

Abstract
Tumors exploit immunoregulatory checkpoints to attenuate T cell responses as a means of circumventing immunologic rejection. By activating the inhibitory costimulatory pathway of Programmed Death 1 (PD1)/PDL1 which provides tumor cells an escape mechanism from immune surveillance, Programmed Death Ligand1 (PDL1)+ tumors hamper activated tumor-specific T cell functions and render them functionally exhausted. To overcome the inhibitory costimulatory effects of PDL1 on the adoptively transferred T cells, we sought to convert PD1 to a T cell costimulatory receptor by exchanging its transmembrane and cytoplasmic tail with CD28 and 4-1BB signaling domains (PD1-CD28-4-1BB, PD1-ACR), anticipating the genetically modified effector T lymphocytes expressing PD1-ACR would exhibit enhanced functional attributes. And the results showed that PD1-ACR expressed T cells retained the ability to bind PDL1, resulting in T cell activation as evidenced by the elevated activity of phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), the augmentation of cytokine secretion and the increased proliferative capacity. Moreover, when systemically administered in the mouse model of glioblastoma metastases, PD1-ACR T cells localized at the area of U87 invasive tumor, which results in suppressed tumor growth and enhanced survival of mice with established U87 glioblastoma. Together, these data demonstrated that PD1-ACR has a high potential to serve as a novel strategy to overcome PDL1 mediated immunosuppression of T cells for cancer therapy.
Keywords: Programmed Death Ligand 1, phosphoinositide 3-kinase, cytotoxicity, activating chimeric receptor
Introduction
By employing a variety of countermeasures including expressing inhibitory ligands for the receptors on T cells, tumor impairs the functional integrity of tumor specific T cells [1,2]. Indeed, one inhibitory receptor ligand, Programmed Death Ligand 1 (PDL1), has been identified as a poor prognostic indicator for several tumor types including myeloma, pancreatic, renal, ovarian and breast cancers [3,4]. The interaction between PDL1 and PD1 on the surface of activated T cells results in the activation of phosphatase SHP-1 and leads to the inhibition of TCR signaling through decreased CD3ξ and Zap-70 phosphorylation [5,6]. Within the tumor microenvironment, the PD1/PDL1 interaction induces T cell exhaustion, which manifests as the step-wise loss of cytolytic activity, proliferative capacity, cytokine secretion (INF-γ, IL-2, TNF-α) and finally T cell apoptosis [7,8]. While the PD1 or PDL1 blockade by monoclonal antibodies can restore the function of T cells and improve the anti-tumor efficacy of adoptively transferred T cells in vivo [9].
On the contrary to the expression of inhibitory ligands, T cell stimulatory molecule coordinated ligands are often absent from the tumor microenvironment, which leads to the inactivation of co-stimulatory signal (signal 2). Costimulatory signaling molecule CD28 not only enhances the proliferation and the differentiation of naive T cells, but the expression of downstream regulators that impact on T-cell proliferation, differentiation and effector functions [10,11]. These events are crucial for the effector T-cell function and the establishment of long-term memory. Unfortunately, CD28 ligands (CD80 and CD86) are often absent in the tumor microenvironment [12]. As a consequence, T cells become anergic or are eliminated by programmed cell death when they are exposed to antigens [13]. And the co-stimulatory molecules 4-1BB (CD137; TNFRS9), as an inducible costimulatory molecule on activated T cells that can be triggered by 4-1BBL expressed on activated APCs [12,13], enhances primary T-cell responses and the maintenance of memory T cells by providing “late-acting signals” and the survival of effector and memory T cells through upregulation of Bcl-XL [14,15]. The functions of CD28 and 4-1BB suggest some complementary effects between CD28 and 4-1BB-mediated costimulation.
To convert tumor PDL1 to a ligand that transmits a CD28 and 4-1BB costimulatory signal to the effector T cells, we generated a combinatorial antigen receptor, PD1-CD28-4-1BB activating chimeric receptor (PD1-ACR). Its effects on T-cell proliferation, cytokine secretion, T-cell survival and tumor elimination was investigated in tumor-bearing immunodeficient mice. As a result, PD1-ACR retains the ability to bind PDL1 resulting in the costimulation of primary T cells, as evidenced by the elevation of phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) activation, the augmentation of cytokine secretion, the increased proliferative capacity and the persistent activation of T cells. PD1-ACR T cells may cause on-target toxicity and tumor regressions in mice after therapy with gene engineered T cells.
Materials and methods
Cells and reagents
The U87, T98G and 293T cell lines were purchased from American Type Culture Collection (ATCC). Peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors and patients. Informed consent was obtained from the participants in accordance with the study protocols approved by the Institutional Review Board of Anhui University of Science & Technology. All cells were maintained in the RPMI1640 culture media (HyClone Laboratories, Logan, UT) supplemented with 10% (v/v) fetal bovine serum (HyClone Laboratories) and 2 mM l-glutamine (Invitrogen, Carlsbad, CA). Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. Phospho-Akt (Ser473) and Akt antibodies were purchased from Cellular Signaling (Beverly, MA), β-actin were purchased from Sigma (St. Louis, MO).
Construction of modified activating chimeric receptor
The PD1-activating chimeric receptor gene cassette which encodes a human PD1 extracellular segment including signal peptide (Uniprot: Q15116) denoted as PD1 in combination with the CD137 (4-1BB) intracellular costimulatory domain (Uniprot: Q07011) was fused in frame with CD28 transmembrane and intracellular domains (Uniprot: P10747). As the construct has the activation of T-cell function, hereafter was denoted as the PD1-CD28-4-1BB activating chimeric receptors (PD1-ACR). A (G4S) 3 peptide was used to link each functional fragment to a spacer domain derived from IgG4-Fc (Uniprot Database: P01861) comprising either ‘Hinge-CH2-CH3’, ‘Hinge-CH3’ or ‘Hinge-only’ sequence. Codon-optimized nucleotide sequences encoding each transgene were synthesized (SANGON Biological Engineering Technology Co., Ltd., Shanghai, China).
Vector construction and preparation of PD1-ACR encoding lentivirus
PD1-ACR was cloned into EcoR I and BamH I sites of lentiviral vector pCDH-EF1-T2A-puro (System Biosciences, CA). The new vector was named pCDH-PD1-ACR. Lentiviral PD1-ACR was generated by calcium phosphate co-transfection of HEK293T packaging cells with three plasmids, pVSV-G, pCMV-dR8.9 and pCDH-PD1-ACR, using the FugeneTM lipofection system (Roche) as described previously [15,16]. The supernatants of transfected cells containing lentiviral particles were filtered (with 0.22 μm filters) and concentrated by ultracentrifugation. The titers were determined using the QuickTiter™ Lentivirus Quantitation Kit (Cell BioLabs). Supernatants with a titer of >107 infectious units/mL were collected and stored at -80°C.
