APOE4-TR mice exhibit a compromised BBB compared with apoE3-TR mice by increased activation of NFκB and secretion of MMP-9 through a proinflammatory cytokine cyclophilin A (CypA)-mediated pathway in capillary pericytes (17). Because our results demonstrated that the inhibitory effect of apoE4 on pericyte migration is less efficient compared with apoE3, it may be an alternative mechanism behind BBB dysfunction, as seen in apoE4-TR mice.
Significant progress has been made toward understanding why APOE4 represents a strong risk factor for Alzheimer disease, which is a progressive neurodegenerative disease characterized by the presence of amyloid-β (Aβ) deposition and neurofibrillary tau tangles within the CNS (2, 4, 7). Studies have shown that apoE4 exhibits both loss of function and gain of function properties compared with apoE3 (2). Although the most consistent finding that differentiates apoE4 from apoE3 is their respective roles in brain Aβ metabolism (43–45), we have shown that apoE isoforms differentially regulate pericyte migration. Epidemiological studies have shown that cerebrovascular disturbances caused by diabetes, atherosclerosis, stroke, hypertension, transient ischemic attacks, microvessel pathology, and smoking all increase the risk for Alzheimer disease (46). Therefore, an increased migratory phenotype by apoE4 in pericytes may play a role in Alzheimer disease pathogenesis by modulating cerebrovascular functions.
In addition, our results demonstrate not only enhanced cellular migration upon depletion of apoE in pericytes but also increased cell adhesion accompanied by morphological changes. ApoE knockdown cells exhibit a significant expansion of actin filaments compared with the compact phenotype of control cells, as demonstrated by phalloidin staining. Consistent with these results, we also found that apoE knockdown significantly increased active RhoA levels compared with control cells. Activation of RhoA results in a more dynamic actin cytoskeleton, rendering the cell more mobile (47), thereby explaining the enhanced migratory and adhesive qualities of pericytes upon genetic depletion of apoE. Furthermore, we found that apoE knockdown increased fibronectin levels in pericytes, which might also influence cellular mobility (48). Although it remains elusive how apoE reduces RhoA activity and increases the fibronectin level, the pathway likely depends on a major apoE receptor, LRP1. By LRP1 knockdown or blocking LRP1 function with its antagonist RAP, the migration was prevented both in control and apoE-depleted pericytes. Although LRP1 is an endocytotic receptor for more than 40 ligands, LRP1 influences several intracellular signaling pathways by coupling with other cell surface receptors (49, 50). LRP1 regulates multiple downstream targets that are involved with basic cellular functions, such as adhesion and the internal structure of the cell (51). Therefore, apoE may compete with other ligands for LRP1 binding or prevent LRP1 interaction with signaling receptors, resulting in a reduction of pericyte mobility. The other possibility is that the binding of apoE to LRP1 directly initiates the inactivation of the RhoA pathway and/or reduction of fibronectin, although further studies are needed to fully evaluate this mechanism.
In summary, our results provide novel evidence toward the roles of apoE isoforms in suppressing pericyte mobility (apoE3 > apoE4). Pericyte-derived apoE likely regulates the RhoA signaling pathway in the same cell or neighboring cells in an autocrine or paracrine manner. Further studies aimed at defining the precise mechanisms and pathological significance of apoE-dependent mobility of pericytes within the brain in response to stress or injury might allow the development of new therapeutic strategies for cerebrovascular and neurodegenerative diseases.
This work was supported by National Institutes of Health Grants P50AG016574, R01AG027924, R01AG035355, R01AG046205, and P01 NS074969 (to G. B.). This work was also supported by a grant from the Alzheimer's Association and a grant from the Cure Alzheimer's Fund (to G. B.) and a Young Investigator Grant from the Alex Lemonade Stand Foundation (to T. K.).
- BBB
- blood-brain barrier
- TR
- targeted replacement
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- RAP
- receptor-associated protein.
