

Background: The role of EPHB4 blood pressure regulation was not previously known.
Results: Male but not female smooth muscle cell-specific Ephb4 gene knock-out mice were hypotensive; bi-directional signaling between EFNBs and EPHB4 modulated small artery contractility.
Conclusion: EPHB4 in vascular smooth muscle cells regulates blood pressure.
Significance: A new mechanism of blood pressure regulation has been discovered.
Keywords: cell biology, gene knockout, hormone, hypertension, vascular smooth muscle cells, EPHB4 kinases, GRIP1, hypotension, sex hormones
Abstract
EPH kinases are the largest family of receptor tyrosine kinases, and their ligands, ephrins (EFNs), are also cell surface molecules. This work presents evidence that EPHB4 on vascular smooth muscle cells (VSMCs) is involved in blood pressure regulation. We generated gene KO mice with smooth muscle cell-specific deletion of EPHB4. Male KO mice, but not female KO mice, were hypotensive. VSMCs from male KO mice showed reduced contractility when compared with their WT counterparts. Signaling both from EFNBs to EPHB4 (forward signaling) and from EPHB4 to EFNB2 (reverse signaling) modulated VSMC contractility. At the molecular level, the absence of EPHB4 in VSMCs resulted in compromised signaling from Ca2+/calmodulin-dependent protein kinase II (CaMKII) to myosin light chain kinase (MLCK) to myosin light chain, the last of which controls the contraction force of motor molecule myosin. Near the cell membrane, an adaptor protein GRIP1, which can associate with EFNB2, was found to be essential in mediating EPHB4-to-EFNB reverse signaling, which regulated VSMC contractility, based on siRNA gene knockdown studies. Our research indicates that EPHB4 plays an essential role in regulating small artery contractility and blood pressure.
Introduction
EPH kinases are the largest family of receptor tyrosine kinases. According to sequence homology, EPHs can be classified into A and B subfamilies. There are nine members in the EPHA subfamily and six members in the EPHB subfamily. The ligands of EPH kinases, ephrins (EFNs),2 are also cell surface molecules (1). There are nine EFNs divided into A and B subfamilies. Interactions among EPHs and EFNs are promiscuous. One EPH can interact with multiple EFNs and vice versa. In general, EPHA members bind preferentially to EFNA members, and EPHB members bind preferentially to EFNB members (2, 3). The preferred ligand of EPH4 is EFNB2, but the latter binds most EPHB subfamily members.
EPHs and EFNs play important roles in the development and function of many tissues and organs, such as the central nervous system (2, 4), immune system (3, 5–13), and digestive system (14). They are also vital in many biological processes, such as bone metabolism, angiogenesis, insulin secretion (15), chemotaxis (16), kidney glomerular filtration (17), ionic homeostasis of vestibular endolymph fluid in the inner ear (18), etc. There are a few studies showing that some EPHs and EFNs are expressed in vascular smooth muscle cells (VSMCs) and play a role in VSMC migration (19–21). However, until our recent publications, there was no documented evidence that these molecules are involved in blood pressure (BP) regulation.
In 2012, we published two articles reporting novel observations that EFNB6 and EFNB1 modulate BP in mice (22, 23). The salient findings are as follows. Systolic pressure (SP) and diastolic pressure (DP) in male and female EPHB6 KO mice are comparable with those in their WT counterparts, but castrated KO mice have higher SP and DP than WT (23), indicating that sex hormones act in concert with EPHB6 in controlling BP. We have identified VSMCs as one of the target tissues of EPHB6 in regulating BP, as small arteries from castrated EPHB6 KO mice show increased contractility in response to phenylephrine (PE) stimulation (23). Adrenal gland chromaffin cells are another target tissue on which EPHB6 and androgens exert their BP-regulating effects (23). Smooth muscle cell (SMC)-specific EFNB1 KO mice also have increased BP in a sex hormone-dependent way, similar but not identical to EPHB6 KO mice (22). Stimulating EFNB1 in VSMCs with solid-phase Ab diminishes VSMC contractility, supporting the concept that EPHB6 reverse signaling reduces VSMC contractility.
Given the new finding that the EPHB6 and EFNB1 KO leads to hypertension based on our earlier studies, we wondered whether other members of the EPH/EFN system play similar roles in BP control. To address this question, we generated floxed Ephb4 mice, and subsequently bred them to achieve SMC-specific deletion of EPHB4. To our total surprise, we found that the male EPHB4 KO mice were hypotensive. A series of mechanistic studies at the cellular and molecular levels was conducted. The results and significance of our unexpected findings are presented and discussed here.
Materials and Methods
Generation of SMC-specific Ephb4 Gene Knock-out Mice
A PCR fragment amplified with a primer set (5′-GCCCTTAAAGGACCGACTTC-3′ and 5′-GCCTAACGCTGGAGAAAGTG-3′) based on the Ephb4 genomic sequence was used as a probe to isolate a genomic bacterial artificial chromosome DNA clone 4M20 from the 129/sv mouse bacterial artificial chromosome genomic library RPCI-22. Targeting vectors were constructed by recombination and routine cloning methods, using a 12-kb Ephb4 genomic fragment from clone 4M20 (illustrated in Fig. 1A). The final targeting fragments for Ephb4 were excised from its cloning vector backbone by BamHI and electroporated into ES cells. After G418 selection, the FRT-flanked Neo/TK cassette was eliminated by subsequent transient transfection of ES cells with a flippase expression vector. The targeting scheme is shown in Fig. 1A. These genetic engineering steps in ES cells resulted in two net insertions in the Ephb4 gene: a 118-bp LoxP-containing sequence (5′-AGTACGGGCCCAAGCTGGCCGCCCTAGGGGCGCGCCTGCAGATAACTTCGTATAATGTATGCTATACGAAGTTATGATATCAAGCTTATCGATACCGTCGAAGCTTGCTAGCGGTACC-3′) at position 26,061 (based on the sequence of AL671478.9 in GenBank), and a 151-bp LoxP- plus FRT-containing sequence (5′-GGCCGCCCTAGGGGCGCGCCTGCAGATAACTTCGATAATGTATGCTATACGAAGTTATGGATCGAAGTTCCTATTTCTAAAAAGTATAGGAACTTCTTAAGGCCACCGCGGCCGAACGCTAGAGCTTGTCGACGGTACCTAACTTCCTAGG-3′) at position 28,713, 756 bp upstream and 1,285 bp downstream of Ephb4 Exon 1, respectively.
FIGURE 1.

