

Abstract
Tumors of the Ocular Surface clinically manifest with a very wide spectrum and include several forms of epithelial, stromal, caruncular, and secondary tumors. As a group, these tumors are seen commonly in the clinical practice of a comprehensive ophthalmologist, cornea specialist, and an ocular oncologist. This review is aimed to discuss the common tumors of the ocular surface and emphasize on their clinical diagnosis and appropriate management.
Keywords: Conjunctiva, cornea, lymphoma, melanoma, ocular surface, ocular surface squamous neoplasia, tumor
Tumors of the ocular surface have a wide clinical spectrum and include several forms of epithelial, stromal, caruncular, and secondary tumors [Table 1]. As a group, these tumors are seen commonly in the clinical practice of a comprehensive ophthalmologist, cornea specialist, and an ocular oncologist. This review is aimed to discuss the common tumors of the ocular surface, their clinical diagnosis, and management.
Table 1.
Classification of tumors of the ocular surface

Epithelial Tumors of the Ocular Surface
Epithelial tumors of the ocular surface can be nonmelanocytic or melanocytic [Table 2].
Table 2.
Epithelial tumors of the ocular surface
Nonmelanocytic epithelial tumors
Squamous papilloma
Squamous papilloma occurs in both children and adults with variable presentation.[1] In children, it results from infection of the conjunctival epithelium with human papilloma virus (HPV) 6, 11 or 16.[3] It could be solitary or multiple, and sessile or pedunculated. It becomes confluent in extreme cases to form massive papillomatosis. It appears most often as a pink or red mass with fleshy frond or finger-like projections in the inferior fornix, most common medially. It is also seen in bulbar conjunctiva but rarely in the cornea.
In adults, clinically, it may resemble squamous cell carcinoma (SCC). It may be associated with HPV infection and immunocompromised status.[4] Usually, unilateral and solitary, it is seen at the limbus, encroaching the cornea as it grows. It can also arise in the caruncle. It has a lighter pink color than the childhood variety. Rarely, it can be pigmented particularly in those with dark skin.[5] Squamous papilloma is reported to have low malignant potential. Occasionally, as a variant, it can assume an inverted growth pattern, which has a greater tendency towards malignant transformation into transitional cell carcinoma, SCC, or mucoepidermoid carcinoma.[2,6,7] Histopathologically, the lesion shows numerous vascularized papillary fronds lined by acanthotic epithelium [Fig. 1].
Figure 1.
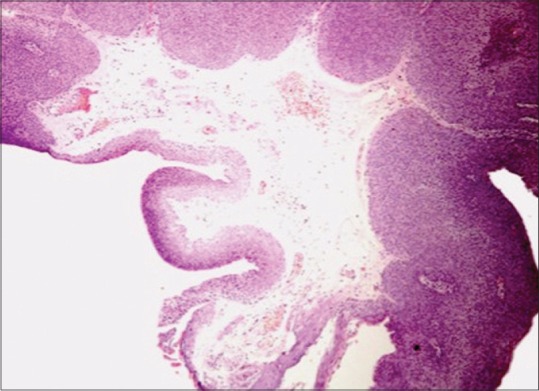
Microphotograph of squamous papilloma (OM ×4). The lesion shows numerous vascularized papillary fronds lined by acanthotic epithelium
Surgical excision by the “no-touch technique” followed by cryotherapy is the treatment of choice.[8] Other reported treatment modalities include laser, dinitrochlorobenzene immunotherapy, interferon alpha 2b (IFN α-2b), and topical mitomycin C (MMC) drops.[9,10,11,12,13] Recent reports show significant role of oral cimetidine in treating recalcitrant and recurrent conjunctival papillomatosis. It enhances the immune system by inhibiting certain T-cell functions.[14]
Inverted papilloma
The papilloma may invaginate inward into the underlying conjunctiva and substantia propria to present as a mixed inverted exophytic papilloma.[15] Rarely, it appears as solid or cystic solitary nodule at the limbus, plica semilunaris, and tarsal conjunctiva. Treatment is by local excision.[16]
Conjunctival pseudoepitheliomatous hyperplasia
It is a benign reactive inflammatory proliferation of the epithelial cells, which simulates carcinoma clinically and histopathologically.[1] It occurs as a conjunctival lesion secondary to irritation by concurrent or preexisting stromal inflammation such as pterygium, pinguecula, allergic conjunctivitis, and foreign body. It appears as an elevated leukoplakic pink lesion in the limbal area [Fig. 2]. Histopathologically, it is characterized by massive acanthosis, hyperkeratosis, and parakeratosis of the conjunctival epithelium. Complete excision and additional cryotherapy would constitute optimal management, as difficulty prevails in clinically and histologically differentiating the lesion from low-grade SCC.
Figure 2.
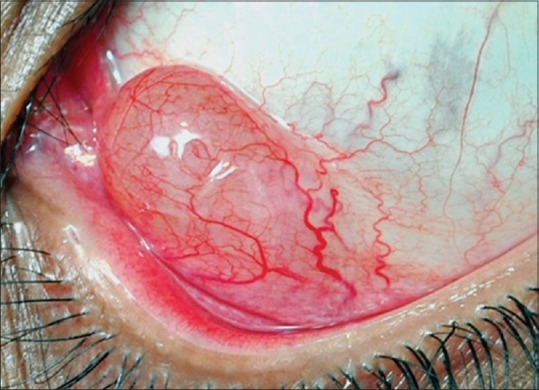
Conjunctival inclusion cyst, a smooth translucent lesion with turbid fluid
Keratoacanthoma
It is a variant of conjunctival pseudoepitheliomatous hyperplasia. Though it is a benign lesion, some believe that it may represent an abortive malignancy that rarely progresses to SCC. An elevated mass with hyperkeratosis or leukoplakia is the usual presentation.[1,17,18,19] Onset as well as the progression is rapid. Treatment is by complete excision and cryotherapy.
Dacryoadenoma
Dacryoadenoma is a rare condition affecting children and young adults. It appears as a translucent and fleshy pink lesion in the bulbar, forniceal or palpebral conjunctiva.[1,2,20] It arises from the surface epithelium and proliferates inward into the stroma and develops glandular lobules similar to those seen in normal lacrimal glands but with abundant goblet cells. Complete excision is the treatment of choice.
Conjunctival epithelial cyst
Conjunctival epithelial cyst can be congenital or acquired.[1,2] There are two subtypes-inclusion cysts and ductal cysts. Inclusion cysts are further classified as spontaneous or posttraumatic. These cysts typically are smooth translucent lesions containing clear fluid. The contents may be turbid, containing epithelial debris seen layered like pseudohypopyon [Fig. 2]. Ductal cysts are lined by two layers of the epithelium and may contain secretory material. The cysts may remain stable and asymptomatic, and rarely undergo spontaneous resolution. Excision is the treatment of choice for a cyst that enlarges or becomes symptomatic.
Hereditary benign intraepithelial dyskeratosis
This is a rare autosomal dominant condition of the conjunctiva and other mucous membranes.[1,2,21,22] It is specifically seen among the inbred Caucasians, African-Americans, and Native Americans known as Haliwa Indians but, is also seen in the population of other descent. The lesion generally presents in the first decade of life as an elevated hyperemic and fleshy V-shaped plaque on the nasal and temporal bulbar conjunctiva and limbus [Fig. 3]. It may be asymptomatic or, can cause redness and discomfort. Severe form can lead to corneal vascularization, opacification, and loss of vision.[2,22] Smaller symptomatic lesions are treated conservatively with lubricants and topical steroids while larger lesions undergo local resection with ocular surface reconstruction. It carries no risk of malignancy, but recurrence is common. Hence, complete excision with clear margins is warranted. Histopathologically, there is marked acanthosis and hyperkeratosis of the conjunctival epithelium with prominent dyskeratosis.
Figure 3.
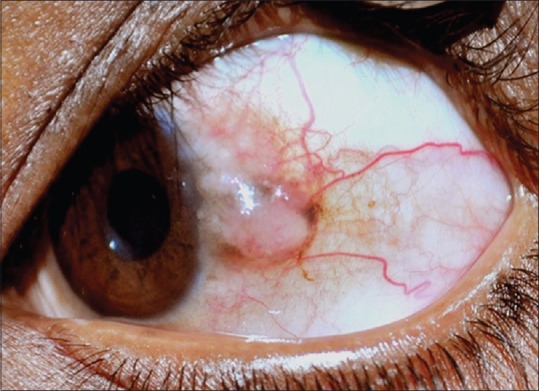
Pseudoepitheliomatous hyperplasia. Elevated leukoplakic pink lesion in the temporal limbal area with apparent feeder vessels and pigmentation. Note that it closely mimics a nodular ocular surface squamous neoplasia
Conjunctival keratotic plaque
This leukoplakic lesion that may develop in the bulbar conjunctiva is usually in the interpalpebral region. There is thickening and keratinization of conjunctival epithelium characterized mainly by acanthosis, hyperkeratosis, and parakeratosis. Dyskeratosis is always absent. It has little or no malignant potential.
Actinic keratosis
Seen commonly as a focal leukoplakic lesion occurring at the interpalpebral area presenting as flat, white plaque sometimes with a frothy covering, actinic keratosis progresses very gradually and shows no tendency towards aggressive growth.[1,2,23] Clinically, it may often be indistinguishable from conjunctival intraepithelial neoplasia (CIN) [Fig. 4]. Rose bengal staining of the surface of the lesion tips the clinical suspicion in favor of CIN. Actinic keratosis is a relative indication for surgical excision and supplemental cryotherapy. Close observation of the lesion until progression is documented is a reasonable option.
Figure 4.