References
- 1. Mahley R. W. (1988) Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630 [DOI] [PubMed] [Google Scholar]
- 2. Bu G. (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hui D. Y., Harmony J. A., Innerarity T. L., Mahley R. W. (1980) Immunoregulatory plasma lipoproteins: role of apoprotein E and apoprotein B. J. Biol. Chem. 255, 11775–11781 [PubMed] [Google Scholar]
- 4. Kanekiyo T., Xu H., Bu G. (2014) ApoE and Aβ in Alzheimer's disease: accidental encounters or partners? Neuron 81, 740–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swertfeger D. K., Bu G., Hui D. Y. (2002) Low density lipoprotein receptor-related protein mediates apolipoprotein E inhibition of smooth muscle cell migration. J. Biol. Chem. 277, 4141–4146 [DOI] [PubMed] [Google Scholar]
- 6. Ishigami M., Swertfeger D. K., Granholm N. A., Hui D. Y. (1998) Apolipoprotein E inhibits platelet-derived growth factor-induced vascular smooth muscle cell migration and proliferation by suppressing signal transduction and preventing cell entry to G1 phase. J. Biol. Chem. 273, 20156–20161 [DOI] [PubMed] [Google Scholar]
- 7. Liu C. C., Liu C. C., Kanekiyo T., Xu H., Bu G. (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schächter F., Faure-Delanef L., Guénot F., Rouger H., Froguel P., Lesueur-Ginot L., Cohen D. (1994) Genetic associations with human longevity at the APOE and ACE loci. Nat. Genet. 6, 29–32 [DOI] [PubMed] [Google Scholar]
- 9. Morikawa M., Fryer J. D., Sullivan P. M., Christopher E. A., Wahrle S. E., DeMattos R. B., O'Dell M. A., Fagan A. M., Lashuel H. A., Walz T., Asai K., Holtzman D. M. (2005) Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol. Dis. 19, 66–76 [DOI] [PubMed] [Google Scholar]
- 10. Xu Q., Bernardo A., Walker D., Kanegawa T., Mahley R. W., Huang Y. (2006) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilhelmus M. M., Otte-Höller I., Davis J., Van Nostrand W. E., de Waal R. M., Verbeek M. M. (2005) Apolipoprotein E genotype regulates amyloid-β cytotoxicity. J. Neurosci. 25, 3621–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bergers G., Song S. (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol. 7, 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Winkler E. A., Bell R. D., Zlokovic B. V. (2011) Central nervous system pericytes in health and disease. Nat. Neurosci. 14, 1398–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zlokovic B. V. (2013) Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol. 70, 440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Göritz C., Dias D. O., Tomilin N., Barbacid M., Shupliakov O., Frisén J. (2011) A pericyte origin of spinal cord scar tissue. Science 333, 238–242 [DOI] [PubMed] [Google Scholar]
- 16. Nishitsuji K., Hosono T., Nakamura T., Bu G., Michikawa M. (2011) Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem. 286, 17536–17542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bell R. D., Winkler E. A., Singh I., Sagare A. P., Deane R., Wu Z., Holtzman D. M., Betsholtz C., Armulik A., Sallstrom J., Berk B. C., Zlokovic B. V. (2012) Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao J., Fu Y., Liu C. C., Shinohara M., Nielsen H. M., Dong Q., Kanekiyo T., Bu G. (2014) Retinoic acid isomers facilitate apolipoprotein E production and lipidation in astrocytes through the retinoid X receptor/retinoic acid receptor pathway. J. Biol. Chem. 289, 11282–11292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krul E. S., Tikkanen M. J., Schonfeld G. (1988) Heterogeneity of apolipoprotein E epitope expression on human lipoproteins: importance for apolipoprotein E function. J. Lipid Res. 29, 1309–1325 [PubMed] [Google Scholar]
- 20. Li Y., Lu W., Bu G. (2003) Essential role of the low density lipoprotein receptor-related protein in vascular smooth muscle cell migration. FEBS Lett. 555, 346–350 [DOI] [PubMed] [Google Scholar]
- 21. Song H., Li Y., Lee J., Schwartz A. L., Bu G. (2009) Low-density lipoprotein receptor-related protein 1 promotes cancer cell migration and invasion by inducing the expression of matrix metalloproteinases 2 and 9. Cancer Res. 69, 879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Castoreno A. B., Smurnyy Y., Torres A. D., Vokes M. S., Jones T. R., Carpenter A. E., Eggert U. S. (2010) Small molecules discovered in a pathway screen target the Rho pathway in cytokinesis. Nat. Chem. Biol. 6, 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Warshawsky I., Bu G., Schwartz A. L. (1993) 39-kD protein inhibits tissue-type plasminogen activator clearance in vivo. J. Clin. Invest. 92, 937–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shinohara M., Petersen R. C., Dickson D. W., Bu G. (2013) Brain regional correlation of amyloid-β with synapses and apolipoprotein E in non-demented individuals: potential mechanisms underlying regional vulnerability to amyloid-β accumulation. Acta Neuropathol. 125, 535–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fagan A. M., Holtzman D. M., Munson G., Mathur T., Schneider D., Chang L. K., Getz G. S., Reardon C. A., Lukens J., Shah J. A., LaDu M. J. (1999) Unique lipoproteins secreted by primary astrocytes from wild type, apoE−/−, and human apoE transgenic mice. J. Biol. Chem. 274, 30001–30007 [DOI] [PubMed] [Google Scholar]
