

Abstract
Microvascular hyperpermeability followed by burn injury is the main cause of shock, and cardiovascular collapse can result if the condition is treated improperly. Our previous studies demonstrated that granulocyte/macrophage colony-stimulating factor (GM-CSF) clearly reduces microvascular permeability and protects microvessels against burn injury. However, the mechanism underlying the protective function of GM-CSF on burn-injured microvessels remains unknown. This study aimed to investigate the effect and mechanism of GM-CSF on endothelial cells after exposure to burn serum. We demonstrated that GM-CSF reduced post-burn endothelial “capillary leak” by inhibiting the activity of RhoA and maintaining the membrane localization of VE-cadherin. Membranous VE-cadherin enhances adherens junctions between endothelial cells and co-localizes with and activates VEGFR2, which protect cells from burn serum-induced apoptosis. Our findings suggest that the protective mechanism of GM-CSF on burn serum-injured endothelial monolayer hyperpermeability is achieved by strengthening cell adherens junctions and improving cell viability.
Keywords: Granulocyte/macrophage colony-stimulating factor, burn, permeability, RhoA
Introduction
The increasing permeability of microvessels is the most important pathological manifestation after burn injury, and this manifestation is evident during the entire disease course [1]. In the early stage of burn, extensive systemic endothelial capillary leakage and subsequent interstitial edema directly lead to dysfunction in multiple organs and hypovolemic shock [2]. In the middle and later stages when wound healing is the main concern, increased wound swelling, secretion, and infection are also induced by local microvascular hyperpermeability [3]. Therefore, a clear understanding of the mechanism associated with burn-induced hyperpermeability and the development of potential therapies to attenuate this process is essential to cure burn patients. Previous studies suggested that inflammatory biochemical mediators in the serum of burn patients, such as cytokines, kinins, and histamines, disrupt inter-endothelial junction assembly [4], which ultimately results in microvascular hyperpermeability.
Granulocyte⁄macrophage colony-stimulating factor (GM-CSF), a cytokine with pleiotropic functions, protects and enhances the function of various cells, including keratinocytes, macrophages, endothelial cells, and fibroblasts [5]. These cells are necessary for accelerating wound healing; thus, GM-CSF was successfully utilized to accelerate epithelization, vascularization, and contraction of different types of wounds [6,7]. Our research team has applied GM-CSF to rats with deep partial-thickness burn wounds and demonstrated that increased angiogenesis is associated with GM-CSF treatment [8]. More interestingly, we also discovered that the anti-leakage function of microvessels in burn wounds was significantly improved after GM-CSF treatment. However, the exact cytobiological mechanisms governing this phenomenon have not been studied.
In this study, we aimed to elucidate the anti-leakage functions of GM-CSF on burn serum-injured endothelial cells and to identify the potential mechanisms that mediate this process.
Materials and methods
Burn serum preparation
Sex-matched Sprague-Dawley rats (220 to 250 g) were provided by the Experimental Animal Research Laboratory at Sun Yat-sen University in China. The rats were anesthetized with an intraperitoneal injection of 3 ml/kg chloral hydrate (10%). A dorsal area equivalent to 30% of the total body surface area was shaved and exposed to an electrically heated brass rod for 8 seconds with a constant pressure and temperature (1 kg, 80°C), producing a clearly defined deep partial thickness burn wound [8,9]. In the control (sham) group, animals were shaved and subjected to the same but unheated brass rod. Arterial blood was collected six hours after burning or sham treatment, and serum was obtained by centrifugation at 6,000 rpm for 20 minutes. Treatment of the animals was conducted in strict accordance with the Care and Use of Laboratory Animals of the National Institutes of Health. The Committee on the Ethics of Animal Experiments of Sun Yat-sen University approved the protocol.
Cell culture and treatment
Human dermal microvascular endothelial cells (HDMVECs) purchased from ATCC (ATCC® PCS-110-010™) were cultured in DMEM (Sigma, St Louis, MO, USA) containing 10% fetal bovine serum (Sigma). After achieving confluence, cells were starved in serum-free DMEM for 4 hours prior to treatment. Burn serum-induced injury to HDMVECs was achieved using 30% burn serum (diluted in PBS), and cells treated with 30% normal serum (sham group) were used as controls. To investigate the protective function of GM-CSF, GM-CSF (R&D, Minneapolis, MN, USA) reconstituted in sterile PBS containing 0.1% bovine serum albumin (BSA) [10] was premixed in DMEM for 1 hour prior to adding burn serum or normal serum.