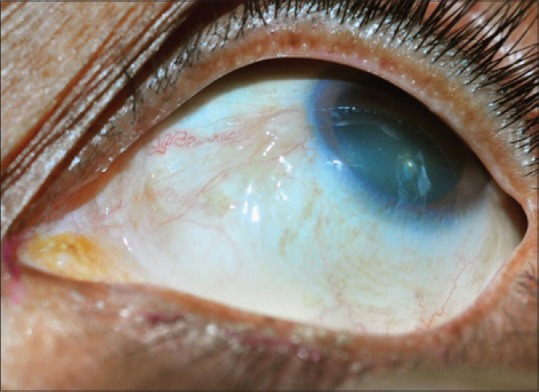
Actinic keratosis, a focal leukoplakic lesion seen in the interpalpebral area. It can easily be misdiagnosed as pinguecula
Ocular surface squamous neoplasia
Ocular surface squamous neoplasia (OSSN) is an encompassing term for precancerous and cancerous epithelial lesions of the conjunctiva and cornea. It includes the spectrum of dysplasia, CIN, and invasive SCC.[1,24,25] Previously used terms for this condition include intraepithelial epithelioma, Bowens disease, and Bowenoid epithelioma.[26] CIN accounts for 39% of all premalignant and malignant lesions of the conjunctiva and for 4% of all conjunctival lesions.[27] Unlike CIN, incidence of invasive SCC is of much lesser frequency, varying from 0.02 to 3.5/100,000 population.[29] About 75% occur in men, 75% are diagnosed in older patients and over 75% occur at the limbus.[2,27] Factors associated with the development of OSSN are exposure to sunlight, HPV type 16 infections, and immunocompromised status.[2,24] Studies have shown interaction of AIDS with ultraviolet-induced damage and HPV infection. Systemic associations of the development of OSSN include xeroderma pigmentosum and Papillon-Leferve syndrome.
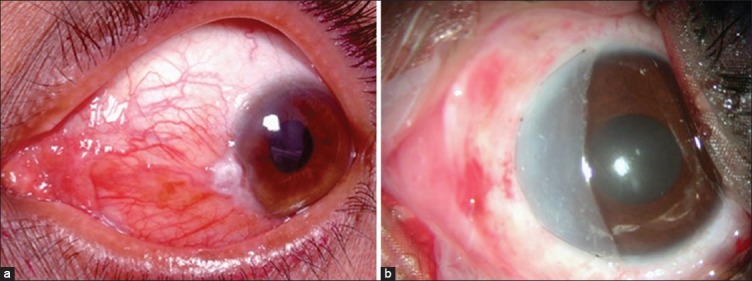
Ocular surface squamous neoplasia is mostly unilateral and is seen in middle-aged and older patients. Rarely, it is bilateral in immunosuppressed patients. It often presents with redness and ocular irritation. Vision is usually unaffected unless it encroaches the center of the cornea. The tumor appears as fleshy or nodular, sessile minimally elevated lesion with surface keratin, feeder vessels, and secondary inflammation.[1,2,24,28] Rose bengal staining is helpful in the diagnosis and delineation of the tumor extent [Fig. 5d]. The tumor may extend for a variable distance into the adjacent corneal epithelium and appear as a subtle wavy, advancing, gray, superficial opacity that may be relatively avascular or may have fine blood vessels. Whereas some lesions may present as diffuse gelatinous or papilliform form usually encroaching the cornea [Fig. 5]. Primary corneal dysplasia affects the corneal epithelium with minimal limbal involvement.[28,29,30] Primary SCC of the cornea is rare.
Figure 5.
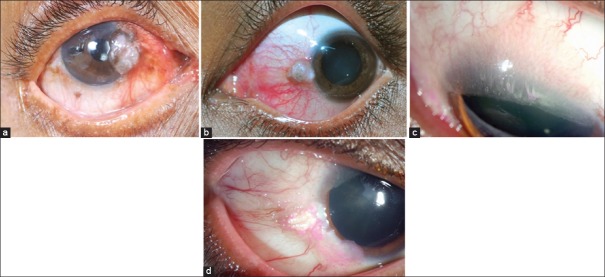
Ocular surface squamous neoplasia. (a) Elevated nasal limbal mass involving the cornea with abundant keratin and feeder vessels. (b) Pigmented variant seen as a nodular mass. (c) Diffuse, elevated, papilliform lesion involving the entire ocular surface with intrinsic vessels. (d) Gelatinous type with episcleral feeder vessels with speckled rose bengal staining
There are no consistent clinical criteria for distinguishing CIN from invasive SCC. Leukoplakia is usually absent or minimal in CIN; extensive leukoplakia raises the suspicion of malignancy. The presence of feeder vessels and intrinsic vascularity favors SCC. Although greater thickness is believed to be a sign of malignant transformation, there are thick tumors that remain within the epithelium. The nodular lesion causes suspicion of invasive SCC. A diffuse lesion can masquerade as chronic conjunctivitis[31] [Table 3]. It is also important to examine the tarsal conjunctiva after everting the eyelid of patients with OSSN to detect contiguous or multifocal involvement of the tarsal conjunctiva.
Table 3.
Morphological types of OSSN

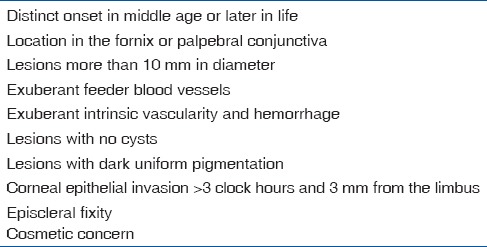
Advanced cases can infiltrate the cornea and sclera to have the intraocular extension.[32] Rarely, the tumor may extend into the orbit causing proptosis. The tumor can metastasize to the regional lymph nodes and rarely distant metastasis may occur. Aggressive variants include spindle cell squamous carcinoma, mucoepidermoid carcinoma, and adenoid SCC.[1,2,24,33,34] According to the American Joint Committee on Cancer (AJCC)-tumor, node, and metastasis (TNM) classification, SCC is classified depending on the size, tumor location, and extent of involvement [Table 4].
Table 4.
AJCC-TNM classification of SCC of conjunctiva
Histopathologically, mild CIN (dysplasia) is characterized by partial thickness replacement of the epithelium by anaplastic cells that lack normal maturation. Severe CIN is characterized by full thickness replacement of the epithelium by similar cells. Both the variants show a characteristic abrupt demarcation between affected epithelium and normal epithelium. Invasive SCC shows a breach in the basement membrane of basal epithelium and is typically a fairly well-differentiated neoplasm composed of abnormal epithelial cells with mitotic activity and keratin. Occasionally, it can be poorly differentiated and shows bizarre, pleomorphic cells, numerous mitotic figures, acanthosis, and dyskeratosis[2] [Fig. 6].
Figure 6.
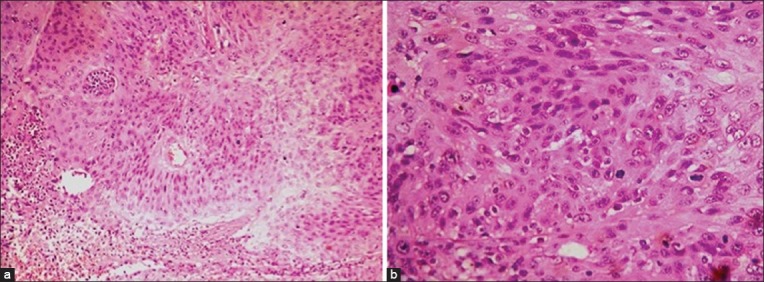
Histopathology of ocular surface squamous neoplasia (OSSN). (a) Microphotograph of OSSN showing abnormal epithelial cells with mitotic activity (OM ×10). (b) Seen at higher magnification (OM ×40)
Complete but gentle surgical excision using a technique without touching the tumor called the “no-touch” technique [Table 5] is the treatment of choice.[1,23,24,25,26,27,28,29,30,31,32,33,34,35] The steps of surgical excision include:
Table 5.
Tips to surgical excision of OSSN

Conjunctival incision is made approximately 4 mm outside the clinically determined tumor margin. The incision incorporates full thickness conjunctiva and tenon's fascia
Dissection is carried out in the episcleral plane (if there is no episcleral adhesion) to reach the limbus
Thin lamellae of the tumor-free sclera, 0.2 mm in depth including 2.0 mm outside the adherent conjunctival mass are removed if scleral fixity is noted
Absolute alcohol is applied with cotton-tipped applicator to the involved cornea to allow for controlled corneal epitheliectomy 2 mm outside the corneal component
The corneal epithelium is scrolled off to the limbus using a controlled sweeping motion with a beaver blade
The entire tumor is removed in one piece without touching the tumor by excising it along the limbus
Cryotherapy, double freeze-thaw cycle, is applied to the edge of the remaining bulbar conjunctiva and the scleral base if there was episcleral adhesion. Limbal cryotherapy should be limited to 6 clock hours
Excision is followed by direct closure of the conjunctiva or with amniotic membrane graft [Fig. 7].
Figure 7.
Primary treatment of ocular surface squamous neoplasia. (a) Nodular lesion with abundant keratin at the limbus. (b) Immediate postoperative appearance following complete excision with margin clearance, double freeze cryotherapy and ocular surface reconstruction with amniotic membrane transplantation with tissue glue
Reported recurrence rate of OSSN is 15–52%. Lee and Hirst reported a 17% recurrence after excision of conjunctival dysplasia, 40% after excision of CIN and 30% for SCC of the conjunctiva.[24] However, with the protocol-based technique as described above, the recurrence rate can be limited to <5%.
Plaque brachytherapy is used to control gross or microscopic residual tumors. More extensive orbital invasion requires orbital exenteration.[2,44]
Conjunctival intraepithelial neoplasia and mild forms of SCC can be treated with topical MMC 0.02–0.04% [Tables 6 and 7].[1,2,24,25,36,37,38] There are several protocols [Table 6] but a dosage of 0.04%, qid, 4-days-a-week for 4 weeks works best in our experience. Associated local complications include conjunctival hyperemia, punctate corneal erosions, and inadvertent prolonged use may lead to scleral melt. Interferon alpha 2b is currently most accepted and favorable form of treatment for OSSN. It is less toxic with fewer complications compared to topical MMC. Their beneficial role includes immunomodulation, anti-proliferative, and anti-viral. The reported success rate is 83% with topical IFN 1 million IU administered 4 times daily for 6–12 months.[39,40] Intralesional injection of IFN α-2b of 3 million IU to 10 million IU combined with 1 million IU topical IFN is widely used as a primary modality (immunotherapy) for unresectable extensive tumors.[39,40,41] It also helps in reducing the tumor size (immunoreduction), ideally followed by complete excision of the residual tumor. Similarly it plays a major role in immunosuppressed patients with OSSN, with a high rate of recurrence.[42] The long-term use of topical IFN helps to prevent recurrence by way of immunomodulation.[41] The most common complication is transient flu-like syndrome, whereas local complications are minimal[36,37,38,39,40] [Table 8]. Other available drugs are 5-fluorouracil and cidofovir.[43]
Table 6.
Protocol for topical MMC

Table 7.
Indications for topical chemotherapy in noninvasive OSSN

Table 8.
