

Abstract
The number of diabetic patients presenting to burn services is predicted to increase significantly over the next decades. Diabetes mellitus represents an independent risk factor for sustaining burn injuries and mediates alterations to key physiological systems including the vascular, renal, nervous, gastrointestinal and immune system. The effects of the pathophysiological permutations need to be carefully considered during both the acute as well as the long-term rehabilitation phase of injury. The purpose of the first part of this review is to outline the metabolic permutations observed in diabetes mellitus pertinent to the clinical presentation and management of burn patients.
Keywords: Diabetes, burn, management
Introduction
Diabetes Mellitus (DM) is one of the largest global health problems of the 21st Century. It is anticipated that the number of Americans diagnosed with the disease will continue to increase by 165% from the year 2000 to 2050 [1]. DM represents a spectrum of metabolic disorders characterised by chronic hyperglycaemia, resulting either from endogenous insulin insufficiency/defective production or from diminished effectiveness at peripheral receptors [2]. There are several different subtypes of DM with the commonest being type 1 and 2.
Type 1 DM (T1DM), which results from the autoimmune destruction of insulin-secreting pancreatic β-cells, has a typically acute juvenile onset and requires lifelong insulin treatment [3]. Despite being the subject of intensive study over the last decades, the causes of T1DM are still not fully understood; nevertheless a combination of environmental and genetic factors has been associated with disease pathogenesis. The chromosomal loci believed to influence T1DM susceptibility can be broadly categorised into those relating to immune function [including the Human Leukocyte Antigen (HLA) region], insulin expression (polymorphisms in promoter gene regions) and β-cell function [including the protein tyrosine phosphatase, non-receptor type 22 gene] (PTPN22) [4].
Type 2 DM (T2DM) is characterised by excessive insulin secretion, tissue insulin resistance and subsequent β-cell dysfunction. It tends to present in later life and affected patients are frequently diagnosed on the basis of diabetic complications [3]. The pathogenesis of type 2 DM has a strong environmental component with obesity being one of the most important modifiable risk factors [5,6]. Genetics are also believed to play an important role in the pathogenesis of T2DM; over 36 genes have already been linked to disease development, many of which are associated with differing degrees of beta cell dysfunction. Of particular note, the transcription factor 7 like 2 (TCF7L2) gene nearly doubles the risk of developing T2DM most likely through the pathway, which regulates proglucagon gene expression in enteroendocrine cells [7]. The American Diabetes Association (ADA) diagnostic criteria for diabetes mellitus are shown in Table 1 [3].
Table 1.
American Diabetes Association (ADA) diagnostic criteria for diabetes mellitus [3]
| Test | Diagnostic levels | Comments |
|---|---|---|
| Fasting plasma glucose (FPG) level | ≥ 126 mg/dl (7.0 mmol/l) | Fasting is defined as no caloric intake for at least 8 hours |
| 2 hour plasma glucose following oral glucose tolerance test (OGTT) | ≥ 200 mg/dl (11.1 mmol/l) | Test to be performed using glucose load with equivalent of 75 g glucose dissolved in water |
| Random plasma glucose level | ≥ 200 mg/dl (11.1 mmol/l) | To be used in presence of symptoms of hyperglycemia or hyperglycemic crisis |
| Glycosylated hemoglobin (HbA1c) | ≥ 6.5% | To be performed using a standardized assay |
Fasting as well as plasma glucose levels following an oral glucose tolerance test are widely used tests for the initial diagnosis of diabetes; nevertheless in patients with severe classic hyperglycemic symptoms/hyperglycemic crisis, a random plasma level is considered adequate. Glycosylated haemoglobin (HbA1c), although widely used as a marker of chronic glycemia (reflecting levels over a 2-3 month period), is currently considered valid in diagnostic terms provided it is performed in a standardized manner.
Similarities between the metabolic features of diabetes mellitus and stress induced hyperglycaemia (SIH) of acute illness
The hormonal profile of diabetic patients has multiple facets, which augment the primary perturbations in glucose metabolism.
In T1DM, in addition to the lack insulin secretion, there are increased levels of glucagon [8], which exacerbate hyperglycaemia by reducing hepatic glucose uptake and increasing glucose release [9]. Elevated levels of fasting catecholamines, especially found in poorly controlled DM, contribute to hyperglycaemia by stimulating glucagon production and impairing the action of insulin [10,11]. Additionally, enhanced lipolysis (in the presence of high cortisol levels) predisposes towards increased concentrations of circulating free fatty-acids (FFAs) and ketone body formation, which can lead to ketoacidosis [12-15].
Type 2 DM is characterised by marked insulin resistance with underlying mechanisms including the deranged expression of liver enzymes and changes in signalling pathways (e.g. c-Jun amino terminal kinase) due to the increased release of TNF-alpha and IL-6 by adipose tissue macrophages [16]. Additionally, the elevated levels of fatty acid metabolites (including diacylglycerol) decrease insulin receptor signalling by activating the phosphorylation of insulin receptor substrates 1 and 2 [17].
Furthermore, under normal physiological conditions, insulin plays an important role in the upregulation of protein synthesis by enhancing the uptake of amino acids into muscle. In DM, the low levels and tissue insensitivity to circulating insulin (especially in poorly controlled patients), mediates an enhancement in proteolysis causing disturbances in nitrogen balance [18].
Burn injuries are associated with a profound hypermetabolic response. Metabolic features include increased energy expenditure, a negative nitrogen balance as well as stress-induced hyperglycaemia and decreased peripheral insulin sensitivity. Key mediators for these derangements include the augmented secretion of catabolic hormones (cortisol and catecholamines), the suppression of endogenous activity of anabolic agents (growth hormone and testosterone) as well as cytokine release (interleukins 1, 6 and tumour necrosis factor-TNF) [18-27].
It is striking that the metabolic profile of diabetes mellitus mirrors changes occurring in critical illness (e.g. hyperglycaemia, insulin tissue insensitivity, negative nitrogen balance), hence burn patients with pre-existing diabetes mellitus may be theoretically subjected to a ‘second hit’ phenomenon. In other words, they may be prone to enhanced metabolic disturbances due to the combined effects of the acute burn injury and the premorbid diabetic pathophysiology. It is interesting to review the different mechanisms by which DM affects the different physiological systems, since this forms the basis for investigating a potential ‘second hit’ phenomenon. The latter will be further elucidated in the second part of this work, which focuses on morbidity and mortality in this cohort of burn patients.
Key physiological system alterations in diabetes mellitus
Vascular system
A variety of biochemical derangements have been identified as contributing factors to end organ tissue damage in diabetes. These include the utilisation of excess glucose along the polyol pathway, the stimulation of the diacylglycerol-protein C kinase pathway as well as the non-enzymatic glycosylation of tissues (Figure 1) [28].
Figure 1.