Generation of mice with SMC-specific Ephb4 null mutation. A, scheme of Ephb4f/f mouse generation. Bold lines represent the left and right arms of genomic sequences used in gene targeting. LoxP and FRT sites are represented by empty and gray small arrowheads, respectively. The hatched box represents a genomic region from which probes were produced for Southern blotting analysis. B, Southern blotting analysis of tail DNA of floxed Ephb4 mice. Tail DNA was digested with EcoRV/BclI or BamHI/EcoRI, and then hybridized with the 5′ probe or 3′ probe, respectively. For the 5′ probe (upper panel), the 11.4-kb band was derived from the WT allele, and the 9.1-kb band was derived from the mutated allele. For the 3′ probe (lower panel), the 12.6-kb band was derived from the WT allele, and the 7.0-kb band was derived from the mutated allele. C, Ephb4 mRNA deletion in mesenteric arteries of Ephb4 KO mice. RNA was extracted from mesenteric arteries and spleens from WT and Ephb4 KO mice and analyzed by RT-qPCR for Ephb4 mRNA levels. β-Actin mRNA levels were used as an internal control. Samples in RT-qPCR were in triplicate, and means ± S.E. of Ephb4 signal/β-actin signal ratios are shown. The experiment was conducted twice; a representative one is shown. D, EPHB4 protein deletion in EPHB4 KO VSMCs according to immunoblotting. VSMCs from Ephb4 KO and WT mice were cultured for 4 days and then harvested. Cell lysates were analyzed for EPHB4 protein expression by immunoblotting. Spleens from KO and WT mice were used as controls for tissue specificity. The experiment was conducted twice; a representative one is shown.
The targeted ES cell clones were injected into C57BL/6 blastocysts. Chimeric male mice were mated with C57BL/6 females to establish mutated Ephb4 allele germ line transmission. Southern blotting with probes corresponding to 5′ and 3′ sequences outside the targeting region, as shown in Fig. 1A (hatched squares), was used to screen and confirm the gene targeting and the successful removal of the Neo/TK cassette in ES cells and eventually in mouse tail DNA.
Mice with floxed Ephb4 allele(s) were named Ephb4f/f (loxP insertions in both alleles). They were backcrossed with C57BL/6 for 10 generations and then mated with smooth muscle myosin heavy chain promoter-driven Cre transgenic mice in the C57BL/6 background (smMHC-Cre-IRES-eGFP transgenic mice (24)) to obtain SMC-specific EPHB4 KO mice. They are referred to as EPHB4 KO mice or KO mice in the text below.
PCR was used for routine genotyping of the floxed allele(s) and the Cre transgene. Primers 5′-GCCCTTAAAGGACCGACTTC-3′ (forward) and 5′-GCCTAACGCTGGAGAAAGTG-3′ (reverse) amplified a 271-bp fragment from the floxed allele and a 133-bp fragment from the WT allele. Primers 5′-CCA ATT TAC TGA CCG TAC ACC-3′ (forward) and 5′-GTT TCA CTA TCC AGG TTA CGG-3′ (reverse) amplified a 310-bp fragment of the Cre transgene. The PCR cycling condition was as follows for both types of PCR: 4 min at 95 °C followed by 34 cycles of 15 s at 94 °C, 30 s at 58 °C, and 60 s at 72 °C, with a final incubation at 72 °C for 10 min.
Reverse Transcription-Quantitative PCR (RT-qPCR)
Ephb4 and Grip1 mRNA levels in VSMCs were measured by RT-qPCR. Total RNA from VSMCs was extracted with TRIzol® (Invitrogen) and reverse-transcribed with iScriptTM cDNA synthesis kit (Bio-Rad, Mississauga, Canada). The primers used are listed in Table 1. iQTM SYBR® Green supermix (Bio-Rad) was employed in qPCR. The cycling conditions for all four molecules were as follows: 2 min at 50 °C, 2 min at 95 °C followed by 25–35 cycles of 10 s at 94 °C, 20 s at 58 °C, and 20 s at 72 °C. β-Actin mRNA levels served as internal controls. Samples were measured in duplicate, and the data are expressed as signal ratios of test gene mRNA/β-actin mRNA.
TABLE 1.
qPCR primer sequences
| Gene | qPCR primer sequences |
PCR product | |
|---|---|---|---|
| Sense sequence | Antisense sequence | ||
| bp | |||
| β-Actin | 5′-TCGTACCACAGGCATTGTGATGGA-3′ | 5′-TGATGTCACGCACGATTTCCCTCT-3′ | 200 |
| Ephb4 | 5′-CTACGTCTCTAACCTCCCATCT-3′ | 5′-GCTGGTCACCCTTTCTCTTT-3′ | 100 |
| Grip1 | 5′-ACAAGTCCCGTCCGGTTGTGATAA-3′ | 5′-TCTATCAGCAGCGTGGCTTCTTGT-3′ | 181 |
BP Measurement by Radiotelemetry
Mice were implanted surgically with TA11PA-C10 telemetry sensors (Data Sciences International, St. Paul, MN) in the left carotid artery for direct measurement of arterial pressure and heart rate (HR), as described previously (22, 23). For radio transmitter implantation and castration/ovariectomy, the mice were anesthetized with isoflurane (2% isoflurane with 0.75 liter/min O2 flow). Radiotelemetry was started 1 week after transmitter implantation. The raw data were processed with the Dataquest A.R.T-Analysis program (25) and are presented as hourly means ± S.E.
Ex Vivo Vessel Constriction
Mice between 18 and 20 weeks of age were used for all in vitro studies. They were euthanized with pentobarbital (400 mg/kg of body weight, intraperitoneal) at the end of in vivo studies or for tissue retrieval. Vessel constriction was studied ex vivo, as described previously (22, 23). Three measurements from three segments per mouse and two mice per group were pooled to construct concentration-response curves with S.E.
VSMC Isolation
Mouse VSMCs were isolated, as described by Golovina and Blaustein (26), with modifications (22, 23).
Measurement of VSMC Contractility
VSMC contractility was measured as described previously (22, 23). In some experiments, an EPHB4 kinase inhibitor NVP-BHG712 (0.5 μm; Tocris Bioscience, Bristol, United Kingdom) was added in the last 4 h of culture.