Topical chemotherapeutic agents for OSSN

Ocular surface squamous neoplasia has a good prognosis. With the modern techniques, the local recurrence rate is about 5% and regional metastasis of 2%.[1,2,45,46] Prognosis is worse in mucoepidermoid or spindle cell variants and in immunosuppressed patients.[1]
Melanocytic Epithelial Tumors
Conjunctival melanocytic nevus
Melanocytic tumors of the conjunctiva have a wide spectrum [Table 9]. Conjunctival nevus usually becomes apparent in the first to the second decade of life as a group of small nests of pigmented epithelial cells in the basal layer of the epithelium. As the cells migrate into the underlying stroma in the second to third decade, the nevus progresses to become the compound nevus. Further migration occurs, and cells reside in the stroma as sub-epithelial nevus during the third and fourth decades. Nevus is more commonly seen in Caucasians (89%) than Africans (6%) and Asians (5%).[45] Although most conjunctival nevi are pigmented (84%), some may be amelanotic or partially pigmented (16%).[2,46]
Table 9.
Melanocytic epithelial tumors

Presentation
Conjunctival nevi are mostly located near the limbus in the interpalpebral area (72%). Other locations are the caruncle, semilunar folds, fornix, tarsus, and cornea.[46,47] Characteristic clear cysts strongly support the diagnosis.[48] They may also clinically demonstrate feeder vessels (64%) and intrinsic vascularity (77%).[48] [Fig. 8a] It can vary in size, color, and location. Conjunctival nevus can increase in size in growing young children, during puberty, pregnancy, and sun exposure.[48,49] Malignant transformation was estimated to be <1%.[47,50] Sudden increase in size, alteration in color, and increased thickness with prominent feeder vessels indicates malignant transformation. Irregular and diffuse growth pattern poses a diagnostic confusion with primary acquired melanosis (PAM), melanoma, lymphoma, and pigmented OSSN. Histopathologically, a conjunctival nevus is composed of nests of benign melanocytes in the stroma near the basal layers of the epithelium [Fig. 8b].[47,50] Positive immunostaining for HMB-45 and Ki-67 are useful adjuncts in differentiating benign melanocytic lesion from suspected malignant entities.[51]
Figures 8.
(a) Conjunctival nevus with intralesional cysts and feeder vessels. (b) Microphotograph of a subepithelial nevus showing clumps of melanocytes with no cellular atypia (OM ×40)
Treatment
Periodic (annual) observation with slit lamp measurements and serial photographs is the management of choice. If excision is performed for cosmesis or suspected growth, it is preferable not to leave any residual lesion [Table 10].
Table 10.
Indications for excision of a conjunctival nevus
Ocular melanocytosis
Ocular melanocytosis is a congenital pigmentary condition of the periocular skin, sclera, orbit, meninges, and soft palate. It appears as irregular patches of scleral and episcleral pigmentation varying in color from brown to gray [Fig. 9]. Typically, there is no conjunctival pigment. It can involve the underlying uveal tract. Since it is episcleral involvement, it does not move with manipulation of the conjunctiva. This condition imparts a 1 in 400 risk for the development of uveal melanoma.[2,52]
Figure 9.
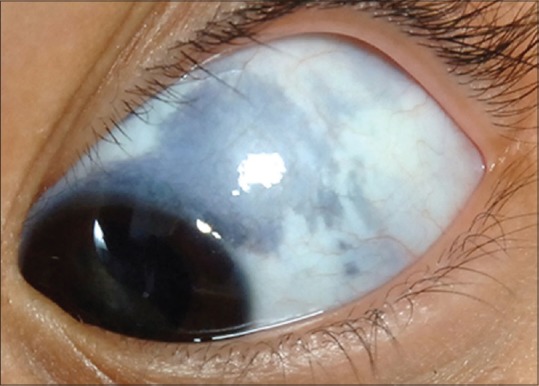
Ocular melanocytosis showing episcleral pigmentation
If associated with the dermal component, it is known as oculodermal melanocytosis or nevus of Ota [Fig. 7b and c]. It is mandatory that all patients with oculodermal melanocytosis undergo fundus examination to exclude uveal melanocytosis or melanoma. Associated hairline pigmentation predisposes them for meningeal melanoma and palate pigmentation to esophageal melanoma, therefore, these signs should be elicited with appropriate referrals when needed.
Complexion-related conjunctival pigmentation
Complex-related conjunctival pigmentation is a relatively common bilateral, flat, diffuse conjunctival pigmentation.[1,2] It is more concentrated in the limbus, often for 360°, with variable pigmentation at the perilimbal bulbar conjunctiva and cornea [Fig. 8]. Uncommonly, it may also involve the fornix and rarely the palpebral conjunctiva. Periodic observation is recommended.
Primary acquired melanosis
Reese noted the tendency of a certain type of acquired conjunctival pigmentation to evolve into melanoma and named it precancerous melanosis. Zimmerman replaced the term with benign acquired melanosis, which was further modified by WHO as PAM in 1980.[50]
Etiology
Sunlight exposure may play a role in the development of PAM. It has also been seen in patients with neurofibromatosis raising suspicion that it may have a developmental relationship to the neural crest.[52]
Presentation
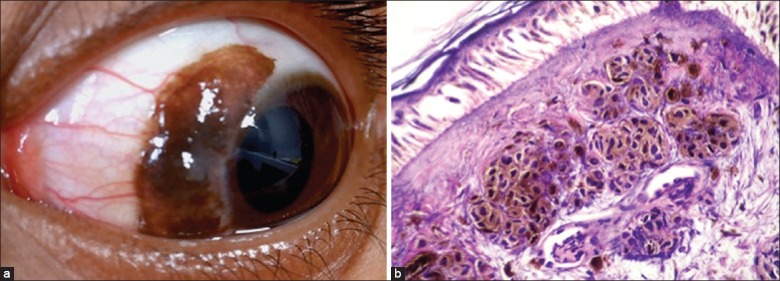
Primary acquired melanosis usually manifests in the middle age as unilateral, superficial, solitary, patchy, diffuse or multifocal pigmentation of the bulbar, fornicial and palpebral conjunctiva, and cornea [Fig. 10a]. Occasionally PAM can be amelanotic.[1,47,54] In PAM without atypia, there occurs melanin pigmentation of the basal epithelium with or without hyperplasia of cytological benign melanocytes. This may progress to cytological atypical melanocytes to form PAM with atypia, where there is an increased risk of developing melanoma [Fig. 10b]. Studies show that among those with atypia, 13% develop melanoma.[1,47]
Figure 10.
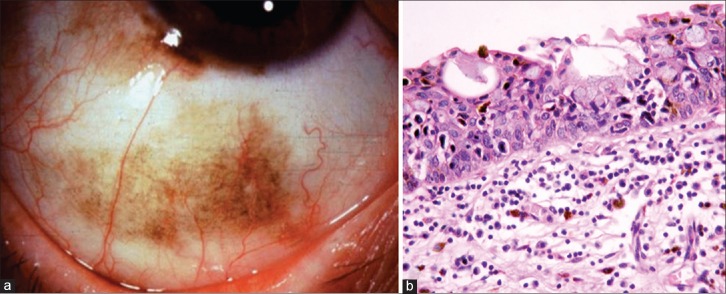
Primary acquired melanosis (PAM). (a) Diffuse flat pigmentation of the bulbar conjunctiva in an elderly male. (b) Microphotograph of PAM with cellular atypia (OM ×10)
Clinically, larger the extent of PAM, greater the risk of malignant transformation.
Treatment
Management strategies are [Table 11]:
Table 11.
Indications for biopsy of PAM[2]
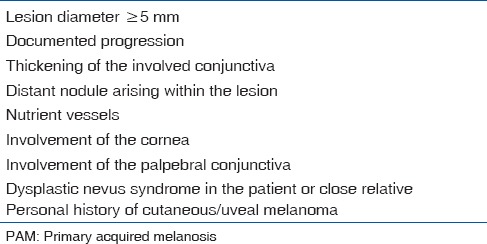
Observation for PAM without atypia
Cryotherapy for PAM with atypia <3 clock hours
Excision with excision edge cryotherapy for PAM >3 clock hours
Topical MMC for diffuse PAM with atypia.
Conjunctival melanoma
Conjunctival melanoma is most common in light-skinned individuals. It usually presents in the middle-aged or elderly.[55] It has no predilection for gender. It arises from PAM in about 75%, preexisting nevus in 20%, and de-novo in 5%.[1,2] Other risk factors are dysplastic nevus syndrome, neurofibromatosis, and xeroderma pigmentosum.[53] Sunlight exposure is also suggested as a cause, but that fails to explain the occurrence of melanoma in the fornices and palpebral conjunctiva.
Presentation
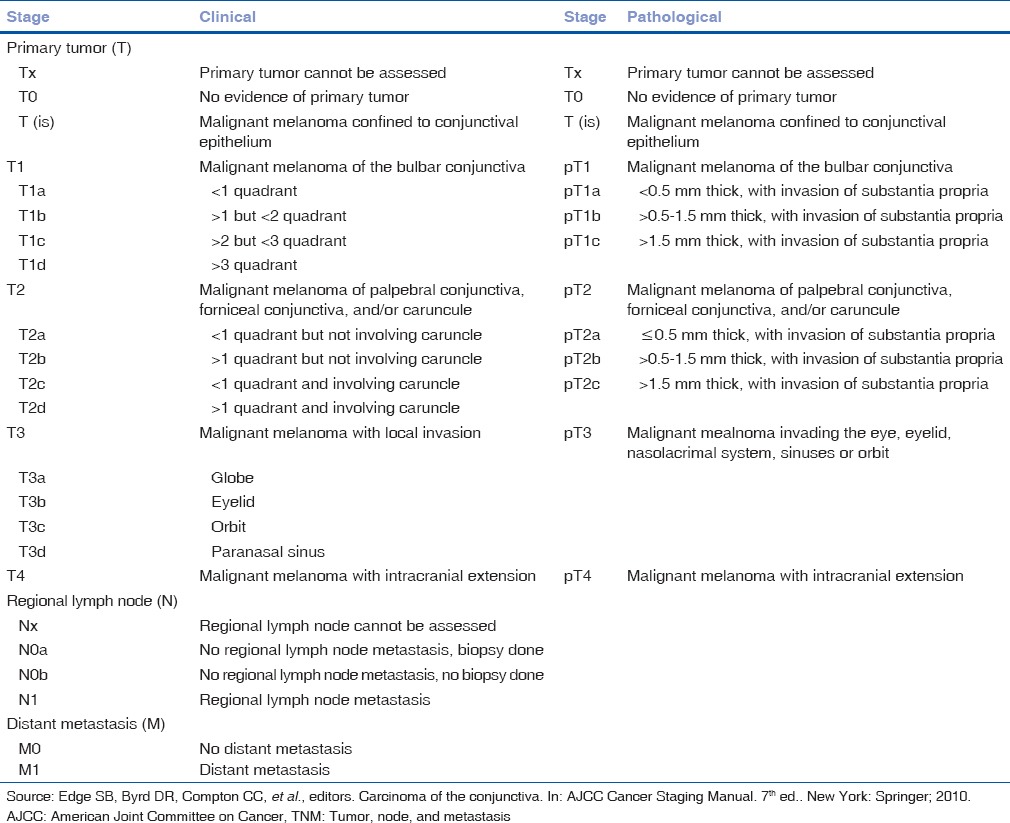
Conjunctival melanoma appears as a pigmented fleshy mass located in the bulbar, fornicial or palpebral conjunctiva [Fig. 11a]. As a variant, it may appear as diffuse or multifocal with ill-defined margins particularly if arising from PAM. It occasionally originates in the forniceal and palpebral conjunctiva.[54,55,56,57,58] It may extend to cover the cornea or even arise as a primary corneal tumor. Conjunctival melanoma may rarely develop secondary to continuous touch from an eyelid margin melanoma (implantation melanoma).[59] Melanoma can be sparsely pigmented or amelanotic. It is typically amelanotic, fleshy, and vascular when it recurs after prior excision.[2] Conjunctival melanoma is classified according to the AJCC-TNM classification [Table 12].