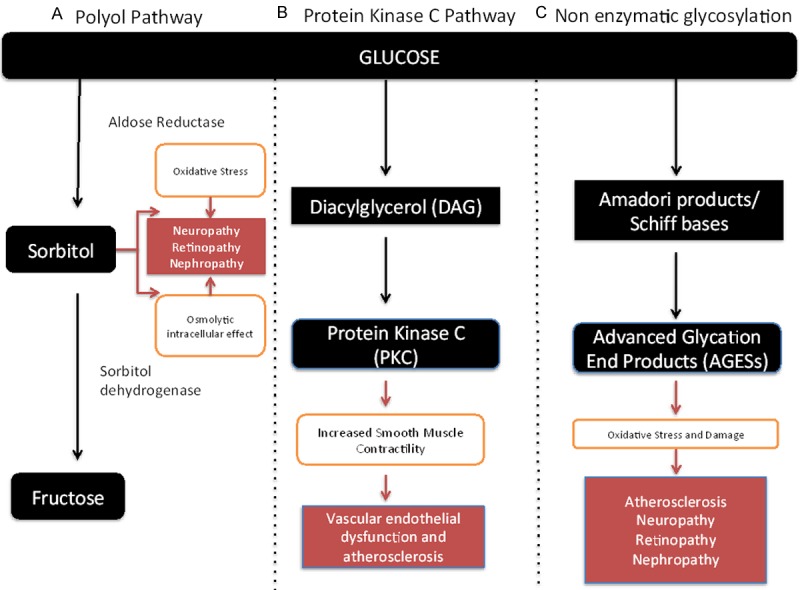
Pathways involved in end organ tissue damage in diabetes mellitus [28]. A. Polyol pathway-Increased blood glucose is converted via sorbitol into fructose. Due to the slow absorption of sorbitol, its accumulation has a significant intracellular osmolytic effect as well as (via interaction with the inositol pathway) increases cellular oxidative stress. B. Protein Kinase C pathway-Glucose handling via the glycolysis pathway leads to an increase in the intermediate product diacylglycerol (DAG). This activates intracellular signalling via protein kinase C and this leads to a variety of cellular effects (including increased smooth muscle contractility, altered calcium homeostasis and sensitivity to growth factors) contributing to vascular dysfunction. C. Non-enzymatic glycosylation pathway-Glucose interacts with reactive amino groups in cellular proteins and forms advanced glycosylation end products (AGEs) via intermediate compounds (Amadori products and Schiff bases). The AGEs interact with fixed tissue elements and circulating cells and lead via a variety of mechanisms to oxidative stress/cellular damage contributing to diabetic complications.
Macrovasculature
Atherosclerosis is more prevalent in diabetic patients with underlying contributing factors relating to abnormalities in the vessel wall, circulating cells/factors and blood flow.
Vessel wall
Increased non enzymatic glycosylation of lipoprotein leads to the accumulation of cholesterol ester in ‘foam’ macrophages [29]; the modified lipoproteins result in the formation of autoantibodies and the ensuing lipoprotein immune complexes activate the endothelium and smooth muscle cells contributing to accelerated atheromatous plaque formation [30].
Circulating cells
Platelets in diabetes are characterised by reduced deformability and propensity to adhesion/aggregation; the latter is due to the enhanced release of α granule contents and hypersensitivity to aggregating agents (including collagen and arachidonic acid) [31]. Furthermore, red blood cells are more prone to oxidative stress contributing to a reduced cellular life span [32-36] and their decreased oxygen affinity has profound effects on tissues by limiting the supply of oxygen [37-40].
Impaired rheological characteristics
Plasma viscosity is increased considerably in diabetic patients, which is thought to promote atherosclerosis and thrombosis in blood vessels [41-43]. Additionally, the elevated levels of coagulation factors (including fibrinogen and factors 7, 8), decreased levels of protein C and S and the impaired fibrinolytic activity contribute towards a pro-coagulant state in the vasculature [28].
The three main macrovascular disease manifestations include coronary artery, cerebrovascular and peripheral vascular disease. The increased risk of diabetic patients from macrovascular complications needs to be carefully considered during the acute and rehabilitative stage of burn injuries. Meticulous attention must be given to careful fluid management in order to balance the risk of under-resuscitation versus cardiac overload (incidence of congestive heart failure is 2-5 times higher in diabetics) [44]. The risk of cerebrovascular accidents needs to be addressed with attention to normotension, normovolaemia and thromboprophylaxis. The implications of impaired peripheral vasculature concern both the acute stage of healing (impaired perfusion of tissues) as well as the mobilisation in later phases of recovery (decreased exercise tolerance).
Microvasculature
Microvascular complications are particularly prominent sequelae of chronic DM, caused by prolonged exposure of a wide variety of cells to high levels of glucose in the circulation via a variety of pathways shown in Figure 1. Chronic intracellular hyperglycaemia of endothelial and mesangial cells, coupled with hypercoagulability secondary to increased platelet and adhesion, causes progressive narrowing, microthrombus formation and eventual occlusion of vascular lumina. This leads to ischemia and dysfunction of the affected tissues with common microvascular complications including nephropathy and peripheral neuropathy [45-47].
Renal system/diabetic nephropathy
Diabetic nephropathy is a chronic, progressive condition characterized by increasing urinary albumin excretion, hypertension and declining glomerular filtration rate. It is a relatively late complication of T1DM, but often established at the time of diagnosis of T2DM. Nephropathy is a marker for other diabetes related complications, including cardiovascular disease and cerebrovascular accidents [48-50] and can lead to end stage renal insufficiency necessitating renal replacement therapy or transplantation [51,52]. The implications of diabetic renal impairment are multiple and apart from the need for careful fluid management include:
Pharmacokinetic/dynamic considerations
Careful administration and monitoring of medications (especially those with a narrow therapeutic range excreted by the kidneys) is paramount in order to avoid deteriorations of renal function in diabetic patients. This becomes more pertinent given the complex effect of the burn injury on drug metabolism; for instance the ‘ebb’ phase of the metabolic response is characterised by a reduction in renal perfusion/glomerular filtration rate (GFR) whereas during the subsequent ‘flow’ phase, the GFR is considerably increased [53]. Furthermore, comorbid conditions including obesity can further impact upon pharmacological strategies; the comparatively higher fat content in obese individuals mediates an increased volume of distribution for lipophilic drugs mandating the need for close liaison between clinicians and pharmacists in order to determine optimal drug dosing [54].
Blood product replacement
Anaemia is one of the common sequelae of diabetic renal disease; judicious transfusion practices are recommended in this subgroup of patients especially those who are candidates for renal transplantation. Administration of transfusions renders patients prone to immune sensitisation, which can limit the range of potential donor organs ahead of renal transplantation [55]. In cases of planned delayed surgery, increasing the dose of erythropoietin administration is a valid option; nevertheless several weeks are needed for the hormone to raise the haematocrit considerably [56,57]. It is clear that decisions regarding the maintenance of haematocrit levels in this cohort of patients need to be made in close liaison with renal physicians.