Modification and expansion of PD1-ACR specific T cells
PBMCs were isolated using Ficoll-Paque (GE Healthcare, Uppsala, Sweden) and cultured in RPMI-1640 supplemented with 10% FCS. The PBMCs were activated with 100 ng/ml Anti-CD3 (Nordic Biosite, Täby, Sweden) and 100 IU/ml IL-2 (Proleukin, Novartis, Basel, Switzerland) for 2 days to selectively stimulate T cells. Activated T cells were transduced with the corresponding lentiviral particles at a multiplicity of infection (MOI) of ~5 for 4 hours at 37°C in the presence of 10 μg/ml protamine sulphate as previously described [17]. Transduced T cells were expanded in RPMI 1640 supplemented with 10% FCS. PD1-ACR expression was confirmed by confocal laser scanning microscope (CLSM) and IHC. Cell surface structures were detected with scanning electron microscope (SEM).
ACR mediated T cells activation and evaluation of ACR expression
When the highly purified CD8+ T cells transduced with PD1-ACR activated, they could express several kinds cytokines. ELISA was performed to measure the secretion of IFN-γ, IL-2 and TNF-α with the corresponding antibody sets (Duoset; R&D, Abingdon, UK). To correct for the different transduction efficiencies (determined using flow cytometry), data were normalized as [mean cytokine level]/% transduced T-cells × 100%.
Western blot was conducted to verify the expression of PD1-ACR protein. T cells (2.0 × 106) were lysed in 150 μl lysis buffer and centrifuged. Cell lysates were then denatured under the reducing condition and electrophoresed using 12% acrylamide resolving gels. The expression of CD28 chain-containing fusion receptors was detected by goat anti-human CD28 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). In some experiments, the T cells were treated with the PI3 kinase inhibitor LY294002 and then detected with western blot describe in Ref 18.
Functional analysis of PD1-ACR T cells
Cytotoxicity assay was conducted using a method previously described in Ref 19. Briefly, the cytokine release assays were performed by coculturing T cells with target cells U87 (Human malignant glioma cell lines, 89.7% expression of PDL1 antigen) according to the effector target ratio 40:1, 20:1, 10:1 and 5:1 respectively. Per well in 96-well-plate in a final volume of 100 μl T-cell media for the indicated time at 37°C in 5 % CO2, non-transduced (NT) T cells were used as control of effector T cells, and FBS was utilized throughout the assays to exclude any contribution of complement-mediated cytotoxicity. After 24 hr, supernatants were assayed for the production of IL-2, IFN-γ and TNF-α using ELISA (R&D Systems, Minneapolis, MN). PD1-ACR-dependent killing rate was calculated by subtracting the percentage of the target cell killing obtained with U87 cells. The percentage of the control T cells killing target cells was the same as above cytotoxicity assay.
To analyze mRNA expression levels of the target molecules, the effector T cells were collected into Trizol (Invitrogen, Carlsbad, CA) and RNA was purified according to the manufacturers’ instructions. cDNA synthesis and RT-PCR was performed using a One-Step RT-PCR System with Platinum Taq (Invitrogen, Carlsbad, CA). The primers for β-actin, IL-2, IFN-γ, and TNF-α are as follows: β-actin (623bp)-For (CCTAAGGCCAACCGTGAAAAG) and Rev (TCTTCATGGTGCTA GGAGCCA); IL-2 (222bp)-For (GAATGGAATTAATAATTACAAGAATCC) and Rev (TGTTTCAGATCCCTTTAGTTCCAG); IFN-γ (180 bp)-for (CTGATTTCAACTTCTTTGGCT) and Rev (TATCCGCTACATCTGAA TGA); TNF-α (325 bp)-For (CAGAGGGAAAGATTCCCCAG) and Rev (CCTTGGTCTGGTAGGAGACG). All primer pairs were designed so that amplification occurs across an intron thereby allowing the distinction between the amplification from the genomic DNA and the cDNA.
In vivo antitumor assays
Xenograft tumor mouse model was established by abdominal subcutaneous injection of 5×104 U87 cells into 6-week-old female BALB/cA-nude mice (Chinese Academy of Science Anhui Experimental Animal Center) as described previously [20,21]. All animal work was conducted at the Anhui University of Science & Technology Animal Facility and Use Committee in accordance with the institutional guideline. About 12-15 days after inoculation of the U87 cells, the mice were assigned to different groups (15 in each group) and injected with a total of 1.0 × 107 different T cells per recipient (PD1-ACR T cells, control T cells, and control PBS) systemically to the tail vein. No preconditioning was administered, nor any cytokine or antibody to support the function of the infused T cells. When the tumor volume reached > 1500 mm3, these mice were killed before the animals exhibited clinical symptoms to avoid unnecessary pain and discomfort according to the ethical animal guidelines of Anhui University of Science & Technology.
Xenograft tumor mouse model was established by abdominal subcutaneous injection of 5 × 104 U87 cells into 6-week-old female BALB/cA-nude mice (Chinese Academy of Science Anhui Experimental Animal Center) as described previously [20,21]. All animal work was conducted at the Anhui University of Science & Technology Animal Facility and Use Committee in accordance with the institutional guideline. About 12-15 days after inoculation of the U87 cells, the mice were assigned to different groups (15 in each group) and injected with a total of 1.0 × 107 different T cells per recipient (PD1-ACR T cells, control T cells, and control PBS) systemically to the tail vein. No preconditioning was administered, nor any cytokine or antibody to support the function of the infused T cells. When the tumor volume reached > 1500 mm3, these mice were killed before the animals exhibited clinical symptoms to avoid unnecessary pain and discomfort according to the ethical animal guidelines of Anhui University of Science & Technology.
Statistical analyses
SPSS 17.0 was used to calculate the statistical significance. ANOVA was used to identify the possible difference among different treatment groups. Once the difference was confirmed, Student’s t test was applied to calculate the significance in the difference between two treatment groups (p values). All p values < 0.05 were considered statistically significant. Data are presented as means ± standard error of mean (SEM), unless otherwise noted.
Results
Design of a human PD1-ACR
The extracellular domain sequence of human PD1 (aa 1-167) including the signaling peptide was designed to be fused to the transmembrane and the intracellular sequences of human CD28 (aa 141-220) and 4-1BB (aa 214-255) costimulatory domain (Figure 1A). When this activating chimeric receptor expressed in effector T cells, it would provide a signal to activate T cells effectively via the costimulation of CD28 and 4-1BB upon engagement of PDL1+ tumor cells (Figure 1B). The PD1-ACR cDNA was ligated into pCDH-EF1-T2A-puro (System Biosciences, CA) under the human EF-1 promoter (Figure 1C and 1D) and a high titer of the third generation CDH-PD1-ACR lentivirus was prepared (Figure 1E).
Figure 1.