Lentiviral transduction
To generate more stable overexpression transfectants, endothelial cells were transfected with lentiviral particles containing RhoA-expressing vectors (Cyagen Biosciences, Guangzhou, China). Briefly, cells were seeded in 6-well plates at a density of 2 × 105 cells/well. Then, after achieving 50% confluency, cells were transfected with the lentivirus (MOI = 20) and polybrene (5 μg/ml). After an 8-h infection period, the lentivirus-containing culture medium was replaced by fresh medium, and G418 (1 mg/ml, Sigma) was used to select stable clones. The efficiency of RhoA overexpression was determined by immunoblot analysis.
Evaluation of the barrier integrity of endothelial cells
HDMVECs (1 × 105/well) were grown as monolayers in fibronectin-coated transwell cell culture inserts (BD Biosciences, Franklin Lakes, NJ, USA). Cells were exposed to 30% normal serum, 30% burn serum, 30% burn serum + BSA, or 30% burn serum + GM-CSF. Next, 5 ml of FITC-albumin (5 mg/mL) was added to the luminal (upper) chambers, and the medium in the abluminal (lower) chambers were collected and analyzed using a luminescence spectrometer (Perkin Elmer, Beaconsfield, United Kingdom) at excitations of 494 nm and 520 nm.
To further investigate the integrity of endothelial cells in different conditions, transendothelial electrical resistance (TEER) was detected using a Millicell electrical resistance apparatus (Endohm-6 and EVOM, World Precision Instruments, Sarasota, FL, USA) [11]. The TEER value of the cell-free transwell insert was used as background resistance for all resistance measurement comparisons.
Immunofluorescence analysis
Cells were fixed with 4% paraformaldehyde at room temperature for 20 minutes. After being washed three times, cells were permeabilized with 0.1% Triton X-100 (Sigma) and incubated with 5% BSA for 30 minutes to block non-specific antigen. Then, cells were incubated with monoclonal anti-VE-cadherin (1:250, Abcam, Cambridge, UK) and anti-β-catenin (1:200, Abcam) antibodies simultaneously overnight at 4°C. After washing with PBS, cells were incubated with secondary antibody conjugates (Alexa Fluor 594 anti-mouse and Alexa 488 anti-goat) (Abcam) in the dark for 1 hour. In addition, 4’6-diamidino-2 phenylindole (Sigma) was used for nuclear staining. Fixed cells were observed under a fluorescence microscope (BX51 WI Olympus, Allen, PA, USA), and images were obtained in five randomly selected fields. Negative control experiments were performed by omitting the primary antibody.
Immunoblotting analysis
RIPA buffer (Invitrogen, Carlsbad, CA, USA) was used for total cellular protein extractions. A membrane/cytosol protein extraction kit (Beyotime Biotechnology, Shanghai, China) was used for cell membrane and cytosolic protein extraction. Equal amounts of protein were separated by SDS-PAGE and transferred to nitrocellulose membranes (Amersham, Chalfont, UK) for immunoblotting. The membranes were incubated with primary antibodies for 2 hours. The following primary antibodies were used: VE-cadherin (1:500), β-catenin (1:1000), p-ROCK (1:1000), ROCK (1:800) (all from Abcam), p-MLK, MLK (1:500), p-Akt (1:500), Akt (1:1000) (all from Cell Signaling, Danvers, MA, USA), α-tubulin (1:1000), and β-actin (1:500) (all from Santa Cruz, CA, USA). Bound primary antibodies were visualized with appropriate secondary antibodies conjugated to horseradish peroxidase (HRP) and developed with an enhanced chemiluminescence kit (GE Healthcare, Piscataway, NJ, USA). Densitometric analysis was performed using Image J software (National Institutes of Health, Bethesda, MD, USA).
Co-immunoprecipitation assay
Pre-cooled RIPA buffer containing protease inhibitors (Roche, Indianapolis, IN, USA) was used to lyse cells. Lysates were precleared by incubation with protein A cross-linked to agarose (Sigma) followed by incubation with anti-VE-cadherin antibodies. Immunocomplexes were collected by centrifugation and then immunoblotted with antibodies raised against β-catenin (1:1000) or VEGFR2 (1:1000). The blots were subsequently incubated with an appropriate HRP-conjugated secondary antibody (1:5000) and detected using an enhanced chemiluminescence reagent.
Small GTPase activity assay
Activities of small GTPases, including RhoA, Cdc42, and Rac1, were assessed using a Rho assay kit and a Rac/Cdc42 assay kit (Millipore, Billerica, MA, USA), following the manufacturer’s standard instructions. Briefly, cells were grown to approximately 70% confluency and serum starved overnight. Agarose beads conjugated with a glutathione S-transferase (GST)-tagged Rho-binding domain of Rhotekin were used to pull down endogenous RhoA-GTP from cell lysates. To pull down Cdc42-GTP and Rac1-GTP, agarose beads conjugated with a GST-tagged p21 binding domain of PAK1 was used. The active forms of RhoA, Cdc42, or Rac1 were assessed by Western blotting using individual GTPases. RhoA, Cdc42, or Rac1 activity was normalized to the corresponding total protein.