Nervous system/diabetic neuropathy
Diabetic neuropathy affects 30-50% of people with established diabetes [58-60]. Its pathophysiology is thought to result from the combined effects of microangiopathy and direct osmotic axonal damage from elevated levels of glucose [61]. Diabetic distal symmetrical sensory polyneuropathy (DPN) is the most relevant manifestation; it classically presents with a “glove and stocking” distribution of sensory loss and is a major contributor towards ulceration as well as burn injuries to the lower limb [62]. The loss of normal neurofeedback contributes to arch flattening and leads to abnormal pressure distribution in the feet. Sympathetic autonomic neuropathy results in dryness and skin fissuring and contributes to increased susceptibility to trauma and infection. Neuropathic arthropathy (Charcot’s joints) and oedema are further contributory factors towards tissue injury, which can lead to limb amputation [63,64].
Neuropathic pain symptoms are quite common in diabetes affecting up to one third of patients [65,66]. Burn injuries can be associated with severe symptoms of pain and pruritus, which in the context of established neuropathy, can present the burns team with unique management challenges. Given the emerging evidence implicating neuropathic mechanisms in post burn sensory disturbances, it appears prudent for burn teams to utilise therapeutic agents acting on the central nervous system (including gabapentin or pregabalin) early on for symptom control in this subgroup of patients [67,68].
Gastrointestinal system
Longstanding hyperglycaemia in diabetes mellitus causes enteric nerve damage and dysregulation of vagal activity resulting in abnormal motility, gastric paresis, and bacterial overgrowth [69]. Clinical manifestations of abnormal motility include diarrhea, nausea and vomiting, which can precipitate significant additional fluid and electrolyte abnormalities.
Poor glycaemic control is also strongly associated with gastrointestinal infections such as oral candidiasis, which can be exacerbated by the antibiotic polypharmacy often practised in the management of burn patients. Consequent symptoms of dysphagia and odynophagia can lead to on-going poor nutrition, with implications on delayed wound healing and subsequent rehabilitation [70]. Fluid replacement, judicious antibiotic therapy and nutritional support should therefore be carefully tailored in the care of the diabetic burn patient. Additionally, obesity (a frequent association with type 2 diabetes) is associated with a higher incidence of gastroesophageal reflux disease, large residual gastric volumes and comparatively lower pH secretions; these factors predispose patients to gastric ulceration/bleeding as well as aspiration pneumonitis and underlie the importance of gastroprophylaxis in this patient cohort [71-73].
Immune system
Many components of the immune (both innate and adaptive) system show significant perturbations in patients with diabetes accounting for the increased susceptibility to infective complications.
Innate immune system
Complement system deficiencies along with a diminished activity of natural killer cells are prominent in diabetes mellitus [74,75]. Furthermore, significant disturbances in the activity of polymorphonuclear leucocytes are observed; these include reduced chemotaxis, adherence, phagocytosis, and bactericidal activity of neutrophils, monocytes and macrophages [76-81].
Adaptive immune system
Lymphocytes responsible for the production of antibodies against pathogens show decreased mitogenic responses. In addition, the production of interleukin 2 (which is vital in sustaining the post-injury inflammatory response via T cell activation) is significantly reduced [82,83]. Interestingly, insulin administration appears to increase the activity of adenosine triphosphate and uptake of glucose in lymphocytes [84], a finding, which highlights the importance of good glycemic control in minimising the effects of diabetic metabolism on immune function.
Healing in diabetics
Wound healing comprises three overlapping phases: inflammation, proliferation and remodelling [85,86].
Diabetes exerts a detrimental effect on wound healing via extrinsic and intrinsic mechanisms. The term ‘intrinsic’ refers to mechanisms relating to the abnormal expression/activity of local growth factors and wound healing constituents. ‘Extrinsic’ parameters include peripheral vascular disease (resulting in decreased oxygen supply to tissues), neuropathy (disruption of neurogenic control of small vessels interfering with the inflammatory response) as well as infection and oedema [87-89].
Inflammation
The defects in neutrophil chemotaxis and function, result in defective tissue debridement and impaired secretion of a variety of growth factors and cytokines important in wound healing [90-92]. The pattern of macrophages is also altered in DM. Under normal circumstances, activation shows two distinctive phenotypes: classical (caM), which predominates in the initial inflammatory phase and alternative activation (aaM), which predominates in the proliferative stage of healing [93]. In diabetic wounds, there is insufficient caM in the early stage but excessive aaM in the later proliferative phase alongside the predominance of a T helper 2 (TH2) over T helper 1 (TH1) cytokine response. These factors are believed to play a significant role in stalling diabetic wounds in an abnormal inflammatory state (comprising an increased number of inflammatory cells, albeit a marked absence of growth on cellular level) and hindering the transition into the proliferative stage [94,95].
Proliferation
This phase is characterised by fibroplasia, neovascularisation and epithelialisation involving fibroblasts, endothelial cells and keratinocytes.
Fibroplasia
Diabetes mellitus is associated with excessive production of advanced glycation end products, which interferes with normal extracellular matrix deposition; additionally, fibroblasts show impaired proliferation and premature senescence. As a result, diabetic wounds have decreased levels of glycosaminoglycans as well as collagen (which is also characterised by an abnormal molecular structure) contributing to a lower wound breaking strength [90,94,96-100].
Neovascularisation
This is a vital process implicated in healing and comprises endothelial proliferation, migration and capillary formation regulated by angiogenic factors including angiopoeitins [101]. Angiopoeitin 2 (Ang-2) is considered as an angiogenesis ‘starting mediator’, which initially is present in high concentrations but gradually in the course of angiogenesis returns to normal levels [102]. Another important growth factor involved in new vessel formation is vascular endothelial growth factor (VEGF), which promotes endothelial cell proliferation and migration [103]. There is normally a synergistic action between Ang-2 and high levels of VEGF inducing ‘sprouting’ angiogenesis, whereas absence of VEGF results in capillary regression in the early stages [104]. The regulation of neovascularisation in deep partial thickness scalds in diabetic rates has revealed marked impairment in wound healing at 2 weeks following injury with inhibition of vascularisation at the wound edges due to a sustained abnormally high expression of Ang-2 and downregulation of VEGF between day 14-21 post injury [105]. Insulin has been found to stimulate human microvascular endothelial cell migration and tube formation [95], a finding, which further highlights the impact of good glycaemic control on diabetic wound healing.
Epithelialisation
Epithelialisation is a process, which is attenuated in DM [106]; porcine burn models suggest that low levels of insulin growth factor 1 (IGF-1) and tumour growth factor beta (TGF-β) in the first week post injury are most likely implicated in this abnormality [90,107].
IGF-1 has been shown to induce chemoattractant activity in endothelial cell lines, stimulate keratinocyte and fibroblast proliferation and re-epithelialisation [108], whilst TGF-beta is thought to be important in chemoattraction of monocytes, keratinocytes, fibroblasts and induction of these cells to release further growth factors [109,110].
Remodelling
This phase is characterised by cessation of fibroblast proliferation/collagen production as well as diminishing vascularisation and myofibroblast mediated wound contraction.
Important factors contributing to disturbances in the remodelling phase of diabetic wounds include: increased levels of proteolytic enzymes reduced activity of growth factors due to non enzymatic glycosylation as well as the imbalance between metalloproteases and their tissue inhibitors (responsible for collagen remodelling) [85,90,111,112].