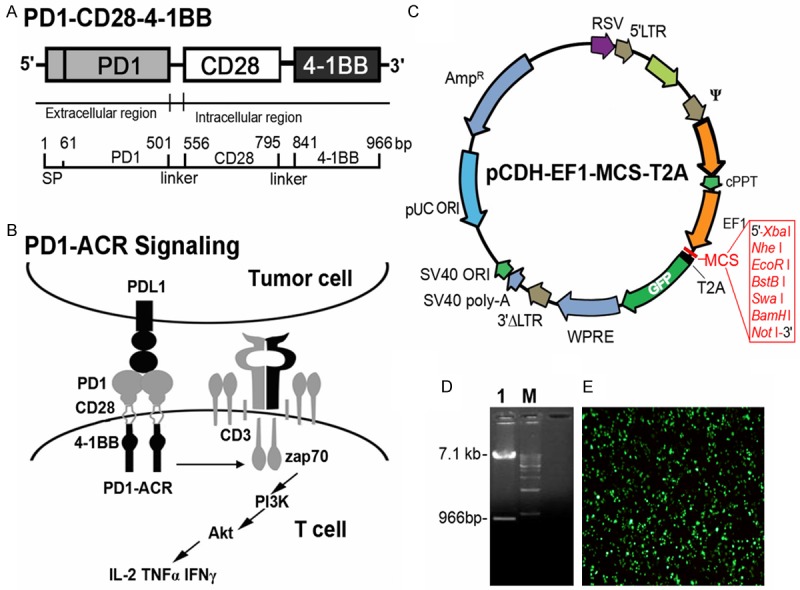
Schematic representation of the PD1-ACR. A. Schematic representation of the PD1-ACR molecule, which contains the truncated extracellular domain of PD1 and the transmembrane and the cytoplasmic signaling domains of CD28 and 4-1BB. B. Schematic representation of the interaction between the T-PDL1+ tumor cell and the PD1-ACR platform. C. The structure of pCDH-EF1-T2A-GFP lentiviral vector. D. pCDH-EF1-T2A-PD1-ACR-GFP digest with restriction enzyme. M: 1 kb DNA ladder; 1: pCDH-EF1-T2A-PD1-ACR-GFP digest with EcoR I and BamH I. E. The packaging and expression of pCDH-EF1-T2A-PD1-ACR-GFP lentivirus vector in 293T cells (× 100).
Expression of PD1-ACRs in human lymphoblastoid cells
To verify the functional activity of the truncated extracellular domain of PD1 as the binding moiety in the ACRs, the lentiviral CDH-PD1-ACR was transfected into anti-CD3 and IL-2 activated PBMCs under the optimized conditions. CD3+ T cells were sorted by flow cytometry and the expression of the PD1-ACR constructs was examined by RT-PCR using primers specific for the PD1-ACR. The primer pair was designed for the amplification to occur across PD1 and CD28, allowing the distinction between the endogenous PD1 and the PD1-ACR chimeric molecule. And only anti-CD3 and IL-2 activated T cells served as the experimental controls. As demonstrated in Figure 2A, a distinct band of approximately 570 bp was amplified from the lentiviral CDH-PD1-ACR T cells, corresponding to the specific transcription of PD1-ACR gene. Moreover, PD1-ACR expression was confirmed by western blot of the extracts from the transduced T-cell populations (Figure 2B). The molecular mass of PD1-ACR was consistent with the efficient protein “cleavage” mediated by the PD1-ACR sequence. And PD1-ACRs were also detected on the surface of the activated cells with increased efficiency 48 h after the transfection by flow cytometry using an CY5-labeled anti-human PD1 antibody (Figure 2C). Moreover, any discernible phenotypic differences between the control and the PD1-ACR T cells were determined by SEM, and the PD1-ACR T cells had no significant changes in cell morphology. However, the surface topography of the PD1-ACR T cells was smoother in comparison to that of the control T cells (Figure 2D, photographs taken at 8500 × magnification).
Figure 2.
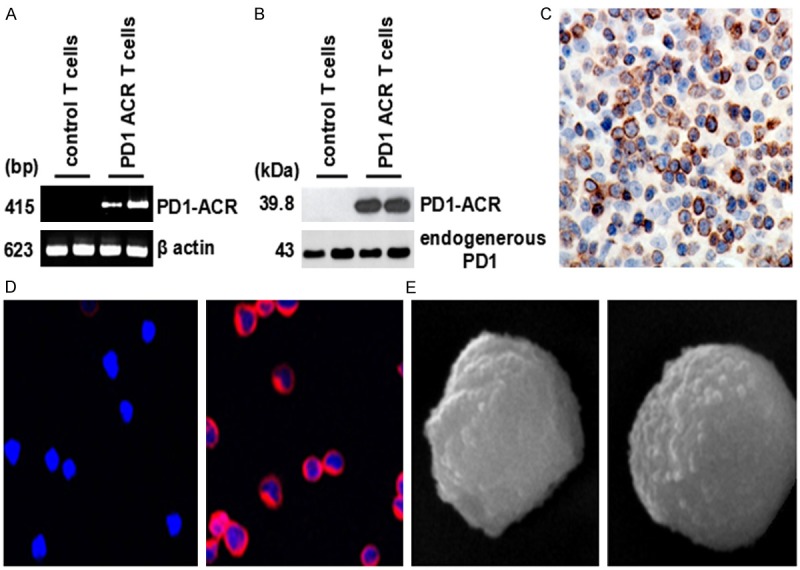
Transduction efficiency of PD1-ACRs and the phenotypic profile of T cells. A. RT-PCR analysis showed the transduction efficiency of the PD1-ACR lentiviral vectors (right) versus a negative control of untransduced T cells (left). B. The expression of ACRs in activated primary T cells transduced with PD1-ACR vectors was assessed by Western blot. C. Confocal laser scanning microscope (CLSM) indicated the percentage of cells positive for PD1-ACR (1000 × magnification) was up to 99.8%. D. The PD1-ACR T cells have no significant change in cell morphology. E. Smoother topography was observed in comparison to the control T cells by SEM (8500 × magnification).
PD1-ACR T cells exhibit enhanced proliferative activity upon PDL1 ligation
Given that the costimulation by CD28 and 4-1BB can effectively promote the proliferation of antigen stimulated T cells, we sought to determine the relative proliferative activity of T cells and their PD1-ACR transduction counterparts. Cell enumeration assays revealed that greater numbers of viable PD1-ACR T cells were presented in cultures than the untransduced T cells post the stimulation by the PDL1+ malignant glioma U87 cell line (Figure 3A, 3B). This difference was statistically significant (p = 0.027), whereas PD1-ACR T cells exhibited cellular attrition over time after the stimulation by the PDL1- T98G tumor cells. To investigate the mechanism underlying the increase in T cell numbers, tritiated thymidine (3H-TdR) incorporation and caspase activation assays were performed on tumor stimulated T cells. And the results showed that, PD1-ACR T cells exhibit about 35%, 123% and 435% increase (p = 0.0411, p < 0.001 and p < 0.0001, respectively) when stimulated by PDL1+ U87 cell lines as compared to PDL1- T98G tumor cell lines at day 3, day 6 and day 12 (Figure 3C). The caspase activation was not statistically different when cells were co-stimulated through the PD1-ACR (Figure 3D).