Figure 11.
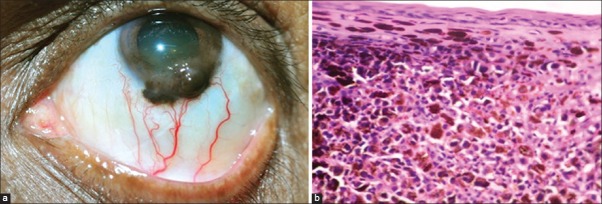
Conjunctival melanoma. (a) Elevated, nodular, pigmented mass at the inferior limbus with extension into the peripheral cornea. Note the presence of feeder vessels and intrinsic vessels. (b) Microphotograph of conjunctival melanoma showing variably pigmented melanocytes with mitotic activity (OM ×40)
Table 12.
AJCC-TNM classification of conjunctival melanoma
Regional metastasis involves preauricular and submandibular lymph nodes. Sentinel lymphangiography makes it possible to accurately remove lymph nodes and is indicated in tumors more than 2 mm thickness.[60] Distant metastasis occurs in the brain, liver, skin, and bone.[55]
Histopathologically, conjunctival melanoma is composed of variably pigmented malignant melanocytes [Fig. 11b].[52] These cells are positive for S-100 protein, tyrosinase, melan-A, HMB-45, HMB-50, and microphthalmia conscription factor[54,61] [Table 12].
Treatment
Treatment of conjunctival melanoma is based on certain established principles:[57,62]
Complete excision in the episcleral plane with 4 mm clinically clear margins
Alcohol keratoepitheliectomy of the corneal epithelial component
Partial lamellar sclerokeratectomy if sclera or corneal stroma are involved
Double freeze-thaw cryotherapy to the excision edge, excision base cryotherapy if sclera is involved and the extent of involvement is <3 clock hours
Postoperative adjuvant plaque brachytherapy if excision base is clinically detected to have been involved >3 clock hours and if the excision base is positive for tumor cells on histopathology. Since conjunctival melanoma is not radiosensitive, brachytherapy is not used as a sole treatment
Extended enucleation with en-bloc excision if the tumor has deep corneal or sclera invasion or intraocular extension
Eyelid sparing exenteration if the tumor extends into the orbit. Proton beam radiotherapy may be used as an alternative and/or adjunct to exenteration
Systemic chemotherapy is administered with combination of IFN and interleukin-2 in disseminated melanoma.
Prognosis
Local recurrence after therapy is as high as 50–70% at 10 years. Overall mortality rate is 25% in 10 years and more than 30% in 15 years.[47,48] Critical thickness that may serve as a prognostic factor, according to various studies implies a value between 0.8 and 4 mm. Conjunctival melanoma AJCC-TNM staging predicts the prognosis and outcome[57] [Table 12].
Stromal Tumors of the Ocular Surface
Vascular stromal tumors
Pyogenic granuloma
Pyogenic granuloma is a misnomer; it is neither pyogenic nor a granuloma, but exhuberant granulation tissue. It is a fibrovascular response to a tissue insult such as surgical or nonsurgical trauma or inflammation. It has rapid onset and progression and presents as fleshy, elevated, red, richly vascular mass [Fig. 12a]. It can be round to ovoid, typically pedunculated, rarely broad-based, and even mushroom shaped. It may be seen in any part of the conjunctiva, limbus, and the cornea.[63,64] Histopathologically, it is composed of granulation tissue with lymphocytes, plasma cells, scattered neutrophils, and numerous small caliber vessels.
Figure 12.
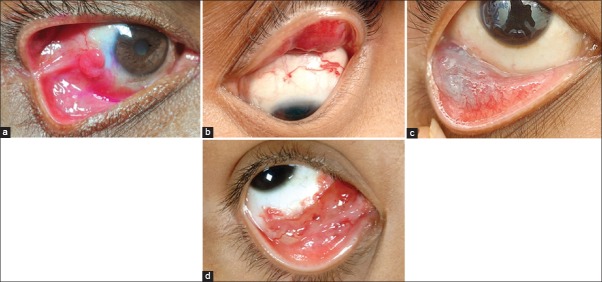
Vascular lesions of the conjunctiva. (a) Reddish pedunculated mass seen in the bulbar conjunctiva, at the site of prior surgical resection of pterygium. (b) Diffuse eyelid hemangioma with a conjunctival component. (c) Diffuse ill-defined bluish lesion seen in the inferior fornix suggestive of the conjunctival varix. (d) Diffuse reddish lesion with multiple dilated cystic spaces is seen in the inferomedial fornix. There are blood filled cysts noticed
Treatment
Often responds to topical steroids when diagnosed early
Excision at the base followed by cauterization or cryotherapy to the excision base is the treatment of choice for larger, unsightly, symptomatic or bleeding pyogenic granuloma
Along with excision, it is optimal to take care of the inciting factor if found to minimize the risk of recurrence. It is usual to find a suture knot or a foreign body at the base if the cause is prior surgery or trauma
Exuberant recurrence can be treated with low dose plaque brachytherapy.
Capillary hemangioma
Capillary hemangioma is common in the eyelids but is less common in the orbit and uncommon in the conjunctiva [Fig. 12b]. Seen as diffuse red elevated lesion, it may present as a small conjunctival component of a predominant eyelid lesion in a neonate. It may uncommonly develop as an acquired lesion in adults.
It presents at birth along with the lid lesion involving the conjunctiva, growing over the initial several months and then regresses spontaneously over several years. Spontaneous regression is often complete by 4–5 years of age. Histologically, it is composed of lobules of proliferating endothelial cells separated by thin fibrous septa.
Management generally is observation until spontaneous regression. Intervention is indicated if the concurrent eyelid component is amblyogenic-is large enough to induce mechanical ptosis or astigmatism. Intervention is also considered if the lesion is unsightly, ulcerates and bleeds and relentlessly progresses. Typical primary intervention is intralesional steroid injection.[1,2] Triamcinolone is used in the maximum dose of 6 mg/kg body weight. In general, one injection results in significant involution of the lesion. Injection may be repeated at 6–8 weeks interval if there is a suboptimal response. Dermatologists favor using high-dose oral steroids over 4–6 weeks. Systemic propranolol 2 mg/kg body weight is being tried as an alternative therapy with encouraging results.[65] If the tumor does not respond to these measures or if there is an indication for an emergent management (as in ulcerated and bleeding lesions), controlled surgical excision or debulking may help.
Cavernous hemangioma
Cavernous hemangioma is a common orbital tumor, but relatively uncommon in the conjunctiva. It appears as red blue lesion in the deep conjunctival stroma.[1,2] Histology shows dilated congested veins separated by connective tissue with smooth muscles in the walls of the blood vessels. Treatment is by surgical excision.
Varix
Varix refers to venous malformation of the conjunctiva that may range from an isolated single channel to dilated complex venous channels [Fig. 12c]. Often, it is the anterior extension of an orbital varix. Management is generally conservative by observation and symptomatic treatment.
Racemose hemangioma
Racemose hemangioma involves loops of dilated arteries and veins communicating directly without the interface of a capillary bed. The lesion is clinically seen as loops of dilated vessels in the conjunctival stroma with no evidence of a stimulus for such vascularization or planned direction. It may be associated with Wyburn-Mason syndrome.[1,2] It is generally observed unless symptomatic or a cosmetic blemish.
Hemangiopericytoma
Hemangiopericytoma is known to be derived from the vascular pericytes but is recently considered to be a vascular entity of the solitary fibrous tumor. It is a very rare conjunctival tumor that presents as an elevated or pedunculated red mass, which is slowly progressive.[66] A wide surgical resection with tumor-free margins is advocated.
Kaposi sarcoma
Kaposi sarcoma was a rare tumor before the AIDS era. It is a malignant tumor seen more frequently in immunocompromised individuals, specifically with HIV infection. Sometimes the conjunctival Kaposi sarcoma is the first sign of immunocompromised status.[67] It clinically appears as single or multifocal vascular red conjunctival lesion, which may become confluent and resemble hemorrhagic conjunctivitis.[67]
Treatment
Lymphangiectasia
When lymphatic channels in the conjunctiva are dilated and prominent, the condition is called lymphangiectasia. There exists a communication with conjunctival veins, and hence these dilated channels may often be filled with blood, termed hemorrhagic lymphangiectasia.[2,70] Surrounding conjunctiva appears edematous and is occasionally associated with sub-conjunctival hemorrhage. It can occur spontaneously or after trauma or inflammation. It is intermittent with a resolution between episodes. No treatment is required unless it is a cosmetic blemish, and the patient is keen on excision.
Lymphangioma
Lymphangioma is a benign tumor of the lymphatic vessels that usually manifests in the first decade of life. It can occur as an isolated conjunctival lesion, but often represents a superficial component of an orbital lymphangioma.[1,2] These are multiloculated lesions with dilated cystic spaces.[71] Those that contain blood are called chocolate cysts [Fig. 12d].
