

Abstract
Background
Histone deacetylase (HDAC) inhibitors combined with other drugs for the treatment of malignant tumors are used more and more widely. In this study, we investigated the effect of trichostatin A (TSA), a HDAC inhibitor, in combination with cisplatin, a cytotoxic chemotherapy agent, on the apoptosis of lung cancer A549 cells.
Methods
A549 cells were treated with TSA alone, cisplatin alone or the two drugs combined. Cell viability and apoptosis were evaluated using a light microscope, methyl thiazolyl tetrazolium (MTT) (3-[4, 5-dimethylthiazol-2-yl] −2, 5-diphenyltetrazolium bromide) assay and Hochst33258 staining. Moreover, Western blot analysis was employed to examine the alterations of apoptosis protein: cellular Fas-associated death domain-like interleukin-1 β converting enzyme inhibitory protein (cFLIP) and caspase-8 in A549 cells in response to the different exogenous stimuli.
Results
Compared with single-agent treatment, the co-treatment of A549 cells with TSA and cisplatin synergistically inhibited cell proliferation, induced apoptosis, and increased the inhibition rate. Treatment with TSA and cisplatin led to a significant decrease of cFLIP expression. Furthermore, the treatment of A549 cells with TSA and cisplatin resulted in a significant decrease of pro-caspase-8 and a significant increase of caspase-8.
Conclusions
Synergistic anti-tumor effects are observed between cisplatin and TSA in lung cancer cells. The combination of TSA with cisplatin may be a more effective method in human lung cancer treatment.
Keywords: Caspase-8, cellular Fas-associated death domain-like interleulin-1 β converting enzyme inhibitory protein (cFLIP), cisplatin, Trichostatin A (TSA)
Introduction
Reports conducted by the World Health Organization show that, in many countries, lung cancer incidence is highest among malignant tumors, resulting annually in more than one million deaths worldwide.1,2 Despite recent advances in diagnosis and multimodality therapies, prognosis remains poor with five-year survival rates of only 16% for all stages.3 New therapeutic methods are needed for lung cancer patients.
In recent years, the importance of epigenetic alterations has been appreciated in cancer development,4 including the role of abnormal DNA methylation and histone acetylation on aberrant silencing of multiple tumor suppressor genes in a diversity of human cancers.5,6 Histone deacetylase (HDAC) inhibitors, which interfere with the function of histone deacetylase, are emerging as potent anticancer agents as a result of their effective anti-proliferative activity in a wide variety of tumors, mediated by mitotic defects through the aberrant acetylation of histone and non-histone proteins.7 It has been reported that trichostatin A (TSA), a potent specific inhibitor of HDAC, is able to lead to cell growth arrest, differentiation, and/or apoptosis in a number of cancers.8–10 Evidence also suggests that TSA may have a promising therapeutic effect on cancer cells when combined with radiotherapy or chemotherapy.11,12
Cisplatin is a kind of anticancer drug containing platinum. It is a broad anticancer spectrum, whose strong function, synergistic effect with various antitumor drugs, and lack of cross resistance, make it one of the most commonly used drugs in combination chemotherapy. Cisplatin places two links with single chain DNA or cross-links with double chain DNA, and inhibits the process of DNA replication in cancer cells, leading to cell apoptosis. Cisplatin belongs to non-specific cell cycle drugs, and causes damage to the cell membrane structure. However, after long-term use of cisplatin, most patients develop a resistance to it. Cisplatin also has a relatively high number of side effects.
Studies have found that TSA can enhance the sensitivity of cisplatin resistant lung cancer cells to cisplatin.13 Another study found that, in gastrointestinal malignant tumors, prostate, breast, and ovarian cancers, TSA combined with cisplatin has a significantly higher inhibitory effect on cell proliferation than single drug treatment groups.14–16 Compared with single drug groups, the HDAC inhibitor combined with cisplatin had an increased acetylation level. Thus, the following questions are raised: Are there synergistic effects when TSA is combined with cisplatin in lung carcinoma cells? Can TSA enhance the sensitivity of lung carcinoma cells to cisplatin, so as to reduce the clinical dosage of cisplatin?
In this light, the present study was designed to investigate antitumor activity by combining TSA with cisplatin on lung adenocarcinoma A549 cells, and its inner mechanism. Our results demonstrated that TSA in combination with cisplatin could be valuable in the treatment of lung cancer.