Figure 3.
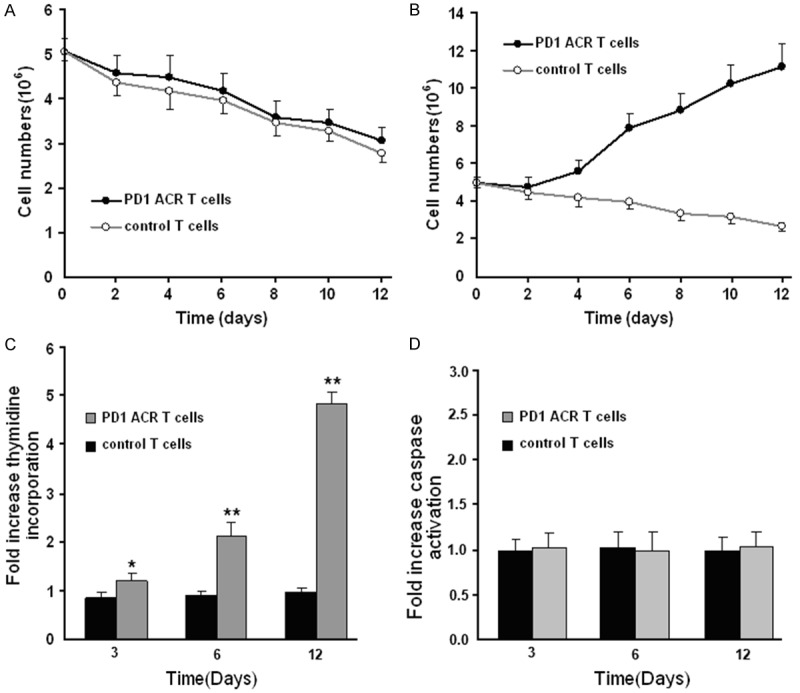
PD1-ACR T cells exhibited augmented proliferation upon engagement of PDL1+ tumor cells. A. CTL responders cocultured with T98G stimulators. B. CTL responders cocultured with U87 stimulators were counted. C. Proliferation at day 3, day 6 and day 12 by thymidine incorporation when cocultured with PDL1+ U87 cells as compared to PDL1- T98G cells. D. Apoptosis was assessed at day 3, day 6 and day 12 by CD3 gating and the CaspSCREEN flow cytometric apoptosis detection kit. All data are the ratio CTL responders cocultured with U87/T98G stimulators. Replicate experiments were represented as a mean (± SEM) fold increase (n = 3) in the caspase activation when cocultured with PDL1+ U87 cells as compared to PDL1- T98G cells. *, p = 0.0212 and **, p < 0.001.
Cytokine release stimulated by tumor cells with various levels of PDL expression
The PD1-CAR T cells with different levels of PDL expression were cocultured with various tumor cell lines at a ratio of 1:1 for 24 h to further explore their effector functions. The supernatants were analyzed for the secretion levels of IL-2, interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α). PD1-ACR T-cells showed an increase in the secretion levels of IL-2, IFN-γ and TNF-α when cocultured with PDL+ MB231 and U87 cells. These increases were statistically significant (2.4-fold increase for IL-2 (p = 0.0442, n = 3), 2.7-fold increase for IFN-γ (p = 0.0290, n = 3) and TNF-α (p < 0.0001, n = 3)) in comparison to the control T cells in multiple experiments (Figure 4A). Meanwhile, the secretion of these cytokines from the PD1-ACR T-cells were at much lower levels when cocultured with PDL- MCF-7 and T98G cell lines, which was not statistically different from the control T cells (Figure 4A). Cytokine mRNA in control T-cells cocultured with various tumor cell lines for 24 h kept wery low level, but those in PD1-ACR T cells cocultured with various tumor cell lines were higher, especially much higher when cocultured with PDL1+ tumor cells (Figure 4B and 4C). All these data demonstrated that the costimulation of T cells through the PD1-ACR binding PDL1 expressing on target cells can induce T cell activation, synthesis and secretion of cytokines like IFN-γ, IL-2 and TNF-α.
Figure 4.
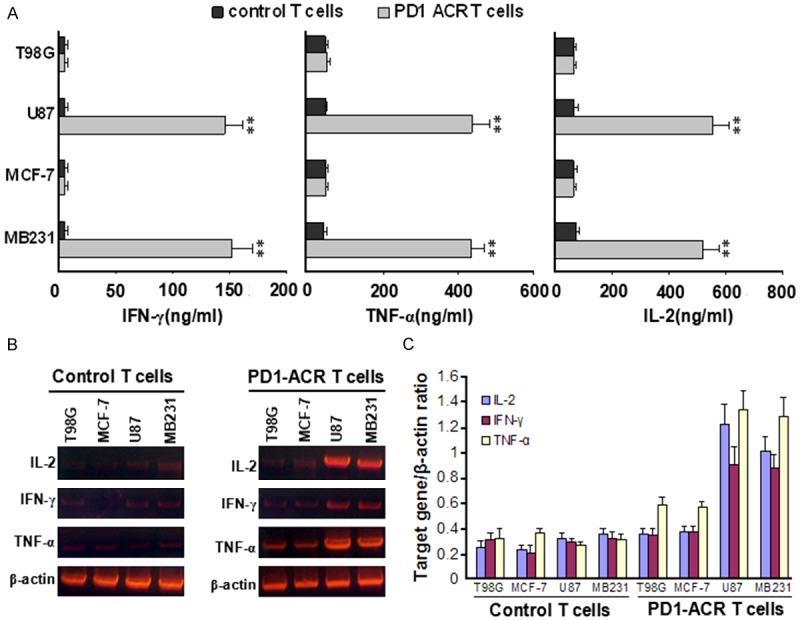
Cytokine secretions and mRNA level of T-cells cocultured with various tumor cell lines for 24 h. A. Supernatants were collected after 24 h of coculture and the secretion of cytokines were assessed by ELISA. Data showed representative results from at least 3 independent experiments. Unpaired, two-tailed Student’s t-test was used to calculate the p values. **, p < 0.001 using the unpaired Student’s t-test comparing the PD1-ACR T cells to the control T cells. B. Cytokine mRNA level in T-cells cocultured with various tumor cell lines for 24 h. C. Cytokine mRNA level/β-actin mRNA level in T-cells cocultured with various tumor cell lines for 24 h.