It histopathologically appears as nonencapsulated, irregular mass composed of numerous cyst-like channels that contain clear fluid, blood, or a combination of the two. The ectatic channels are lined by somewhat attenuated endothelial cells. These channels are separated by loose connective tissue that contains aggregates of small lymphocytes, sometimes forming lymph follicle.
Treatment
Lymphoproliferative Tumors
They are of three major types of conjunctival lymphoproliferative lesions, varying from benign to malignant and present as a spectrum, but may appear identical clinically:
Reactive lymphoid hyperplasia
Atypical lymphoid hyperplasia
Conjunctival lymphoma.
There is increasing emphasis that many conjunctival lymphomas may be low-grade B-cell lymphomas of the mucosa-associated lymphoid tissue type. In a third of patients, conjunctival lymphoma manifests with coexisting systemic lymphoma.[74,75] Patients usually present with a conjunctival mass. They may also present with nonspecific irritation, ptosis, epiphora, blurred vision, proptosis, and diplopia.
Conjunctival lymphoproliferative lesions appear as diffuse, slightly elevated pink mass, resembling smoked salmon. Seen mostly in the bulbar conjunctiva and fornix [Fig. 13a]. Some appear in the caruncle and plica, but very rarely in the palpebral conjunctiva.[1,75] In the unilateral cases chance of systemic lymphoma is 17%, and if bilateral the chance is 47%. Systemic lymphoma occurs in 15% of patients at 5 years and 28% in 10 years.[69]
Figure 13.
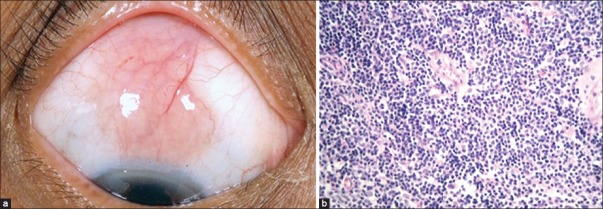
Conjuctival lymphoma. (a) Superior bulbar conjunctiva shows typical salmon pink mass with cork-screw vessels. (b) Microphotograph of conjunctival lymphoma (OM ×40). Note the monomorphic appearance of cells
Histopathologically, conjunctival lymphoproliferative tumors are composed of solid sheets of lymphocytes, with overlap between benign reactive lymphoid hyperplasia, atypical lymphoid hyperplasia, and malignant lymphoma [Fig. 13b]. Benign reactive lymphoid hyperplasia is generally polymorphic, with well-differentiated lymphocytes and plasma cells, while lymphoma tends to be more monomorphic and poorly differentiated. Most are non-Hodgkin's B-cell lymphomas whereas Hodgkins and T-cell lymphoma affect the conjunctiva rarely.[2,74] Immunohistochemistry may be helpful in determining the cell types.
Prognostic factors for developing systemic lymphoma are fornicial or mid-bulbar location, multifocality, and bilaterality. There are several treatment options.[1,2,74,75,76]
Treatment
Excision biopsy
Cryotherapy
Low-dose external beam radiotherapy
Local injection of IFN α
Brachytherapy
Chemotherapy if associated with systemic involvement.
Fibrous Tumors
Fibrous tumors manifest generally as slowly progressive acquired white stromal tumors in adults. These could be a well-circumscribed lesion or multi-nodular. Common fibrous tumors affecting the ocular surface are fibrous histiocytoma and nodular fasciitis.
Fibrous Histiocytoma
It can be benign, locally aggressive, and malignant, seen in adults. Presents commonly as well-circumscribed to diffuse amelanotic mass, often in the limbus and frequently extends to involve the cornea. Seen deep in the conjunctiva and attached to the sclera.[1,77] It has yellow color owing to the presence of histiocytes. Malignant transformation is extremely rare. Benign fibrous histiocytoma is completely excised while malignant fibrous histiocytoma may need radical excision with clear margins.
Nodular Fasciitis (Pseudosarcomatous Fasciitis)
It is thought to arise from the tenon's capsule.[1,2] It presents in the varied age group ranging from 3 to 81 years as a solitary white episcleral enlarging nodule at the limbus or over the sclera anterior to the insertion of one of the rectus muscle. The nodule may grow quickly and show signs of inflammation. Treatment is by complete surgical excision.
Neural Tumors
Simple neuroma
Simple neuroma is soft mucosal neural tumors that appear in the conjunctiva and other mucous membrane in patients with MEN-2b. All such patients have prominent corneal nerves.[1,2] Clinically, these lesions appear as pink-yellow and grow over time. There is significant association with medullary carcinoma of the thyroid.[2]
Schwannoma
Schwannoma of the ocular surface are rare and can occur in any part of the conjunctiva. These present as pink or yellow elevated slow-growing lesion and may have mildly dilated conjunctival or episcleral nutrient vessels.[1,78] Treatment is by complete excision along with the tumor capsule.
Granular Cell Tumor
Granular cell tumor, also known as myoblastoma, is extremely rare. Originally thought of having a striated muscle origin, recent suggestions are that it is of neural derivative, Schwann cell origin.[1] It is seen as a pink elevated smooth mass of the conjunctival stroma. Pseudoepitheliomatous hyperplasia of the overlying conjunctival epithelium is a recognized feature of this tumor.[79] Treatment is by complete excision.
Histiocytic Tumors
Xanthoma
Xanthoma presents as a yellow subepithelial mass on the epibulbar surface. In xanthoma disseminatum, multiple limbal lesions are found in both the eyes.[2,80]
Xanthogranuloma
Xanthogranuloma occurs as a solitary orange-pink stromal mass usually near the limbus in adults.[2,81] There are isolated case reports of juvenile xanthogranuloma of the conjunctiva in children. Histopathologically, histiocytes admixed with Touton's giant cell confirms the diagnosis. It is managed by observation for spontaneous resolution, topical or systemic steroids for cases that do not resolve, and excision in recurrent cases.
Reticulohistiocytoma
Reticulohistiocytoma is a benign conjunctival lesion that is often a part of the systemic disorder known as multicentric reticulohistiocytosis. It appears as a single painless mass localized to the cornea and limbus with evidence of systemic disease.[82] Treatment is by complete excision.
Myxoid Tumors
Myxoid tumors are rare benign stromal tumors, manifesting as slowly growing, asymptomatic freely movable unilateral, soft, pink white lesions usually seen in the temporal bulbar conjunctiva.[1,83] These can be associated with Carney complex. The presence of these lesions should prompt the evaluation of cardiac myxoma-a life-threatening condition. Histologically, they are hypocellular and are composed of stellate and spindle-shaped cells interspersed in the loose stroma.
Myogenic Tumors
Rhabdomyosarcoma
The occurrence of rhabdomyosarcoma in the conjunctiva alone is rare - it generally has an orbital component. Most commonly the embryonal type manifests with a conjunctival component[84,85] Botryoid rhabdomyosarcoma may be seen in the conjunctival fornices.[86] Rhabdomyosarcoma presents as a pink, rapidly growing vascular conjunctival mass [Fig. 14]. It may appear as a pedunculated soft tissue mass but occasionally swelling and erythema precede the visible tumor. Complete surgical excision with protocol-based adjuvant chemotherapy and radiotherapy is the treatment of choice for rhabdomyosarcoma localized to the conjunctiva.[84]
Figure 14.
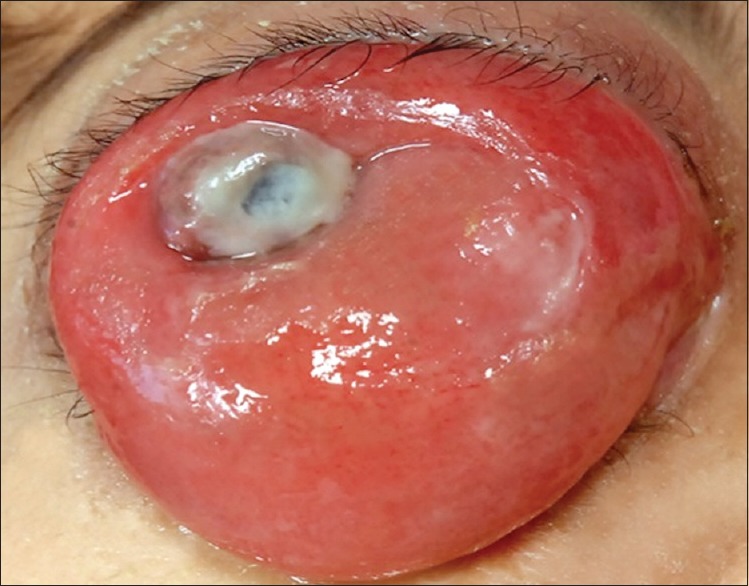
Conjunctival extension of orbital rhabdomyosarcoma
Lipomatous Tumors
Lipoma and liposarcoma
Lipoma is usually seen in adults. It appears as yellow pink stromal mass. Liposarcoma is clinically similar to lipoma but histopathology reveals neoplastic stellate lipid cells and signet ring cells have been observed.[1,2,87]
Chroistoma
Choristoma is a congenital lesion representing the excess of normal tissue in an abnormal location. It is considered a simple choristoma when composed of one type of tissue and a complex choristoma when a combination of displaced tissue is involved. Epibulbar choristomas are generally found in children, dermoid and dermolipoma being most common.[2] Usual locations are cornea, limbus, and episclera. Epibulbar choristomas can be associated with eyelid and uveal coloboma, Goldenhar syndrome or organoid nevus syndrome[88] [Fig. 15a].
Figure 15.