TUNEL sstaining
To evaluate cell apoptosis in different conditions, a terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay was performed using the FragEL™ DNA Fragmentation Detection Kit (EMD Millipore). Briefly, cells grown on slides were equilibrated with the terminal deoxynucleotidyl transferase (TdT) equilibration buffer and subsequently incubated with the labeling reaction mixture. The slides were examined under a microscope (BX51 WI Olympus), and cells stained with brown were identified as apoptosis. Apoptotic cells were counted in five randomly selected high-power fields (magnification, × 400), and the apoptosis index was defined as the ratio of apoptotic cells to total cells.
MTT assay
MTT assay was used to assess the cell viability. Briefly, cells (2 × 103/well) were seeded in 96-well plates and then incubated with the MTT solution (5 mg/ml, Sigma) until a purple precipitate was visible. MTT-formazan crystals were solubilized with DMSO (Sigma), and then measured with a microplate reader at a wavelength of 570 nm corrected to 650 nm. Values were normalized to controls.
Statistical analyses
Data in this study are expressed as the mean ± SD. Significant differences between the sham control and each experimental group were compared using one-way ANOVA with Bonferroni/Dunnett post-hoc tests. The differences between experimental groups were compared using the unpaired Student’s t-test or one-way ANOVA followed by post hoc analysis with the Bonferroni test. All statistical analyses were performed using SPSS 18.0 software (SPSS, Chicago, IL, USA), and p < 0.05 was considered to be statistically significant.
Results
GM-CSF attenuates burn serum-induced endothelial monolayer hyperpermeability
When compared with endothelial monolayers exposed to serum from sham rats (normal serum), endothelial monolayers exposed to serum from burn rats (burn serum) displayed a significant increase in permeability in a time-dependent manner (Figure 1A). In addition, the TEER value of endothelial monolayers was decreased in a time-dependent fashion upon exposure to the burn serum (Figure 1B). After GM-CSF treatment, endothelial monolayer leakage was significantly decreased (Figure 1C), and the TEER value increased (Figure 1D); these effects were dependent on the concentration of GM-CSF. For example, 40 ng/ml GM-CSF was the most effective concentration in this study, and we used this concentration in the following experiments.
Figure 1.
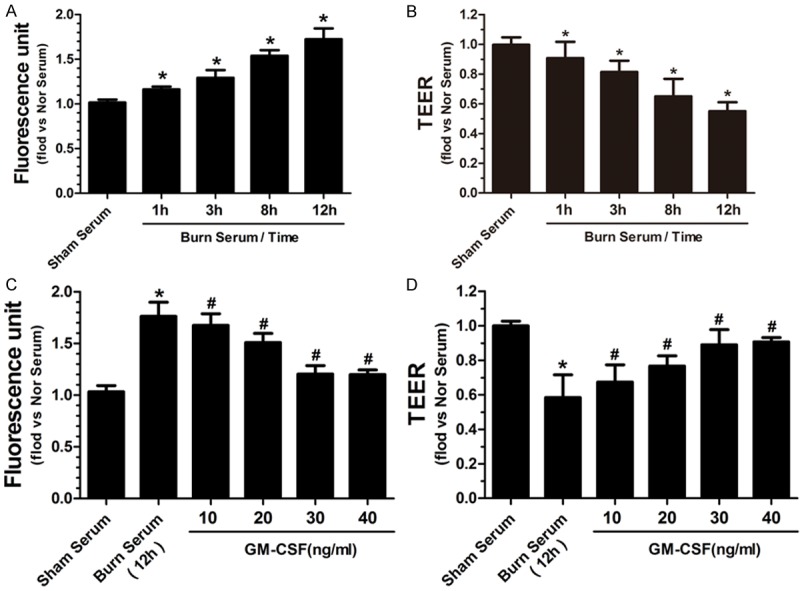
Burn serum increased the permeability of endothelial monolayers to FITC-albumin in a time-dependent manner (A). Burn serum reduced TEER values of endothelial monolayers in a time-dependent manner (B). (*p < 0.05 compared with no serum). GM-CSF reduced burn serum-induced cell leakage (C) and enhanced TEER values (D) in a dose-dependent fashion, and 40 ng/ml was the most effective concentration (*p < 0.05 compared with sham serum, #p < 0.05 compared with burn serum).