Graded levels of PI3K/Akt activation by PD1-ACRs
The PD1-ACR T cell was selectively bound to the PDL1+ target cells, indicating that CD28-CD137 (4-1BB) costimulatory domain of PD1-ACR does not sterically interfere with PD1 ligand binding. Given that the PD1-ACR and PDL1 ligation would result in the activation of canonical CD28 and 4-1BB signaling which mediates the PI3K/Akt pathway. Using mouse anti-human CD3ζ (anti-human CD3 monoclonal antibody, eBioscience) and recombinant human PDL1-Fc Chimera (SinoBiological, Beijing, China) for 24 h respectively, cell lysates were analyzed at multiple time points by Western blot. Akt phosphorylation was slightly increased by CD3 stimulation, while further increased by either the CD3/PDL1-Fc Chimera costimulation or the PDL1-Fc Chimera stimulation (four fold higher than the control T cells by the stimulation of OKT3 alone). Akt phosphorylation was also greater in PDL+ U87 tumor cells (Figure 5). To further confirm the importance of the PI3K/Akt activation for PD1-ACR, we examined whether the proliferative activity and cytokines expression of T cells were inhibited by LY294002 in PD1-ACR T cells with Western blot (Figure 5A). As LY29400 nearly abolishes Akt phosphorylation under all of these conditions. These data demonstrated that the PD1-ACR was capable of binding PDL1 and transmitting a CD28 and 4-1BB costimulatory signal. And the ability of the PD1-ACR to induce Akt phosphorylation was independent on the TCR-CD3 signaling. Western blot revealed that some PARP cleavage could be detected in PD1-ACR T cells treated PDL1+ U87 cells, but not in PD1-ACR T cells treated PDL1- T98G cells (Figure 5B). Cleavage of the principal executioner caspase, caspase-3 was not detected (Figure 5B). Moreover, PD1-ACR T cells could effectively induced apoptosis and necrosis of PDL1+ cells using flow cytometry (Figure 5C), the apoptosis and necrosis of PDL1+ cells were raised after 48h treatment with PD1-ACR T cells at the ratio 1:10 (PDL1+ U87 cells: PD1-ACR T cells).
Figure 5.
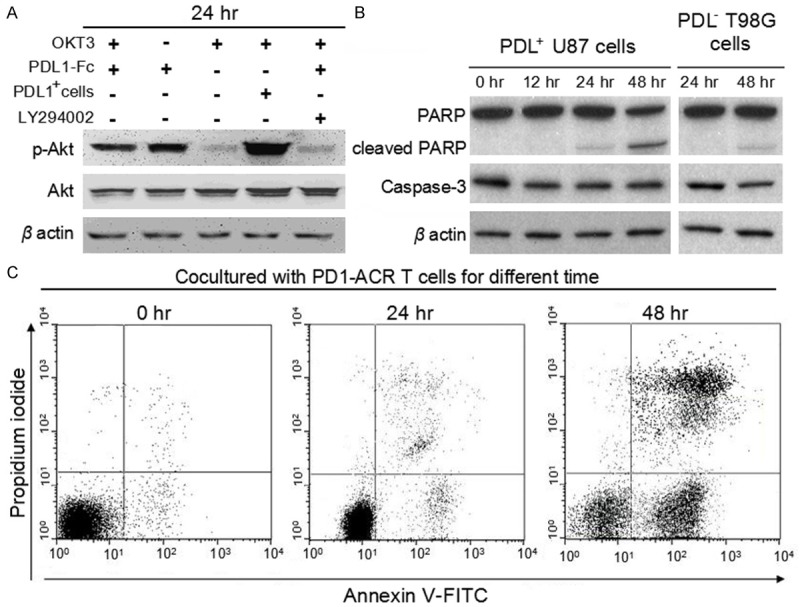
The binding of PDL1 to PD1 upregulated the PI3K/Akt-dependent signaling in PD1-ACR T cells and effectively induced apoptosis and necrosis of PDL1+ U87 cells. A. PD1-ACR T cells sustained higher Akt phosphorylation (ser437) following the stimulation by the PDL1-Fc Chimera or the PDL+ tumor cells. When PD1-ACR T cells were cocultured with OKT3 and PDL1-Fc Chimera in the presence of 10 μmol/L Ly29400 for 2 h, the ratio of phospho-Akt to the total protein level returned to that before stimulation. B. PDL1+ U87 cells and PDL- T98G cells were treated with PD1-ACR T cells with the ratio 1:10 for the specified time intervals and cell lysates were prepared. Western blots for caspase 3 and PARP were shown with β-actin as a loading control. C. The apoptosis and necrosis of PDL1+ U87 cells were raised after 48 h treatment with PD1-ACR T cells at the ratio 1:10 (PDL1+ U87 cells: PD1-ACR T cells ) using annexin V-FITC and PI double-staining.
Activity of the PD1-ACR T cells specifically targeting and killing PDL1+ tumor cells
To confirm the specificity of our PD1-ACR T cells, we examined the cytotoxic activity of PD1-ACR+ T lymphocytes and PD1-ACR- T cells (the control T cells) to different target cell lines with different endogenously expressing PDL1 levels including H1299 (93% PDL1+), HCC38 (43% PDL1+), HCC1143 (5% PDL1+), AU565 (PDL1-) and MCF-7 (PDL1-). After 24 hrs, 51Cr release assays showed that the anti-tumor activity was proportional to the levels of PDL1 antigen expression. As anticipated, the PD1-ACR T cells had little activity against the PDL1− tumor cell lines (AU565/MCF-7) (Figure 6A). The control T cells have very poor ability to kill all kinds of tumor cells (Figure 6B). Above results revealed that the PD1-ACR transduced primary human T cells could express PD1-ACR, which was capable to redirect PDL1 signaling functionally to CD28 and 4-1BB costimulation. And then, PD1-ACR T cells effectively eliminated PDL1+ target cell lines (H1299/MDA-MB231/U87), while no effect on the PDL1− cell lines (MCF-7/AU565/T98G).
Figure 6.
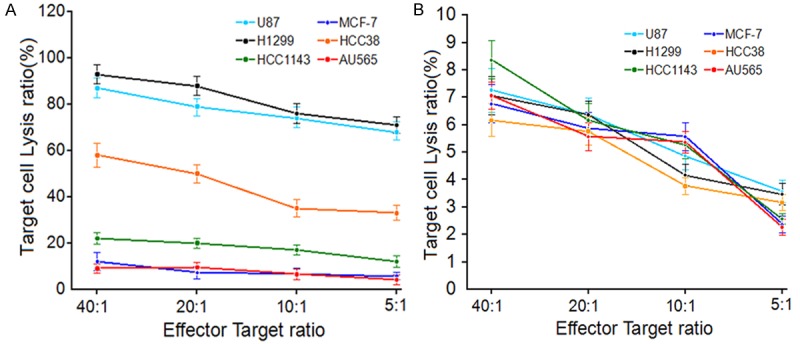
PD1-ACR T cells lyse PDL1+ tumor cells. A. Specif 23ic cytotoxicity of PD1-ACR T cells against different tumor cell lines with different endogenous PDL1 expressing level. B. Cytotoxicity of the control T cells against different tumor cell lines with different level of PDL1 antigen expression. Data represented mean values of triplicate wells ± SEM.