Diabetes mellitus as a risk factor for burn injuries
Diabetes mellitus represents a significant risk factor for burn injuries due to a variety of reasons. Peripheral neuropathy and retinopathy result in decreased tactile sensation as well as visual impairment. This implies that diabetics are less able to detect and avoid sources of burn injury hence are more susceptible to sustaining injuries and presenting to medical care in a delayed manner with deeper burns. Additionally, gait abnormalities stemming from neuropathy and pre-existing amputated limbs can predispose an individual to injuries by increasing the risk of falling and limiting the ability to remove oneself from a source of injury.
The ability of the skin to dissipate heat energy relies on passive conduction to lateral and deep tissues as well as dispersion via the increase in blood flow in the affected tissues [113]. Studies have confirmed that diabetes is characterised by diminished heat energy transfer to the surrounding skin as well as a weaker hyperaemic response [114]. The conductive properties of skin in DM are reduced by virtue of a comparatively thinner dermal and thicker subcutaneous fat layer [115] as well as physiological disturbances in vasoregulation [116].
The main vasodilating factors include nitric oxide, substance P, calcitonin gene-related peptide and prostaglandins [116-119]. Diabetes (as well as ageing) makes the skin more dependent on nitric oxide for vasodilation, whereas the ability to vasoconstrict remains unaltered [113,117,120-122]. Interestingly, the nitric oxide pathway is impaired in diabetes most likely due to a combination of reduced L-arginine in endothelial cells as well as diminished production of nitric oxide/bioavailability due to the presence of high concentrations of free radicals in vascular endothelial cells [116,123].
Conclusion
Diabetes mellitus is responsible for a host of physiological disturbances affecting most bodily systems and represents a significant risk factor for sustaining burn injuries. The effects of diabetes as a premorbid condition have an impact on both the acute phase of the injury as well as long-term rehabilitation. Recognition of the exact pathophysiological permutations is paramount in planning appropriate management strategies to improve the standard of care in this cohort of burn patients.
Disclosure of conflict of interest
None.
References
- 1.Foster GD, Makris AP, Bailer BA. Behavioral treatment of obesity. Am J Clin Nutr. 2005;82:230S–235S. doi: 10.1093/ajcn/82.1.230S. [DOI] [PubMed] [Google Scholar]
- 2.Holt R CC, Flyvbjerg A, Goldstein B, George K, Alberti MM. Textbook of Diabetes: John Wiley & Sons. 2011. The Classification and Diagnosis of Diabetes Mellitus. [Google Scholar]
- 3.ADA American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 2013;36:67S–74S. [Google Scholar]
- 4.Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2:a007732. doi: 10.1101/cshperspect.a007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schienkiewitz A, Schulze MB, Hoffmann K, Kroke A, Boeing H. Body mass index history and risk of type 2 diabetes: results from the European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Am J Clin Nutr. 2006;84:427–433. doi: 10.1093/ajcn/84.1.427. [DOI] [PubMed] [Google Scholar]
- 6.Gregg EW, Cheng YJ, Narayan KM, Thompson TJ, Williamson DF. The relative contributions of different levels of overweight and obesity to the increased prevalence of diabetes in the United States: 1976-2004. Prev Med. 2007;45:348–352. doi: 10.1016/j.ypmed.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 7.Das SK, Elbein SC. The Genetic Basis of Type 2 Diabetes. Cellscience. 2006;2:100–131. doi: 10.1901/jaba.2006.2-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seino Y, Ikeda M, Kurahachi H, Taminato T, Sakurai H, Goto Y, Inoue Y, Kadowaki S, Mori K, Imura H. Failre of suppress plasma glucagon concentrations by orally administered glucose in diabetic patients after treatment. Diabetes. 1978;27:1145–1150. doi: 10.2337/diab.27.12.1145. [DOI] [PubMed] [Google Scholar]
- 9.Rizza R, Verdonk C, Miles J, Service FJ, Gerich J. Effect of intermittent endogenous hyperglucagonemia on glucose homeostasis in normal and diabetic man. J Clin Invest. 1979;63:1119–1123. doi: 10.1172/JCI109404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolli G, Cartechini MG, Compagnucci P, Malvicini S, De Feo P, Santeusanio F, Angeletti G, Brunetti P. Effect of metabolic control on urinary excretion and plasma levels of catecholamines in diabetics. Horm Metab Res. 1979;11:493–497. doi: 10.1055/s-0028-1092768. [DOI] [PubMed] [Google Scholar]
- 11.Rizza RA, Cryer PE, Haymond MW, Gerich JE. Adrenergic mechanisms for the effects of epinephrine on glucose production and clearance in man. J Clin Invest. 1980;65:682–689. doi: 10.1172/JCI109714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miles JM, Haymond MW, Nissen SL, Gerich JE. Effects of free fatty acid availability, glucagon excess, and insulin deficiency on ketone body production in postabsorptive man. J Clin Invest. 1983;71:1554–1561. doi: 10.1172/JCI110911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Avogaro A, Valerio A, Gnudi L, Maran A, Miola M, Duner E, Marescotti C, Iori E, Tiengo A, Nosadini R. The effects of different plasma insulin concentrations on lipolytic and ketogenic responses to epinephrine in normal and type 1 (insulin-dependent) diabetic humans. Diabetologia. 1992;35:129–138. doi: 10.1007/BF00402544. [DOI] [PubMed] [Google Scholar]
- 14.Dinneen S, Alzaid A, Miles J, Rizza R. Effects of the normal nocturnal rise in cortisol on carbohydrate and fat metabolism in IDDM. Am J Physiol. 1995;268:E595–603. doi: 10.1152/ajpendo.1995.268.4.E595. [DOI] [PubMed] [Google Scholar]
- 15.Schade DS, Eaton RP, Standefer J. Modulation of basal ketone body concentration by cortisol in diabetic man. J Clin Endocrinol Metab. 1978;47:519–528. doi: 10.1210/jcem-47-3-519. [DOI] [PubMed] [Google Scholar]
