

Abstract
Primary vitreoretinal lymphoma (PVRL) is an uncommon, but potentially fatal intraocular malignancy, which may occur with or without primary central nervous system lymphoma (PCNSL). Considered to be a subset of PCNSL, it is mostly of diffuse large B-cell type. The diagnosis of PVRL poses a challenge not only to the clinician, but also to the pathologist. Despite aggressive treatment with chemotherapy and/or radiotherapy, relapses or CNS involvement are common.
Keywords: Intraocular, lymphoma, retina, vitreous
Lymphoma is a malignant neoplasm derived from monoclonal proliferations of B- or T-lymphocytes and rarely, natural killer cells. Ocular lymphoma is classified based on the tissue involved as intraocular, orbital and adnexal. Intraocular lymphoma is a rare subset of primary central nervous system lymphoma (PCNSL) and is characterized by an aggressive clinical course.[1] Most intraocular lymphomas are retino-vitreal, while ocular manifestation of systemic lymphoma occurs commonly in the uvea.[2,3,4] There is also a rare subset of primary uveal lymphoma (PUL).[2,3,4]
Given its rarity, primary intraocular lymphoma (PIOL) is often misdiagnosed and inappropriately managed, with no established standards for its diagnosis and therapy.[4] PIOL is a challenge not only to an ophthalmologist, but also to a pathologist. The tendency of PIOL to masquerade as infective and inflammatory conditions and the complexity associated with acquisition and preservation of tissue samples makes the diagnosis difficult and thus delays appropriate management.[5] We aim to review the available literature on PIOL and discuss the salient aspects in it's clinical presentation, diagnosis and management.
Historical Perspective
The earliest mention of intraocular lymphoma dates back to Triebenstein's report on primary choroidal lymphoma in 1920.[6] Three decades later Cooper and Riker, followed by Givner described this disease as “malignant lymphoma of the uveal tract.”[7,8] Klingele and Hogan later renamed it as “ocular reticulum sarcoma.”[9] It was in 1968 that Vogel et al. described “reticulum cell sarcoma” involving the retina.[10] After the terminology “reticulum cell sarcoma” was shown to be a misnomer and with the advent of the World Health Organisation (WHO) lymphoma classification, this entity was named as “intraocular lymphoma.”[11,12] Subsequently, in early 1980s, reports in the literature described associations between intraocular lymphoma and PCNSL and the entity PIOL was established.[13,14] Recently, the nomenclature “primary vitreoretinal lymphoma (PVRL)” was proposed in lieu of PIOL (to denote it as a distinct entity from PUL) and the same is currently being used in the literature.[2,4]
Nomenclature and Classification
The WHO classification of tumors correlates clinical features with distinct morphology, immunophenotype and genetics.[15] Intraocular lymphoma represents a heterogeneous group of malignancies located in different tissues within the eye, each of which have different morphological, immunophenotypical and genetic features, and completely different clinical courses.[2,4,16] It has been suggested that intraocular lymphomas be referred to based on their location and whether they are primary or secondary to CNS lymphoma.[4] Thus, PIOL can be classified as PVRL and PUL. PVRL may involve the retina with or without involvement of the vitreous or the optic nerve. PUL may be further sub-classified as choroidal, ciliary body or iridial. Further characterization should be based on the WHO classification.[15]
Definition
With advances in the understanding of intraocular lymphoma and recent concepts in the literature, PVRL is currently defined as “a subset of PCNSL and a heterogeneous group of malignant lymphocytic neoplasms affecting the retina with or without involvement of the vitreous or the optic nerve, and without evidence of brain or cerebrospinal fluid (CSF) involvement.” Many patients, however, develop intracranial involvement either concomitantly or subsequently, and once there is manifest CNS involvement, the entity is denoted as PCNSL.
Epidemiology and Risk Factors