- 26. Fagan A. M., Younkin L. H., Morris J. C., Fryer J. D., Cole T. G., Younkin S. G., Holtzman D. M. (2000) Differences in the Aβ40/Aβ42 ratio associated with cerebrospinal fluid lipoproteins as a function of apolipoprotein E genotype. Ann. Neurol. 48, 201–210 [PubMed] [Google Scholar]
- 27. Bu G., Marzolo M. P. (2000) Role of rap in the biogenesis of lipoprotein receptors. Trends Cardiovasc. Med. 10, 148–155 [DOI] [PubMed] [Google Scholar]
- 28. Ridley A. J. (2001) Rho GTPases and cell migration. J. Cell Sci. 114, 2713–2722 [DOI] [PubMed] [Google Scholar]
- 29. Etienne-Manneville S., Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 30. Kovac A., Erickson M. A., Banks W. A. (2011) Brain microvascular pericytes are immunoactive in culture: cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J. Neuroinflamm. 8, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Faulkner J. R., Herrmann J. E., Woo M. J., Tansey K. E., Doan N. B., Sofroniew M. V. (2004) Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 24, 2143–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Silver J., Miller J. H. (2004) Regeneration beyond the glial scar. Nat. Rev. Neurosci. 5, 146–156 [DOI] [PubMed] [Google Scholar]
- 33. Seitz A., Kragol M., Aglow E., Showe L., Heber-Katz E. (2003) Apolipoprotein E expression after spinal cord injury in the mouse. J. Neurosci. Res. 71, 417–426 [DOI] [PubMed] [Google Scholar]
- 34. Jha A., Lammertse D. P., Coll J. R., Charlifue S., Coughlin C. T., Whiteneck G. G., Worley G. (2008) Apolipoprotein E epsilon4 allele and outcomes of traumatic spinal cord injury. J. Spinal Cord Med. 31, 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Senger D. R., Davis G. E. (2011) Angiogenesis. Cold Spring Harb. Perspect. Biol. 3, a005090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stratman A. N., Malotte K. M., Mahan R. D., Davis M. J., Davis G. E. (2009) Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 114, 5091–5101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carmeliet P. (2005) Angiogenesis in life, disease and medicine. Nature 438, 932–936 [DOI] [PubMed] [Google Scholar]
- 38. Pencheva N., Tran H., Buss C., Huh D., Drobnjak M., Busam K., Tavazoie S. F. (2012) Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 151, 1068–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zlokovic B. V. (2011) Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat. Rev. Neurosci. 12, 723–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fernández-López D., Faustino J., Daneman R., Zhou L., Lee S. Y., Derugin N., Wendland M. F., Vexler Z. S. (2012) Blood-brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. J. Neurosci. 32, 9588–9600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iadecola C. (2013) The pathobiology of vascular dementia. Neuron 80, 844–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dore-Duffy P., Owen C., Balabanov R., Murphy S., Beaumont T., Rafols J. A. (2000) Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc. Res. 60, 55–69 [DOI] [PubMed] [Google Scholar]
- 43. Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R. B., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., Goate A. M., Bales K. R., Paul S. M., Bateman R. J., Holtzman D. M. (2011) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reiman E. M., Chen K., Liu X., Bandy D., Yu M., Lee W., Ayutyanont N., Keppler J., Reeder S. A., Langbaum J. B., Alexander G. E., Klunk W. E., Mathis C. A., Price J. C., Aizenstein H. J., DeKosky S. T., Caselli R. J. (2009) Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 106, 6820–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bales K. R., Liu F., Wu S., Lin S., Koger D., DeLong C., Hansen J. C., Sullivan P. M., Paul S. M. (2009) Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. de la Torre J. C. (2002) Alzheimer disease as a vascular disorder: nosological evidence. Stroke 33, 1152–1162 [DOI] [PubMed] [Google Scholar]
- 47. Wennerberg K., Rossman K. L., Der C. J. (2005) The Ras superfamily at a glance. J. Cell Sci. 118, 843–846 [DOI] [PubMed] [Google Scholar]
- 48. Pankov R., Yamada K. M. (2002) Fibronectin at a glance. J. Cell Sci. 115, 3861–3863 [DOI] [PubMed] [Google Scholar]
- 49. Kanekiyo T., Bu G. (2014) The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer's disease. Front. Aging Neurosci. 6, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Herz J., Strickland D. K. (2001) LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lillis A. P., Van Duyn L. B., Murphy-Ullrich J. E., Strickland D. K. (2008) LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918 [DOI] [PMC free article] [PubMed] [Google Scholar]