

Abstract
Background
Specific host-parasite systems often embody a particular co-distribution phenomenon, in which the parasite’s phylogeographic pattern is dependent on its host. In practice, however, both congruent and incongruent phylogeographic patterns between the host and the parasite have been reported. Here, we compared the population genetics of the plateau zokor (Eospalax baileyi), a subterranean rodent, and its host-associated flea species, Neopsylla paranoma, with an aim to determine whether the two animals share a similar phylogeographic pattern.
Results
We sampled 130 host-parasite pairs from 17 localities in the Qinghai-Tibet Plateau (QTP), China, and sequenced a mitochondrial DNA (mtDNA) segment (~2,500 bp), including the complete COI and COII genes. We also detected 55 zokor and 75 flea haplotypes. AMOVA showed that the percentage of variation among the populations of zokors constituted 97.10%, while the within population variation was only 2.90%; for fleas, the values were 85.68% and 14.32%, respectively. Moreover, the flea Fst (fixation index) values were significantly smaller than in zokor. Although the Fst values between zokors and fleas were significantly and positively correlated (N =105, R =0.439, p =0.000), only a small amount (R2= 0.19) of the flea Fst variations could be explained by the zokor Fst variations. The two animals showed very distinct haplotype network structures from each other while co-phylogenetic analyses were unable to reject the hypothesis of an independence of speciation events.
Conclusions
Zokors and fleas have very distinct population genetic patterns from each other, likely due to the influence of other sympatrically-distributed vertebrates on the transmission of fleas.
Keywords: Zokor, Flea, Population genetics, Fixation index, Host switching
Background
Comparative phylogeography aims to compare geographical patterns of evolutionary subdivision across multiple co-distributed species or species complexes, making it a powerful tool for elucidating the demographic and historical nature of intraspecific evolution [1],[2]. Host-parasite systems represent a specific co-distribution phenomenon, in which the parasite’s phylogeographic pattern is typically dependent on their host; comparative investigations of host-parasite systems have thus become one of the most promising directions of comparative phylogeography [3],[4]. Several studies have shown congruent phylogeographic patterns between parasites and their hosts, such as nematodes (Heligmosomoides polygyrus) and field mice (Apodemus sylvaticus) [5] or mite (Spinturnix myoti) and bat (Myotis punicus) [6]. In contrast, other studies have shown either incongruent (e.g. lice (Polyplax arvicanthis) and rodents (Rhabdomys spp.) [7]) or somewhat incongruent (e.g. nematode (Heligmosomoides spp.) and mice (Apodemus spp.) [8]) phylogeographic patterns between parasites and their hosts. Moreover, many features of the host and its parasite can influence the overall phylogeographic or genealogic congruence, such as the intimacy of the interaction between the two organisms, the scale of vertical transmission of the symbionts through the host generations, and differences in life history traits (population sizes, generation times, and migrating abilities between the hosts and the parasites, etc.) [9]-[12].
Fleas belong to the order Siphonaptera; all species are obligatory blood-feeding parasites of endothermic vertebrates, including mammals and birds [13]. The life cycles of fleas consist of four stages: egg, larva, inactive pupa, and adult. All of these stages are highly dependent on the nests or burrows of their hosts. However, as a typical ectoparasite, all four stages can be viewed as free-living phases. Moreover, adult fleas may also leave their hosts and migrate onto new hosts [13],[14], which potentially negatively influences the congruence of the phylogeographic pattern between fleas and their hosts. Gomez-Diaz et al.[15] compared the phylogeography of fleas (Xenopsylla gratiosa) and shearwaters (Calonectris spp.) and showed that neither genetic distances among host populations nor their spatial distribution explained the patterns of genetic variability observed in the fleas, likely due to a local adaptation to the sympatric host species or to a parasitic exchange during mixing among breeding populations of the hosts. In addition, Jones and Britten [16] compared the genetic structure between prairie dogs (Cynomys ludovicianus) and fleas (Oropsylla hirsuta); interestingly, the prairie dogs, but not the fleas, showed a significant isolation-by-distance pattern. Moreover, the estimated rates of gene flow among the flea colonies were higher than those among host colonies. Furthermore, these authors argued that other sympatric prairie mammals might be involved in dispersing the fleas, thus resulting in a lack of concordance between the population genetic structures of the host and ectoparasite.