Primary malignant intraocular neoplasms are relatively rare, with uveal melanoma and retinoblastoma constituting the bulk.[17] PVRL is a rare malignancy whose exact incidence is not known.[4,18] PCNSL with ocular involvement and PVRL are estimated to constitute 1% of non-Hodgkin's lymphoma, 1% of all intracranial tumors, and < 1% of all intraocular tumors.[19] In recent times, a 30-fold increase in the incidence of PCNSL has been documented, which can be attributed to increased prevalence of immunodeficiency and immunosuppression, and may be, to improved diagnostic acumen.[1,20,21] PVRL is usually a disease of adulthood with few cases being reported in infants and teenagers.[1,4,18,22,23,24] Most patients of PVRL are older than 40 years with the usual age of onset being in the late 50s and 60s.[1] Although most reports describe PVRL to involve females more commonly than males, there is no convincing gender predilection.[1,25,26] No geographic or racial variations have been described in PVRL. Immunodeficiency and immunosuppression are considered as risk factors for development of PVRL.[1]
Etiopathogenesis
The exact etiology of PVRL stands unclear with several hypotheses being suggested. These include role of infectious agents, the chemokine hypothesis, selective homing of malignant lymphocytes to the eye and clonal transformation of local polyclonal inflammatory cells. Infectious agents such as Epstein-Barr virus (EBV), human herpes virus-8 (HHV-8) and Toxoplasma gondii are suspected to be involved in the pathogenesis of PVRL.[27,28] EBV transforms human B-lymphocytes, eventually leading to continuous proliferation, while HHV-8 viral genome expresses genes responsible for inhibition of apoptosis, cell cycle entry and angiogenesis with proliferation of B-lymphocytes.[21,29] The role of T. gondii in PVRL in unknown. However, It is possible that genotoxicity from the immunological response to ocular T. gondii infection leads to PVRL development.[28]
Chemokines and chemokine receptors selective for B-lymphocytes were identified in retinal pigment epithelium (RPE) and malignant B-cells, respectively.[30] It has been suggested that B-cell chemokines may be involved in the pathogenesis of PVRL by selectively attracting lymphoma cells to RPE from the choroidal circulation.[30] Another hypothesis on infiltration of malignant lymphocytes from systemic circulation into the eye and brain suggests that permissive retinal endothelial cell receptors and lack of robust immune surveillance may allow preferential entry of malignant lymphocytes to the retina than the choroid with subsequent clonal proliferation in the eye.[2,31,32]
Primary vitreoretinal lymphoma can develop subsequent to infectious uveitis.[28] This opens up another hypothesis that initial polyclonal proliferation of inflammatory lymphocytes may undergo mutagenesis leading to a monoclonal proliferation characteristic of a PVRL. Endoantigens from a noninfectious uveitis may lead up to the same phenomenon.[32]
Clinical Features
Diagnosis of PVRL is based on clinical features that help suspect it, investigative tools that help differentiate it from simulating conditions, and finally histopathology that confirms the diagnosis.[32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62] In 64–83% cases, PVRL is bilateral.[4] It frequently masquerades as chronic posterior uveitis [Fig. 1]. Insidious onset and delay in diagnosis are common.[4] Vitreous floaters may be noticed by the patient long before PVRL is suspected. These are often attributed to normal degenerative changes or vitritis.[4] Steroids are prescribed for suspected vitritis, which may temporarily benefit the patient and thus mask and delay the diagnosis of PVRL. Anterior segment inflammation is usually absent.[4,33] Keratic precipitates and cells may be seen in the anterior chamber but infiltrating cells are usually confined to the vitreous cavity.[4,34] Pseudohypopyon is a rare presentation. Sheets and clusters of lymphoma cells may be seen in the vitreous. These cells are larger than the inflammatory cells and do not cluster with the reactive cells resulting in an “aurora borealis” appearance from cells lining along the vitreous fibriles.[4] A mild to moderate haze may be seen, which, at times is accentuated at the periphery or superiorly.[4,18] Lymphoma cells may proliferate along the Bruch's membrane under the RPE. These may be seen as creamy lesions with yellow-white infiltrates deep to the retina [Figs. 2 and 3].[18] Islands of pigment float on these deposits which with early or diffuse involvement gives rise to a characteristic “leopard-skin” appearance.[4] RPE atrophy and subretinal fibrosis may result from spontaneous resolution.[36] Optic nerve, contiguous sclerochoroidal, extrascleral and orbital involvement are rare, but may occur.[37,38]
Figure 1.
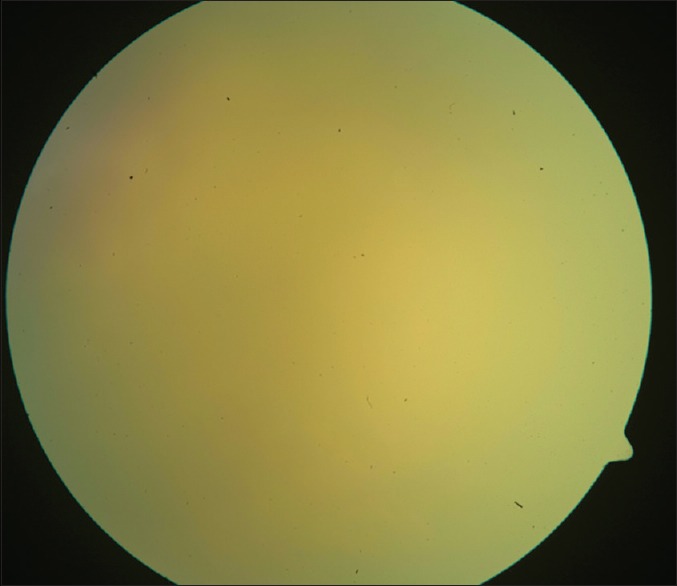
Fundus picture displaying a diffuse vitreous haze
Figure 2.
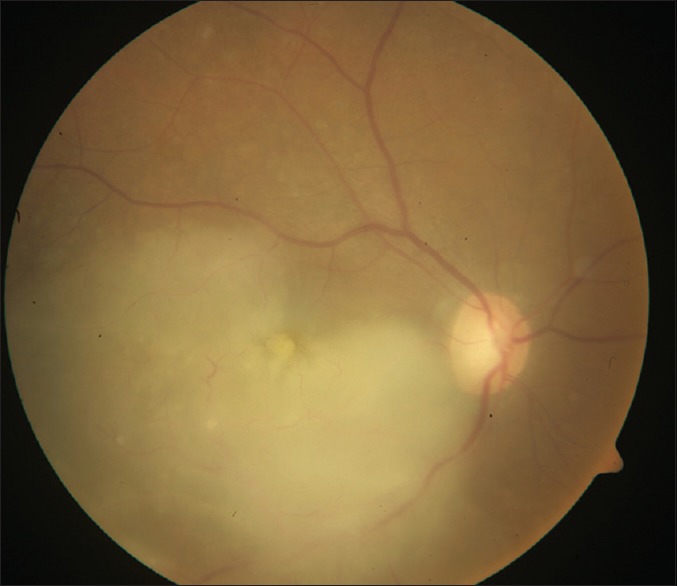
Fundus picture vitreous haze with yellowish subretinal deposits and surface hemorrhages
Figure 3.