Ca22+ Flux Measurements
PE-stimulated Ca2+ flux in VSMCs was measured by fluorescence microscopy (21, 22). Briefly, VSMCs were incubated in DMEM containing 10% FCS and 5 μm Fura-2-AM for 30 min at 37 °C. The cells were rinsed twice in warm DMEM containing 15% FCS without the dye, and then cultured in the same medium at 37 °C for 30 min to remove extracellular dye. Afterward, the cells were placed in Ca2+-free Hanks' balanced salt solution at 37 °C, stimulated with PE (20 μm), and imaged for 60 s at a rate of 1 picture per 3 s under a Zeiss fluorescence microscope. Excitation wavelengths were switched between 340 nm and 380 nm, and emission wavelength was 510 nm. Signals from more than 15 randomly selected cells were recorded, and the results were expressed as ratios of fluorescence intensity at 510 nm excited by 340 nm versus 380 nm.
Immunoblotting
VSMCs from the aorta and mesenteric arteries of WT and KO mice were cultured for 3–4 days. In some experiments, the EPHB4 kinase inhibitor NVP-BHG712 (0.5 μm) was added for the indicated duration. The VSMCs were stimulated with PE (20 μm) for 3 s and then lysed by immunoprecipitation assay buffer, which contained PhosSTOP and protease inhibitor mixtures (Roche Applied Science, Meylan, France). Immunoblotting of phosphorylated myosin light chain (MLC) and total MLC, as well as phosphorylated and total MLC kinase (MLCK), has been described before (22, 23). Phosphorylated and total MLC phosphatase (MYPT) and phosphorylated and total Ca2+/calmodulin-dependent protein kinase II (CaMKII) of the VSMCs were measured with rabbit anti-MLCK mAb (clone EP1458Y; Abcam, Cambridge, UK), rabbit anti-phospho-MLCK Ab (Invitrogen), rabbit anti-MYPT Ab (2634; Cell Signaling, Danvers, MA), rabbit anti-phospho-MYPT Ab (5163; Cell Signaling), rabbit anti-CaMKII (phospho-Thr-286) Ab (ab32678; Abcam), and rabbit anti-CaMKII Ab (ab52476; Abcam). For α1-adrenergic receptor (α1-AR) detection, VSMCs were cultured and lysed as described above without stimulation. Rabbit Ab against α1-AR (ab3462; Abcam) was used for blotting. All the Abs were used at the manufacturers' recommended dilutions. Signals were detected with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Burlington, Ontario, Canada).
siRNA Transfection
siRNAs of Grip1 and negative control siRNAs were synthesized by Integrated DNA Technologies (Coralville, IA). Their sequences have already been described in our previous publication (23). VSMCs were cultured in wells coated with recombinant human IgG Fc-tagged EPHB4 (EPHB4-Fc; 2 μg/ml during coating) or normal human IgG (NHIgG; 2 μg/ml during coating) as control for 3 days, and then in medium free of antibiotics for an additional 24 h. They were then transfected with a mix of three pairs of siRNAs of Grip1 (for each pair, the final concentration was 10 nm), as described before (23).
Ethics Statement
All studies were approved by the Animal Protection Committee (Le Comité institutionnel d'intégration de la protection des animaux) of the CRCHUM.
Statistical Analysis
All results were expressed as means ± S.E. Data were statistically analyzed by analysis of variance or Student's t-tests. p values of <0.05 were considered statistically significant.
Results
Generation and Characterization of SMC-specific Ephb4 Conditional Gene KO Mice
To investigate the function of EPHB4 in BP regulation, we generated Ephb4 floxed mice, with LoxP sites flanking exon 1 of Ephb4 (Fig. 1A). Germline transmission of the mutated gene was confirmed by Southern blotting of tail DNA (Fig. 1B). With the 5′ probe, the floxed allele showed a 9.1-kb EcoRV/BclI band, and the WT allele showed an 11.4-kb EcoRV/BclI band. With the 3′ probe, the floxed allele showed a 7.0-kb BamHI/EcoRI band, and the WT allele showed a 12.6-kb BamHI/EcoRI band. These floxed mice were first backcrossed to the C57BL/6 background for nine generations, and then crossed with transgenic mice expressing smooth muscle myosin heavy chain promoter-driven Cre recombinase (smMHC-Cre-IRES-eGFP transgenic mice (24)) to achieve SMC-specific deletion of EPHB4. The deletion of Ephb4 at the mRNA level in vascular smooth muscles, but not in spleen cells, was confirmed by RT-qPCR (Fig. 1C). The deletion of EPHB4 at the protein level in VSMCs, but not in spleen cells, was further confirmed by immunoblotting (Fig. 1D).
EPHB4 Deletion in SMC Causes Hypotension in Males
Our previous study showed that BP is significantly heightened in castrated EPHB6 KO, but not in uncastrated or female EPHB6 KO mice, when compared with their WT counterparts, indicating that EPHB6 acts in concert with sex hormones in BP regulation (22). We further demonstrated that one of the tissues responsible for this phenotype is VSMC (22). As EPHB4 is another notable member of the EPHB subfamily, we wondered whether SMC-specific deletion of EPHB4 would result in a similar BP phenotype. BP and HR of EPHB4 KO mice were measured by radiotelemetry. The means ± S.E. values of hourly SP, DP, mean arterial pressure, and HR are presented in Fig. 2. Surprisingly, male KO mice presented significantly reduced SP and mean arterial pressure with normal HR when compared with WT mice (Fig. 2A). On the other hand, SP, DP, and mean arterial pressure of KO females were comparable with those of their WT counterparts, although the former showed reduced HR (Fig. 2B). BP and HR of both KO and WT mice, either males or females, all showed expected circadian fluctuations.
FIGURE 2.

Male EPHB4 KO mice were hypotensive. BP and HR were measured for 72 h by radiotelemetry, starting at least 7 days after radio transmitter implantation. The number of mice per group and their mean age at the time of BP measurement are indicated. Means ± S.E. of hourly BP and HR during the 72-h period are presented. Shaded areas represent dark periods. MAP, mean arterial pressure. The statistical significance of differences between BP and HR of the experimental groups was evaluated by analysis of variance, and p values are indicated. A, BP and HR of males. B, BP and HR of females.
Decreased Vessel Contractility in Male EPHB4 KO Mice ex Vivo
BP is a function of cardiac output and blood vessel flow resistance, which is determined mainly by small arteries. As KO mice only had EPHB4 deletion on SMC, we focused our attention on small artery contractility, which determines small arterial tone.
PE-triggered Contractility of Mesenteric Arteries from KO and WT Mice Was Assessed ex Vivo
Vessel contractility of male KO (Fig. 3A) but not female KO (Fig. 3C) mice was significantly lower than that of their WT counterparts, corroborating the hypotensive phenotype in male KO mice. Removal of the vessel endothelium did not change the phenotype of KO vessels (Fig. 3, B and D), indicating that the reduced contractility of male KO vessels is not caused by cytokines or nitric oxide produced by the endothelium.