Neopsylla paranoma (Ctenophthalmidae) is a Qinghai-Tibet Plateau (QTP) endemic flea [17]. The main host of N. paranoma is the plateau zokor (Eospalax baileyi), a rodent also endemic to the QTP. During a recent survey (Chen et al., Chinese Journal of Vector Biology and Control, in press), N. paranoma comprised 70% of the total fleas collected from trapped zokors. This species is reported as parasitizing sympatric animals (Himalayan marmots (Marmota himalayana), hamsters (Cricetulus longicaudatus), dipodids (Allactaga sibirica), plateau pikas (Ochotona curzoniae), and some birds), but comprise a very small proportion (<1%) of the total fleas collected from each of these hosts [18]. It should be mentioned that, as a typical subterranean rodent, plateau zokors spend almost all of their life in underground burrows with rather limited dispersal. Moreover, as a solitary species, zokors of the same gender rarely come into contact with each other; only during the mating season do males and females meet in temporary burrows for a very short time period (several minutes) [19]. Hence, unlike in the shearwaters and prairie dogs mentioned above, zokor-associated fleas have limited opportunities to transfer among zokor populations and between zokors and other sympatric animals. As a result, we hypothesized that the phylogeographic patterns of fleas would largely reflect the phylogeographic history of the zokors. Here, we sampled both zokors and fleas, and using mitochondrial DNA sequences, aimed to establish whether there was a high congruence in the phylogeographic patterns between the two species.
Results
General information
A total of 130 zokor-flea pairs were collected from 17 locations (Table 1, including the GPS coordinates; Figure 1). In 12 locations, 7~12 pairs were collected. In the other 5 locations, only 1~4 pairs were collected because of a low parasitism rate for the fleas. The concatenated mitochondrial DNA (mtDNA) sequences of the zokors were 2,502 bp in length, successively consisting of complete sequences of six genes: tRNA-Cys, tRNA-Tyr, COI (cytochrome c oxidase subunit I), tRNA-Ser, tRNA-Asp, and COII (cytochrome c oxidase subunit II). The main segments were the COI and COII genes, which have 1,545 bp and 684 bp, respectively. The concatenated mtDNA sequences of the fleas were 2,407 bp in length, successively consisting of complete sequences of five genes: tRNA-Cys, tRNA-Tyr, COI, tRNA-Leu, and COII. Moreover, the main segments were also COI and COII genes, which have 1,536 bp and 681 bp, respectively.
Table 1.
Sampling sites and haplotype distribution of zokors and fleas
| Population | County | Longitude | Latitude | Elevation | Sample size | Haplotype distribution | |
|---|---|---|---|---|---|---|---|
| /E° | /N° | /m | Zokor | Flea* | |||
| BM | Banma | 100.5639 | 33.12453 | 3705 | 7 | Zh1~Zh3 | Fh1~Fh4 |
| DT | Datong | 101.7891 | 37.15203 | 2988 | 12 | Zh4~Zh11 | Fh5~Fh13 |
| GD | Guide | 101.5539 | 36.29937 | 3119 | 1 | Zh12 | Fh14 |
| GH | Gonghe | 99.73498 | 37.03382 | 3209 | 10 | Zh13, Zh14 | Fh15~Fh22 |
| GN2 | Guinan | 100.4618 | 35.57738 | 3306 | 10 | Zh15~Zh20 | Fh23~Fh26 |
| GN3 | Guinan | 101.3012 | 35.76745 | 3302 | 4 | Zh21 | Fh27~Fh29 |
| HL | Hualong | 102.2971 | 36.18848 | 3185 | 10 | Zh22~Zh26 | Fh30~Fh37 |
| HN | Henan | 101.5598 | 34.77488 | 3552 | 2 | Zh27, Zh28 | Fh38 |
| HY | Huangyuan | 101.0782 | 36.65460 | 3043 | 10 | Zh29~Zh35 | Fh39~Fh48 |
| HZ | Huzhu | 102.2560 | 37.03535 | 2857 | 10 | Zh36~Zh38 | Fh49~Fh55 |
| JZ1 | Jiuzhi | 101.4916 | 33.25988 | 3741 | 8 | Zh39 | Fh56~Fh58 |
| JZ2 | Jiuzhi | 100.8221 | 33.60497 | 3847 | 4 | Zh40, Zh41 | Fh59~Fh61 |
| MY | Menyuan | 101.8255 | 37.32348 | 2715 | 8 | Zh42, Zh43 | Fh62 |
| QL1 | Qilian | 100.1932 | 38.10408 | 3213 | 10 | Zh44~Zh46 | Fh63~Fh66 |
| QL2 | Qilian | 100.5255 | 37.65958 | 3566 | 12 | Zh47~Zh52 | Fh67~F69, Fh64 |
| XH | Xinghai | 99.9187 | 35.85298 | 3566 | 11 | Zh53, Zh54 | Fh70~Fh74 |
| ZK | Zeku | 100.9617 | 35.23765 | 3428 | 1 | Zh55 | Fh75 |
*Fh25 was shared by GN2, GN3, and XH; Fh64 was shared by QL1 and QL2.
Figure 1.
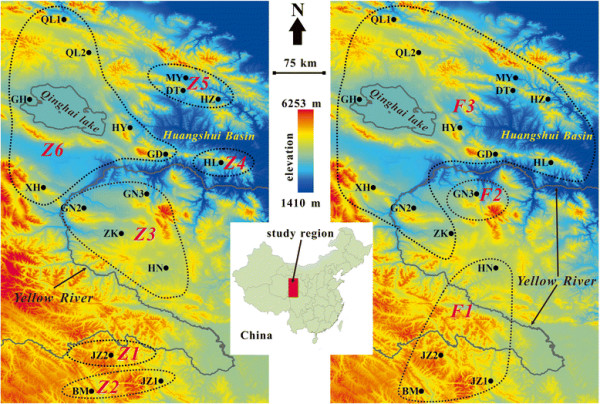
Phylogeographic distributions of phylogenetic clades of zokor (left) and flea (right) populations.