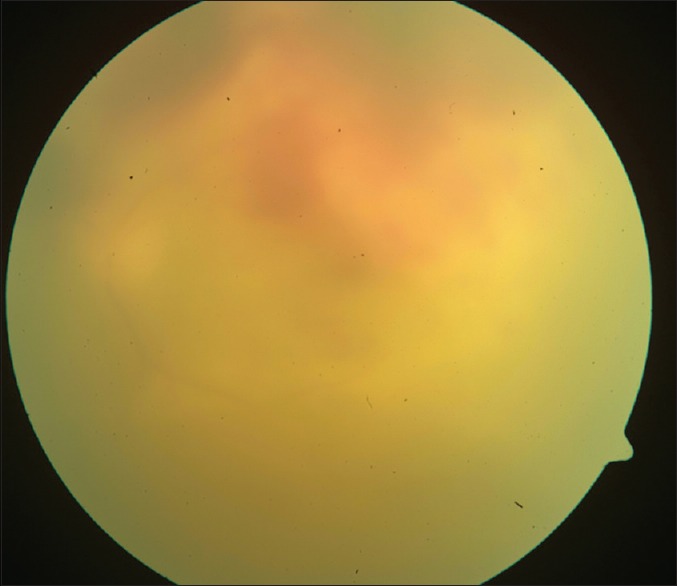
All ill-defined yellowish grey, subretinal elevated lesion
Intraocular lymphoma can be distinguished clinically from uveitis based on two principal features: (1) Lymphoma cells increase by a proliferation in situ rather than by the amplification and recruitment of inflammatory cells that occurs in uveitis; (2) in B-cell PVRL there is predominance of interleukin-10 (IL-10) cytokine, presumably elaborated by the lymphoma cells.[32] It acts as a growth factor for B lymphocytes, along with other mediators, and is also anti-inflammatory, which may stifle immune defenses against the tumor cells and produce the typical “quiet” eye of PVRL.[32] In contrast, the inflammatory milieu in uveitis with high levels of IL-6 is associated with breakdown of vitreous structure with stranding and focal vitreous opacities.[32] There are exceptions to these stereotypical patterns that can complicate clinical diagnosis.[32] Higher degrees of reactive inflammation may occur when there are more reactive T-cell lymphocytes. The presence of iritis and keratic precipitates seem to have racial variation -25% in the Japanese population versus 75% in the French.[32] The most useful signs for distinguishing lymphoma from simulating conditions were better vision, less anterior chamber flare, fewer cases with posterior synechiae, and less optic disc swelling, epiretinal membrane or retinal vasculitis.[44]
Concomitant CNS involvement is present in 16–34% of patients at the time of diagnosis of PVRL [Fig. 4].[18] Subsequent PCNSL occurs in 42–92% patients of PVRL within a mean interval of 8–29 months.[18] Hemiparesis, ataxia and new-onset seizures are the common presentation of CNS involvement.[37,39]
Figure 4.
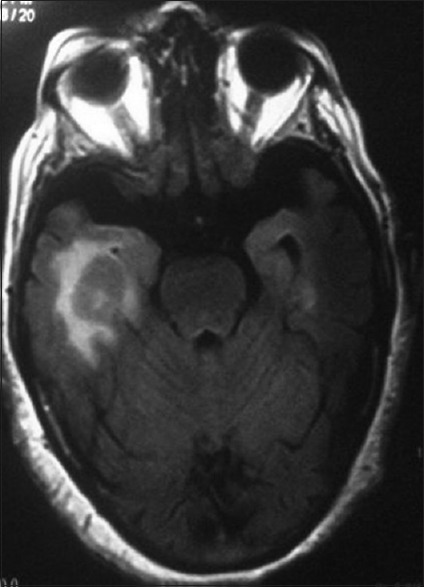
Magnetic resonance imaging showing a homogenously enhancing solid mass lesion abutting right temporal horn of lateral ventricle and a small enhancing lesion in the left temporal lobe
Diagnostic Tests
Although clinical examination and ocular imaging are of help in the diagnosis of PIOL, the gold standard for its diagnosis is a demonstration of neoplastic lymphocytes in the eye.
Optical coherence tomography
Direct infiltration of the retina by lymphoma cells with focal proliferation creates a semi-opaque interface that appears homogenous on optical coherence tomography (OCT). Lymphomatous infiltration can be seen as hyper-reflective signals in the form of dots, bands and nodules at or above the level of RPE with spectral-domain OCT [Fig. 5].[40,41] However, these hyper-reflective signals need to be differentiated from those seen in age-related macular degeneration or diabetic macular edema.[42,43]
Figure 5.
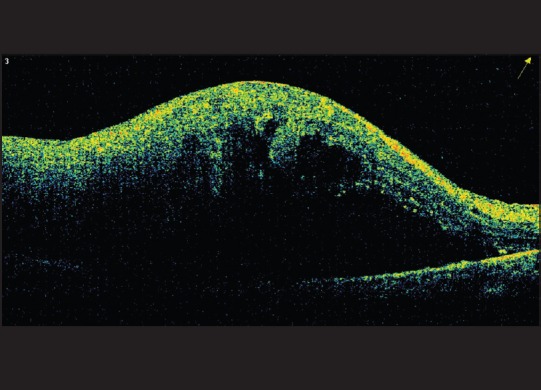
Optical coherence tomography showing elevated neurosensory retina with dense subretinal shadowing
Fundus fluorescein angiography and indocyanine green angiography
Fundus fluorescein angiography (FFA) shows early and late hypofluorescence in cases with outer retinal involvement [Fig. 6].[32] Punctuate hyperfluorescent window defects, round hypofluorescent lesions and vasculitis were observed by Cassoux et al. in their study on 44 patients with PVRL.[33] Leakage of fluorescein along retinal veins and periarteriolar staining have been demonstrated in PVRL.[42] In the early phase of PVRL, small hypofluorescent lesions are seen on indocyanine green angiography (ICGA) while such lesions become less apparent in the later phases.[18] FA and ICGA, when used together, have a positive- and negative-predictive value of 89% and 85% respectively.[44]
Figure 6.