FIGURE 3.

Reduced contractility of mesenteric arteries from male EPHB4 KO mice. A–D, segments of the third-order branch of the mesenteric artery with (A and C) or without (B and D) endothelium were stimulated with PE. A single cumulative concentration-response curve to PE (1 nm to 100 μm) was obtained. Maximal tension (Emax) was determined by challenging the vessels with physiological saline containing 127 mm KCl. Vessel contractility is expressed as the percentage of Emax. Data from two mice (with three arterial segments tested for each mouse, i.e. six determinants per group) were pooled, and means ± S.E. are reported. Contractility differences were analyzed by paired Student's t tests, and p values are indicated. *, p < 0.05. A, contractility of mesenteric arteries with endothelium from male KO and WT mice. B, contractility of mesenteric arteries without endothelium from male KO and WT mice. C, contractility of mesenteric arteries with endothelium from female KO and WT mice. D, contractility of mesenteric arteries without endothelium from female KO and WT mice.
Both Forward and Reverse Signaling between EPHB4 and Its Ligands Is Responsible for Regulating VSMC Contractility
Because EPHB4 deletion is SMC-specific, and male mice had reduced BP and vasoconstriction, we conducted further experiments at the cellular level to understand the underlying mechanisms. As shown in Fig. 4A, VSMCs from male KO mice indeed presented decreased contractility on PE stimulation. VSMCs from male mice were then used in all subsequent cellular and molecular studies.
FIGURE 4.

VSMCs stimulated by both EPHB4 forward and EPHB4 reverse signaling show increased contractility. VSMCs from male WT and KO mice were cultured for 4 days. In some experiments, the VSMCs were cultured in wells coated with EFNB2-Fc (2 μg/ml for coating), EPHB4-Fc (2 μg/ml for coating), or NHIgG (2 μg/ml for coating, as control for EPHB4-Fc), as indicated. VSMCs were stimulated with 20 μm PE at 37 °C and imaged every min for 15 min. All experiments were conducted 3 times independently. Means ± S.E. of percentage of contraction of 15–30 cells in a representative experiment are shown. Data at 10 and 15 min of 3 independent experiments are summarized and expressed as bar graphs (means ± S.E.) on the left. The data were analyzed by paired Student's t test (*, p < 0.05). WT, WT VSMC; KO, KO VSMC. A, decreased contractility of VSMCs from male EPHB4 KO mice. VSMCs were cultured in plain wells. B, forward signaling through EPHB4 increases WT VSMC contractility. WT VSMCs were cultured in wells coated with EFNB2-Fc. KO VSMCs in coated wells were used as negative controls. WT VSMCs cultured in wells coated with NHIgG were used as additional controls, and their mean contractility is presented as a thin line without S.E. to facilitate viewing. C, EPHB4 inhibitor NVP-BHG712 suppresses WT VSMC contractility. WT VSMCs were cultured in plain wells in the presence of NVP-BHG712 (0.5 μm) or vehicle for the last 4 h of culture. Their contractility after PE stimulation was measured. D, EPHB4 inhibitor NVP-BHG712 does not affect KO VSMC contractility. KO VSMCs were cultured in plain wells in the presence of NVP-BHG712 (0.5 μm) or vehicle for the last 4 h of culture. Their contractility after PE stimulation was measured. E, VSMCs cultured in the presence of EPHB4 inhibitor NVP-BHG712 show normal proliferation. VSMCs from WT male mice were cultured for 2–5 days in the presence of NVP-BHG712 at 0.5 μm as indicated. Cell number per well of 24-well plate at days 2 and 5 was enumerated. Means ± S.E. of cell counting of three wells are shown. The experiment was conducted twice; data from a representative experiment are shown. No significant difference was found between NVP-BHG712- and vehicle-treated cells (Student's t test). F, reverse signaling triggered by EPHB4 increases WT VSMC contractility. WT VSMCs were cultured in wells coated with EPHB4-Fc. In one of the groups, soluble EFNB2-Fc (2 μg/ml) was added to culture to block interaction between solid-phase EPHB4-Fc and cell surface EFNB2. WT VSMCs cultured in wells coated with NHIgG were used as an additional control, and their mean contractility is presented as a thin line without S.E. to facilitate viewing.
EPHB4 and its ligands (mainly EFNBs) are all expressed in VSMCs (22). EPHB4 and its cell surface ligands EFNBs can trigger signaling in both directions, i.e. from EFNBs to EPHB4 (forward signaling) and from EPHB4 to EFNBs (reverse signaling). We wondered which direction is essential in regulating VSMC contractility. When WT VSMCs were cultured in wells coated with recombinant EFNB2 (in the form of EFNB2 tagged with human IgG Fc (EFNB2-Fc) for triggering forward signaling), the preferred ligand of EPHB4, their contractility on PE stimulation was significantly increased when compared with WT VSMCs cultured in wells coated with control normal human IgG (NHIgG) (Fig. 4B). On the other hand, EPHB4 KO VSMCs did not respond to solid-phase EFNB2-Fc (Fig. 4B, thin line), and their contractility was no different from that of WT VSMCs treated with NHIgG. This indicates that the major receptor receiving the solid-phase EFNB2 stimulation is EPHB4, and that enhanced EPHB4 forward signaling increases VSMC contractility. To verify this finding via a different approach, we treated WT male VSMCs with an EPHB4 inhibitor, NVP-BHG712, at 0.5 μm for 4 h and found that it could also inhibit VSMC contractility (Fig. 4C), rendering the WT VSMC phenotype similar to that of EPHB4 KO VSMCs. Because the inhibitor did not suppress the contractility of VSMCs from the KO mice (Fig. 4D), it means that inhibition occurs mainly via EPHB4. Such inhibition is not due to general cytotoxicity of the inhibitor, as VSMCs cultured in 0.5 μm NVP-BHG712 up to 5 days proliferated at a similar rate as those cultured in vehicle (Fig. 4E).
To assess whether reverse signaling through EPHB4 ligands could also regulate VSMC contractility, we cultured WT VSMCs in wells coated with recombinant EPHB4-Fc, which could trigger reverse signaling via EFNBs. Such treatment also enhanced VSMC contractility on PE stimulation (Fig. 4F), and this enhancement could be specifically neutralized by soluble recombinant EFNB2-Fc, indicating that reverse signaling is mainly mediated by EFNB2. It is worth mentioning that cross-linked EPHB4-Fc or EFNB2-Fc, in the absence of PE, had no effect on VSMC contractility (data not shown).