Methods
Materials
Human lung adenocarcinoma A549 cells were obtained from the central laboratory of the Shandong Provincial Hospital affiliated to Shandong University. Cellular Fas-associated death domain-like interleukin-1 β converting enzyme inhibitory protein (cFLIP) antibody (Proteintech Company, 10394-1-AP, Chicago, IL, USA) and caspase-8 antibody (Proteintech Company, 13423-1-AP) were stored at −20°C. Horseradish peroxidase labeled Goat anti rabbit IgG (H + L) (Proteintech Company, SA00001-2) was stored at −20°C.
Cell culture
A549 cell line was cultured in RPMI 1640 (Gibco, Brussels, Belgium) containing 100 U/mL penicillin and 100 μg/mL streptomycin and supplemented with 10% calf blood serum (Sijiqing Laboratories, Hangzhou, China) at 37°C in a humidified atmosphere with 5% CO2.
Drug preparation and cell grouping
One milligram of TSA (Sigma, St Louis, MO, USA, T8552-1MG) was dissolved in sterile dimethyl sulfoxide (DMSO) solution; the storage concentration was 1g/L, stored at −20°C, and diluted to the required concentration with fresh culture medium before each experiment. Cisplatin (Qi lu Pharmaceutical Co, Ltd, Shandong, China) was dissolved in sterile culture medium; the storage concentration was 100 g/L, stored at −20°C, and diluted to the required concentration with fresh culture medium before each experiment. The cell groups were categorized as: 1, blank control group; 2, TSA 250 nmol/L group; 3, cisplatin 10ug/mL group; and 4, TSA 250 nmol/L + cisplatin 10ug/mL group.
Morphological changes of the cells
After treatment with the drug for 24 hours, the cells’ morphology was observed to have changed. Pictures were taken with an inverted phase contrast microscope (Olympus, Tokyo, Japan) and it was found that there were differences between the groups.
Assay inhibition rate using methyl thiazolyl tetrazolium method (MTT)
The effects of TSA, cisplatin, and TSA/cisplatin, on A549 cell growth were measured by methyl thiazolyl tetrazolium (MTT) assay. The cells were chosen in the log growth phase (5 × 104 cells/mL) and cultured overnight in 96-well plates (200μL/well). The cells were incubated with: 1, blank control; 2, TSA 250 nmol/L; 3, cisplatin 10ug/mL; and 4, TSA 250 nmol/L + cisplatin 10ug/mL. Three replicate wells were conducted at each condition in all treatment groups. After the cells were cultured at 37°C in a humidified atmosphere with 5% CO2 for 24 hours, 20 μL (5 mg/mL) MTT (Amresco Company, Hamilton, ON, Canada) was added to each well and then the cells were blended and incubated at 37°C for four hours. The supernatant was then removed and 200 μL DMSO was added to each well. The absorbance value (A value) was quantified at 490 nm using a Microplate Reader (Spectra Max M2, CA, USA) and the average of each group was calculated. The inhibitory rate of cell growth was calculated as [1-Atest group/Acontrol group)] ×100%. The growth curve was drawn using time as abscissa and inhibition rate as ordinate. At least three replicate experiments were performed with three wells per concentration.
Determination of apoptosis by Hoechst 33258 staining
The cells were cultured overnight on an axenic cover-slide in 24-well plates, making a fusion level of approximately 70%, then the cells were divided into four groups and treated with: 1, blank control; 2, TSA 250 nmol/L; 3, cisplatin 10ug/mL; and 4, TSA 250 nmol/L + cisplatin 10ug/mL.
After the drug treatment (as described above), cells were fixed with fixing solution for five minutes. The cells were then washed three times with phosphate buffered saline (PBS) and incubated in Hoechst 33258 solution (Sigma, USA) for five minutes, after which the cells were washed again three times with PBS. Apoptosis was viewed under fluorescent microscopy.
Western blotting analysis
Cells in different groups were treated with 1, blank control; 2, TSA 250 nmol/L; 3, cisplatin 10ug/mL; and 4, TSA 250 nmol/L + cisplatin (10 μg/mL), for 24 hours. The whole protein was then extracted using a Nuclear and Cytoplasmic Protein Extraction Kit (Proteintech Company) and the concentration was analyzed by Bradford way. Equivalent cellular proteins were isolated and resolved in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked in a PBS buffer containing 10% non-fat dried milk, and the primary antibody solution was added and incubated overnight at 4°C. Secondary antibody binding was carried out at room temperature for one hour. The PVDF membranes were rinsed three times for five minutes each. Chemiluminescence detection was carried out with the ECL plus Western Blotting detection system to detect the cFLIP, pro-caspase-8, and caspase-8 expression.