PD1-ACR T cells inhibited the growth of PDL1 expressing tumor cells in vivo
In order to further investigate the local antitumor effects mediated by the PD1-ACR T cells, we developed a xenograft model by inoculating the PDL1+ U87 cell line in the flank of the BALB/cA-nude mice (Chinese Academy of Science Anhui Experimental Animal Center). The tumor-loaded mice received the injection of the PD1-ACR+ T cells and the control T cells (2.5 × 106 per dose) on days 8, 10, 12 and 14 for a total of 4 injections. U87 cell tumor sizes in mice with PD1-ACR+ T cells treatment started to shrink three weeks after adoptive cell transfer. In contrast, the control T cells did not affect the U87 tumor growth. Statistical analysis of the tumor growth curves revealed significant differences between the PD1-ACR+ group and the control group (p < 0.05, n = 3, Figure 7A). Progressive tumor growth was also observed in the control group, while the PDL1+ U87 tumors regressed in the mice treated with PD1-ACR T cells. As shown in Figure 7B, the mice treated with PD1-ACR T cells exhibited the greatest survival rate at the end of the study (75%), compared with the mice treated with control T cell (20%) and PBS (0%). These results indicated the anti-PDL1+ tumor therapeutic potential of PD1-ACR T cells in vivo.
Figure 7.
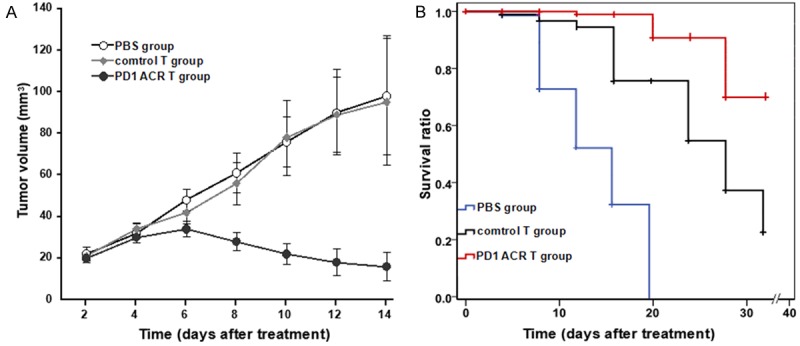
In vivo anti-tumor activity of the PD1-ACR+ T cells. PDL1+ U87 cells and PDL1- T98G cells were used for the xenograft mouse model. PDL1 bearing BALB/cA-nude mice received different treatments: group 1, PD1-ACR+ T cells; group 2, control T cells; group 3, PBS. Data represented mean values of the tumor volumes with n = 15 for each group (mm3 ± SEM). A. Tumor size was measured weekly. B. Kaplan-Meier survival analysis of PDL1+ U87 cell xenograft models. control T cell and PBS treatment were ineffective in this model (p = 0.23), whereas PD1-ACR+ T cell treatment improved survival compared to control T cell and PBS treatment (p = 0.012, p < 0.001 respectively).
In vivo localization of T cells to the tumor
Sequential tumor specimens from mice were stained for HE to confirm that PDL1 was expressed by the primary glioma cells and the PD1-ACR T cells in the glioma tumor tissue (Figure 8A and 8B). Next, we analyzed PDL1 expression in paraffin-embedded tumor tissue sections. The IHC staining showed moderate PDL1 expression, which was consistent with those observed in the human tumor (Figure 8C). The in vivo targeting of PD1-ACR+ T cells upon PDL+ cells in the tumor-loaded mice was evaluated by the overall localization of PD1 by immunohistochemistry and morphology. Immunohistochemical results showed PD1 expression in U87 tumor tissue (Figure 8D), which suggested that the PD1-ACR T cells had the ability to specifically target PDLl+ tumor tissue in vivo. Together, these results indicated that PD1-ACR+ T cells exhibited robust antitumor effects by specifically killing target cells.
Figure 8.
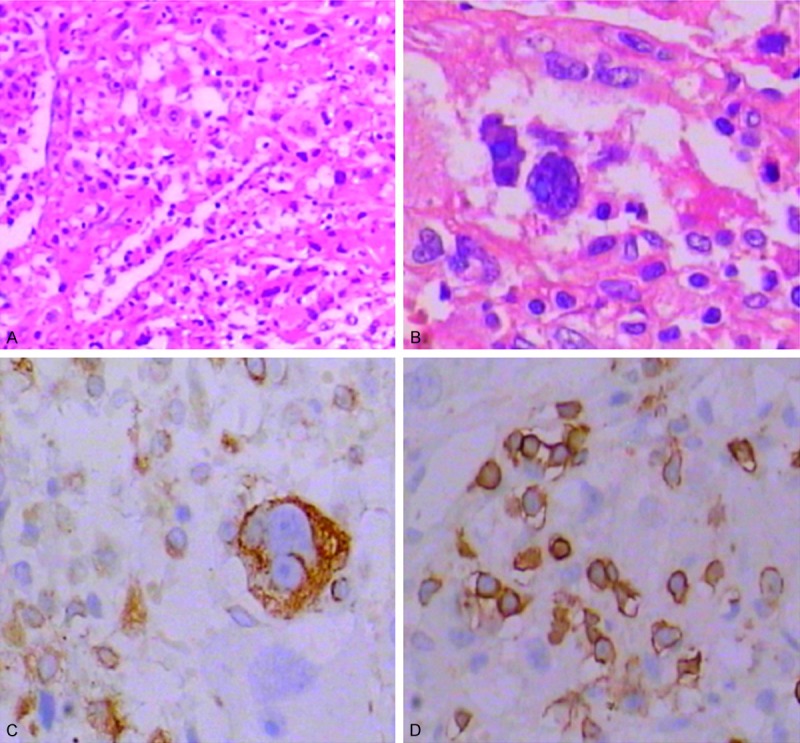
Sequential tumor specimens from mice stained for HE and IHC. A. HE staining of the U87 tumor tissue (× 100). B. HE staining of the U87 tumor tissue (× 400). C. Moderate staining of the PDL1 in DCIS (× 400). D. Strong PD1-Fc staining of the T cells in the U87 metastases tissue (× 400).
Discussion
Constitutive endogenous PD1 upregulated expression has been observed by infiltrating T lymphocytes in solid tumors [22,23], while tumors frequently co-opt immunoregulatory signaling pathways to evade immune recognition and elimination. PDL1/2-PD1 interactions contribute to effector T cell exhaustion and clonal deletion as a mechanism of maintaining tolerance in normal immune physiology [3-7]. Tumor microenvironment PDL1/2 expression, therefore, is a significant obstacle against achieving therapeutic responses for adoptive immunotherapy with either tumor specific cytotoxic T cells (CTL), tumor infiltrating lymphocytes (TIL) or chimeric antigen receptor (CAR) redirected T cells [24,25]. Additionally, the absent or aberrant expression of MHC molecules by malignant cells possibly precludes the recognition by autologous MHC-restricted T cells [1,2,24,25]. Our work focused on the development of an engineering strategy to equip T cells with the ability to circumvent PDL1-PD1 signaling by converting PDL1 to a ligand for T cell costimulation. And the PD1-CD28-4-1BB (PD1-ACR) is shown to convert PDL1 mediated inhibition to costimulation of T cells.