GM-CSF strengthens endothelial cell adherens junctions of after burn serum damage
Given that adherens junctions, which are formed by membrane-localized VE-cadherin and its intracellular partner β-catenin, play an important role in controlling paracellular permeability to circulating leukocytes and solutes [12,13], we thus investigated the presence and location of VE-cadherin and β-catenin. The results indicated that in control group, wherein endothelial cells were treated with normal serum, VE-cadherin and β-catenin colocalized at the cell-cell borders in a continuous pattern (Figure 2A1-4 ). Upon treatment with burn serum, cell-cell contacts were lost or clearly reduced. In addition, the expression of cell surface-associated VE-cadherin and the accumulation of β-catenin bound to VE-cadherin were significantly diminished (Figure 2A5-8 ). This distribution pattern was not altered upon treatment with BSA, the solution that was used to dilute GM-CSF powder (Figure 2A9-12 ). Importantly, when burn serum-damaged cells were treated with GM-CSF, cell-cell contacts resumed an uninterrupted pattern, wherein VE-cadherin associates with β-catenin to form distinct adhesive structures (Figure 2A3-16 ).
Figure 2.

The cell membrane distribution of VE-cadherin (A1, 5, 9, 13) and β-catenin (A2, 6, 10, 14). Nuclei were stained with DAPI (A3, 7, 11, 15). Co-localization (merged images) of VE-cadherin and β-catenin (A4, 8, 12, 16). (Scale bar = 20 μm). Quantitative determination of the association between VE-cadherin and β-catenin by Co-IP assay. Burn serum caused a dissociation of β-catenin from VE-cadherin, and this effect was reversed by GM-CSF (B). Cellular distribution of VE-cadherin was quantified by immunoblot (C). Burn serum induced a shift in VE-cadherin localization from the membrane to the cytosol fraction. GM-CSF attenuated this change and maintained the membrane expression of VE-cadherin (D, E), (*p < 0.05).
Furthermore, immunoblotting was used to accurately quantify VE-cadherin distribution and its association with β-catenin. The results indicate that VE-cadherin was mainly expressed at cell membrane in normal conditions, whereas burn serum reduced this expression pattern. GM-CSF pretreatment attenuated burn serum-induced changes and restored the membrane expression of VE-cadherin (Figure 2B). Regarding the association of VE-cadherin and β-catenin, we also observed that burn serum clearly reduced VE-cadherin and β-catenin binding, whereas GM-CSF maintained VE-cadherin and β-catenin binding, even in the presence of burn serum (Figure 2C, 2D).
GM-CSF inhibits burn serum-induced cell death by enhancing the association between VE-cadherin and VEGFR2
VEGFR2 associates with VE-cadherin at cell–cell contacts [14], and this interaction modulates cell survival through activating the phosphatidylinositol (PI) 3-kinase/Akt signaling pathway [15,16]. Thus, we detected VE-cadherin and VEGFR2 binding in various conditions. VEGFR2 dissociated from VE-cadherin after exposure to burn serum, whereas GM-CSF restored this association (Figure 3A). Regarding the activation of the PI3K/Akt signaling pathway, we examined Akt phosphorylation and found that pAkt expression was reduced in burn serum conditions, and this effect was not altered after BSA treatment. Conversely, GM-CSF pretreatment increased pAkt levels in endothelial cells, even in response to burn serum-damaged conditions (Figure 3B). To further understand the protective effect of GM-CSF on endothelial cell survival, TUNEL and MTT analysis were used to evaluate cell apoptosis and cell viability, respectively. A significant increase in apoptotic cells and a more rapid descending rate of cell survival were noted in endothelial cells exposed to burn serum. Significantly, GM-CSF treatment not only reduced the number of apoptotic cells but also enhanced cell viability under burn serum conditions (Figure 3C, 3D).
Figure 3.

GM-CSF enhanced the burn serum-diminished association of VE-cadherin and VEGFR2 (A). Low pAkt expression was observed after burn serum treatment, and the levels were increased to approximately normal levels when cells were pre-treated with GM-CSF (B). The number of apoptotic cells (shown in brown) increased after burn serum exposure, whereas the number of cells was unaltered by BSA treatment and attenuated by GM-CSF treatment (C) (Scale bar = 50 μm). GM-CSF increased cell viability in burn serum conditions as measured by the MTT assay (D). (*p < 0.05).