Contractility-related Signaling Events Affected by EPHB4 in VSMCs
The reduced contractility of KO VSMCs in response to PE could be due, in theory, to reduced α1-AR expression. However, based on immunoblotting, male WT and KO VSMCs had similar levels of α1-AR (Fig. 5A). Activation of AR leads to Ca2+ flux, which is an event controlling contractility. Again, PE-triggered Ca2+ flux showed no differences between male WT and KO VSMCs (Fig. 5B).
FIGURE 5.
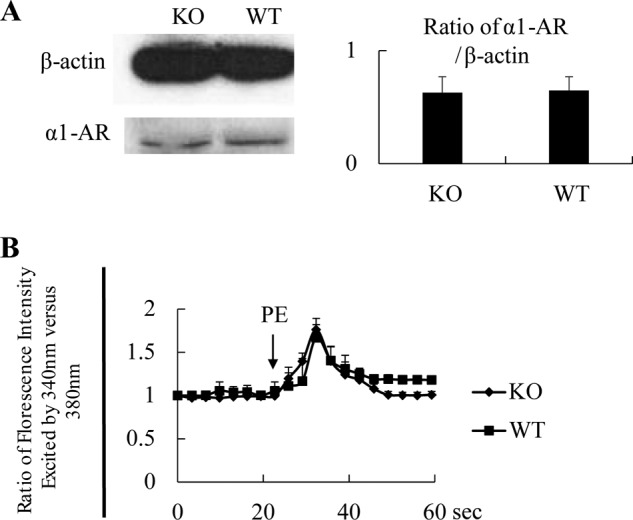
AR and Ca2+ flux in KO VSMCs. A, normal α1-AR expression in EPHB4 KO VSMCs according to immunoblotting. VSMCs from male Ephb4 KO and WT mice were cultured for 4 days and then harvested. Cell lysates were analyzed for α1-AR expression by immunoblotting. β-Actin levels were used as internal controls. A representative immunoblot is shown. Densitometry data from 2 independent experiments were pooled and presented as a bar graph on the right with means ± S.E. indicated. No statistically significant differences between KO and WT VSMCs were found according to Student's t test. B, normal Ca2+ flux in VSMCs from EPHB4 KO mice. VSMCs from male Ephb4 KO or WT mice were cultured for 4 days and loaded with Fura2. They were then placed in Hanks' balanced salt solution without Ca2+ at 37 °C and stimulated with PE (20 μm). The arrow indicates the time point at which PE was added. The ratio of emissions at 510 nm triggered by 340 nm versus 380 nm excitation in each cell was registered every 3 s for 1 min. The experiments were conducted 3 times. Means ± S.E. of the ratio of more than 15 randomly selected VSMCs in a representative experiment are illustrated. No statistically significant differences between the KO and WT groups were found according to Student's t test.
We then suspected that the reduced contractility of male KO VSMCs was a result of decreased VSMC Ca2+ responsiveness, which is regulated by MLC phosphorylation. We tested MLC phosphorylation at different time points after PE stimulation and found that it occurred very quickly (within seconds) and then subsided (data not shown). We thus chose 3 s as a testing point as it was technically the earliest time point possible. KO VSMCs from males indeed manifested a significantly lower degree of MLC phosphorylation than their WT counterparts (Fig. 6A). Inhibition of EPHB4 kinase activity in VSMCs by NVP-BHG712 also repressed MLC phosphorylation (Fig. 6B). MLC is phosphorylated by MLCK. This kinase is phosphorylated at Ser-1760 by CaMKII or protein kinase A, and such phosphorylation reduces its kinase enzymatic activity (26). We found that MLCK phosphorylation at Ser-1760 was increased in male KO VSMCs when compared with WT counterparts (Fig. 6C). The kinase enzymatic activity of CaMKII is controlled by its phosphorylation at Thr-286; the phosphorylated CaMKII presented higher activity to phosphorylate MLCK (27, 28). In male KO VSMCs, such CaMKII phosphorylation was increased (Fig. 6D), corroborating the increased MLCK phosphorylation. Thus, deletion of EPHB4 compromised the signaling from CaMKII to MLCK to MLC in VSMCs, with enhanced CaMKII phosphorylation as the upstream event. On the other hand, in VSMCs, phosphorylation of MYPT, a phosphatase that also regulates MLC phosphorylation, was not influenced by EPHB4 deletion in VSMCs (Fig. 6E).
FIGURE 6.

MLC, MLCK, CaMKII, and MYPT phosphorylation of VSMC from WT and KO mice. VSMCs from male KO and WT mice were cultured for 4 days and then stimulated with 20 μm PE for 3 s and immediately lysed. Total (T-) and phosphorylated (P-) MLC, MLCK, and CaMKII were analyzed by immunoblotting. Three independent experiments were conducted. Immunoblotting images from representative experiments are illustrated. The signal ratios of phosphorylated versus total MLC, MLCK, CaMKII, and MYPT were quantified by densitometry. Densitometry data from the 3 independent experiments were pooled and are presented as bar graphs (means ± S.E. of relative intensity) in the panels on the left. Paired Student's t tests were employed for statistical analysis. * indicates p < 0.05. A, decreased MLC phosphorylation in VSMC from EPHB4 KO mice. B, decreased MLC phosphorylation in VSMCs after EPHB4 inhibitor treatment. WT VSMCs were cultured in the presence of NVP-BHG712 (0.5 μm) for the last 4 h of culture. The controls were treated with vehicle (dimethyl sulfoxide (DMSO)) at the same dilution and duration. C, increased MLCK phosphorylation in VSMCs from EPHB4 KO mice. D, increased CaMKII phosphorylation in VSMCs from EPHB4 KO mice. E, MYPT phosphorylation remains unchanged in VSMCs from EPHB4 KO mice.
Identification of GRIP1 as a Component in the EPHB4-triggered Reverse Signaling Leading to Increased VSMC Contractility
To understand how reverse signaling is transmitted through EFNBs after EPHB4 triggering in VSMCs, we used siRNA to knock down the expression of an adaptor protein, GRIP1, which is known to associate with EFNBs (29). The effectiveness of mRNA and protein knockdown was verified by RT-qPCR (Fig. 7A) and immunoblotting (Fig. 7B), respectively. VSMCs from WT males were cultured in wells coated with recombinant EPHB4-Fc to invoke EFNB reverse signaling. With Grip1 siRNA transfection, the contraction-enhancing effect of EFNB reverse signaling (mainly through EFNB2, as demonstrated in Fig. 3D) was eliminated (Fig. 7C). Grip1 siRNA transfection in WT VSMCs without solid-phase EPHB4-Fc stimulation did not affect their contractility (Fig. 7D), indicating that GRIP1 is only functional in the presence of EPHB4-triggered reverse signaling.