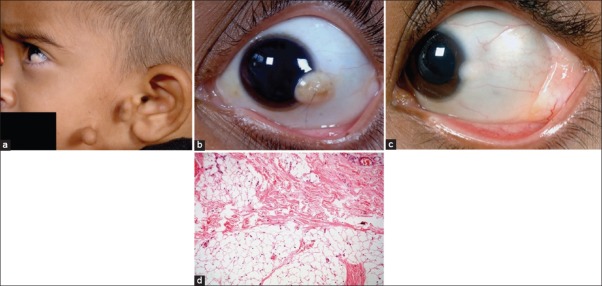
Counjunctival choristoma. (a) Preauricular tags in a patient with limbal dermoid suggestive of Goldenhar syndrome. (b) Limbal dermoid with hair follicle on the surface. (c) Dermolipoma manifesting as a yellow-white smooth lesion in the lateral fornix with a co-existing limbal dermoid. (d) Microphotograph of dermolipoma (OM ×10) showing conjunctival epithelium lining its surface and sub-epithelial tissue with collagenous connective tissue and adipose elements
Dermoid
Epibulbar dermoid is a well-circumscribed yellow white solid lesion involving the corneoscleral limbus. It most commonly occurs at the inferotemporal limbus and has fine white hairs that are best seen with slit lamp biomicroscopy [Fig. 15b and c]. It is not uncommon to see them associated with Goldenhar syndrome [Fig. 15a]. Rarely, it can extend to the central cornea or be located in the other quadrants.[2] In addition to becoming a cosmetic blemish, can cause severe astigmatism and amblyopia in some cases. Histopathologically, epibulbar dermoid is a simple choristomatous malformation that consists of dense fibrous tissue lined by conjunctival epithelium with deeper dermal elements including hair follicles and sebaceous glands [Fig. 15d].
Treatment
Observation alone is preferred if the dermoid is small and does not cause visual symptoms. Larger demoids can be excised by lamellar keratosclerectomy with amniotic membrane grafting if the defect is superficial or closure using lamellar or full thickness corneal or sclerocorneal graft if the defect is deep or full thickness. While the cosmetic appearance does improve with surgery, the astigmatic error and visual acuity may not change significantly unless the child is treated early.
Dermolipoma
Although dermolipoma is congenital and present at birth, it typically remains asymptomatic for years and may not be detected until adulthood. The lesion presents as a pale yellow, soft, fluctuant, mass protruding from the orbit through the conjunctival fornix superotemporally[2] [Fig. 15c]. Unlike herniated orbital fat, dermolipoma may show the fine white hair on its surface. Histopathologically, it is lined by conjunctival epithelium on its surface, and sub-epithelial tissue has variable quantities of collagenous connective tissue and adipose elements. Pilosebaceous units and lacrimal gland tissue may occasionally be present.
Treatment
No treatment is required unless for cosmetic considerations or in symptomatic patients with exuberant hair growth over the lesion. Visible portion of the dermolipoma may be debulked, and the ocular surface reconstructed with amniotic membrane graft.
Caruncular Tumors and Cysts
The caruncle is a unique anatomic structure containing elements of both conjunctiva and skin. The lesions occurring in the caruncle are similar to those that occur in mucous membranes and cutaneous structures.[1,2,89] By histopathological analysis, 95% of the caruncular lesions are benign, and 5% are malignant.[90] The most common lesions in the caruncle include papilloma and nevus. [Fig. 16a] Other lesions of caruncle include pyogenic granuloma, inclusion cysts, sebaceous hyperplasia, sebaceous adenoma, and oncocytoma. Oncocytoma is a benign tumor that occurs more commonly in the lacrimal or salivary glands. It probably arises from the accessory lacrimal gland tissue in the caruncle.[91] Malignant tumors occurring rarely in the caruncle are SCC, melanoma, lymphoma, and sebaceous carcinoma.[89] [Fig. 16a and b] Treatment includes either observation or local resection, depending upon the diagnosis.
Figure 16.
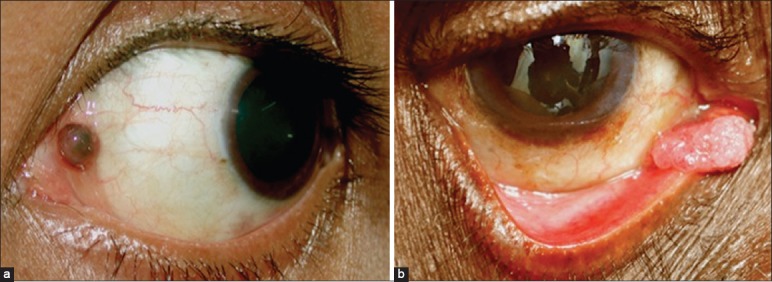
Caruncular tumors. (a) Pigmented caruncular nevus. (b) Squamous cell carcinoma arising from the caruncle
Metastatic and Secondary Tumors
Metastatic tumors
Metastatic tumors are rare, but conjunctival metastasis can occur from breast carcinoma, cutaneous melanoma, or other primary tumors. They appear as one or more fleshy pink vascularised conjunctival stromal tumors. Metastatic melanoma to the conjunctiva is usually pigmented.[1,2,92]
Leukemic infiltrate
Conjunctival leukemic infiltrate is associated with acute myeloid leukemia. It is often an early sign of relapse of previously treated disease. It may be unilateral or bilateral with focal or diffuse lesions in the bulbar or palpebral conjunctiva.[2] It has a spectrum of presentation, ranging from subconjunctival hemorrhage to direct infiltration of the tissue with leukemic cells but often manifests as a firm, nontender, pink smooth mass associated with hemorrhage.[92] It has the tendency to appear in the perilimbal tissue near the cornea.
Secondary tumors
The conjunctiva can be secondarily involved by tumors of adjacent structures, particularly by direct extension from the tumor of the eyelid. Intraocular and orbital tumors may also extend into the conjunctiva. Most important is the sebaceous gland carcinoma of the eyelid which can exhibit pagetoid invasion and direct invasion into the conjunctival epithelium.[1,2] Uveal melanoma can extend extrasclerally into the subconjunctival tissues. Rhabdomyosarcoma of the orbit in children occasionally presents first with its conjunctival component.
Conclusion
Tumors of the ocular surface have a wide clinical spectrum. It is possible to make an accurate clinical diagnosis in most cases. Benign tumors and choristomas are excised only if there is a cosmetic or functional concern. Malignant tumors generally need complete excision with clear margins and excision edge cryotherapy. Prognosis for local tumor control is excellent with protocol-based management.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Shields CL, Shields JA. Tumors of the conjunctiva and cornea. Surv Ophthalmol. 2004;49:3–24. doi: 10.1016/j.survophthal.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Shields JA, Shields CL. An Atlas and Textbook. 2nd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2008. Eyelid, Conjunctival and Orbital Tumors; pp. 250–445. [Google Scholar]
- 3.Lass JH, Jenson AB, Papale JJ, Albert DM. Papillomavirus in human conjunctival papillomas. Am J Ophthalmol. 1983;95:364–8. doi: 10.1016/s0002-9394(14)78307-2. [DOI] [PubMed] [Google Scholar]
- 4.Lass JH, Grove AS, Papale JJ, Albert DM, Jenson AB, Lancaster WD. Detection of human papillomavirus DNA sequences in conjunctival papilloma. Am J Ophthalmol. 1983;96:670–4. doi: 10.1016/s0002-9394(14)73426-9. [DOI] [PubMed] [Google Scholar]
- 5.Kremer I, Sandbank J, Weinberger D, Rotem A, Shapiro A. Pigmented epithelial tumours of the conjunctiva. Br J Ophthalmol. 1992;76:294–6. doi: 10.1136/bjo.76.5.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Streeten BW, Carrillo R, Jamison R, Brownstein S, Font RL, Zimmerman LE. Inverted papilloma of the conjunctiva. Am J Ophthalmol. 1979;88:1062–6. doi: 10.1016/0002-9394(79)90417-3. [DOI] [PubMed] [Google Scholar]
- 7.Jakobiec FA, Harrison W, Aronian D. Inverted mucoepidermoid papillomas of the epibulbar conjunctiva. Ophthalmology. 1987;94:283–7. doi: 10.1016/s0161-6420(87)33459-1. [DOI] [PubMed] [Google Scholar]
- 8.Harkey ME, Metz HS. Cryotherapy of conjunctival papillomata. Am J Ophthalmol. 1968;66:872–4. doi: 10.1016/0002-9394(68)92803-1. [DOI] [PubMed] [Google Scholar]
- 9.Bosniak SL, Novick NL, Sachs ME. Treatment of recurrent squamous papillomata of the conjunctiva by carbon dioxide laser vaporization. Ophthalmology. 1986;93:1078–82. doi: 10.1016/s0161-6420(86)33634-0. [DOI] [PubMed] [Google Scholar]
- 10.Jackson WB, Beraja R, Codère F. Laser therapy of conjunctival papillomas. Can J Ophthalmol. 1987;22:45–7. [PubMed] [Google Scholar]
- 11.Petrelli R, Cotlier E, Robins S, Stoessel K. Dinitrochlorobenzene immunotherapy of recurrent squamous papilloma of the conjunctiva. Ophthalmology. 1981;88:1221–5. doi: 10.1016/s0161-6420(81)34869-6. [DOI] [PubMed] [Google Scholar]
- 12.Lass JH, Foster CS, Grove AS, Rubenfeld M, Lusk RP, Jenson AB, et al. Interferon-alpha therapy of recurrent conjunctival papillomas. Am J Ophthalmol. 1987;103:294–301. [PubMed] [Google Scholar]
- 13.Hawkins AS, Yu J, Hamming NA, Rubenstein JB. Treatment of recurrent conjunctival papillomatosis with mitomycin C. Am J Ophthalmol. 1999;128:638–40. doi: 10.1016/s0002-9394(99)00221-4. [DOI] [PubMed] [Google Scholar]
- 14.Shields CL, Lally MR, Singh AD, Shields JA, Nowinski T. Oral cimetidine (Tagamet) for recalcitrant, diffuse conjunctival papillomatosis. Am J Ophthalmol. 1999;128:362–4. doi: 10.1016/s0002-9394(99)00265-2. [DOI] [PubMed] [Google Scholar]