Data analysis
Data is presented as x ± s and was analyzed using the Student's t-test, χ2 test, and analysis of variance by SPSS 13.0 software. P < 0.05 was considered to be statistically significant.
Results
Trichostatin A (TSA) combined with cisplatin changed the cell morphology
As shown in Figure 1, A549 cells treated with TSA alone for 24 hours, compared with the control group, became larger or finger like, and the cell gap widened. The cells treated with cisplatin alone shrank and became small and round. Both groups had visible suspended death cells. When treated with TSA combined with cisplatin, cell density decreased significantly and death cells obviously increased.
Figure 1.

Morphological change of cells induced by trichostatin A (TSA) and cisplatin (×200). (a) Control; (b) TSA 250 nmol/L; (c) cisplatin 10μg/mL; (d) TSA 250 nmol/L+ cisplatin 10μg/mL.
TSA in combination with cisplatin produced synergistic inhibition on A549 cells
The inhibitory effects of cisplatin alone with different concentrations (3∼20ug/ml) and combined with TSA were determined over time by MTT assay. As illustrated in Figure 2, cisplatin significantly inhibited cell proliferation in a dose-dependent manner. The specific inhibition rates of different cisplatin concentration and cisplatin combined with TSA are shown in Table 1. The cell apoptosis rate increased with the increase of cisplatin concentration. Notably, TSA in combination with cisplatin caused a greater inhibitory effect on A549 cells than single-drug therapy (P < 0.05). The results are repeatable. These data suggest that TSA-cisplatin combination treatment yielded synergistic inhibition on A549 cells.
Figure 2.
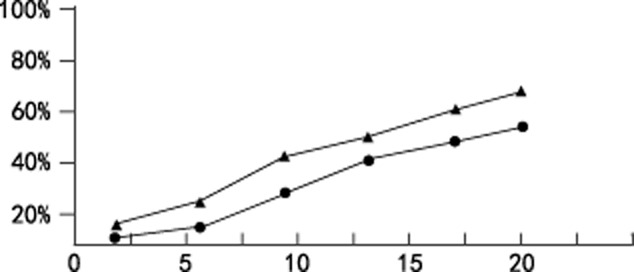
Inhibitory effects of cisplatin or cisplatin and trichostatin A (TSA) on A549 cell proliferation.  , cisplatin;
, cisplatin;  , cisplatin+TSA.
, cisplatin+TSA.
Table 1.
The specific inhibition rates of different cisplatin concentrations
| Cis (umol/L) | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2.1 | 3.8 | 5.3 | 7.67 | 9 | 13 | 16.67 | 20 | |
| Inhibition rate | ||||||||
| Cis | 5.45 | 7.4 | 13.68 | 27.83 | 28.73 | 44.58 | 52.24 | 51.12 |
| Cis+TSA | 12.3 | 18.9 | 26.42 | 35.68 | 46.72 | 49.65 | 60.25 | 61.92 |
The cell apoptosis rate increased with the increase of cisplatin (Cis) concentration. Notably, trichostatin A (TSA) in combination with cisplatin caused a greater inhibitory effect on A549 cells than single-drug therapy (P < 0.05).
TSA in combination with cisplatin induced apoptosis of A549 cells
To examine the morphological changes of A549 cells, Hoechst 33258 staining was employed after the administration of drugs for 24 hours. As shown in Figure 3, TSA, cisplatin alone, and in combination led to marked morphological changes, such as chromatin condensation, nuclear fragmentation, and apoptotic bodies, which were in accordance with typical morphological characteristics of cells undergoing apoptosis. Compared with single treatment, the combination of TSA with cisplatin elicited a much more evident occurrence of apoptosis. These findings suggest that these agents exerted their inhibitory effects on A549 cells, mainly via triggering the apoptotic pathway.
Figure 3.