FIGURE 7.

GRIP1 mediates EPHB4 reverse signaling in controlling VSMC contractility. A, effective Grip1 mRNA knockdown by siRNA. Cultured WT VSMCs were transfected with a mixture of three pairs of Grip1 siRNAs or control siRNA, as indicated. After a 24-h culture, the cells were harvested, and the mRNA expression was determined by RT-qPCR, samples being in duplicate. The experiments were conducted twice, and the data (four determinants for each group) were pooled and expressed as means ± S.E. of the ratios of the target gene signal versus the β-actin signal. The data were analyzed by Student's t test. *, p < 0.05. B, effective GRIP1 protein knockdown by siRNA. The cells as described in panel A were analyzed by immunoblotting for GRIP1 protein expression. β-Actin was used as loading control. Experiments were conducted 3 times, and representative ones are shown. C, GRIP1 knockdown by siRNAs partially reverses the enhancing effect of solid-phase EPHB4-Fc in VSMC contractility. VSMCs from WT males were cultured in wells coated with recombinant EPHB4-Fc (2 μg/ml during coating). After 2 days, they were transfected with siRNAs targeting Grip1 mRNA, or with control siRNA. On day 4 of culture, they were stimulated with PE (20 μm), and their percentage of contraction was recorded. Means ± S.E. of percentages are reported. The thin lines represent the mean percentage of contraction of VSMCs cultured in wells coated with NHIgG (2 μg/ml) without siRNA transfection (for a better visual effect, the S.E. of each time point in this control is omitted). All experiments were conducted 3 times independently. Means ± S.E. of percentage of contraction of 15–30 cells of a representative experiment are shown. The data at 10 and 15 min of 3 independent experiments are summarized and expressed as bar graphs (means ± S.E.) on the left. The data were analyzed by paired Student's t test (*, p < 0.05). D, GRIP1 knockdown in VSMCs without EPHB4 reverse signaling does not affect VSMC contractility. WT VSMCs were cultured in plain wells and transfected with Grip1 or control siRNA as described for panel C. Their contractility upon PE stimulation was measured. No significant differences were observed according to Student's t test.
Discussion
Our recent work demonstrates that deletion of several EPH/EFN members in VSMCs, such as EPHB6 and EFNB1, leads to hypertension in mouse models, revealing a previously unknown BP control system (22, 23). In support of our findings that deletion of certain members of EPH/EFN families could cause hypertension, a recent publication by Carlstrom and co-workers (30) demonstrated that mice with deletion of EPHA4 intracellular tail are also hypertensive, although in that case, abnormalities in the kidney are considered the cause of the observed hypertensive phenotype. Quite unexpectedly, we found in the present study that deletion of a member of the EPH/EFN system, EPHB4, in VSMCs resulted in hypotension. This effect of EPHB4 deletion was achieved in its own right without affecting the expression of other EPHBs and EFNBs (data not shown). Such phenotype was only apparent in males, suggesting that the phenotype is affected by sex hormones. Obviously, a tremendous amount of mechanistic study needs to be conducted to elucidate how sex hormones in concert with EPHB4 modulate VSMC contractility. There is no evidence that sex hormones change the expression levels of EPHB4 and EFNB family members in VSMCs.3 We found that cell surface estrogen receptor GPR30 is critical for the concerted effect of EFNB3 and estrogen in regulating VSMC contractility.3 As EFNB3 and EFNB2 belong to the same EFN subfamily, and the latter is the preferred ligand of EPHB4, it will be interesting to explore whether GRP30 is also involved in mediating the differential contractility responses of male versus female KO VSMCs. Our present work serves as a starting point for a full understanding of the complex interaction among EPHs/EFNs, sex hormones, and BP regulation. The co-existence of opposing effects of these EPH/EFN molecules in VSMC contractility and BP regulation is reminiscent of the Yin and Yang of many other cellular and molecular events, such as kinases/phosphatases, osteoblasts/osteoclasts, calcium influx through L-type and T-type calcium channels, outflux through BK channels, etc. The co-existence of these cells/molecules with opposite functions likely results in better homeostatic control of different biological processes: in this case, the fine tuning of vascular tone, and consequently, BP. Their opposite effects will need to be modulated to achieve this fine tuning. We found that sex hormones are among these modulators with regard to EPHB4 functions in BP regulation.
For EPHB4, its effect on VSMC constriction could be reached by both forward and reverse signaling, and EFNB2 is the main ligand mediating the two-way signaling, consistent with the fact that EFNB2 is the major ligand for EPHB4 (31). This suggests that EFNB2 deletion in VSMCs might also lead to reduced VSMC contractility and hypotension. Further experimentation by our group has proved that this prediction is generally correct.4 The bi-directional vasoconstriction-enhancing effect of EPHB4 and EFNB2 also implies that inhibiting EPHB4 kinase activity could achieve a BP-lowering effect. We proved, in principle, in vitro that a Novartis EPHB4 kinase inhibitor NVP-BHG712 (32, 33) could effectively reduce VSMC contractility. Although NVP-BHG712 was developed as an EPHB4 kinase inhibitor, it is not mono-specific, and it inhibits the kinase activity of other EPH kinase family members as well (32). Therefore, the action we observed in VSMCs is likely the sum of effects it has on all the concerned EPH kinases, some of which may have effects opposite to EPHB4 in terms of modulating VSMC contractility. With that said, we noticed that NVP-BHG712 did not further reduce EPHB4 KO VSMC contractility (Fig. 4D), suggesting that the major impact of this inhibitor is via EPHB4.
It is worth mentioning that EPHB4 plays a critical role in angiogenesis (34), and thus is a potential drug target in tumor therapy. Many pharmaceutical and biotechnology companies have been developing EPHB4 inhibitors for such an application (32, 33). They will certainly benefit from our current findings by becoming aware of the BP-lowering potential of EPHB4 inhibitors as possible side effects.
We noticed that in female KO mice, HR was significantly lower than in their WT counterparts, although their BP was in the normal range. As BP is a tightly regulated event with multiple feedback regulations, it is not surprising that reduced HR does not translate into lower BP. The reason why HR is affected by SMC-specific EPHB4 deletion is not clear. One possible explanation is leaky Cre expression in pacing cells controlling the cardiac rhythm; reduced EPHB4 kinase activity in these cells in turn affects cardiac rhythm. We, however, did not detect a reduction of EPHB4 expression in the right atrium of the KO mice, but this is probably due to the very sparse presence of the pacing cells in the right atrium and even in the sinus nodes and atrioventricular node. Using a different approach, we found that EPHB4 kinase inhibitors could quickly and drastically reduce the HR in WT mice (35), suggesting a critical role of EPHB4 in controlling heart rhythm. Further investigation of this aspect is warranted.