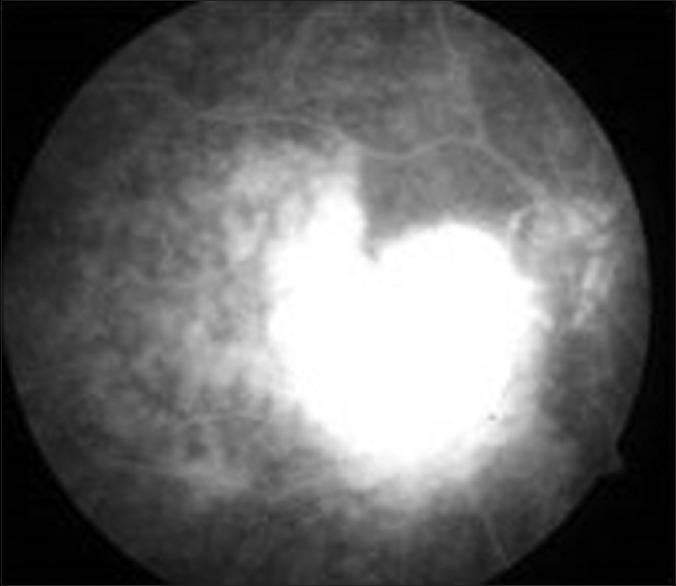
Early patchy hyperfluoresence in the area of the lesion with late leakage seen on fundus fluorescein angiography
Fundus autofluorescence
Fundus autofluorescence (FAF) is a noninvasive technique that can demonstrate a topographic map of the lipofuscin distribution in the RPE cells. One of the characteristic FAF findings in eyes with PVRL is a granular pattern consisting of hyperautofluorescent spots surrounded by a hypoautofluorescent ring.[45] Lymphomatous infiltration in the sub-RPE space alters RPE metabolism leading to hyperautofluorescence.[41] On the other hand, lymphomatous infiltration above the RPE may block autofluorescence from the RPE cells resulting in a granular pattern of hypoautofluorescent spots surrounded by hyperautofluorescent rings.[41] Hypofluorescent spots (leopard spots) seen on FFA correspond to hyperautofluorescent spots on FAF.[41,45] Whitish areas of retinal infiltration appear hypoautofluorescent on FAF and hyperfluorescent on FFA.[45]
Ultrasound B-scan
Although ultrasonological findings are not specific for PVRL, it can be used in cases where posterior segment is difficult to visualize.[18] Findings on ultrasound include vitreous debris, retinal detachment, elevated chorioretinal lesions, and widening of the optic nerve shadow.[18]
Vitreous sampling
Although lymphoma cells are typically located between the RPE and the Bruch's membrane, they may invade the vitreous. Vitreous biopsy and pars plana vitrectomy (PPV) are the two methods practiced to sample the vitreous. Vitreous biopsy essentially samples the core vitreous resulting in the collection of 1–2 ml of undiluted specimen while PPV targets the core and cortical vitreous resulting in collection of 50–100 ml of diluted vitrectomy cassette fluid.[46] In order to ensure adequate and maximum cellular viability, samples need to be transported to the laboratory without delay, preferably within 30 min after sampling. Although many prefer to use a fixative when transportation delay is expected, it is suggested that the sample be fixed immediately.[47] The authors use a fixative containing ethanol, methanol and propranolol in a 8:1:1 ratio. Others prefer Shandon cytofix or HEPES-glutamic acid buffer mediated organic-solvent protection effect (HOPE) solution.[18,46] In addition to maximizing the sample volume, PPV also improves patient's vision by clearance of vitreous debris. The greater cellularity of PPV as compared to core vitreous biopsy also permits performance of additional tests such as immunohistochemistry, DNA extraction for monoclonality and polymerase chain reaction (PCR) studies.[46] Tumor seeding through the sclerotomy port to the epibulbar space is a potential complication of vitreous biopsy.[46,48]
Neuroimaging, cerebrospinal fluid examination and brain biopsy
Since PVRL is a subset of PCNSL, it is crucial that CNS lesions be ruled out with the help of contrast enhanced magnetic resonance imaging (MRI). CNS lesions may be uni- or multi-focal that appear hypodense on T1-weighted and hyperdense on T2-weighted MRI.[18,49] Identification of lymphoma cells in CSF in a patient with concurrent ocular involvement obviates the need for tissue diagnosis of ocular lymphoma and thus spares the patient from an invasive procedure such as vitrectomy. Although the yield of lymphoma cells in CSF is low, up to 25% patients with CNS lesions have a positive CSF cytology.[11,50] Image-guided stereotactic brain biopsy needs to be performed in patients with suspicious MRI lesions but a negative CSF cytology.
Light microscopy
Although malignant lymphoid cells can be visualized using a Papanicolou or Hematoxylin-eosin stain, Giemsa or Diff-Quick stains are preferred since they outline the characteristics of lymphoma cells better. PVRL cells are tightly packed in the sub-RPE space and appear as large lymphoid cells with scanty basophilic cytoplasm and large nuclei which may be round, oval, bean-shaped or hyper-segmented with prominent nucleoli and mitoses [Fig. 7].[52,53,54] Inflammatory cells and necrotic lymphoma cells may be seen in the background.[51,55]
Figure 7.
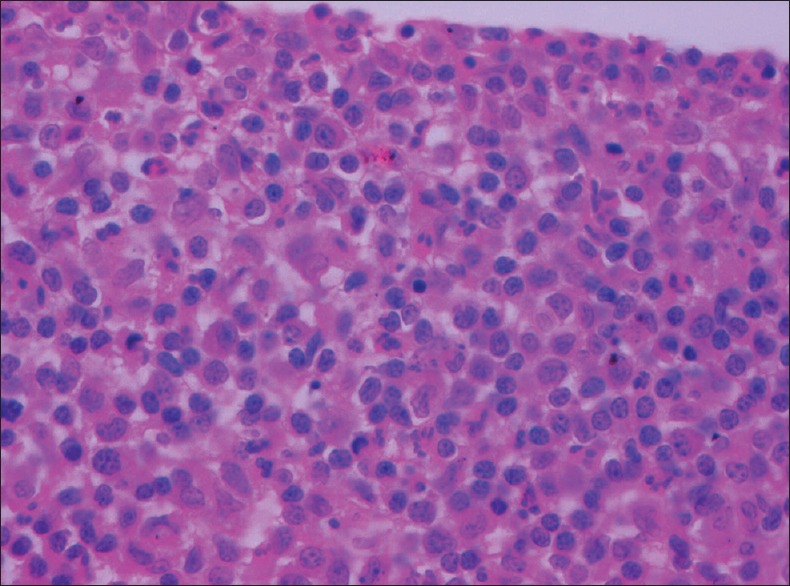
Vitreous biopsy showing large cells with scanty to moderate cytoplasm and vesicular nuclei having prominent nucleoli (H and E, ×400)
Immunohistochemistry and flow cytometry
Demonstration of monoclonality, either as kappa or lambda chain restriction, in the lymphoid cells supports the diagnosis of PVRL. Most PVRL are of B-cell origin thus expressing CD19, CD20 and CD22 [Fig. 8].[56] Concomitant expression of MUM1 and Bcl6 has also been reported.[55] Rare cases of anaplastic large cell and T-cell types of PVRL have been described.[57,58] T-cell lymphomas express CD3 and thus m (fake differentiation from a reactive infiltrate difficult. In such cases, PCR studies for T-cell receptor gene rearrangements help in making the diagnosis of T-cell type PVRL.