From the 130 zokor sequences, a total of 287 variable sites and 55 haplotypes (Zh1~Zh55) were detected. No haplotype was shared by two or more populations. For the 130 flea sequences, a total of 157 variable sites and 75 haplotypes (Fh1~Fh75) were detected. Of these, there were two haplotypes shared by two (Fh64, shared by QL1 and QL2) and three (Fh25, shared by GN2, GN3, and XH) populations, respectively. The distributions of the haplotypes are listed in Table 1.
Population structure
AMOVA (Analyses of Molecular Variance, based on all 17 locations) showed that, for zokors, the percentage of variation between populations constituted 97.10%, while the within population variation constituted only 2.90%. For fleas, the percentage of variation among populations and within populations constituted 85.68% and 14.32%, respectively. All these AMOVA tests were highly significant (p = 0.000).
The Fst (fixation index) values and geographic distances among the 15 zokor/flea populations are listed in Table 2. A Mantel test showed that the correlation of (R2) the zokor Fst with the geographic distance was 15.84% (P = 0.000), while the flea Fst values were also significantly correlated with geographic distance (p = 0.000). However, the R2 value was 46.20%, much higher than that of the zokors. A Wilcoxon Signed Ranks Test revealed that the flea Fst values were significantly smaller than zokor Fst (N =105, Z = −7.712, p =0.000). In addition, a Spearman correlation analysis showed that the Fst values of zokors and fleas were also significantly and positively correlated (N =105, R =0.439, p =0.000).
Table 2.
Geographic distance (upper-right triangular) and genetic distance ( Fst ) (lower-left triangular; zokor, before the comma; flea, after the comma) among 15 populations (for the other 2 which had only one sample each were not considered)
| BM | DT | GH | GN2 | GN3 | HL | HN | HY | HZ | JZ1 | JZ2 | MY | QL1 | QL2 | XH | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BM | — | 460.59 | 440.12 | 272.21 | 300.90 | 375.26 | 204.91 | 394.41 | 460.57 | 87.80 | 58.45 | 479.83 | 553.40 | 503.09 | 308.34 |
| DT | 0.98,0.94 | — | 183.07 | 211.48 | 159.78 | 116.23 | 264.66 | 84.05 | 43.50 | 432.79 | 403.28 | 19.31 | 176.10 | 125.26 | 221.02 |
| GH | 0.99,0.96 | 0.98,0.63 | — | 174.25 | 198.68 | 247.67 | 299.89 | 126.95 | 224.30 | 448.18 | 392.96 | 188.39 | 125.45 | 98.61 | 132.02 |
| GN2 | 0.98,0.98 | 0.97,0.71 | 0.98,0.40 | — | 78.86 | 179.09 | 133.90 | 131.77 | 228.37 | 273.95 | 221.26 | 229.11 | 281.36 | 231.09 | 57.87 |
| GN3 | 0.99,0.77 | 0.98,0.78 | 1.00,0.77 | 0.39,0.80 | — | 101.28 | 112.63 | 100.47 | 164.77 | 278.77 | 243.89 | 178.98 | 277.43 | 221.11 | 125.29 |
| HL | 0.97,0.97 | 0.96,0.60 | 0.96,0.68 | 0.97,0.81 | 0.97,0.79 | — | 170.60 | 120.98 | 94.11 | 333.31 | 316.82 | 132.88 | 283.10 | 227.13 | 217.61 |
| HN | 0.97,0.90 | 0.97,0.93 | 0.99,0.95 | 0.43,0.99 | 0.66,0.53 | 0.96,0.96 | — | 213.10 | 258.68 | 168.21 | 146.52 | 283.90 | 389.20 | 333.33 | 191.24 |
| HY | 0.96,0.92 | 0.94,0.47 | 0.62,0.12 | 0.96,0.26 | 0.95,0.72 | 0.91,0.51 | 0.94,0.90 | — | 113.27 | 378.55 | 339.14 | 99.70 | 178.92 | 121.85 | 136.98 |