Naïve and memory T cells downregulate endogenous CD28 expression and differentiate to effector T cells as a consequence of antigen activation [12,13]. However, a chimeric antigen receptors (CAR) containing combined 4-1BB and CD28 signaling domains exhibited augmented proliferative responses, cytokine release, antigen-elicited Granzyme B, expression of pro-survival proteins and cytolytic activity as compared to more moderate responses using chimeric antigen receptors expressing only 4-1BB or CD28 signaling domains [13,14]. Similarly, we demonstrated the functionality of PD1-ACR T cells by analyzing the upregulation of PI3K/pAkt pathway activity, production of inflammatory cytokines (IL-2, IFN-γ and TNF-α) and proliferation in response to PDL+ cell lines. PD1-ACR T cells proliferated in response to PDL+ target cells. And this proliferation was likely due to the PD1-ACR T cells going through multiple rounds of the rapid expansion protocol prior to being used in the proliferation assays. Our PD1-ACR T cells exhibited a PDL1 dependent reconstitution of CD28 and 4-1BB signaling, resulting in increased IFN-γ, TNF-α, IL-2, proliferation and cytolytic activity. CD28 and 4-1BB costimulation occurs preferentially when T cells encounter professional antigen presenting cells that express B7-1 and -2, while PD1-ACR shifts the negative costimulation event to the positive costimulation and augments of CTL costimulation. Based on the restricted expression of both tumor antigen and PDL1, we therefore anticipate that the effect of PD1-ACR signaling will be more profound when effector T cells are present in the tumor microenvironment.
In vitro cytolytic activity was enhanced through PD1-ACR costimulation, while addressing the effect of the PD1-ACR T cells on the anti-tumor activity is an important extension of this work that needs to be examined. We hypothesize that PD1-ACR can serve to promote the anti-PDL+ tumor activity of adoptively transferred T cells expressing either endogenous TCR specific for tumor antigens or the co-expression of this PD1-ACR, with the expectation that the costimulatory impact of the PD1-ACR will be further enhanced. Our findings indicated that PD1-ACR T cells exhibited a significantly higher cytotoxicity than CTLs. Although these results suggests that ACR-based immunotherapies may be intrinsically superior to CTL approaches, TCR is dependent on endogenous proteins for the transduction of an activatory signal, unlike ACR molecules, which contain CD28 and 4-1BB signaling domains and the CD3ζ chain of the T-cell receptor (TCR) signaling complex [26,27]. It should also be noted that TCR preferentially signal through ZAP70 tyrosine kinases, which may affect the strength of the transduced signal [25-27].
Next, we assessed the antineoplastic potential of PD1-ACR T cells in the xenograft models of human PDL1+ U87 and PDL1- T98G human malignant glioma cell lines in NSG mice respectively. We showed that the PD1-ACR T cells can effectively control the growth of aggressive U87 cells, but not that of the slow-growing T98G cells. The clearance of U87 tumor cells suggests that U87 cells are sensitive to the cytotoxic potential of PD1-ACR T cells in vivo, which are capable of eliminating a large number of targets upon engagement. It also implies that the lack of response to TCR cells is caused by factors that are not directly related to their cytolytic functions. For example, the contact between CTL cells and PDL1+ U87 target cells could alter signaling pathways in CTL cells that are critical for their sustained activation through regulating PI3K/Akt signaling pathway negatively and inducing immune cell tolerance.
We also sought to determine whether PD1-ACR T cells exhibit preferential anticancer cytotoxicity. To this aim, we injected PD1-ACR T cells into metastases of malignant glial cells U87 which express PDL1. In this system, only T cells bearing the PD1-targeting ACR were effective, while it is unclear to what extent the relative density of PDL1 molecules on U87 cells could have biased these results.
The superiority of PD1-ACR T cells for antitumor potential over CTLs was demonstrated when the target was PDL1+ tumor. More importantly, we found that PD1-ACR T cells also controlled the growth of some PDL1- T98G malignant glial metastases xenografts to a certain extent in immunocompromised mice, when given systemically or locally. But CTL cells could not effectively control the metastases of malignant glial U87 and T89G cell growth. Metastasis tumor grows in mice continuously and the mice died eventually.
Conclusion
In summary, our findings support the utility of the PD1-ACR as a genetic engineering platform to address tumor mediated immune suppression through PDL1 in tumor microenvironment. The data demonstrated that CD28 and 4-1BB signals are functionally additive when combined within a single ACR. The improved PD1-ACR T-cell activation is at least in part dependent on the activation of the Akt pathway. Future experiments will address the immunobiology of this PD1-ACR in CD4+ and CD8+ T cells in vitro respectively, as well as in CD4+ and CD8+ T cells in the context of in vivo tumor adoptive therapy with applicability to ACR redirected T cells. Meanwhile, we should observe the efficacy of PD1-ACR T cells in different PDL1+ tumor tissues. Furthermore, incorporation of more potent costimulatory domains into the ACR-molecule, better gene transfer methods and provision of T-cell-specific cytokines are expected to improve the activation of the engineered T cells and prolong their persistence in vivo. As we learn more about the mechanisms responsible for immune activation, ACR-modified T cells will likely play an increasing role in cellular therapy of cancer.
Acknowledgements
The authors are grateful for the Open Research Fund Program of the State Key Laboratory of Virology of China (no.2014KF004), science and technology supporting in Huai’an (HAS2013010) and the start up funds in science research from Huai’an Second People‘s Hospital (YK201217) and Postdoctoral fund of Huai’an Second People’s Hospital.
Disclosure of conflict of interest
None.