Figure 8.
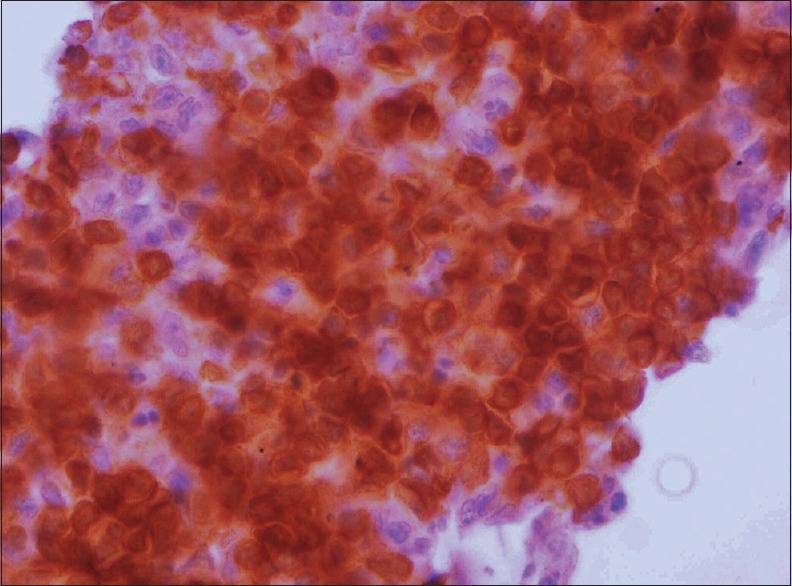
CD20-positivity in tumor cells (CD20, ×400)
Other laboratory tests and genetics of primary vitreoretinal lymphoma
Analysis of IL ratios may help in the diagnosis of lymphoma. Malignant B-cells express high levels of IL-10 while inflammatory cells express high levels of IL-6.[59,60,61] Blood tests for human immunodeficiency virus, complete blood count and test to rule out the causes of uveitis may be useful. The only chromosomal translocation reported in PVRL is t (14;18) involving the bcl2 gene.[21,62]
Differential Diagnosis
The differential diagnosis of PVRL, a challenging masquerade, includes a variety of infectious and noninfectious lesions such as viral retinitis, retinochoroidal toxoplasmosis, syphilitic retinitis, Whipple's disease, sarcoidosis, tuberculosis, Behcet's disease, idiopathic uveitis, endophthalmitis and metastasis.
Treatment
Treatment modalities for PVRL could be intravitreal injection, systemic chemotherapy and radiotherapy, used alone or in appropriate combination. Treatment protocols for PVRL have not been validated. It has not been conclusively demonstrated whether ocular therapy has an impact on the overall patient survival.[4] The international PCNSL collaborative group recommended systemic therapy for PVRL with CNS involvement and a local therapy for disease localized to the eye.[4] Local treatment in the form of intravitreal methotrexate, intravitreal rituximab or low-dose (30–35 Gy) stereotactic external beam radiotherapy to the eye are recommended for PVRL involving a single eye.[11] Intravitreal methotrexate 400 microgram in 0.1 ml in an intensive induction-consolidation-maintenance regimen of 25 injections delivered over 1-year is considered optimal but carries risks of keratopathy and maculopathy, as well as drug resistance.[45] Fewer and widely spaced-out intravitreal methotrexate or rituximab perhaps are the less toxic alternatives.[45] Bilateral ocular disease without concurrent systemic involvement can also be treated with local treatment as per the recommendation of the International PCNSL collaborative group.[4] Systemic chemotherapy in addition to intravitreal therapy or external beam radiation has been suggested for a bilateral disease by the British Neurooncology Society.[4,11] In relapsed or refractory cases, intense chemotherapy with thiotepa, busulfan, and cyclophosphamide combined with hemopoietic stem cell rescue improved a 5-year survival probability to 62% and 5-year event free survival probability to 43.7%.[63]
Prognosis
No consistent data exists on the mortality involved in PVRL due to the diverse patient populations and rarity of this disease.[18] Reports in literature describe mortality rates ranging widely from 9% to 81%.[18] A multicenter study which included 221 patients from 7 different countries recently concluded that ocular therapy improved local tumor control, but had no effect on survival.[64] This study was, however, uncontrolled and retrospective. In another study, a survival advantage was seen in patients diagnosed before CNS involvement (60 months) as compared to the ones diagnosed with CNS involvement (35 months).[65]
Conclusion
Primary vitreoretinal lymphoma is a rare clinical entity that is often misdiagnosed as simulating infectious or inflammatory conditions. High index of clinical suspicion, especially when there is suboptimal response to standard treatment with steroids or recurrence in suspected vitritis/uveits, or to conventional antibiotics in suspected infectious etiology, followed by prompt vitreous/retinal/subretinal biopsy or vitrectomy helps make an early diagnosis and initiate specific treatment. Baseline screening and periodic evaluation for PCNSL helps identify cases with high-risk of mortality and initiate aggressive systemic therapy. The role of the ophthalmologist is mainly to suspect the diagnosis, coordinate with an ocular oncologist and a retina specialist to procure adequate material by an appropriate technique for pathological examination, to collaborate with a pathologist to confirm the diagnosis and then on with an oncologist to help plan treatment, and finally to monitor the patient for response and treatment-related complications. Apart from the ophthalmologist, a retina specialist and an ocular oncologist being aware of clinical manifestations and protocol for diagnosis and management of PVRL, the pathologist should be sensitive to the difficulties involved in obtaining and processing tissue samples. Despite early initiation of treatment, mortality is high with PVRL associated with PCNSL.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Mochizuki M, Singh AD. Epidemiology and clinical features of intraocular lymphoma. Ocul Immunol Inflamm. 2009;17:69–72. doi: 10.1080/09273940902957305. [DOI] [PubMed] [Google Scholar]