GM-CSF inhibited burn serum-induced RhoA activation and subsequent ROCK and MLK Phosphorylation
The Rho family of GTPases, including RhoA, Rac1, and Cdc42, interact with and activate downstream effector proteins when bound to GTP; these GTPases, have been implicated in the regulation of cellular junctional complexes [17]. Therefore, we utilized a GTP-pull-down assay to identify which Rho GTPases were responsible for GM-CSF function. As shown in Figure 4A and 4B, burn serum significantly increased active (GTP-bound) RhoA, Rac1, and Cdc42, whereas the addition of BSA did not later the activation of these proteins. GM-CSF reduces active RhoA; however, it had no effect on Rac1 and Cdc42 activation. To demonstrate that the RhoA signaling pathway is involved in GM-CSF function, we constructed recombinant lentiviral particles encoding a constitutively active form of RhoA (Figure 4C) to investigate whether ROCK and MLK, the downstream effector proteins of RhoA, are altered accordingly. As shown in Figure 4D and 4E, ROCK and MLK phosphorylation increased after burn serum treatment, whereas phosphorylation was not altered by the addition of BSA. GM-CSF notably reversed these changes and reduced pROCK and pMLK expression, restoring approximately normal expression levels. Importantly, GM-CSF did not decrease pROCK and pMLK expression in RhoA-overexpressing endothelial cells.
Figure 4.

Burn serum markedly increased the levels of activated RhoA, Rac1, and Cdc42 in endothelial cells. GM-CSF treatment reduced active RhoA, not Rac1 or Cdc42 (A, B). The efficiency of RhoA expression in endothelial cells was determined by immunoblotting (C). GM-CSF inhibited burn serum-stimulated ROCK and MLK phosphorylation, and these effects were inhibited by RhoA overexpression (D, E). (*p < 0.05).
RhoA overexpression attenuates the function of GM-CSF in adherens junction maintenance
To further clarify the important role of RhoA in the protective function of GM-CSF, we assessed VE-cadherin/β-catenin junctional complex formation in RhoA-overexpressing endothelial cells. Our results indicate that cell-cell contacts were disrupted and that cell membrane-associated VE-cadherin and β-catenin expression was decreased in burn serum-treated cells (Figure 5A1-4 ); these effects were reversed by GM-CSF (Figure 5A5-8 ). However, in RhoA-overexpressing endothelial cells, VE-cadherin and β-catenin was not present at cell-cell contacts, even in the presence of GM-CSF (Figure 5A13-16 ). Immunoblotting also indicated that VE-cadherin expression in the cell membrane and its association with β-catenin (Figure 5B) were decreased upon exposure to burn serum. GM-CSF restored the membrane localization of VE-cadherin and increased VE-cadherin/β-catenin complex formation. in normal endothelial cells but not in RhoA-overexpressing cells (Figure 5C, 5D). Additionally, RhoA overexpression remarkably inhibited GM-CSF-induced reductions in FITC-albumin diffusion and increased TEER values under burn serum conditions (Figure 5E, 5F).
Figure 5.

Burn serum caused a loss of cell surface-associated VE-cadherin and β-catenin expression (A1-4). GM-CSF prevented the loss of VE-cadherin and β-catenin membrane expression (A5-8); these effects were unaltered by scrambled siRNA (A9-12) and reversed by RhoA overexpression (A13-16). GM-CSF enhanced VE-cadherin and β-catenin binding in the burn serum conditions. RhoA overexpression reduced this binding, even in the presence of GM-CSF (B). GM-CSF increased the membrane expression of VE-cadherin after exposure to burn serum, which was inhibited by RhoA overexpression (C, D). RhoA overexpression attenuated GM-CSF function by reducing FITC-albumin diffusion and increasing TEER values upon burn serum treatment (E, F) (*p < 0.05).
RhoA overexpression inhibits the anti-apoptotic function of GM-CSF by inhibiting the association of VE-cadherin and VEGFR2
Based on our previous results indicating that RhoA overexpression reduced the membrane expression of VE-cadherin, which regulates cell survival via associations with VEGFR2, we hypothesized that RhoA plays an important role in the anti-apoptotic function of GM-CSF. Using co-immunoprecipitation assay, we found that the association between VE-cadherin and VEGFR2 was significantly reduced (Figure 6A) and pAkt expression was decreased in RhoA-overexpressing cells (Figure 6B), even in the presence of GM-CSF. TUNEL and MTT analysis also demonstrated that RhoA overexpression rendered GM-CSF ineffective. In addition, in RhoA-overexpressing cells, the number of apoptotic cells increased, and the cell survival rate decreased, even after GM-CSF treatment (Figure 6C, 6D).
Figure 6.

GM-CSF enhanced the association between VE-cadherin and VEGFR2 in burn serum conditions, and this effect was not observed in RhoA-overexpressing cells (A). RhoA overexpression decreased pAkt expression, even with GM-CSF treatment (C). RhoA overexpression attenuated the GM-CSF functions in the reduction of cell apoptosis and increased cell viability after burn serum damaged (C, D) (*p < 0.05).