References
- 1.Uchiyama M, Jin X, Matsuda H, Bashuda H, Imazuru T, Shimokawa T, Yagita H, Niimi M. An agonistic anti-BTLA mAb (3C10) induced generation of IL-10-dependent regulatory CD4+ T cells and prolongation of murine cardiac allograft. Transplantation. 2014;97:301–319. doi: 10.1097/01.TP.0000438204.96723.8b. [DOI] [PubMed] [Google Scholar]
- 2.Dietze KK, Zelinskyy G, Liu J, Kretzmer F, Schimmer S, Dittmer U. Combining regulatory T cell depletion and inhibitory receptor blockade improves reactivation of exhausted virus-specific CD8+ T cells and efficiently reduces chronic retroviral loads. PLoS Pathog. 2013;9:e1003798. doi: 10.1371/journal.ppat.1003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martner A, Thorén FB, Aurelius J, Hellstrand K. Immunotherapeutic strategies for relapse control in acute myeloid leukemia. Blood Rev. 2013;27:209–216. doi: 10.1016/j.blre.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Badoual C, Combe P, Gey A, Granier C, Roussel H, De Guillebon E, Oudard S, Tartour E. PD-1 and PDL-1 expression in cancer: significance and prognostic value. Med Sci (Paris) 2013;29:570–572. doi: 10.1051/medsci/2013296005. [DOI] [PubMed] [Google Scholar]
- 5.Bekiaris V, Sedý JR, Macauley MG, Rhode-Kurnow A, Ware CF. The inhibitory receptor BTLA controls γδ T cell homeostasis and inflammatory responses. Immunity. 2013;39:1082–1094. doi: 10.1016/j.immuni.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, Speiser DE, Rufer N. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest. 2013;123:1044–1056. doi: 10.1172/JCI65325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int Immunol. 2009;21:1065–1077. doi: 10.1093/intimm/dxp072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guerin LR, Prins JR, Robertson SA. Regulatory T-cells and immune tolerance in pregnancy: a new target for infertility treatment? Hum Reprod Update. 2009;15:517–535. doi: 10.1093/humupd/dmp004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berghoff AS, Ricken G, Widhalm G, Rajky O, Hainfellner JA, Birner P, Raderer M, Preusser M. PD1 (CD279) and PD-L1 (CD274, B7H1) expression in primary central nervous system lymphomas (PCNSL) Clin Neuropathol. 2014;33:42–49. doi: 10.5414/np300698. [DOI] [PubMed] [Google Scholar]
- 10.Ankri C, Cohen CJ. Out of the bitter came forth sweet: Activating CD28-dependent costimulation via PD-1 ligands. Oncoimmunology. 2014;3:e27399. doi: 10.4161/onci.27399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Zhao C, Peng Q, Shi J, Gu G. Expression levels of CD28, CTLA-4, PD-1 and Tim-3 as novel indicators of T-cell immune function in patients with chronic hepatitis B virus infection. Biomed Rep. 2014;2:270–274. doi: 10.3892/br.2014.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koorella C, Nair JR, Murray ME, Carlson LM, Watkins SK, Lee KP. Novel Regulation of CD80/CD86-induced Phosphatidylinositol 3-Kinase Signaling by NOTCH1 Protein in Interleukin-6 and Indoleamine 2,3-Dioxygenase Production by Dendritic Cells. J Biol Chem. 2014;289:7747–7762. doi: 10.1074/jbc.M113.519686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan L, Cui H, Xiao H, Zhang Q. Anergic pulmonary tuberculosis is associated with contraction of the Vd2+T cell population, apoptosis and enhanced inhibitory cytokine production. PLoS One. 2013;8:e71245. doi: 10.1371/journal.pone.0071245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vinay DS, Kwon BS. 4-1BB (CD137), an inducible costimulatory receptor, as a specific target for cancer therapy. BMB Rep. 2014;47:122–129. doi: 10.5483/BMBRep.2014.47.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chacon JA, Pilon-Thomas S, Sarnaik AA, Radvanyi LG. Continuous 4-1BB co-stimulatory signals for the optimal expansion of tumor-infiltrating lymphocytes for adoptive T-cell therapy. Oncoimmunology. 2013;2:e25581. doi: 10.4161/onci.25581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang G, Wang L, Cui H, Wang X, Zhang G, Ma J, Han H, He W, Wang W, Zhao Y, Liu C, Sun M, Gao B. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep. 2014;4:3571. doi: 10.1038/srep03571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell-mediated Tumor Eradication. Mol Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu L, Martin TD, Vazeux R, Unutmaz D, KewalRamani VN. Functional evaluation of DC-SIGN monoclonal antibodies reveals DC-SIGN interactions with ICAM-3 do not promote human immunodeficiency virus type 1 transmission. J Virol. 2002;6:5905–5914. doi: 10.1128/JVI.76.12.5905-5914.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stärck L, Scholz C, Dörken B, Daniel PT. Costimulation by CD137/4-1BB inhibits T cell apoptosis and induces Bcl-xL and c-FLIP(short) via phosphatidylinositol 3-kinase and AKT/protein kinase B. Eur J Immunol. 2005;35:1257–1266. doi: 10.1002/eji.200425686. [DOI] [PubMed] [Google Scholar]
- 21.Lee do Y, Choi BK, Lee DG, Kim YH, Kim CH, Lee SJ, Kwon BS. 4-1BB signaling activates the t cell factor 1 effector/β-catenin pathway with delayed kinetics via ERK signaling and delayed PI3K/AKT activation to promote the proliferation of CD8+ T Cells. PLoS One. 2013;8:e69677. doi: 10.1371/journal.pone.0069677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin GH, Snell LM, Wortzman ME, Clouthier DL, Watts TH. GITR-dependent regulation of 4-1BB expression: implications for T cell memory and anti-4-1BB-induced pathology. J Immunol. 2013;190:4627–4639. doi: 10.4049/jimmunol.1201854. [DOI] [PubMed] [Google Scholar]
- 24.Tomita Y, Yuno A, Tsukamoto H, Senju S, Yoshimura S, Osawa R, Kuroda Y, Hirayama M, Irie A, Hamada A, Jono H, Yoshida K, Tsunoda T, Kohrogi H, Yoshitake Y, Nakamura Y, Shinohara M, Nishimura Y. Identification of CDCA1-derived long peptides bearing both CD41 and CD8+ T-cell epitopes: CDCA1-specific CD4+ T-cell immunity in cancer patients. Int J Cancer. 2014;134:352–366. doi: 10.1002/ijc.28376. [DOI] [PubMed] [Google Scholar]
- 25.Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A, Laghi L, Allavena P, Mantovani A, Marchesi F. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin Cancer Res. 2014;20:2147–2158. doi: 10.1158/1078-0432.CCR-13-2590. [DOI] [PubMed] [Google Scholar]
- 26.Fedorov VD, Sadelain M, Kloss CC. Novel approaches to enhance the specificity and safety of engineered T cells. Cancer J. 2014;20:160–165. doi: 10.1097/PPO.0000000000000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu B, Chen W, Evavold BD, Zhu C. Accumulation of Dynamic Catch Bonds between TCR and Agonist Peptide-MHC Triggers T Cell Signaling. Cell. 2014;157:357–368. doi: 10.1016/j.cell.2014.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monserrat J, Sánchez MÁ, de Paz R, Díaz D, Mur S, Reyes E, Prieto A, de la Hera A, Martínez-A C, Alvarez-Mon M. Distinctive patterns of naïve/memory subset distribution and cytokine expression in CD4 T lymphocytes in ZAP-70 B-chronic lymphocytic patients. Cytometry B Clin Cytom. 2014;86:32–43. doi: 10.1002/cyto.b.21120. [DOI] [PubMed] [Google Scholar]