- 16.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 17.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 18.Ballian N, Rabiee A, Andersen DK, Elahi D, Gibson BR. Glucose metabolism in burn patients: the role of insulin and other endocrine hormones. Burns. 2010;36:599–605. doi: 10.1016/j.burns.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 19.Gauglitz GG, Herndon DN, Jeschke MG. Insulin resistance postburn: underlying mechanisms and current therapeutic strategies. J Burn Care Res. 2008;29:683–694. doi: 10.1097/BCR.0b013e31818481ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kagan RJ. Metabolism and nutrition in the burned patient. Nutr Clin Pract. 1991;6:1–2. doi: 10.1177/011542659100600101. [DOI] [PubMed] [Google Scholar]
- 21.Herndon D. Total Burn Care. New York: W.B Saunders; 2002. [Google Scholar]
- 22.Ding X, Price SR, Bailey JL, Mitch WE. Cellular mechanisms controlling protein degradation in catabolic states. Miner Electrolyte Metab. 1997;23:194–197. [PubMed] [Google Scholar]
- 23.Fang CH, James HJ, Ogle C, Fischer JE, Hasselgren PO. Influence of burn injury on protein metabolism in different types of skeletal muscle and the role of glucocorticoids. J Am Coll Surg. 1995;180:33–42. [PubMed] [Google Scholar]
- 24.Hart DW, Wolf SE, Chinkes DL, Gore DC, Mlcak RP, Beauford RB, Obeng MK, Lal S, Gold WF, Wolfe RR, Herndon DN. Determinants of skeletal muscle catabolism after severe burn. Ann Surg. 2000;232:455–465. doi: 10.1097/00000658-200010000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jahoor F, Desai M, Herndon DN, Wolfe RR. Dynamics of the protein metabolic response to burn injury. Metabolism. 1988;37:330–337. doi: 10.1016/0026-0495(88)90132-1. [DOI] [PubMed] [Google Scholar]
- 26.Wolfe RR, Durkot MJ, Allsop JR, Burke JF. Glucose metabolism in severely burned patients. Metabolism. 1979;28:1031–1039. doi: 10.1016/0026-0495(79)90007-6. [DOI] [PubMed] [Google Scholar]
- 27.Zeineh RA, Paulissian EB, Dubin A, Plzak LF, Boswick JA Jr, Kukral JC. Increased protein anabolism in burn patients with negative nitrogen balance. Surg Forum. 1970;21:69–71. [PubMed] [Google Scholar]
- 28.Kamal K, Powell RJ, Sumpio BE. The pathobiology of diabetes mellitus: implications for surgeons. J Am Coll Surg. 1996;183:271–289. [PubMed] [Google Scholar]
- 29.Lopes-Virella MF, Klein RL, Lyons TJ, Stevenson HC, Witztum JL. Glycosylation of low-density lipoprotein enhances cholesteryl ester synthesis in human monocyte-derived macrophages. Diabetes. 1988;37:550–557. doi: 10.2337/diab.37.5.550. [DOI] [PubMed] [Google Scholar]
- 30.Virella G, Munoz JF, Galbraith GM, Gissinger C, Chassereau C, Lopes-Virella MF. Activation of human monocyte-derived macrophages by immune complexes containing low-density lipoprotein. Clin Immunol Immunopathol. 1995;75:179–189. doi: 10.1006/clin.1995.1069. [DOI] [PubMed] [Google Scholar]
- 31.Winocour PD, Perry DW, Kinlough-Rathbone RL. Hypersensitivity to ADP of platelets from diabetic rats associated with enhanced fibrinogen binding. Eur J Clin Invest. 1992;22:19–23. doi: 10.1111/j.1365-2362.1992.tb01930.x. [DOI] [PubMed] [Google Scholar]
- 32.McMillan DE, Utterback NG, La Puma J. Reduced erythrocyte deformability in diabetes. Diabetes. 1978;27:895–901. doi: 10.2337/diab.27.9.895. [DOI] [PubMed] [Google Scholar]
- 33.MacRury SM, Small M, Anderson J, MacCuish AC, Lowe GD. Evaluation of red cell deformability by a filtration method in type 1 and type 2 diabetes mellitus with and without vascular complications. Diabetes Res. 1990;13:61–65. [PubMed] [Google Scholar]
- 34.Le Devehat C, Vimeux M, Bondoux G, Khodabandehlou T. Red blood cell aggregation in diabetes mellitus. Int Angiol. 1990;9:11–15. [PubMed] [Google Scholar]
- 35.Ziegler O, Guerci B, Muller S, Candiloros H, Mejean L, Donner M, Stoltz JF, Drouin P. Increased erythrocyte aggregation in insulin-dependent diabetes mellitus and its relationship to plasma factors: a multivariate analysis. Metabolism. 1994;43:1182–1186. doi: 10.1016/0026-0495(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 36.Murakami K, Kondo T, Ohtsuka Y, Fujiwara Y, Shimada M, Kawakami Y. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism. 1989;38:753–758. doi: 10.1016/0026-0495(89)90061-9. [DOI] [PubMed] [Google Scholar]
- 37.Watala C. Altered structural and dynamic properties of blood cell membranes in diabetes mellitus. Diabet Med. 1993;10:13–20. doi: 10.1111/j.1464-5491.1993.tb01990.x. [DOI] [PubMed] [Google Scholar]
- 38.Kung CM, Tseng ZL, Wang HL. Erythrocyte fragility increases with level of glycosylated hemoglobin in type 2 diabetic patients. Clin Hemorheol Microcirc. 2009;43:345–351. doi: 10.3233/CH-2009-1245. [DOI] [PubMed] [Google Scholar]
- 39.Lippi G, Mercadanti M, Aloe R, Targher G. Erythrocyte mechanical fragility is increased in patients with type 2 diabetes. Eur J Intern Med. 2012;23:150–153. doi: 10.1016/j.ejim.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Stanescu M, Z G, D I. Effect of glucose and insulin upon human erythrocyte membrane ATPases. Rom J Biophys. 2002;12:117–128. [Google Scholar]
- 41.Memeh CU. Differences between plasma viscosity and proteins of types 1 and 2 diabetic Africans in early phase of diabetes. Horm Metab Res. 1993;25:21–23. doi: 10.1055/s-2007-1002038. [DOI] [PubMed] [Google Scholar]
- 42.Zioupos P, Barbenel JC, Lowe GD, MacRury S. Foot microcirculation and blood rheology in diabetes. J Biomed Eng. 1993;15:155–158. doi: 10.1016/0141-5425(93)90048-4. [DOI] [PubMed] [Google Scholar]
- 43.Brown CD, Zhao ZH, Berweck S, de Alvaro F, Chan S, Friedman EA. Effects of alloxan-induced diabetes on hemorheology in rabbits. Horm Metab Res. 1992;24:254–257. doi: 10.1055/s-2007-1003307. [DOI] [PubMed] [Google Scholar]
- 44.Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29–34. doi: 10.1016/0002-9149(74)90089-7. [DOI] [PubMed] [Google Scholar]