We have established that EPHB4 deletion compromises the CaMKII/MLCK/MLC signaling cascade in VSMCs, with increased CaMKII phosphorylation as the most upstream culpable event. Close to the cell membrane, we have identified GRIP1, which is known to associate with EFNB2, as an essential component in the reverse signaling of VSMCs with regard to contractility. GRIP1 knockdown resulted in reduced VSMC contractility in the presence of EFNB2 cross-linking. Molecular events between GRIP1 and CaMKII will need to be further elucidated for full characterization of the signaling pathway from EFNB2 to motor molecules (myosins). We previously identified GRIP1 as a key molecule mediating EFNB1 reverse signaling; its knockdown results in increased VSMC contractility after EFNB1 cross-linking (23). This indicates that the function of GRIP1 is versatile, depending on the EFNBs it associates with. Such versatile effects of GRIP1 could be attributed to the fact that GRIP1 functions as a scaffold and interacts with multiple types of signaling molecules; thus, its function is context-dependent. For example, GRIP1 could interact with glucocorticoid receptors and function as a glucocorticoid receptor co-activator or repressor depending on the context (36). Under physiological conditions in VSMCs, GRIP1 probably acts as a node receiving opposite inputs from various EFNBs, and its final function is based on the sum of these positive and negative inputs, with regard to VSMC contractility, and consequently, BP.
Our current findings have revealed the existence of a previously undocumented BP-lowering mechanism. Such new knowledge will lead to a better understanding of BP regulation mechanisms. Moreover, as EPHB4 inhibitors are being developed as cancer drugs for their anti-angiogenesis effect, our findings have raised timely caution with this previously unknown effect on lowering BP.
This work was supported by Canadian Institutes of Health Research Grants MOP57697, MOP69089, and MOP123389 (to J. W.), MOP97829 (to H. L.), MOP14496, (to E. T.) and ISO106797 (to J. T.). This work was also funded by grants from the Heart and Stroke Foundation of Quebec, the Natural Sciences and Engineering Research Council of Canada (203906-2012), the Juvenile Diabetes Research Foundation (17-2013-440), and the J.-Louis Lévesque Foundation (to J. W.).
Y. Wang, E. Thorin, H. Luo, J. Tremblay, J. L. Lavoie, Z. Wu, J. Peng, S. Qi, and J. Wu, unpublished observations.
Y. Wang and J. Wu, unpublished observations.
- EFN
- ephrin
- VSMC
- vascular smooth muscle cell
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- MLCK
- MLC kinase
- MLC
- myosin light chain
- MYPT
- MLC phosphatase
- AR
- adrenergic receptor
- NHIgG
- normal human IgG
- PE
- phenylephrine
- BP
- blood pressure
- SP
- systolic pressure
- DP
- diastolic pressure
- HR
- heart rate
- RT-qPCR
- reverse transcription-quantitative PCR.
References
- 1. Eph Nomenclature Committee (1997) Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell 90, 403–404 [DOI] [PubMed] [Google Scholar]
- 2. Pasquale E. B. (2008) Eph-ephrin bidirectional signaling in physiology and disease. Cell 133, 38–52 [DOI] [PubMed] [Google Scholar]
- 3. Wu J., Luo H. (2005) Recent advances on T-cell regulation by receptor tyrosine kinases. Curr. Opin. Hematol. 12, 292–297 [DOI] [PubMed] [Google Scholar]
- 4. Flanagan J. G., Vanderhaeghen P. (1998) The ephrins and Eph receptors in neural development. Annu. Rev. Neurosci. 21, 309–345 [DOI] [PubMed] [Google Scholar]
- 5. Luo H., Charpentier T., Wang X., Qi S., Han B., Wu T., Terra R., Lamarre A., Wu J. (2011) Efnb1 and Efnb2 proteins regulate thymocyte development, peripheral T cell differentiation, and antiviral immune responses and are essential for interleukin-6 (IL-6) signaling. J. Biol. Chem. 286, 41135–41152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luo H., Wan X., Wu Y., Wu J. (2001) Cross-linking of EphB6 resulting in signal transduction and apoptosis in Jurkat cells. J. Immunol. 167, 1362–1370 [DOI] [PubMed] [Google Scholar]
- 7. Luo H., Wu Z., Qi S., Jin W., Han B., Wu J. (2011) Ephrinb1 and Ephrinb2 are associated with interleukin-7 receptor α and retard its internalization from the cell surface. J. Biol. Chem. 286, 44976–44987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo H., Yu G., Tremblay J., Wu J. (2004) EphB6-null mutation results in compromised T cell function. J. Clin. Invest. 114, 1762–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luo H., Yu G., Wu Y., Wu J. (2002) EphB6 crosslinking results in costimulation of T cells. J. Clin. Invest. 110, 1141–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu G., Luo H., Wu Y., Wu J. (2003) Ephrin B2 induces T cell costimulation. J. Immunol. 171, 106–114 [DOI] [PubMed] [Google Scholar]
- 11. Yu G., Luo H., Wu Y., Wu J. (2003) Mouse ephrinB3 augments T-cell signaling and responses to T-cell receptor ligation. J. Biol. Chem. 278, 47209–47216 [DOI] [PubMed] [Google Scholar]
- 12. Yu G., Luo H., Wu Y., Wu J. (2004) EphrinB1 is essential in T-cell-T-cell co-operation during T-cell activation. J. Biol. Chem. 279, 55531–55539 [DOI] [PubMed] [Google Scholar]
- 13. Yu G., Mao J., Wu Y., Luo H., Wu J. (2006) Ephrin-B1 is critical in T-cell development. J. Biol. Chem. 281, 10222–10229 [DOI] [PubMed] [Google Scholar]
- 14. Batlle E., Henderson J. T., Beghtel H., van den Born M. M., Sancho E., Huls G., Meeldijk J., Robertson J., van de Wetering M., Pawson T., Clevers H. (2002) β-Catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 111, 251–263 [DOI] [PubMed] [Google Scholar]