- 2.Coupland SE, Damato B. Understanding intraocular lymphomas. Clin Experiment Ophthalmol. 2008;36:564–78. doi: 10.1111/j.1442-9071.2008.01843.x. [DOI] [PubMed] [Google Scholar]
- 3.Cao X, Shen D, Callanan DG, Mochizuki M, Chan CC. Diagnosis of systemic metastatic retinal lymphoma. Acta Ophthalmol. 2011;89:e149–54. doi: 10.1111/j.1755-3768.2009.01797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan CC, Rubenstein JL, Coupland SE, Davis JL, Harbour JW, Johnston PB, et al. Primary vitreoretinal lymphoma: A report from an International Primary Central Nervous System Lymphoma Collaborative Group symposium. Oncologist. 2011;16:1589–99. doi: 10.1634/theoncologist.2011-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sen HN, Bodaghi B, Hoang PL, Nussenblatt R. Primary intraocular lymphoma: Diagnosis and differential diagnosis. Ocul Immunol Inflamm. 2009;17:133–41. doi: 10.1080/09273940903108544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Triebenstein O. Ein beitrag zur frage der aleukämischen augenveränderungen. Klin Monatsbl Augenheilkd. 1920;64:825–36. [Google Scholar]
- 7.Cooper EL, Riker JL. Malignant lymphoma of the uveal tract. Am J Ophthalmol. 1951;34:1153–8. doi: 10.1016/0002-9394(51)90691-5. [DOI] [PubMed] [Google Scholar]
- 8.Givner I. Malignant lymphoma with ocular involvement; a clinico-pathologic report. Am J Ophthalmol. 1955;39:29–32. doi: 10.1016/0002-9394(55)92650-7. [DOI] [PubMed] [Google Scholar]
- 9.Klingele TG, Hogan MJ. Ocular reticulum cell sarcoma. Am J Ophthalmol. 1975;79:39–47. doi: 10.1016/0002-9394(75)90453-5. [DOI] [PubMed] [Google Scholar]
- 10.Vogel MH, Font RL, Zimmerman LE, Levine RA. Reticulum cell sarcoma of the retina and uvea. Report of six cases and review of the literature. Am J Ophthalmol. 1968;66:205–15. doi: 10.1016/0002-9394(68)92065-5. [DOI] [PubMed] [Google Scholar]
- 11.Chan CC, Sen HN. Current concepts in diagnosing and managing primary vitreoretinal (intraocular) lymphoma. Discov Med. 2013;15:93–100. [PMC free article] [PubMed] [Google Scholar]
- 12.Char DH, Ljung BM, Miller T, Phillips T. Primary intraocular lymphoma (ocular reticulum cell sarcoma) diagnosis and management. Ophthalmology. 1988;95:625–30. doi: 10.1016/s0161-6420(88)33145-3. [DOI] [PubMed] [Google Scholar]
- 13.Char DH, Ljung BM, Deschênes J, Miller TR. Intraocular lymphoma: Immunological and cytological analysis. Br J Ophthalmol. 1988;72:905–11. doi: 10.1136/bjo.72.12.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qualman SJ, Mendelsohn G, Mann RB, Green WR. Intraocular lymphomas. Natural history based on a clinicopathologic study of eight cases and review of the literature. Cancer. 1983;52:878–86. doi: 10.1002/1097-0142(19830901)52:5<878::aid-cncr2820520523>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 15.Jaffe ES, Harris NL, Stein H, Vardman JS. Pathology and Genetics. 4th ed. Lyon: IARC Press; 2008. Tumours of hematologic and lymphoid tissues 2007. World Health Organization. Classification of Tumours. [Google Scholar]
- 16.Coupland SE, Chan CC, Smith J. Pathophysiology of retinal lymphoma. Ocul Immunol Inflamm. 2009;17:227–37. doi: 10.1080/09273940903168696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eagle RC., Jr The pathology of ocular cancer. Eye (Lond) 2013;27:128–36. doi: 10.1038/eye.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sagoo MS, Mehta H, Swampillai AJ, Cohen VM, Amin SZ, Plowman PN, et al. Primary intraocular lymphoma. Surv Ophthalmol. 2014;59:503–16. doi: 10.1016/j.survophthal.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Bardenstein DS. Intraocular lymphoma. Cancer Control. 1998;5:317–25. doi: 10.1177/107327489800500403. [DOI] [PubMed] [Google Scholar]
- 20.Schabet M. Epidemiology of primary CNS lymphoma. J Neurooncol. 1999;43:199–201. doi: 10.1023/a:1006290032052. [DOI] [PubMed] [Google Scholar]
- 21.Chan CC. Molecular pathology of primary intraocular lymphoma. Trans Am Ophthalmol Soc. 2003;101:275–92. [PMC free article] [PubMed] [Google Scholar]
- 22.Chan SM, Hutnik CM, Heathcote JG, Orton RB, Banerjee D. Iris lymphoma in a pediatric cardiac transplant recipient: Clinicopathologic findings. Ophthalmology. 2000;107:1479–82. doi: 10.1016/s0161-6420(00)00172-x. [DOI] [PubMed] [Google Scholar]
- 23.Wender A, Adar A, Maor E, Yassur Y. Primary B-cell lymphoma of the eyes and brain in a 3-year-old boy. Arch Ophthalmol. 1994;112:450–1. doi: 10.1001/archopht.1994.01090160024009. [DOI] [PubMed] [Google Scholar]
- 24.Sobrin L, Dubovy SR, Davis JL, Murray TG. Isolated, bilateral intraocular lymphoma in a 15-year-old girl. Retina. 2005;25:370–3. doi: 10.1097/00006982-200504000-00021. [DOI] [PubMed] [Google Scholar]
- 25.Cassoux N, Merle-Beral H, Leblond V, Bodaghi B, Miléa D, Gerber S, et al. Ocular and central nervous system lymphoma: Clinical features and diagnosis. Ocul Immunol Inflamm. 2000;8:243–50. doi: 10.1076/ocii.8.4.243.6463. [DOI] [PubMed] [Google Scholar]