- 45.Giannini C, Dyck PJ. Basement membrane reduplication and pericyte degeneration precede development of diabetic polyneuropathy and are associated with its severity. Ann Neurol. 1995;37:498–504. doi: 10.1002/ana.410370412. [DOI] [PubMed] [Google Scholar]
- 46.Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–2890. doi: 10.1172/JCI118746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 48.Tuomilehto J, Borch-Johnsen K, Molarius A, Forsen T, Rastenyte D, Sarti C, Reunanen A. Incidence of cardiovascular disease in Type 1 (insulin-dependent) diabetic subjects with and without diabetic nephropathy in Finland. Diabetologia. 1998;41:784–790. doi: 10.1007/s001250050988. [DOI] [PubMed] [Google Scholar]
- 49.Mann JF, Gerstein HC, Pogue J, Bosch J, Yusuf S. Renal insufficiency as a predictor of cardiovascular outcomes and the impact of ramipril: the HOPE randomized trial. Ann Intern Med. 2001;134:629–636. doi: 10.7326/0003-4819-134-8-200104170-00007. [DOI] [PubMed] [Google Scholar]
- 50.Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR, Ukpds G. Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64) Kidney Int. 2003;63:225–232. doi: 10.1046/j.1523-1755.2003.00712.x. [DOI] [PubMed] [Google Scholar]
- 51.Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28:164–176. doi: 10.2337/diacare.28.1.164. [DOI] [PubMed] [Google Scholar]
- 52.Deshpande AD, Harris-Hayes M, Schootman M. Epidemiology of diabetes and diabetes-related complications. Phys Ther. 2008;88:1254–1264. doi: 10.2522/ptj.20080020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weinbren MJ. Pharmacokinetics of antibiotics in burn patients. J Antimicrob Chemother. 1999;44:319–327. doi: 10.1093/jac/44.3.319. [DOI] [PubMed] [Google Scholar]
- 54.Goutos I, Sadideen H, Pandya AA, Ghosh SJ. Obesity and burns. J Burn Care Res. 2012;33:471–482. doi: 10.1097/BCR.0b013e318247959b. [DOI] [PubMed] [Google Scholar]
- 55.Hunter J, Heetun M, Sinha S. Finding the perfect match: the living donor paired exchange system. Br J Hosp Med (Lond) 2014;75:202–206. doi: 10.12968/hmed.2014.75.4.202. [DOI] [PubMed] [Google Scholar]
- 56.Krishnan M. Preoperative care of patients with kidney disease. Am Fam Physician. 2002;66:1379, 1471–1476. [PubMed] [Google Scholar]
- 57.New JP, Aung T, Baker PG, Yongsheng G, Pylypczuk R, Houghton J, Rudenski A, New RP, Hegarty J, Gibson JM, O’Donoghue DJ, Buchan IE. The high prevalence of unrecognized anaemia in patients with diabetes and chronic kidney disease: a population-based study. Diabet Med. 2008;25:564–569. doi: 10.1111/j.1464-5491.2008.02424.x. [DOI] [PubMed] [Google Scholar]
- 58.Candrilli SD, Davis KL, Kan HJ, Lucero MA, Rousculp MD. Prevalence and the associated burden of illness of symptoms of diabetic peripheral neuropathy and diabetic retinopathy. J Diabetes Complications. 2007;21:306–314. doi: 10.1016/j.jdiacomp.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 59.Gregg EW, Sorlie P, Paulose-Ram R, Gu Q, Eberhardt MS, Wolz M, Burt V, Curtin L, Engelgau M, Geiss L 1999-2000 national health and nutrition examination survey. Prevalence of lower-extremity disease in the US adult population > 40 years of age with and without diabetes: 1999-2000 national health and nutrition examination survey. Diabetes Care. 2004:1591–1597. doi: 10.2337/diacare.27.7.1591. [DOI] [PubMed] [Google Scholar]
- 60.Shaw JE, Zimmet PZ. The epidemiology of diabetic neuropathy. Diabetes Review. 1999:245–253. [Google Scholar]
- 61.Feldman EL. Oxidative stress and diabetic neuropathy: a new understanding of an old problem. J Clin Invest. 2003;111:431–433. doi: 10.1172/JCI17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shaw JE, Zimmet PZ, Gries FA, Ziegler D. Textbook of Diabetic Neuropathy. New York: Stuttgart/New York: Thieme; 2003. [Google Scholar]
- 63.Apelqvist J, Larsson J, Agardh CD. Long-term prognosis for diabetic patients with foot ulcers. J Intern Med. 1993;233:485–491. doi: 10.1111/j.1365-2796.1993.tb01003.x. [DOI] [PubMed] [Google Scholar]
- 64.Pecoraro RE, Reiber GE, Burgess EM. Pathways to diabetic limb amputation. Basis for prevention. Diabetes Care. 1990;13:513–521. doi: 10.2337/diacare.13.5.513. [DOI] [PubMed] [Google Scholar]
- 65.Vinik AI, Park TS, Stansberry KB, Pittenger GL. Diabetic neuropathies. Diabetologia. 2000;43:957–973. doi: 10.1007/s001250051477. [DOI] [PubMed] [Google Scholar]
- 66.Vinik A, Mitchell B. Clinical manifestations and measurement of somatic neuropathy. Diabetes Review. 1997;7:312–325. [Google Scholar]
- 67.Goutos I. Neuropathic mechanisms in the pathophysiology of burns pruritus: redefining directions for therapy and research. J Burn Care Res. 2013;34:82–93. doi: 10.1097/BCR.0b013e3182644c44. [DOI] [PubMed] [Google Scholar]
- 68.Ahuja RB, Gupta GK. A four arm, double blind, randomized and placebo controlled study of pregabalin in the management of post-burn pruritus. Burns. 2013;39:24–29. doi: 10.1016/j.burns.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 69.Yoshida MM, Schuffler MD, Sumi SM. There are no morphologic abnormalities of the gastric wall or abdominal vagus in patients with diabetic gastroparesis. Gastroenterology. 1988;94:907–914. doi: 10.1016/0016-5085(88)90546-x. [DOI] [PubMed] [Google Scholar]
- 70.Wolosin JD, Edelman SV. Diabetes and the Gastrointestinal Tract. Clincial Diabetes. 2000;18:4. [Google Scholar]
- 71.Anand G, Katz PO. Gastroesophageal reflux disease and obesity. Rev Gastroenterol Disord. 2008;8:233–239. [PubMed] [Google Scholar]
- 72.Vaughan RW, Bauer S, Wise L. Volume and pH of gastric juice in obese patients. Anesthesiology. 1975;43:686–689. doi: 10.1097/00000542-197512000-00021. [DOI] [PubMed] [Google Scholar]
- 73.Vaughan RW, Engelhardt RC, Wise L. Postoperative hypoxemia in obese patients. Ann Surg. 1974;180:877–882. doi: 10.1097/00000658-197412000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Negishi K, Gupta S, Chandy KG, Waldeck N, Kershnar A, Buckingham B, Charles MA. Interferon responsiveness of natural killer cells in type I human diabetes. Diabetes Res. 1988;7:49–52. [PubMed] [Google Scholar]