Discussion
Post-burn microvascular hyperpermeability is associated with large fluid shifts and edema formation in both damaged and non-damaged tissues [3,18]. The presence of edema in a local burn wound can increase the inflammatory exudation and delay wound healing, and systemic edema can directly cause or exacerbate multiple organ dysfunction and can even cause shock and/or death [2]. Thus, inhibiting microvascular hyperpermeability is the most important task when treating burns.
GM-CSF, a cytokine with pleiotropic bioactive functions, has received increasing attention given its efficacy in hematopoietic progenitor cell mobilization and skin cell activation [19]. In recent years, GM-CSF has been successfully employed in the treatment of poorly healing wounds of diverse etiologies [6,7]. Our previous study on burns demonstrated that GM-CSF stabilizes microvessels and decreases vascular permeability after burn injury in addition to promoting microvessel formation [8]. Therefore, we hypothesized that GM-CSF function as an anti-leakage agent to protect endothelial cells from burn damage. In this study, we exposed endothelial cells to serum from burned rats to mimics in vivo microvascular injury [20] and demonstrated that GM-CSF considerably reduced endothelial hyperpermeability and enhanced its barrier integrity after burn serum treatment. Next, we further investigated the mechanism underlying the anti-leakage function of GM-CSF. Previous studies have demonstrated that, various inflammatory mediators that released into the serum after thermal injury, such as interleukin 6, interleukin 8, tumor necrosis factor α, and transforming growth factor β, disrupt inter-endothelial junction assembly, thereby opening intercellular gaps and causing endothelial hyperpermeability [21]. Adherens junctions, which form through the binding of the membrane portion of VE-cadherin to its intracellular partners, such as β-catenin, primarily exist in endothelial cells and are responsible for maintaining cell-cell interactions [22]. Disruption of these junctional complexes contributes to the loss of endothelial monolayer integrity and consequently creates an endothelial “capillary leak” [23,24]. In this study, burn serum-stimulated cells exhibit a complete loss of or truncated VE-cadherin/β-catenin complexes in cell-cell contacts. GM-CSF pretreatment prevented this loss and maintained VE-cadherin/β-catenin complex localization at cell junctions. To clarify the mechanism by which GM-CSF restored endothelial junctional complex organization, we detected the cellular distribution of VE-cadherin. Studies have demonstrated that membrane VE-cadherin acts as an anchor, recruiting cytoplasmic β-catenin to adhere to membranes and form cadherin-catenin junctional complexes [25,26]. Our previous research also indicated that the loss of membrane localized VE-cadherin directly resulted in detached junctional complexes, thereby causing endothelial hyperpermeability [27 JCP]. In this study, VE-cadherin was translocated from membranes to the cytoplasm after exposure to burn serum. GM-CSF reversed this translocation and maintained the membrane expression of VE-cadherin. More interestingly, membrane localized VE-cadherin interacts with endothelial-specific signaling proteins. For example, VE-cadherin can bind to vascular endothelial growth factor receptor 2 (VEGFR2) [14,28,29] and modulate cell survival through the phosphatidylinositol (PI) 3-kinase/Akt signaling pathway [16]. Therefore, we assessed the binding of VE-cadherin and VEGFR2 as well as downstream Akt phosphorylation. GM-CSF remarkably enhanced the association between VE-cadherin and VEGFR2, activated the PI3K/Akt pathway by phosphorylating Akt, and subsequently protected endothelial cells against burn serum-induced death.
Although these above observations suggested that GM-CSF-mediated improvements in endothelial monolayer leakage after burn serum injury was related to VE-cadherin membrane expression, which enhances adherent junctional complex formation and Akt activation, the exact mechanism by which GM-CSF modulates VE-cadherin membrane expression is unclear. Previous studies have implicated small G-proteins, including RhoA, Rac1, and Cdc42, in the regulation of endothelial permeability by affecting adherens or tight junctions [30,31]. Numerous reports also link the disruption of endothelial junctions by inflammatory cytokines, thrombin, and histamine with Rho/ROCK activation [32-34]. Accordingly, we performed a GTP-pull-down assay to identify whether these small G-proteins are involved GM-CSF function. Our results demonstrated that GM-CSF significantly inhibited RhoA activation, not Rac1 or Cdc42. In addition to the downstream activation of RhoA, ROCK and MLK exhibited a low level of phosphorylation, suggesting that RhoA plays an essential role in GM-CSF function. To further verify our hypothesis, we overexpressed RhoA in burn serum-damaged cells. We noted reductions in adherent junctional complexes, the association between VE-cadherin and VEGFR2, Akt phosphorylation, even after GM-CSF treatment. Additionally, we also found that GM-CSF does not decrease burn serum-induced endothelial hyperpermeability or maintain cell survival in RhoA-overexpressing cells.