- 15. Konstantinova I., Nikolova G., Ohara-Imaizumi M., Meda P., Kucera T., Zarbalis K., Wurst W., Nagamatsu S., Lammert E. (2007) EphA-Ephrin-A-mediated β cell communication regulates insulin secretion from pancreatic islets. Cell 129, 359–370 [DOI] [PubMed] [Google Scholar]
- 16. Salvucci O., de la Luz Sierra M., Martina J. A., McCormick P. J., Tosato G. (2006) EphB2 and EphB4 receptors forward signaling promotes SDF-1-induced endothelial cell chemotaxis and branching remodeling. Blood 108, 2914–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hashimoto T., Karasawa T., Saito A., Miyauchi N., Han G. D., Hayasaka K., Shimizu F., Kawachi H. (2007) Ephrin-B1 localizes at the slit diaphragm of the glomerular podocyte. Kidney Int. 72, 954–964 [DOI] [PubMed] [Google Scholar]
- 18. Dravis C., Wu T., Chumley M. J., Yokoyama N., Wei S., Wu D. K., Marcus D. C., Henkemeyer M. (2007) EphB2 and ephrin-B2 regulate the ionic homeostasis of vestibular endolymph. Hear. Res. 223, 93–104 [DOI] [PubMed] [Google Scholar]
- 19. Foo S. S., Turner C. J., Adams S., Compagni A., Aubyn D., Kogata N., Lindblom P., Shani M., Zicha D., Adams R. H. (2006) Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell 124, 161–173 [DOI] [PubMed] [Google Scholar]
- 20. Shin D., Garcia-Cardena G., Hayashi S., Gerety S., Asahara T., Stavrakis G., Isner J., Folkman J., Gimbrone M. A., Jr., Anderson D. J. (2001) Expression of ephrinB2 identifies a stable genetic difference between arterial and venous vascular smooth muscle as well as endothelial cells, and marks subsets of microvessels at sites of adult neovascularization. Dev. Biol. 230, 139–150 [DOI] [PubMed] [Google Scholar]
- 21. Gale N. W., Baluk P., Pan L., Kwan M., Holash J., DeChiara T. M., McDonald D. M., Yancopoulos G. D. (2001) Ephrin-B2 selectively marks arterial vessels and neovascularization sites in the adult, with expression in both endothelial and smooth-muscle cells. Dev. Biol. 230, 151–160 [DOI] [PubMed] [Google Scholar]
- 22. Luo H., Wu Z., Tremblay J., Thorin E., Peng J., Lavoie J. L., Hu B., Stoyanova E., Cloutier G., Qi S., Wu T., Cameron M., Wu J. (2012) Receptor tyrosine kinase Ephb6 regulates vascular smooth muscle contractility and modulates blood pressure in concert with sex hormones. J. Biol. Chem. 287, 6819–6829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu Z., Luo H., Thorin E., Tremblay J., Peng J., Lavoie J. L., Wang Y., Qi S., Wu T., Wu J. (2012) Possible role of Efnb1 protein, a ligand of Eph receptor tyrosine kinases, in modulating blood pressure. J. Biol. Chem. 287, 15557–15569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xin H. B., Deng K. Y., Rishniw M., Ji G., Kotlikoff M. I. (2002) Smooth muscle expression of Cre recombinase and eGFP in transgenic mice. Physiol. Genomics 10, 211–215 [DOI] [PubMed] [Google Scholar]
- 25. Lavoie J. L., Lake-Bruse K. D., Sigmund C. D. (2004) Increased blood pressure in transgenic mice expressing both human renin and angiotensinogen in the renal proximal tubule. Am. J. Physiol. Renal Physiol. 286, F965–F971 [DOI] [PubMed] [Google Scholar]
- 26. Golovina V. A., Blaustein M. P. (2006) Preparation of primary cultured mesenteric artery smooth muscle cells for fluorescent imaging and physiological studies. Nat Protoc. 1, 2681–2687 [DOI] [PubMed] [Google Scholar]
- 27. Tansey M. G., Word R. A., Hidaka H., Singer H. A., Schworer C. M., Kamm K. E., Stull J. T. (1992) Phosphorylation of myosin light chain kinase by the multifunctional calmodulin-dependent protein kinase II in smooth muscle cells. J. Biol. Chem. 267, 12511–12516 [PubMed] [Google Scholar]
- 28. Pfitzer G. (2001) Invited review: regulation of myosin phosphorylation in smooth muscle. J. Appl. Physiol. 91, 497–503 [DOI] [PubMed] [Google Scholar]
- 29. Brückner K., Pablo Labrador J., Scheiffele P., Herb A., Seeburg P. H., Klein R. (1999) EphrinB ligands recruit GRIP family PDZ adaptor proteins into raft membrane microdomains. Neuron 22, 511–524 [DOI] [PubMed] [Google Scholar]
- 30. Sällström J., Peuckert C., Gao X., Larsson E., Nilsson A., Jensen B. L., Onozato M. L., Persson A. E., Kullander K., Carlström M. (2013) Impaired EphA4 signaling leads to congenital hydronephrosis, renal injury, and hypertension. Am. J. Physiol. Renal Physiol. 305, F71–F79 [DOI] [PubMed] [Google Scholar]
- 31. Cheng N., Brantley D. M., Chen J. (2002) The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 13, 75–85 [DOI] [PubMed] [Google Scholar]
- 32. Martiny-Baron G., Holzer P., Billy E., Schnell C., Brueggen J., Ferretti M., Schmiedeberg N., Wood J. M., Furet P., Imbach P. (2010) The small molecule specific EphB4 kinase inhibitor NVP-BHG712 inhibits VEGF driven angiogenesis. Angiogenesis 13, 259–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wnuk M., Hlushchuk R., Janot M., Tuffin G., Martiny-Baron G., Holzer P., Imbach-Weese P., Djonov V., Huynh-Do U. (2012) Podocyte EphB4 signaling helps recovery from glomerular injury. Kidney Int. 81, 1212–1225 [DOI] [PubMed] [Google Scholar]
- 34. Salvucci O., Tosato G. (2012) Essential roles of EphB receptors and EphrinB ligands in endothelial cell function and angiogenesis. Adv. Cancer Res. 114, 21–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu J., Wang Y., Luo H., Caflisch A., Nevado C., Frugier E., Lafleur K., Zhao H., Huang D. (September 5, 2014) Inhibitors for use in the treatment of hypertension and tachycardia and in regulating blood pressure. European Patent Office, application number EP14183830
- 36. Rogatsky I., Wang J. C., Derynck M. K., Nonaka D. F., Khodabakhsh D. B., Haqq C. M., Darimont B. D., Garabedian M. J., Yamamoto K. R. (2003) Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc. Natl. Acad. Sci. U.S.A. 100, 13845–13850 [DOI] [PMC free article] [PubMed] [Google Scholar]