| HZ | 0.99,0.95 | 0.63,0.15 | 0.99,0.65 | 0.98,0.75 | 0.99,0.77 | 0.97,0.64 | 0.98,0.94 | 0.95,0.48 | — | 424.80 | 402.39 | 49.87 | 217.46 | 168.28 | 247.24 |
| JZ1 | 0.84,0.67 | 0.98,0.95 | 1.00,0.97 | 0.98,0.99 | 1.00,0.79 | 0.98,0.97 | 0.99,0.96 | 0.97,0.93 | 0.99,0.96 | — | 73.09 | 452.00 | 550.14 | 495.96 | 321.81 |
| JZ2 | 0.99,0.42 | 0.98,0.94 | 1.00,0.95 | 0.98,0.99 | 1.00,0.68 | 0.98,0.96 | 0.98,0.92 | 0.96,0.91 | 0.99,0.95 | 1.00,0.62 | — | 422.55 | 502.34 | 450.62 | 262.70 |
| MY | 0.98,0.99 | 0.59,0.53 | 0.99,0.83 | 0.98,0.96 | 0.99,0.80 | 0.96,0.86 | 0.98,1.00 | 0.94,0.65 | 0.84,0.62 | 0.99,1.00 | 0.99,0.99 | — | 167.99 | 120.87 | 236.08 |
| QL1 | 0.98,0.98 | 0.97,0.47 | 0.86,0.80 | 0.98,0.90 | 0.98,0.82 | 0.95,0.80 | 0.97,0.98 | 0.46,0.63 | 0.98,0.53 | 0.99,0.98 | 0.99,0.98 | 0.97,0.69 | — | 57.34 | 250.94 |
| QL2 | 0.98,0.93 | 0.96,0.28 | 0.76,0.49 | 0.97,0.59 | 0.98,0.76 | 0.94,0.54 | 0.97,0.91 | 0.25,0.33 | 0.97,0.29 | 0.98,0.94 | 0.98,0.92 | 0.97,0.37 | 0.61,0.19 | — | 207.62 |
| XH | 0.99,0.97 | 0.98,0.70 | 0.96,0.41 | 0.99,0.14 | 1.00,0.79 | 0.96,0.77 | 0.99,0.97 | 0.70,0.28 | 0.99,0.73 | 1.00,0.98 | 1.00,0.97 | 0.99,0.91 | 0.89,0.87 | 0.84,0.58 | — |
Network and co-phylogenetic results
Both zokors and fleas had very distinct network features. The 55 zokor haplotypes formed into six distinct clades (Z1~Z6, Figure 2), corresponding to six geographic regions (Figure 1). However, the 75 flea haplotypes formed only three clades: F1 included four populations while F2 included only one population and F3 included the other 12 populations related with the majority of the haplotypes (61 of 75) (Figures 1 and 3).
Figure 2.
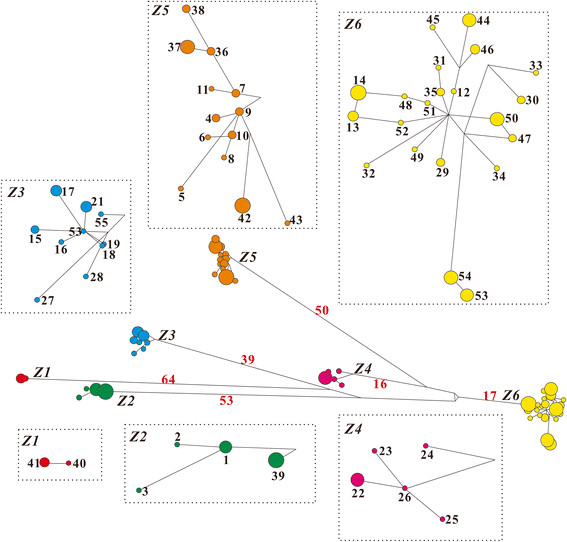
Network structures of zokor haplotypes. Z1~Z6 show the network clades, also see Figure 1 for details. The red numbers show the lengths (number of substitutions) of the main branches.
Figure 3.
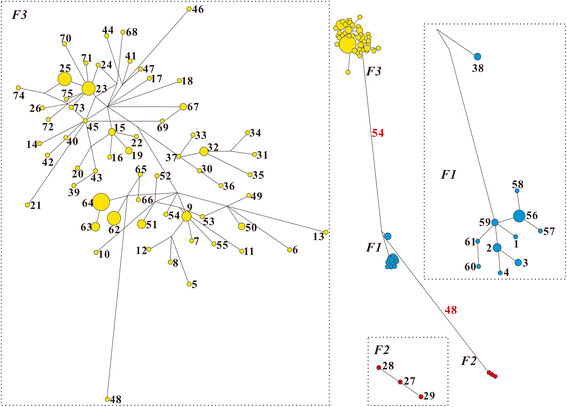
Network structures of flea haplotypes. F1~F3 show the network clades, also see Figure 1 for details. The red numbers show the lengths (number of substitutions) of the main branches.
Co-phylogenetic analyses, based on either the 17 populations (ParaFitGlobal = 0.0156, p = 0.286) or the 15 populations (ParaFitGlobal = 0.0107, p = 0.284), were unable to reject the hypothesis of independence of speciation events. Based on ParaFit1 values, only one (JZ2) of 17 or 15 host-parasite links was significant (p <0.05).
Discussion
Incongruent genetic structure
Our results showed that the zokors and fleas had very distinct population genetic structures from each other. First, the AMOVA results revealed that the variation between the flea populations (85.68%) was considerably lower than that among zokor populations (97.10%). Second, flea Fst values were significantly smaller compared to the zokor Fst (p =0.000). Third, each zokor haplotype was uniquely distributed within a single population; for fleas, however, two haplotypes were shared between two or more populations. Fourth, the co-phylogenetic analyses were unable to reject the hypothesis of independence of speciation events between the two animals.