- 75.Vergani D, Johnston C, B-Abdullah N, Barnett AH. Low serum C4 concentrations: an inherited predisposition to insulin dependent diabetes? Br Med J (Clin Res Ed) 1983;286:926–928. doi: 10.1136/bmj.286.6369.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Delamaire M, Maugendre D, Moreno M, Le Goff MC, Allannic H, Genetet B. Impaired leucocyte functions in diabetic patients. Diabet Med. 1997;14:29–34. doi: 10.1002/(SICI)1096-9136(199701)14:1<29::AID-DIA300>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 77.Llorente L, De La Fuente H, Richaud-Patin Y, Alvarado-De La Barrera C, Diaz-Borjón A, López-Ponce A, Lerman-Garber I, Jakez-Ocampo J. Innate immune response mechanisms in non-insulin dependent diabetes mellitus patients assessed by flow cytoenzymology. Immunol Lett. 2000;74:239–244. doi: 10.1016/s0165-2478(00)00255-8. [DOI] [PubMed] [Google Scholar]
- 78.Hostetter MK. Handicaps to host defense. Effects of hyperglycemia on C3 and Candida albicans. Diabetes. 1990;39:271–275. doi: 10.2337/diab.39.3.271. [DOI] [PubMed] [Google Scholar]
- 79.Katz S, Klein B, Elian I, Fishman P, Djaldetti M. Phagocytotic activity of monocytes from diabetic patients. Diabetes Care. 1983;6:479–482. doi: 10.2337/diacare.6.5.479. [DOI] [PubMed] [Google Scholar]
- 80.Park S, Rich J, Hanses F, Lee JC. Defects in innate immunity predispose C57BL/6J-Leprdb/Leprdb mice to infection by Staphylococcus aureus. Infect Immun. 2009;77:1008–1014. doi: 10.1128/IAI.00976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wysocki J, Wierusz-Wysocka B, Wykretowicz A, Wysocki H. The influence of thymus extracts on the chemotaxis of polymorphonuclear neutrophils (PMN) from patients with insulin-dependent diabetes mellitus (IDD) Thymus. 1992;20:63–67. [PubMed] [Google Scholar]
- 82.Speert DP, Silva J Jr. Abnormalities of in vitro lymphocyte response to mitogens in diabetic children during acute ketoacidosis. Am J Dis Child. 1978;132:1014–1017. doi: 10.1001/archpedi.1978.02120350078016. [DOI] [PubMed] [Google Scholar]
- 83.Kaye WA, Adri MN, Soeldner JS, Rabinowe SL, Kaldany A, Kahn CR, Bistrian B, Srikanta S, Ganda OP, Eisenbarth GS. Acquired defect in interleukin-2 production in patients with type I diabetes mellitus. N Engl J Med. 1986;315:920–924. doi: 10.1056/NEJM198610093151502. [DOI] [PubMed] [Google Scholar]
- 84.Mowat A, Baum J. Chemotaxis of polymorphonuclear leukocytes from patients with diabetes mellitus. N Engl J Med. 1971;284:621–627. doi: 10.1056/NEJM197103252841201. [DOI] [PubMed] [Google Scholar]
- 85.Jeffcoate WJ, Price P, Harding KG International Working Group on Wound Healing and Treatments for People with Diabetic Foot Ulcers. Wound healing and treatments for people with diabetic foot ulcers. Diabetes Metab Res Rev. 2004;20(Suppl 1):S78–89. doi: 10.1002/dmrr.476. [DOI] [PubMed] [Google Scholar]
- 86.Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–1743. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- 87.Edmonds ME. The diabetic foot, 2003. Diabetes Metab Res Rev. 2004;20(Suppl 1):S9–S12. doi: 10.1002/dmrr.458. [DOI] [PubMed] [Google Scholar]
- 88.Pecoraro RE, Ahroni JH, Boyko EJ, Stensel VL. Chronology and determinants of tissue repair in diabetic lower-extremity ulcers. Diabetes. 1991;40:1305–1313. doi: 10.2337/diab.40.10.1305. [DOI] [PubMed] [Google Scholar]
- 89.Reiber GE, Pecoraro RE, Koepsell TD. Risk factors for amputation in patients with diabetes mellitus. A case-control study. Ann Intern Med. 1992;117:97–105. doi: 10.7326/0003-4819-117-2-97. [DOI] [PubMed] [Google Scholar]
- 90.Singer AJ, Taira BR, McClain SA, Rooney J, Steinhauff N, Zimmerman T, Clark RA. Healing of mid-dermal burns in a diabetic porcine model. J Burn Care Res. 2009;30:880–886. doi: 10.1097/BCR.0b013e3181b48a6b. [DOI] [PubMed] [Google Scholar]
- 91.Broughton G 2nd, Janis JE, Attinger CE. The basic science of wound healing. Plast Reconstr Surg. 2006;117:12S–34S. doi: 10.1097/01.prs.0000225430.42531.c2. [DOI] [PubMed] [Google Scholar]
- 92.Park JE, Barbul A. Understanding the role of immune regulation in wound healing. Am J Surg. 2004;187:11S–16S. doi: 10.1016/S0002-9610(03)00296-4. [DOI] [PubMed] [Google Scholar]
- 93.Miao M, Niu Y, Xie T, Yuan B, Qing C, Lu S. Diabetes-impaired wound healing and altered macrophage activation: a possible pathophysiologic correlation. Wound Repair Regen. 2012;20:203–213. doi: 10.1111/j.1524-475X.2012.00772.x. [DOI] [PubMed] [Google Scholar]
- 94.Al-Watban FA, Andres BL. Polychromatic LED therapy in burn healing of non-diabetic and diabetic rats. J Clin Laser Med Surg. 2003;21:249–258. doi: 10.1089/104454703322564451. [DOI] [PubMed] [Google Scholar]
- 95.Ferguson MW, Herrick SE, Spencer MJ, Shaw JE, Boulton AJ, Sloan P. The histology of diabetic foot ulcers. Diabet Med. 1996;13(Suppl 1):S30–33. [PubMed] [Google Scholar]
- 96.Goodson WH 3rd, Hung TK. Studies of wound healing in experimental diabetes mellitus. J Surg Res. 1977;22:221–227. doi: 10.1016/0022-4804(77)90137-8. [DOI] [PubMed] [Google Scholar]
- 97.Black CT, Hennessey PJ, Ford EG, Andrassy RJ. Protein glycosylation and collagen metabolism in normal and diabetic rats. J Surg Res. 1989;47:200–202. doi: 10.1016/0022-4804(89)90107-8. [DOI] [PubMed] [Google Scholar]
- 98.Rowe DW, Starman BJ, Fujimoto WY, Williams RH. Abnormalities in proliferation and protein synthesis in skin fibroblast cultures from patients with diabetes mellitus. Diabetes. 1977;26:284–290. doi: 10.2337/diab.26.4.284. [DOI] [PubMed] [Google Scholar]
- 99.Archer FJ, Kaye R. Aging of diabetic and nondiabetic skin fibroblasts in vitro: life span and sequential growth curves. J Gerontol. 1989;44:M93–99. doi: 10.1093/geronj/44.3.m93. [DOI] [PubMed] [Google Scholar]
- 100.Magre J, Grigorescu F, Reynet C, Caron M, Capony JP, White MF, Picard J, Mirouze J, Capeau J. Tyrosine-kinase defect of the insulin receptor in cultured fibroblasts from patients with lipoatropic diabetes. J Clin Endocrinol Metab. 1989;69:142–150. doi: 10.1210/jcem-69-1-142. [DOI] [PubMed] [Google Scholar]