In summary, GM-CSF is a strong protective cytokines that protects endothelial cells from thermal injury. Our study demonstrates that GM-CSF administration maintains the membrane expression of VE-cadherin by inhibiting the activation of RhoA. Membrane VE-cadherin enhances the integrity of the endothelium by forming junctional complexes with β-catenin and improves cell survival via associations with VEGFR2. The results of our study indicate that GM-CSF can be used as an anti-leakage agent for burn treatment.
Acknowledgements
We would like to acknowledge Jinchang Lu (Department of Musculoskeletal Oncology, the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China) for the help with animal experiment and the expert technical assistance. We would also like to acknowledge the Translational Medicine Laboratory and the Medical Science Experimentation Center of Sun Yat-sen University for providing experimental facilities. This study was supported by the frant from Natural Science Foundation of China (NSFC) (Contract grant number: 81272153).
Disclosure of conflict of interest
None.
References
- 1.Saffle JR, Sullivan JJ, Tuohig GM, Larson CM. Multiple organ failure in patients with thermal injury. Crit Care Med. 1993;21:1673–1683. doi: 10.1097/00003246-199311000-00016. [DOI] [PubMed] [Google Scholar]
- 2.Kremer T, Abe D, Weihrauch M, Peters C, Gebhardt MM, Germann G, Heitmann C, Walther A. Burn plasma transfer induces burn edema in healthy rats. Shock. 2008;30:394–400. doi: 10.1097/SHK.0b013e3181673908. [DOI] [PubMed] [Google Scholar]
- 3.Demling RH. The burn edema process: current concepts. J Burn Care Rehabil. 2005;26:207–227. [PubMed] [Google Scholar]
- 4.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985) 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 5.Mann A, Breuhahn K, Schirmacher P, Blessing M. Keratinocyte-derived granulocyte-macrophage colony stimulating factor accelerates wound healing: Stimulation of keratinocyte proliferation, granulation tissue formation, and vascularization. J Invest Dermatol. 2001;117:1382–1390. doi: 10.1046/j.0022-202x.2001.01600.x. [DOI] [PubMed] [Google Scholar]
- 6.Mery L, Girot R, Aractingi S. Topical effectiveness of molgramostim (GM-CSF) in sickle cell leg ulcers. Dermatology. 2004;208:135–137. doi: 10.1159/000076487. [DOI] [PubMed] [Google Scholar]
- 7.Groves RW, Schmidt-Lucke JA. Recombinant human GM-CSF in the treatment of poorly healing wounds. Adv Skin Wound Care. 2000;13:107–112. [PubMed] [Google Scholar]
- 8.Zhao J, Chen L, Shu B, Tang J, Zhang L, Xie J, Qi S, Xu Y. Granulocyte/macrophage colony-stimulating factor influences angiogenesis by regulating the coordinated expression of VEGF and the Ang/Tie system. PLoS One. 2014;9:e92691. doi: 10.1371/journal.pone.0092691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson JM. Animal models for wound repair. Arch Dermatol Res. 1998;290:S1–S11. doi: 10.1007/pl00007448. [DOI] [PubMed] [Google Scholar]
- 10.Xia L, Dong Z, Zhang Y, Zhang X, Song X, Sun M, Hu Y, Liu S, Wang K, Qu X, Wei F. Interleukin-4 and granulocyte-macrophage colonystimulating factor mediates the upregulation of soluble vascular endothelial growth factor receptor-1 in RAW264.7 cells-a process in which p38 mitogen-activated protein kinase signaling has an important role. J Microbiol Immunol Infect. 2014 doi: 10.1016/j.jmii.2014.06.008. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 11.Shimizu F, Sano Y, Haruki H, Kanda T. Advanced glycation end-products induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood-nerve barrier by stimulating the release of TGF-beta and vascular endothelial growth factor (VEGF) by pericytes. Diabetologia. 2011;54:1517–1526. doi: 10.1007/s00125-011-2107-7. [DOI] [PubMed] [Google Scholar]
- 12.Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol. 1999;19:2286–2297. doi: 10.1161/01.atv.19.10.2286. [DOI] [PubMed] [Google Scholar]
- 13.Navarro P, Caveda L, Breviario F, Mandoteanu I, Lampugnani MG, Dejana E. Catenin-dependent and -independent functions of vascular endothelial cadherin. J Biol Chem. 1995;270:30965–30972. doi: 10.1074/jbc.270.52.30965. [DOI] [PubMed] [Google Scholar]
- 14.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 15.Pece S, Chiariello M, Murga C, Gutkind JS. Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell-cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J Biol Chem. 1999;274:19347–19351. doi: 10.1074/jbc.274.27.19347. [DOI] [PubMed] [Google Scholar]