The results of the network analyses also showed a distinct geographic haplotype distribution pattern between the two species. The JZ2 population, which was an independent clade in zokors, showed a closer relation with its neighboring populations, JZ1 and BM. Also, the 12 populations (DT, GD, GH, HL, GN2, HY, HZ, MY, QL1, QL2, XH, and ZK), which were grouped into four separate clades in zokors, were grouped into a single clade in fleas (Table 1 and Figure 1). These results indicate that fleas have a lower level of genetic differentiation than zokors.
Effects of geographic barriers
The QTP is the largest and highest plateau on Earth. The QTP began a severe uplift during the Pliocene, and the uplift continued until the Quaternary glaciations. As a result, the plateau began to topographically diversify due to the intricate and continual development of new mountains and watercourses [20], which likely became geographical barriers that helped create various genetic structures for endemic animals [21]. It should be mentioned that with different dispersion abilities, each species of animal living on the QTP tends to have very distinct genetic structures. For example, there is a reported absence in distinct genetic structures among populations of a snow finch (Pyrgilauda ruficollis), which is a strong flier and tend to move seasonally. In contrast, a high level of genetic structure was reported for the ground tit (Pseudopodoces humilis; a resident bird species) and a lizard (Phrynocephalus vlangalii), because the two species tend not to disperse (reviewed in [22]).
Like other subterranean rodents (such as the tiny tuco-tuco (Ctenomys minutus) [23]), geographic barriers seem to have played an important role in shaping the phylogeographic structure of zokors: the Yellow River separated Z1 and Z2 from Z3~Z6; the extremely high altitude at and around the JZ2 group isolated the haplotypes in this region into a basal clade Z1, whereas the Huangshui Basin separated Z4, Z5, and Z6 from each other. However, it is noteworthy that distance, rather than physical barriers, seems to have played an important role in shaping the geographic patterns of fleas: the R2 of the IBD effects in fleas (46.20%) was much higher than that of the zokors (15.84%). The most evident example of the different effects of geographic barriers on population genetics occurred between the XH and GN2 groups. The geographic distance between the two populations was only 57.87 km, but the Fst between them was 0.99; as such, they were grouped into two different clades Z3 and Z6. For fleas however, the Fst between the two populations was only 0.14, and subsequently, they were grouped into a monophyletic clade F3. Taken together, these results suggest that the QTP-associated geographic barriers had a strong isolation effect in the genetic variation of zokors; in contrast, this effect was much weaker on fleas.
The third-part flea hosts?
As mentioned above, the differences in life history traits, such as population sizes, generation times, and migrating abilities, between the hosts and the parasites could directly or indirectly influence the interactions between the two organisms [10]-[12]. Although little is known about the ecology of the flea (N. paranoma), much more is known about zokors. Zokors spend most of their lives underground and have a very limited migration ability (only about 66.9 meters per year) [24]. As such, genetic interactions between the populations resulting from zokor migrations would also be very limited. In the current paper, we found that no haplotype was shared by different populations of zokors, further supporting this view. Moreover, zokors live a solitary life except during the mating season [19]. As a result, the migration of fleas via zokor contact should also be very limited. In other words, even if the fleas in our study have very distinct life history traits compared to the zokors that they live on, the phylogeographic features of fleas should still largely depend on the zokors, provided no other vertebrates were involved in this host-parasite system.
Unexpectedly, however, we found that the fleas had lower inter-population genetic variation levels than zokors, suggesting that some other host animals have also mediated their migration and subsequently had a significant influence on their phylogeographic patterns. The degree of association between fleas and hosts varies among different host/flea species [25]. Zhang [18] reported that Himalayan marmots, hamsters, dipodids, plateau pikas, and some birds, all of which are good migrators, could also be infected by N. paranoma. It is puzzling, however, how zokors might exchange their parasites with above-ground animals. Pikas are the most closely related sympatric animals to plateau zokors. In many cases, pikas occupy the burrows of zokors [26], which might provide opportunities of flea exchange between the two species. Moreover, as a key species in the QTP [27], plateau pikas also share habitats with most of the other species in the QTP, as well. We hypothezed that the plateau pikas first serve as a mediator of fleas between zokors and above-ground animals (including birds), and then these above-ground animals help to disperse the fleas among different regions. It should be mentioned that there is a haplotype (Fh25) that is shared by GN2, GN3, and XH, which were separated by the Yellow River. Since a large majority of mammalian animals seem unable to migrate over the Yellow River, we suggest that birds might play a more important role for cross-river flea transmission.
Conclusions
Despite the unique subterranean lifestyle of the zokor, the population genetic patterns of the fleas were distinct from their hosts. Moreover, fleas had a lower level of genetic variation between populations compared to zokors, suggesting other sympatrically distributed vertebrates serve as a mediator for flea transmission.