- 26.Sugita S, Takase H, Sugamoto Y, Arai A, Miura O, Mochizuki M. Diagnosis of intraocular lymphoma with a combination of polymerase chain reaction and cytokine profile in the vitreous fluids. Jpn J Ophthalmol. 2009;53:209–14. doi: 10.1007/s10384-009-0662-y. [DOI] [PubMed] [Google Scholar]
- 27.Chan CC, Shen DF, Whitcup SM, Nussenblatt RB. Detection of human herpesvirus-8 and Epstein-Barr virus DNA in primary intraocular lymphomas. Blood. 1999;93:2749–51. [PubMed] [Google Scholar]
- 28.Sauer TC, Meyers SM, Shen D, Vegh S, Vygantas C, Chan CC. Primary intraocular (retinal) lymphoma after ocular toxoplasmosis. Retin Cases Brief Rep. 2010;4:160–3. doi: 10.1097/ICB.0b013e3181ad3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan LD. Human herpesvirus-8: Kaposi sarcoma, multicentric Castleman disease, and primary effusion lymphoma. Hematology Am Soc Hematol Educ Program. 2013;2013:103–8. doi: 10.1182/asheducation-2013.1.103. [DOI] [PubMed] [Google Scholar]
- 30.Chan CC, Shen D, Hackett JJ, Buggage RR, Tuaillon N. Expression of chemokine receptors, CXCR4 and CXCR5, and chemokines, BLC and SDF-1, in the eyes of patients with primary intraocular lymphoma. Ophthalmology. 2003;110:421–6. doi: 10.1016/S0161-6420(02)01737-2. [DOI] [PubMed] [Google Scholar]
- 31.McCann KJ, Ashton-Key M, Smith K, Stevenson FK, Ottensmeier CH. Primary central nervous system lymphoma: Tumor-related clones exist in the blood and bone marrow with evidence for separate development. Blood. 2009;113:4677–80. doi: 10.1182/blood-2008-09-179366. [DOI] [PubMed] [Google Scholar]
- 32.Davis JL. Intraocular lymphoma: A clinical perspective. Eye (Lond) 2013;27:153–62. doi: 10.1038/eye.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cassoux N, Merle-Beral H, Leblond V, Bodaghi B, Miléa D, Gerber S, et al. Ocular and central nervous system lymphoma: Clinical features and diagnosis. Ocul Immunol Inflamm. 2000;8:243–50. doi: 10.1076/ocii.8.4.243.6463. [DOI] [PubMed] [Google Scholar]
- 34.Coupland SE, Heimann H, Bechrakis NE. Primary intraocular lymphoma: A review of the clinical, histopathological and molecular biological features. Graefes Arch Clin Exp Ophthalmol. 2004;242:901–13. doi: 10.1007/s00417-004-0973-0. [DOI] [PubMed] [Google Scholar]
- 35.Lobo A, Larkin G, Clark BJ, Towler HM, Lightman S. Pseudo-hypopyon as the presenting feature in B-cell and T-cell intraocular lymphoma. Clin Experiment Ophthalmol. 2003;31:155–8. doi: 10.1046/j.1442-9071.2003.00624.x. [DOI] [PubMed] [Google Scholar]
- 36.Dean JM, Novak MA, Chan CC, Green WR. Tumor detachments of the retinal pigment epithelium in ocular/central nervous system lymphoma. Retina. 1996;16:47–56. doi: 10.1097/00006982-199616010-00009. [DOI] [PubMed] [Google Scholar]
- 37.Gill MK, Jampol LM. Variations in the presentation of primary intraocular lymphoma: Case reports and a review. Surv Ophthalmol. 2001;45:463–71. doi: 10.1016/s0039-6257(01)00217-x. [DOI] [PubMed] [Google Scholar]
- 38.Matzkin DC, Slamovits TL, Rosenbaum PS. Simultaneous intraocular and orbital non-Hodgkin lymphoma in the acquired immune deficiency syndrome. Ophthalmology. 1994;101:850–5. doi: 10.1016/s0161-6420(94)31248-6. [DOI] [PubMed] [Google Scholar]
- 39.Hochberg FH, Miller DC. Primary central nervous system lymphoma. J Neurosurg. 1988;68:835–53. doi: 10.3171/jns.1988.68.6.0835. [DOI] [PubMed] [Google Scholar]
- 40.Jang HS, Sepah YJ, Sophie R, Bittencourt MG, Ferraz D, Hanout M, et al. Longitudinal spectral domain optical coherence tomography changes in eyes with intraocular lymphoma. J Ophthalmic Inflamm Infect. 2013;3:59. doi: 10.1186/1869-5760-3-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Egawa M, Mitamura Y, Hayashi Y, Naito T. Spectral-domain optical coherence tomographic and fundus autofluorescence findings in eyes with primary intraocular lymphoma. Clin Ophthalmol. 2014;8:335–41. doi: 10.2147/OPTH.S58114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Folgar FA, Chow JH, Farsiu S, Wong WT, Schuman SG, O’Connell RV, et al. Spatial correlation between hyperpigmentary changes on color fundus photography and hyperreflective foci on SDOCT in intermediate AMD. Invest Ophthalmol Vis Sci. 2012;53:4626–33. doi: 10.1167/iovs.12-9813. [DOI] [PubMed] [Google Scholar]
- 43.Ota M, Nishijima K, Sakamoto A, Murakami T, Takayama K, Horii T, et al. Optical coherence tomographic evaluation of foveal hard exudates in patients with diabetic maculopathy accompanying macular detachment. Ophthalmology. 2010;117:1996–2002. doi: 10.1016/j.ophtha.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 44.Fardeau C, Lee CP, Merle-Béral H, Cassoux N, Bodaghi B, Davi F, et al. Retinal fluorescein, indocyanine green angiography, and optic coherence tomography in non-Hodgkin primary intraocular lymphoma. Am J Ophthalmol. 2009;147:886–94. 894.e1. doi: 10.1016/j.ajo.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 45.Casady M, Faia L, Nazemzadeh M, Nussenblatt R, Chan CC, Sen HN. Fundus autofluorescence patterns in primary intraocular lymphoma. Retina. 2014;34:366–72. doi: 10.1097/IAE.0b013e31829977fa. [DOI] [PubMed] [Google Scholar]