- 16.Gerber HP, McMurtrey A, Kowalski J, Yan MH, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase Akt signal transduction pathway - Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 17.Wojciak-Stothard B, Entwistle A, Garg R, Ridley AJ. Regulation of TNF-alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. J Cell Physiol. 1998;176:150–165. doi: 10.1002/(SICI)1097-4652(199807)176:1<150::AID-JCP17>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 18.Evers LH, Bhavsar D, Mailander P. The biology of burn injury. Exp Dermatol. 2010;19:777–783. doi: 10.1111/j.1600-0625.2010.01105.x. [DOI] [PubMed] [Google Scholar]
- 19.Gorin NC, Coiffier B, Hayat M, Fouillard L, Kuentz M, Flesch M, Colombat P, Boivin P, Slavin S, Philip T. Recombinant human granulocyte-macrophage colony-stimulating factor after high-dose chemotherapy and autologous bone marrow transplantation with unpurged and purged marrow in non-Hodgkin’s lymphoma: a double-blind placebo-controlled trial. Blood. 1992;80:1149–1157. [PubMed] [Google Scholar]
- 20.Stagg HW, Whaley JG, Tharakan B, Hunter FA, Jupiter D, Little DC, Davis ML, Smythe WR, Childs EW. Doxycycline attenuates burn-induced microvascular hyperpermeability. J Trauma Acute Care Surg. 2013;75:1040–1046. doi: 10.1097/TA.0b013e3182aa9c79. discussion 1046. [DOI] [PubMed] [Google Scholar]
- 21.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med. 2009;11:e19. doi: 10.1017/S1462399409001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnittler HJ. Structural and functional aspects of intercellular junctions in vascular endothelium. Basic Res Cardiol. 1998;93(Suppl 3):30–39. doi: 10.1007/s003950050205. [DOI] [PubMed] [Google Scholar]
- 23.Vittet D, Prandini MH, Berthier R, Schweitzer A, MartinSisteron H, Uzan G, Dejana E. Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood. 1996;88:3424–3431. [PubMed] [Google Scholar]
- 24.Tinsley JH, Wu MH, Ma W, Taulman AC, Yuan SY. Activated neutrophils induce hyperpermeability and phosphorylation of adherens junction proteins in coronary venular endothelial cells. J Biol Chem. 1999;274:24930–24934. doi: 10.1074/jbc.274.35.24930. [DOI] [PubMed] [Google Scholar]
- 25.Navaratna D, McGuire PG, Menicucci G, Das A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes. 2007;56:2380–2387. doi: 10.2337/db06-1694. [DOI] [PubMed] [Google Scholar]
- 26.Angst BD, Marcozzi C, Magee AI. The cadherin superfamily: diversity in form and function. J Cell Sci. 2001;114:629–641. doi: 10.1242/jcs.114.4.629. [DOI] [PubMed] [Google Scholar]
- 27.Zhao J, Chen L, Shu B, Tang J, Zhang L, Xie J, Xu Y, Qi S. Angiopoietin-1 protects the endothelial cells against advanced glycation end product injury by strengthening cell junctions and inhibiting cell apoptosis. J Cell Physiol. 2015;230:1895–905. doi: 10.1002/jcp.24920. [DOI] [PubMed] [Google Scholar]
- 28.Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, Resnick N. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci U S A. 2002;99:9462–9467. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.St Croix B, Sheehan C, Rak JW, Florenes VA, Slingerland JM, Kerbel RS. E-cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1) J Cell Biol. 1998;142:557–571. doi: 10.1083/jcb.142.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem. 1998;273:21867–21874. doi: 10.1074/jbc.273.34.21867. [DOI] [PubMed] [Google Scholar]
- 31.Hippenstiel S, Tannert-Otto S, Vollrath N, Krull M, Just I, Aktories K, von Eichel-Streiber C, Suttorp N. Glucosylation of small GTP-binding Rho proteins disrupts endothelial barrier function. Am J Physiol. 1997;272:L38–43. doi: 10.1152/ajplung.1997.272.1.L38. [DOI] [PubMed] [Google Scholar]
- 32.van Nieuw Amerongen GP, Draijer R, Vermeer MA, van Hinsbergh VW. Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ Res. 1998;83:1115–1123. doi: 10.1161/01.res.83.11.1115. [DOI] [PubMed] [Google Scholar]
- 33.Vouret-Craviari V, Boquet P, Pouyssegur J, Van Obberghen-Schilling E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol Biol Cell. 1998;9:2639–2653. doi: 10.1091/mbc.9.9.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng HZ, Zhao KS, Zhou BY, Huang QB. Role of Rho kinase and actin filament in the increased vascular permeability of skin venules in rats after scalding. Burns. 2003;29:820–827. doi: 10.1016/j.burns.2003.08.004. [DOI] [PubMed] [Google Scholar]