Methods
Sampling
During April and May, 2012, individual zokors were sampled using ground arrows [28] from the east of Qinghai Province, China. The geographical information at each sampling site was recorded using an Etrex GPS unit (Garmin, Taiwan). Each of the trapped zokors was immediately put into a plastic bag filled with diethyl ether for ten minutes. The dead fleas were collected and stored in 75% alcohol. Muscle samples from each zokor were collected and fixed in 95% alcohol. The taxonomy of the fleas was identified under light microscopy. All animal work in this study was conducted with ethical approval from the Ethics Committee, Northwest Institute of Plateau Biology, Chinese Academy of Sciences. No specific permissions were required for the locations/activities in this study. The field studies did not involve endangered or protected species.
DNA extracting, amplifying and sequencing
Total DNA of each zokor sample was extracted using standard methods for animal tissue [29]. The sequences of the three mitochondrial DNA (mtDNA) segments were amplified by polymerase chain reaction (PCR) using three pairs of primers (Table 3) designed with reference to the plateau zokor mitochondrial genome (accession No. JN540033.1). Total DNA from each flea was extracted using a spin column kit (DNeasy tissue kit; Qiagen, Germany), according to the manufacturer’s protocol. The sequences of three target mtDNA segments were amplified using three primer pairs (Table 3). Two were designed according to the mitochondrial genome of a mecopteran species (Boreus elegans, NC_015119.1), which was viewed as the closest sister group of the fleas [30], and the other one is according to Whiting [30].
Table 3.
Primers used for amplifying and sequencing mitochondrial segments of zokors and fleas
| Primer pairs | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| zokor1-F/R | CCTCAACAAACAACATAA | TGAAAAGGAAAATAAAAC |
| zokor2-F/R | TTATTTCCCATATCGTTAC | GTTCTCCTGGTTTTAGTTC |
| zokor3-F/R | CAACAAACCTGGAATGAC | GAGAAAGAGGCGAATAAA |
| flea1-F/R | ACGCCCTTTCATTTTTGA | AAGTTTACCTGATTCTTGAG |
| flea2-F/R | TGGTCACCCAGAAGTA | AGAAGGAAGGGCAAT |
| COII-F-leu/R-lys* | TCTAATATGGCAGATTAGTGC | GAGACCAGTACTTGCTTTCAGTCATC |
*According to Whiting (2002) [30].
PCR amplifications were performed in total reaction volumes of 50 μL, containing 10mM Tris–HCl (pH 8.3), 1.5 mM MgCl2, 50 mM KCl, 100 μM of each dNTP, 0.25 μMol each primer (synthesized by Sangon Ltd., China), 0.5 μl of template DNA, and 2.5U Taq DNA polymerase (Takara). The reaction mixtures were denatured at 95°C for 5min and subjected to 35 cycles of 45 s at 95°C, 1 min at 50~55°C (depending on the primer pair), 1.5 min at 72°C, and a final extension step of 7 min at 72°C. PCR products were purified using a CASpure PCR Purification Kit, following the manufacturer’s recommended protocol (Casarray, Shanghai, China). Purified DNA products were sequenced in both directions with the PCR primers on an ABI 373 automated sequencer. Both strands of each segment were sequenced using forward and reverse primers. Sequences were recorded in both strands with an overlap of at least 30%. The sequences of each segment were checked by eye and aligned using CLUSTAL W [31] and refined manually. Finally, the three segments of each species were combined for further analyses.
Genetic variation calculation
Basic polymorphism data (variable sites, haplotype distribution, etc.) were determined using DnaSP (version 5) [32]. The Arlequin (version 3.11) [33] software was used to calculate AMOVA (Analyses of Molecular Variance) and Fst values among populations. We then used ArcGIS software (version 9.3) to calculate the geographic distances among the sampling locations, and used Arlequin to carry out Mantel test [34] to examine isolation-by-distance effects of the two species (1000 permutations were used). We used a non-parametric 2 Related Samples Test (Wilcoxon Signed Ranks Test) in SPSS software (version 20.0) to compare the zokor/flea Fst values. We also used a Spearman correlation in SPSS to test the relationships between Fst values of zokors and fleas. It should be mentioned that, in two locations (GD and ZK) there was only one zokor-flea pair each, which could induce unrealistic Fst estimates such as negative values. Hence, for the analyses relating Fst values, these locations were excluded.
Network and co-phylogenetic analysis
Roehl data files were generated with DnaSP. The haplotype networks of zokors and fleas were constructed with the program NETWORK (version 4.6.1.2). The Median Joining method [35] was used to calculate network structure. Then the drawn networks were saved as bitmap and were introduced into CorelDraw (version X6) to prepare the final feature figures.