- 15.Streeten BW, Carrillo R, Jamison R, Brownstein S, Font RL, Zimmerman LE. Inverted papilloma of the conjunctiva. Am J Ophthalmol. 1979;88:1062–6. doi: 10.1016/0002-9394(79)90417-3. [DOI] [PubMed] [Google Scholar]
- 16.Jakobiec FA, Harrison W, Aronian D. Inverted mucoepidermoid papillomas of the epibulbar conjunctiva. Ophthalmology. 1987;94:283–7. doi: 10.1016/s0161-6420(87)33459-1. [DOI] [PubMed] [Google Scholar]
- 17.Munro S, Brownstein S, Liddy B. Conjunctival keratoacanthoma. Am J Ophthalmol. 1993;116:654–5. doi: 10.1016/s0002-9394(14)73218-0. [DOI] [PubMed] [Google Scholar]
- 18.Schellini SA, Marques ME, Milanezi MF, Bacchi CE. Conjunctival keratoacanthoma. Acta Ophthalmol Scand. 1997;75:335–7. doi: 10.1111/j.1600-0420.1997.tb00791.x. [DOI] [PubMed] [Google Scholar]
- 19.Coupland SE, Heimann H, Kellner U, Bornfeld N, Foerster MH, Lee WR. Keratoacanthoma of the bulbar conjunctiva. Br J Ophthalmol. 1998;82:586. doi: 10.1136/bjo.82.5.584b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakobiec FA, Perry HD, Harrison W, Krebs W. Dacryoadenoma. A unique tumor of the conjunctival epithelium. Ophthalmology. 1989;96:1014–20. doi: 10.1016/s0161-6420(89)32799-0. [DOI] [PubMed] [Google Scholar]
- 21.Yanoff M. Hereditary benign intraepithelial dyskeratosis. Arch Ophthalmol. 1968;79:291–3. doi: 10.1001/archopht.1968.03850040293012. [DOI] [PubMed] [Google Scholar]
- 22.Shields CL, Shields JA, Eagle RC., Jr Hereditary benign intraepithelial dyskeratosis. Arch Ophthalmol. 1987;105:422–3. doi: 10.1001/archopht.1987.01060030142045. [DOI] [PubMed] [Google Scholar]
- 23.Mauriello JA, Jr, Napolitano J, McLean I. Actinic keratosis and dysplasia of the conjunctiva: A clinicopathological study of 45 cases. Can J Ophthalmol. 1995;30:312–6. [PubMed] [Google Scholar]
- 24.Lee GA, Hirst LW. Ocular surface squamous neoplasia. Surv Ophthalmol. 1995;39:429–50. doi: 10.1016/s0039-6257(05)80054-2. [DOI] [PubMed] [Google Scholar]
- 25.Pe’er J. Ocular surface squamous neoplasia. Ophthalmol Clin North Am. 2005;18:1–13, vii. doi: 10.1016/j.ohc.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Pizarello LD, Jakobeic FA. Bowens disease of the conjunctiva: A misnomer. In: Jakobeic FA, editor. Ocular and Adnexal Tumors. Brimingham, AL: Aesculapius; 1978. pp. 553–71. [Google Scholar]
- 27.Shields CL, Demirci H, Karatza E, Shields JA. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology. 2004;111:1747–54. doi: 10.1016/j.ophtha.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 28.Farah S, Baum TD, Conton MR. Tumors of cornea and conjunctiva. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. 2nd ed. Philadelphia: WB Saunders; 2000. pp. 1002–19. [Google Scholar]
- 29.Tunc M, Char DH, Crawford B, Miller T. Intraepithelial and invasive squamous cell carcinoma of the conjunctiva: Analysis of 60 cases. Br J Ophthalmol. 1999;83:98–103. doi: 10.1136/bjo.83.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waring GO, 3rd, Roth AM, Ekins MB. Clinical and pathologic description of 17 cases of corneal intraepithelial neoplasia. Am J Ophthalmol. 1984;97:547–59. doi: 10.1016/0002-9394(84)90371-4. [DOI] [PubMed] [Google Scholar]
- 31.Akpek EK, Polcharoen W, Chan R, Foster CS. Ocular surface neoplasia masquerading as chronic blepharoconjunctivitis. Cornea. 1999;18:282–8. doi: 10.1097/00003226-199905000-00007. [DOI] [PubMed] [Google Scholar]
- 32.Nicholson DH, Herschler J. Intraocular extension of squamous cell carcinoma of the conjunctiva. Arch Ophthalmol. 1977;95:843–6. doi: 10.1001/archopht.1977.04450050121015. [DOI] [PubMed] [Google Scholar]
- 33.Searl SS, Krigstein HJ, Albert DM, Grove AS., Jr Invasive squamous cell carcinoma with intraocular mucoepidermoid features. Conjunctival carcinoma with intraocular invasion and diphasic morphology. Arch Ophthalmol. 1982;100:109–11. doi: 10.1001/archopht.1982.01030030111011. [DOI] [PubMed] [Google Scholar]
- 34.Johnson TE, Tabbara KF, Weatherhead RG, Kersten RC, Rice C, Nasr AM. Secondary squamous cell carcinoma of the orbit. Arch Ophthalmol. 1997;115:75–8. doi: 10.1001/archopht.1997.01100150077013. [DOI] [PubMed] [Google Scholar]
- 35.Shields JA, Shields CL, De Potter P. Surgical management of conjunctival tumors. The 1994 Lynn B. McMahan Lecture. Arch Ophthalmol. 1997;115:808–15. doi: 10.1001/archopht.1997.01100150810025. [DOI] [PubMed] [Google Scholar]
- 36.Poothullil AM, Colby KA. Topical medical therapies for ocular surface tumors. Semin Ophthalmol. 2006;21:161–9. doi: 10.1080/08820530500351694. [DOI] [PubMed] [Google Scholar]
- 37.Majmudar PA, Epstein RJ. Antimetabolites in ocular surface neoplasia. Curr Opin Ophthalmol. 1998;9:35–9. doi: 10.1097/00055735-199808000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Sepulveda R, Pe’er J, Midena E, Seregard S, Dua HS, Singh AD. Topical chemotherapy for ocular surface squamous neoplasia: Current status. Br J Ophthalmol. 2010;94:532–5. doi: 10.1136/bjo.2009.160820. [DOI] [PubMed] [Google Scholar]
- 39.Shields CL, Kaliki S, Kim HJ, Al-Dahmash S, Shah SU, Lally SE, et al. Interferon for ocular surface squamous neoplasia in 81 cases: Outcomes based on the American Joint Committee on Cancer classification. Cornea. 2013;32:248–56. doi: 10.1097/ICO.0b013e3182523f61. [DOI] [PubMed] [Google Scholar]
- 40.Karp CL, Galor A, Chhabra S, Barnes SD, Alfonso EC. Subconjunctival/perilesional recombinant interferon a2b for ocular surface squamous neoplasia: A 10-year review. Ophthalmology. 2010;117:2241–6. doi: 10.1016/j.ophtha.2010.03.052. [DOI] [PubMed] [Google Scholar]
- 41.Kim HJ, Shields CL, Shah SU, Kaliki S, Lally SE. Giant ocular surface squamous neoplasia managed with interferon alpha-2b as immunotherapy or immunoreduction. Ophthalmology. 2012;119:938–44. doi: 10.1016/j.ophtha.2011.11.035. [DOI] [PubMed] [Google Scholar]
- 42.Shields CL, Ramasubramanian A, Mellen PL, Shields JA. Conjunctival squamous cell carcinoma arising in immunosuppressed patients (organ transplant, human immunodeficiency virus infection) Ophthalmology. 2011;118:2133–7.e1. doi: 10.1016/j.ophtha.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 43.Sherman MD, Feldman KA, Farahmand SM, Margolis TP. Treatment of conjunctival squamous cell carcinoma with topical cidofovir. Am J Ophthalmol. 2002;134:432–3. doi: 10.1016/s0002-9394(02)01569-6. [DOI] [PubMed] [Google Scholar]
- 44.Shields JA, Shields CL, Freire JE, Brady LW, Komarnicky L. Plaque radiotherapy for selected orbital malignancies: Preliminary observations: The 2002 Montgomery Lecture, part 2. Ophthal Plast Reconstr Surg. 2003;19:91–5. doi: 10.1097/01.IOP.0000056020.66654.33. [DOI] [PubMed] [Google Scholar]
- 45.Cervantes G, Rodríguez AA, Jr, Leal AG. Squamous cell carcinoma of the conjunctiva: Clinicopathological features in 287 cases. Can J Ophthalmol. 2002;37:14–9. doi: 10.1016/s0008-4182(02)80093-x. [DOI] [PubMed] [Google Scholar]
- 46.Shields CL, Fasiuddin AF, Mashayekhi A, Shields JA. Conjunctival nevi: Clinical features and natural course in 410 consecutive patients. Arch Ophthalmol. 2004;122:167–75. doi: 10.1001/archopht.122.2.167. [DOI] [PubMed] [Google Scholar]
- 47.Shields CL, Demirci H, Karatza E, Shields JA. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology. 2004;111:1747–54. doi: 10.1016/j.ophtha.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 48.Shields CL, Belinsky I, Romanelli-Gobbi M, Guzman JM, Mazzuca D, Jr, Green WR, et al. Anterior segment optical coherence tomography of conjunctival nevus. Ophthalmology. 2011;118:915–9. doi: 10.1016/j.ophtha.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 49.Zamir E, Mechoulam H, Micera A, Levi-Schaffer F, Pe’er J. Inflamed juvenile conjunctival naevus: Clinicopathological characterisation. Br J Ophthalmol. 2002;86:28–30. doi: 10.1136/bjo.86.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zimmerman LE, Sobin LH. No. 24: Histological Typing of Tumors of the Eye and its Adnexa. Geneva: World Health Organization; 1980. International histological classification of tumors; p. 30. [Google Scholar]
- 51.Jakobiec FA, Bhat P, Colby KA. Immunohistochemical studies of conjunctival nevi and melanomas. Arch Ophthalmol. 2010;128:174–83. doi: 10.1001/archophthalmol.2009.394. [DOI] [PubMed] [Google Scholar]
- 52.Shields CL, Kaliki S, Livesey M, Walker B, Garoon R, Bucci M, et al. Association of ocular and oculodermal melanocytosis with the rate of uveal melanoma metastasis: Analysis of 7872 consecutive eyes. JAMA Ophthalmol. 2013;131:993–1003. doi: 10.1001/jamaophthalmol.2013.129. [DOI] [PubMed] [Google Scholar]
- 53.To KW, Rabinowitz SM, Friedman AH, Merker C, Cavanaugh CP. Neurofibromatosis and neural crest neoplasms: Primary acquired melanosis and malignant melanoma of the conjunctiva. Surv Ophthalmol. 1989;33:373–9. doi: 10.1016/0039-6257(89)90014-3. [DOI] [PubMed] [Google Scholar]