- 101.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 102.Fiedler U, Krissl T, Koidl S, Weiss C, Koblizek T, Deutsch U, Martiny-Baron G, Marme D, Augustin HG. Angiopoietin-1 and angiopoietin-2 share the same binding domains in the Tie-2 receptor involving the first Ig-like loop and the epidermal growth factor-like repeats. J Biol Chem. 2003;278:1721–1727. doi: 10.1074/jbc.M208550200. [DOI] [PubMed] [Google Scholar]
- 103.Asahara T, Chen D, Takahashi T, Fujikawa K, Kearney M, Magner M, Yancopoulos GD, Isner JM. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ Res. 1998;83:233–240. doi: 10.1161/01.res.83.3.233. [DOI] [PubMed] [Google Scholar]
- 104.Qiao L, Lu SL, Dong JY, Song F. Abnormal regulation of neo-vascularisation in deep partial thickness scalds in rats with diabetes mellitus. Burns. 2011;37:1015–1022. doi: 10.1016/j.burns.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 105.Liu Y, Petreaca M, Martins-Green M. Cell and molecular mechanisms of insulin-induced angiogenesis. J Cell Mol Med. 2009;13:4492–4504. doi: 10.1111/j.1582-4934.2008.00555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Velander P, Theopold C, Hirsch T, Bleiziffer O, Zuhaili B, Fossum M, Hoeller D, Gheerardyn R, Chen M, Visovatti S, Svensson H, Yao F, Eriksson E. Impaired wound healing in an acute diabetic pig model and the effects of local hyperglycemia. Wound Repair Regen. 2008;16:288–293. doi: 10.1111/j.1524-475X.2008.00367.x. [DOI] [PubMed] [Google Scholar]
- 107.Kratz G, Lake M, Ljungstrom K, Forsberg G, Haegerstrand A, Gidlund M. Effect of recombinant IGF binding protein-1 on primary cultures of human keratinocytes and fibroblasts: selective enhancement of IGF-1 but not IGF-2-induced cell proliferation. Exp Cell Res. 1992;202:381–385. doi: 10.1016/0014-4827(92)90089-q. [DOI] [PubMed] [Google Scholar]
- 108.Roberts AB. Transforming growth factor-beta: activity and efficacy in animal models of wound healing. Wound Repair Regen. 1995;3:408–418. doi: 10.1046/j.1524-475X.1995.30405.x. [DOI] [PubMed] [Google Scholar]
- 109.Bennett NT, Schultz GS. Growth factors and wound healing: Part II. Role in normal and chronic wound healing. Am J Surg. 1993;166:74–81. doi: 10.1016/s0002-9610(05)80589-6. [DOI] [PubMed] [Google Scholar]
- 110.Jude EB, Blakytny R, Bulmer J, Boulton AJ, Ferguson MW. Transforming growth factor-beta 1, 2, 3 and receptor type I and II in diabetic foot ulcers. Diabet Med. 2002;19:440–447. doi: 10.1046/j.1464-5491.2002.00692.x. [DOI] [PubMed] [Google Scholar]
- 111.Blakytny R, Jude E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabet Med. 2006;23:594–608. doi: 10.1111/j.1464-5491.2006.01773.x. [DOI] [PubMed] [Google Scholar]
- 112.Petrofsky JS, McLellan K, Bains GS, Prowse M, Ethiraju G, Lee S, Gunda S, Lohman E, Schwab E. Skin heat dissipation: the influence of diabetes, skin thickness, and subcutaneous fat thickness. Diabetes Technol Ther. 2008;10:487–493. doi: 10.1089/dia.2008.0009. [DOI] [PubMed] [Google Scholar]
- 113.Petrofsky J, Lee H, Trivedi M, Hudlikar AN, Yang CH, Goraksh N, Alshammari F, Mohanan M, Soni J, Agilan B, Pai N, Chindam T, Murugesan V, Yim JE, Katrak V. The influence of aging and diabetes on heat transfer characteristics of the skin to a rapidly applied heat source. Diabetes Technol Ther. 2010;12:1003–1010. doi: 10.1089/dia.2010.0152. [DOI] [PubMed] [Google Scholar]
- 114.McLellan K, Petrofsky JS, Bains G, Zimmerman G, Prowse M, Lee S. The effects of skin moisture and subcutaneous fat thickness on the ability of the skin to dissipate heat in young and old subjects, with and without diabetes, at three environmental room temperatures. Med Eng Phys. 2009;31:165–172. doi: 10.1016/j.medengphy.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 115.Petrofsky J, Bains G, Prowse M, Gunda S, Berk L, Raju C, Ethiraju G, Vanarasa D, Madani P. Does skin moisture influence the blood flow response to local heat? A re-evaluation of the Pennes model. J Med Eng Technol. 2009;33:532–537. doi: 10.1080/03091900902952683. [DOI] [PubMed] [Google Scholar]
- 116.Holowatz LA, Thompson-Torgerson C, Kenney WL. Aging and the control of human skin blood flow. Front Biosci (Landmark Ed) 2010;15:718–739. doi: 10.2741/3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hodges GJ, Sharp L, Clements RE, Goldspink DF, George KP, Cable NT. Influence of age, sex, and aerobic capacity on forearm and skin blood flow and vascular conductance. Eur J Appl Physiol. 2010;109:1009–1015. doi: 10.1007/s00421-010-1441-7. [DOI] [PubMed] [Google Scholar]
- 118.Tew GA, Klonizakis M, Saxton JM. Effects of ageing and fitness on skin-microvessel vasodilator function in humans. Eur J Appl Physiol. 2010;109:173–181. doi: 10.1007/s00421-009-1342-9. [DOI] [PubMed] [Google Scholar]
- 119.McLellan K, Petrofsky JS, Zimmerman G, Lohman E, Prowse M, Schwab E, Lee S. The influence of environmental temperature on the response of the skin to local pressure: the impact of aging and diabetes. Diabetes Technol Ther. 2009;11:791–798. doi: 10.1089/dia.2009.0097. [DOI] [PubMed] [Google Scholar]
- 120.Maloney-Hinds C, Petrofsky JS, Zimmerman G, Hessinger DA. The role of nitric oxide in skin blood flow increases due to vibration in healthy adults and adults with type 2 diabetes. Diabetes Technol Ther. 2009;11:39–43. doi: 10.1089/dia.2008.0011. [DOI] [PubMed] [Google Scholar]
- 121.Petrofsky JS, Bains GS, Prowse M, Mc Lellan K, Ethiraju G, Lee S, Gunda S, Lohman E, Schwab E. The influence of age and diabetes on the skin blood flow response to local pressure. Med Sci Monit. 2009;15:CR325–331. [PubMed] [Google Scholar]
- 122.Petrofsky JS, McLellan K. Galvanic skin resistance--a marker for endothelial damage in diabetes. Diabetes Technol Ther. 2009;11:461–467. doi: 10.1089/dia.2008.0096. [DOI] [PubMed] [Google Scholar]
- 123.Toda N, Imamura T, Okamura T. Alteration of nitric oxide-mediated blood flow regulation in diabetes mellitus. Pharmacol Ther. 2010;127:189–209. doi: 10.1016/j.pharmthera.2010.04.009. [DOI] [PubMed] [Google Scholar]