The optimized versions of Pierre Legendre’s Parafit (i.e. AxParafit) [36] was used to test the co-phylogenetic congruence between zokors and fleas. First, the between group mean distance (Kimura 2-parameter model) matrices based on the 17 populations and 15 populations (GD and ZK excluded) were calculated with MEGA (version 5.2) [37]. The global-fit congruence and individual host-parasite interaction analysis were tested with 100,000 permutations using zokor distance matrix, flea distance matrix, and the zokor-flea association file. Each individual zokor-flea interaction is determined to be significant if either its ParaFit 1 or Parafit 2 p-value <0.05.
Availability of data
The data supporting the results of this article are available in the Dryad repository (http://dx.doi.org/10.5061/dryad.d0v11) [38]. The haplotypes are also available in GenBank (accession numbers: Zh1~Zh55, KJ470899~KJ470953; Fh1~Fh75, KJ470954~KJ471028).
Abbreviations
QTP: The Qinghai-Tibet Plateau
mtDNA: Mitochondrial DNA
COI: Cytochrome c oxidase subunit I
COII: Cytochrome c oxidase subunit II
AMOVA: Analyses of molecular variance
PCR: Polymerase chain reaction
cytb: Cytochrome b
Fst: (Fixation index)
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GL, JS, and TZ conceived the study. FZ carried out the molecular genetic work. GL and JS performed data analysis. HC and XD coordinated sampling of material. GL and JS interpreted the biological context of results. All authors wrote, read and approved the final manuscript.
Contributor Information
Gonghua Lin, Email: lingonghua@gmail.com.
Fang Zhao, Email: zf_lgh@163.com.
Hongjian Chen, Email: mjy70315@163.com.
Xiaogong Deng, Email: xgdeng@nwipb.cas.cn.
Jianping Su, Email: jpsu@nwipb.cas.cn.
Tongzuo Zhang, Email: nwipb@hotmail.com.
Acknowledgements
The research was supported by the National Natural Science Foundation of China (31101628, 81260416 and 31370405) and the West Light Foundation of the Chinese Academy of Sciences to G. Lin (2011–2013). We thank Prof. Mike Bruford in Cardiff University for his valuable comments on the manuscript. We also thank Prof. Huabin Zhao in Wuhan University for his help on laboratory work.
References
- Arbogast B, Kenagy GJ. Comparative phylogeography as an integrative approach to historical biogeography. J Biogeogr. 2001;28:819–825. doi: 10.1046/j.1365-2699.2001.00594.x. [DOI] [Google Scholar]
- Avise JC. Phylogeography: retrospect and prospect. J Biogeogr. 2009;36:3–15. doi: 10.1111/j.1365-2699.2008.02032.x. [DOI] [Google Scholar]
- Toon A, Hughes JM. Are lice good proxies for host history? A comparative analysis of the Australian magpie, Gymnorhina tibicen, and two species of feather louse. Heredity. 2008;101(2):127–135. doi: 10.1038/hdy.2008.37. [DOI] [PubMed] [Google Scholar]
- Kholodova MV. Comparative phylogeography: molecular methods, ecological interpretation. Mol Biol. 2009;43(5):847–854. doi: 10.1134/S002689330905015X. [DOI] [PubMed] [Google Scholar]
- Nieberding C, Morand S, Libois R, Michaux JR. A parasite reveals cryptic phylogeographical history of its host. P Roy Soc B-Biol Sci. 2004;271:2559–2568. doi: 10.1098/rspb.2004.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruyndonckx N, Biollaz F, Dubey S, Goudet J, Christe P. Mites as biological tags of their hosts. Mol Ecol. 2010;19(13):2770–2778. doi: 10.1111/j.1365-294X.2010.04699.x. [DOI] [PubMed] [Google Scholar]
- du Toit N, van Vuuren BJ, Matthee S, Matthee CA. Biogeography and host-related factors trump parasite life history: limited congruence among the genetic structures of specific ectoparasitic lice and their rodent hosts. Mol Ecol. 2013;22(20):5185–5204. doi: 10.1111/mec.12459. [DOI] [PubMed] [Google Scholar]
- Nieberding CM, Durette-Desset MC, Vanderpoorten A, Casanova JC, Ribas A, Deffontaine V, Feliu C, Morand S, Libois R, Michaux JR. Geography and host biogeography matter for understanding the phylogeography of a parasite. Mol Phylogenet Evol. 2008;47:538–554. doi: 10.1016/j.ympev.2008.01.028. [DOI] [PubMed] [Google Scholar]
- Nieberding CM, Olivieri I. Parasites: proxies for host genealogy and ecology? Trends Ecol Evol. 2007;22:156–165. doi: 10.1016/j.tree.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Criscione CD, Poulin R, Blouin MS. Molecular ecology of parasites: elucidating ecological and microevolutionary processes. Mol Ecol. 2005;14(8):2247–2257. doi: 10.1111/j.1365-294X.2005.02587.x. [DOI] [PubMed] [Google Scholar]