- 54.Jakobiec FA, Folberg R, Iwamoto T. Clinicopathologic characteristics of premalignant and malignant melanocytic lesions of the conjunctiva. Ophthalmology. 1989;96:147–66. doi: 10.1016/s0161-6420(89)32920-4. [DOI] [PubMed] [Google Scholar]
- 55.Shields CL, Markowitz JS, Belinsky I, Schwartzstein H, George NS, Lally SE, et al. Conjunctival melanoma: Outcomes based on tumor origin in 382 consecutive cases. Ophthalmology. 2011;118:389–95.e1. doi: 10.1016/j.ophtha.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 56.Folberg R. Melanocytic lesions of the conjunctiva. In: Spencer WH, editor. Ophthalmic Pathology. An Atlas and Textbook. 4th ed. Philadelphia: WB Saunders; 1996. pp. 125–47. [Google Scholar]
- 57.Shields CL, Kaliki S, Al-Dahmash SA, Lally SE, Shields JA. American Joint Committee on Cancer (AJCC) clinical classification predicts conjunctival melanoma outcomes. Ophthal Plast Reconstr Surg. 2012;28:313–23. doi: 10.1097/IOP.0b013e3182611670. [DOI] [PubMed] [Google Scholar]
- 58.Bajaj MS, Pushker N, Kashyap S, Balasubramanya R, Chandra M, Ghose S. Conjunctival malignant melanoma. Orbit. 2003;22:47–53. doi: 10.1076/orbi.22.1.47.14016. [DOI] [PubMed] [Google Scholar]
- 59.Giblin ME, Shields JA, Shields CL, Eagle RC., Jr Primary eyelid malignant melanoma associated with primary conjunctival malignant melanoma. Aust N Z J Ophthalmol. 1988;16:127–31. doi: 10.1111/j.1442-9071.1988.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 60.Cohen VM, Tsimpida M, Hungerford JL, Jan H, Cerio R, Moir G. Prospective study of sentinel lymph node biopsy for conjunctival melanoma. Br J Ophthalmol. 2013;97:1525–9. doi: 10.1136/bjophthalmol-2013-303671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharara NA, Alexander RA, Luthert PJ, Hungerford JL, Cree IA. Differential immunoreactivity of melanocytic lesions of the conjunctiva. Histopathology. 2001;39:426–31. doi: 10.1046/j.1365-2559.2001.01168.x. [DOI] [PubMed] [Google Scholar]
- 62.Shields JA, Shields CL, De Potter P. Surgical management of circumscribed conjunctival melanomas. Ophthal Plast Reconstr Surg. 1998;14:208–15. doi: 10.1097/00002341-199805000-00012. [DOI] [PubMed] [Google Scholar]
- 63.Ferry AP. Pyogenic granulomas of the eye and ocular adnexa: A study of 100 cases. Trans Am Ophthalmol Soc. 1989;87:327–43. [PMC free article] [PubMed] [Google Scholar]
- 64.Cameron JA, Mahmood MA. Pyogenic granulomas of the cornea. Ophthalmology. 1995;102:1681–7. doi: 10.1016/s0161-6420(95)30809-3. [DOI] [PubMed] [Google Scholar]
- 65.Vassallo P, Forte R, Di Mezza A, Magli A. Treatment of infantile capillary hemangioma of the eyelid with systemic propranolol. Am J Ophthalmol. 2013;155:165–70.e2. doi: 10.1016/j.ajo.2012.06.021. [DOI] [PubMed] [Google Scholar]
- 66.Grossniklaus HE, Green WR, Wolff SM, Iliff NT. Hemangiopericytoma of the conjunctiva. Two cases. Ophthalmology. 1986;93:265–7. doi: 10.1016/s0161-6420(86)33755-2. [DOI] [PubMed] [Google Scholar]
- 67.Kurumety UR, Lustbader JM. Kaposi's sarcoma of the bulbar conjunctiva as an initial clinical manifestation of acquired immunodeficiency syndrome. Arch Ophthalmol. 1995;113:978. doi: 10.1001/archopht.1995.01100080028016. [DOI] [PubMed] [Google Scholar]
- 68.Ghabrial R, Quivey JM, Dunn JP, Jr, Char DH. Radiation therapy of acquired immunodeficiency syndrome-related Kaposi's sarcoma of the eyelids and conjunctiva. Arch Ophthalmol. 1992;110:1423–6. doi: 10.1001/archopht.1992.01080220085027. [DOI] [PubMed] [Google Scholar]
- 69.Shields JA, De Potter P, Shields CL, Komarnicky LT. Kaposi's sarcoma of the eyelids: Response to radiotherapy. Arch Ophthalmol. 1992;110:1689. doi: 10.1001/archopht.1992.01080240027019. [DOI] [PubMed] [Google Scholar]
- 70.Awdry P. Lymphangiectasia haemorrhagica conjunctivae. Br J Ophthalmol. 1969;53:274–8. doi: 10.1136/bjo.53.4.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wright JE, Sullivan TJ, Garner A, Wulc AE, Moseley IF. Orbital venous anomalies. Ophthalmology. 1997;104:905–13. doi: 10.1016/s0161-6420(97)30208-5. [DOI] [PubMed] [Google Scholar]
- 72.Spector JA, Zide BM. Carbon dioxide laser ablation for treatment of lymphangioma of the conjunctiva. Plast Reconstr Surg. 2006;117:609–12. doi: 10.1097/01.prs.0000200872.78741.9a. [DOI] [PubMed] [Google Scholar]
- 73.Behrendt S, Bernsmeier H, Randzio G. Fractionated beta-irradiation of a conjunctival lymphangioma. Ophthalmologica. 1991;203:161–3. doi: 10.1159/000310244. [DOI] [PubMed] [Google Scholar]
- 74.Coupland SE, Krause L, Delecluse HJ, Anagnostopoulos I, Foss HD, Hummel M, et al. Lymphoproliferative lesions of the ocular adnexa. Analysis of 112 cases. Ophthalmology. 1998;105:1430–41. doi: 10.1016/S0161-6420(98)98024-1. [DOI] [PubMed] [Google Scholar]
- 75.Shields CL, Shields JA, Carvalho C, Rundle P, Smith AF. Conjunctival lymphoid tumors: Clinical analysis of 117 cases and relationship to systemic lymphoma. Ophthalmology. 2001;108:979–84. doi: 10.1016/s0161-6420(01)00547-4. [DOI] [PubMed] [Google Scholar]
- 76.Eichler MD, Fraunfelder FT. Cryotherapy for conjunctival lymphoid tumors. Am J Ophthalmol. 1994;118:463–7. doi: 10.1016/s0002-9394(14)75797-6. [DOI] [PubMed] [Google Scholar]
- 77.Kim HJ, Shields CL, Eagle RC, Jr, Shields JA. Fibrous histiocytoma of the conjunctiva. Am J Ophthalmol. 2006;142:1036–43. doi: 10.1016/j.ajo.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 78.Perry HD. Isolated episcleral neurofibroma. Ophthalmology. 1982;89:1095–8. doi: 10.1016/s0161-6420(82)34678-3. [DOI] [PubMed] [Google Scholar]
- 79.Ferry AP. Granular cell tumor (myoblastoma) of the palpebral conjunctiva causing pseudoepitheliomatous hyperplasia of the conjunctival epithelium. Am J Ophthalmol. 1981;91:234–8. doi: 10.1016/0002-9394(81)90180-x. [DOI] [PubMed] [Google Scholar]
- 80.Giller RH, Folberg R, Keech RV, Piette WW, Sato Y. Xanthoma disseminatum. An unusual histiocytosis syndrome. Am J Pediatr Hematol Oncol. 1988;10:252–7. [PubMed] [Google Scholar]
- 81.Chaudhry IA, Al-Jishi Z, Shamsi FA, Riley F. Juvenile xanthogranuloma of the corneoscleral limbus: Case report and review of the literature. Surv Ophthalmol. 2004;49:608–14. doi: 10.1016/j.survophthal.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 82.Eagle RC, Jr, Penne RA, Hneleski IS., Jr Eyelid involvement in multicentric reticulohistiocytosis. Ophthalmology. 1995;102:426–30. doi: 10.1016/s0161-6420(95)31005-6. [DOI] [PubMed] [Google Scholar]
- 83.Pe’er J, Hidayat AA. Myxomas of the conjunctiva. Am J Ophthalmol. 1986;102:80–6. doi: 10.1016/0002-9394(86)90213-8. [DOI] [PubMed] [Google Scholar]
- 84.Shields CL, Shields JA, Honavar SG, Demirci H. Clinical spectrum of primary ophthalmic rhabdomyosarcoma. Ophthalmology. 2001;108:2284–92. doi: 10.1016/s0161-6420(01)00840-5. [DOI] [PubMed] [Google Scholar]
- 85.Cameron JD, Wick MR. Embryonal rhabdomyosarcoma of the conjunctiva. A clinicopathologic and immunohistochemical study. Arch Ophthalmol. 1986;104:1203–4. doi: 10.1001/archopht.1986.01050200109062. [DOI] [PubMed] [Google Scholar]
- 86.Polito E, Pichierri P, Loffredo A, Lasorella G. A case of primary botryoid conjunctival rhabdomyosarcoma. Graefes Arch Clin Exp Ophthalmol. 2006;244:517–9. doi: 10.1007/s00417-005-0085-5. [DOI] [PubMed] [Google Scholar]
- 87.Bryant J. Pleomorphic lipoma of the bulbar conjunctiva. Ann Ophthalmol. 1987;19:148–9. [PubMed] [Google Scholar]
- 88.Krema H, El-Bolkainy N. Rapid growth of an epibulbar complex choristoma in organoid nevus syndrome. Can J Ophthalmol. 2013;48:e82–5. doi: 10.1016/j.jcjo.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 89.Kaeser PF, Uffer S, Zografos L, Hamédani M. Tumors of the caruncle: A clinicopathologic correlation. Am J Ophthalmol. 2006;142:448–55. doi: 10.1016/j.ajo.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 90.Luthra CL, Doxanas MT, Green WR. Lesions of the caruncle: A clinicohistopathologic study. Surv Ophthalmol. 1978;23:183–95. doi: 10.1016/0039-6257(78)90155-8. [DOI] [PubMed] [Google Scholar]
- 91.Say EA, Shields CL, Bianciotto C, Eagle RC, Jr, Shields JA. Oncocytic lesions (oncocytoma) of the ocular adnexa: Report of 15 cases and review of literature. Ophthal Plast Reconstr Surg. 2012;28:14–21. doi: 10.1097/IOP.0b013e31822dd236. [DOI] [PubMed] [Google Scholar]
- 92.Lee SS, Robinson MR, Morris JC, Mirtsching BC, Shen D, Chan CC. Conjunctival involvement with T-cell prolymphocytic leukemia: Report of a case and review of the literature. Surv Ophthalmol. 2004;49:525–36. doi: 10.1016/j.survophthal.2004.06.005. [DOI] [PubMed] [Google Scholar]