- 46.Mudhar HS, Sheard R. Diagnostic cellular yield is superior with full pars plana vitrectomy compared with core vitreous biopsy. Eye (Lond) 2013;27:50–5. doi: 10.1038/eye.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitcup SM, Chan CC, Buggage RR, Nussenblatt RB, Byrnes GA, Rubin BI. Improving the diagnostic yield of vitrectomy for intraocular lymphoma. Arch Ophthalmol. 2000;118:446. [PubMed] [Google Scholar]
- 48.Cursiefen C, Holbach LM, Lafaut B, Heimann K, Kirchner T, Naumann GO. Oculocerebral non-Hodgkin's lymphoma with uveal involvement: Development of an epibulbar tumor after vitrectomy. Arch Ophthalmol. 2000;118:1437–40. doi: 10.1001/archopht.118.10.1437. [DOI] [PubMed] [Google Scholar]
- 49.Jack CR, Jr, O’Neill BP, Banks PM, Reese DF. Central nervous system lymphoma: Histologic types and CT appearance. Radiology. 1988;167:211–5. doi: 10.1148/radiology.167.1.3279454. [DOI] [PubMed] [Google Scholar]
- 50.DeAngelis LM. Primary central nervous system lymphomas. Curr Treat Options Oncol. 2001;2:309–18. doi: 10.1007/s11864-001-0024-6. [DOI] [PubMed] [Google Scholar]
- 51.Gonzales JA, Chan CC. Biopsy techniques and yields in diagnosing primary intraocular lymphoma. Int Ophthalmol. 2007;27:241–50. doi: 10.1007/s10792-007-9065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whitcup SM, de Smet MD, Rubin BI, Palestine AG, Martin DF, Burnier M, Jr, et al. Intraocular lymphoma. Clinical and histopathologic diagnosis. Ophthalmology. 1993;100:1399–406. doi: 10.1016/s0161-6420(93)31469-7. [DOI] [PubMed] [Google Scholar]
- 53.Michels RG, Knox DL, Erozan YS, Green WR. Intraocular reticulum cell sarcoma. Diagnosis by pars plana vitrectomy. Arch Ophthalmol. 1975;93:1331–5. doi: 10.1001/archopht.1975.01010020961005. [DOI] [PubMed] [Google Scholar]
- 54.Chan CC, Buggage RR, Nussenblatt RB. Intraocular lymphoma. Curr Opin Ophthalmol. 2002;13:411–8. doi: 10.1097/00055735-200212000-00012. [DOI] [PubMed] [Google Scholar]
- 55.Chan CC, Wallace DJ. Intraocular lymphoma: Update on diagnosis and management. Cancer Control. 2004;11:285–95. doi: 10.1177/107327480401100502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan CC, Gonzalez JA. Primary Intraocular Lymphoma. Hackensack, NJ: World Scientific Publishing Co. Pte. Ltd.; 2007. [Google Scholar]
- 57.Mihaljevic B, Sretenovic A, Jakovic L, Jovanovic MP, Kovacevic D, Rasic D, et al. A case of primary peripheral T-cell type non-Hodgkin lymphoma originating in the iris - Clinicopathological findings. Vojnosanit Pregl. 2010;67:1025–8. [PubMed] [Google Scholar]
- 58.Park CY, Hwang SW, Kim do Y, Huh HJ, Oh JH. Anaplastic large cell lymphoma involving anterior segment of the eye. Korean J Ophthalmol. 2014;28:108–12. doi: 10.3341/kjo.2014.28.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blay JY, Favrot M, Rossi JF, Wijdenes J. Role of interleukin-6 in paraneoplastic thrombocytosis. Blood. 1993;82:2261–2. [PubMed] [Google Scholar]
- 60.Chan CC, Whitcup SM, Solomon D, Nussenblatt RB. Interleukin-10 in the vitreous of patients with primary intraocular lymphoma. Am J Ophthalmol. 1995;120:671–3. doi: 10.1016/s0002-9394(14)72217-2. [DOI] [PubMed] [Google Scholar]
- 61.Cassoux N, Merle-Beral H, Lehoang P, Herbort C, Chan CC. Interleukin-10 and intraocular-central nervous system lymphoma. Ophthalmology. 2001;108:426–7. doi: 10.1016/s0161-6420(00)00401-2. [DOI] [PubMed] [Google Scholar]
- 62.Wallace DJ, Shen D, Reed GF, Miyanaga M, Mochizuki M, Sen HN, et al. Detection of the bcl-2 t (14;18) translocation and proto-oncogene expression in primary intraocular lymphoma. Invest Ophthalmol Vis Sci. 2006;47:2750–6. doi: 10.1167/iovs.05-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soussain C, Choquet S, Fourme E, Delgadillo D, Bouabdallah K, Ghesquières H, et al. Intensive chemotherapy with thiotepa, busulfan and cyclophosphamide and hematopoietic stem cell rescue in relapsed or refractory primary central nervous system lymphoma and intraocular lymphoma: A retrospective study of 79 cases. Haematologica. 2012;97:1751–6. doi: 10.3324/haematol.2011.060434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grimm SA, McCannel CA, Omuro AM, Ferreri AJ, Blay JY, Neuwelt EA, et al. Primary CNS lymphoma with intraocular involvement: International PCNSL Collaborative Group Report. Neurology. 2008;71:1355–60. doi: 10.1212/01.wnl.0000327672.04729.8c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hormigo A, Abrey L, Heinemann MH, DeAngelis LM. Ocular presentation of primary central nervous system lymphoma: Diagnosis and treatment. Br J Haematol. 2004;126:202–8. doi: 10.1111/j.1365-2141.2004.05028.x. [DOI] [PubMed] [Google Scholar]