- Rannala B, Michalakis Y. In: Tangled trees: phylogeny, cospeciation and coevolution. Page RDM, editor. University of Chicago Press, Chicago; 2003. Population genetics and cospeciation: from process to pattern; pp. 120–143. [Google Scholar]
- Morand S, Krasnov BR, Poulin R. Micromammals and macroparasites. Springer-Verlag, Tokyo; 2006. [Google Scholar]
- Krasnov BR. Functional and evolutionary ecology of fleas - a model for ecological parasitology. Cambridge University Press, Cambridge; 2008. [Google Scholar]
- Wu HY, Liu Q, Lu L, Gong ZD, Wang DQ, Yu X, Li C, Liu JY. Fauna sinica, Insecta, Siphonaptera. Science Press, Beijing; 2007. [Google Scholar]
- Gomez-Diaz E, Gonzalez-Solis J, Peinado MA, Page RD. Lack of host-dependent genetic structure in ectoparasites of Calonectris shearwaters. Mol Ecol. 2007;16:5204–5215. doi: 10.1111/j.1365-294X.2007.03539.x. [DOI] [PubMed] [Google Scholar]
- Jones PH, Britten HB. The absence of concordant population genetic structure in the black-tailed prairie dog and the flea, Oropsylla hirsuta, with implications for the spread of Yersinia pestis. Mol Ecol. 2010;19:2038–2049. doi: 10.1111/j.1365-294X.2010.04634.x. [DOI] [PubMed] [Google Scholar]
- Cai LY, Zhan XR, Wu WZ, Li C. The Flea Fauna of Qinghai-Xizang Plateau. Shaanxi Science and Technology Publishing House, Xian; 1997. [Google Scholar]
- Zhang GD. Epidemiological survey of fleas in Qinghai Nan Mountain. Chinese J Vector Biol Control. 1994;3:186, 228. [Google Scholar]
- Zhou WY, Dou FM. Studies on activity and home range of plateau zokor. Acta Theri Sin. 1990;10(1):31–39. [Google Scholar]
- Li JJ, Li BY, Wang FB. The process of uplift of the Qinghai-Xizang Plateau. Geological and ecological studies of Qinghai-Xizang Plateau. Science Press, Beijing; 1981. [Google Scholar]
- Yang S, Dong H, Lei F. Phylogeography of regional fauna on the Tibetan Plateau: a review. Prog Nat Sci. 2009;19:789–799. doi: 10.1016/j.pnsc.2008.10.006. [DOI] [Google Scholar]
- Ci HX, Lin GH, Cai ZY, Tang LZSJP, Liu JQ. Population history of the plateau pika endemic to the Qinghai-Tibetan Plateau based on mtDNA sequence data. J Zool. 2009;279(4):396–403. doi: 10.1111/j.1469-7998.2009.00635.x. [DOI] [Google Scholar]
- Lopes CM, Ximenes SSF, Gava A, de Freitas TRO. The role of chromosomal rearrangements and geographical barriers in the divergence of lineages in a South American subterranean rodent (Rodentia: Ctenomyidae: Ctenomys minutus) Heredity. 2013;111:293–305. doi: 10.1038/hdy.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei WH, Wang QY, Zhou WY, Fan NC. The population dynamics and dispersal of plateau zokors after removing. Acta Their Sin. 1997;17:53–61. [Google Scholar]
- Poulin R, Krasnov BR, Shenbrot GI, Mouillot D, Khokhlova IS. Evolution of host specificity in fleas: is it directional and irreversible? Int J Parasitol. 2006;36:185–191. doi: 10.1016/j.ijpara.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Zhang WG, Jiang XL. A study of commensalism of plateau zokor and plateau pika. Acta Prataculturae Sin. 1998;7(2):43–47. [Google Scholar]
- Smith AT, Foggin JM. The plateau pika (Ochotona curzoniae) is a keystone species for biodiversity on the Tibetan plateau. Anim Conserv. 1999;2:235–240. doi: 10.1111/j.1469-1795.1999.tb00069.x. [DOI] [Google Scholar]
- Han CX, Yang XJ, Wang MC. The integrate management of forest-rodents. Northwest Sci-Tech University of Agriculture and Forestry Press, Xian; 2003. [Google Scholar]
- Sambrook J, Russell DW, Russell DW. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York; 2001. [Google Scholar]
- Whiting MF. Mecoptera is paraphyletic: multiple genes and phylogeny of Mecoptera and Siphonaptera. Zool Scr. 2002;31:93–104. doi: 10.1046/j.0300-3256.2001.00095.x. [DOI] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Mougin FJ, Higgins DG. The Clustal X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acid Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Excoffier LGL, Schneider S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209–220. [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Legendre P, Desdevises Y, Bazin E. A statistical test for host–parasite coevolution. Syst Biol. 2002;51(2):217–234. doi: 10.1080/10635150252899734. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.5061/dryad.d0v11. 10.5061/dryad.d0v11 Lin G, Zhao F, Chen H, Deng X, Su J, Zhang T: Comparative phylogeography of the plateau zokor (Eospalax baileyi) and its host-associated flea (Neopsylla paranoma) in the Qinghai-Tibet Plateau.Dryad Digital Repository 2014, http://dx.doi.org/. [DOI] [PMC free article] [PubMed]