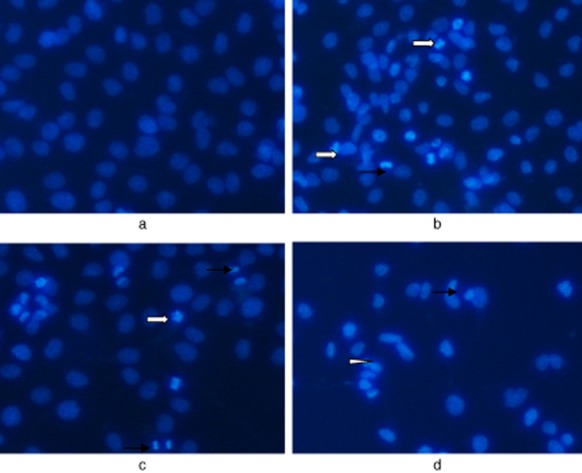
Observation of A549 cell apoptosis staining with Hoechst33258 (×200).  , Condensed;
, Condensed;  , Fragmented nuclei;
, Fragmented nuclei;  , Apoptotic bodies. (a) Control; (b) TSA 250 nmol/L; (c) cisplatin 10μg/mL; (d) TSA 250 nmol/L+ cisplatin 10μg/mL.
, Apoptotic bodies. (a) Control; (b) TSA 250 nmol/L; (c) cisplatin 10μg/mL; (d) TSA 250 nmol/L+ cisplatin 10μg/mL.
TSA in combination with cisplatin decreased the expression of cFLIP
To investigate whether the treatment with cisplatin or TSA alone or as a combination increased cleavage of cFLIP, known to be involved in the apoptotic cascade, Western blot analysis was used to examine the expression of cFLIP in A549 cells following these interventions. The results showed that either single treatment or combination treatment resulted in decreased levels of cFLIP. The level of cFLIP protein in combination treatment cells was lower than that in single treatment cells (Fig 4). These findings indicated that the occurrence of apoptosis in response to these stimuli was through a caspase-dependent signal pathway.
Figure 4.
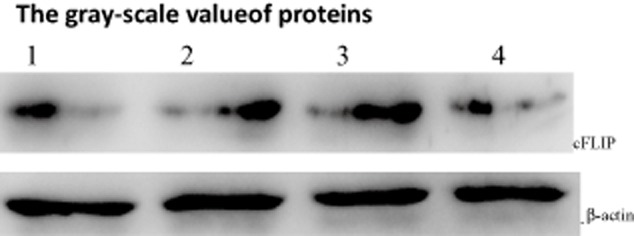
Synergistic effects of trichostatin A (TSA) and cisplatin on Cellular Fas-associated death domain-like interleukin-1 β converting enzyme inhibitory protein (cFLIP) as detected by Western blotting. 1: control; 2: TSA 250 nmol/L; 3: cisplatin 10μg/mL; 4: TSA 250 nmol/L+ cisplatin 10μg/mL. * p < 0.05,**p < 0.01 versus control group.
TSA in combination with cisplatin reduced pro-caspase-8 and increased the expression of caspase-8
Finally, we assessed the level of pro-caspase-8 and caspase-8 in A549 cells treated with TSA or cisplatin alone and their combination for 24 hours. As shown in Figure 5, either single treatment or combination treatment resulted in a decrease of pro-caspase-8 and an increase of caspase-8. The level of caspase-8 protein in combination treatment cells was higher than that in single treatment cells (Fig 5). These findings indicated that the occurrence of apoptosis in response to these stimuli was through a caspase-dependent signal pathway.
Figure 5.
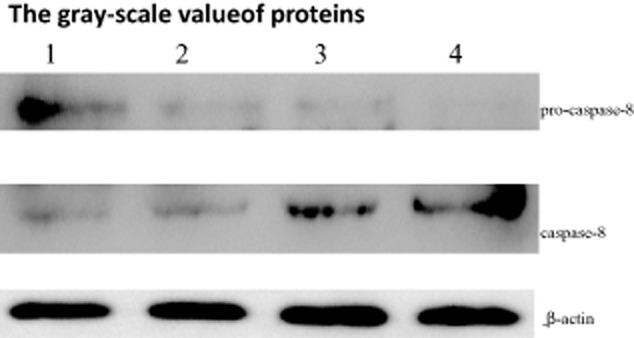
Synergistic effects of trichostatin A (TSA) and cisplatin on caspase-8 as detected by Western blot. 1: control; 2: TSA 250 nmol/L; 3: cisplatin 10μg/mL; 4: TSA 250 nmol/L+ cisplatin 10μg/mL group. * p < 0.05,**p < 0.01 versus control group.
The gray-scale value of cFLIP, pro-caspase-8 and caspase-8 are shown in the bar chart in Figure 6.
Figure 6.
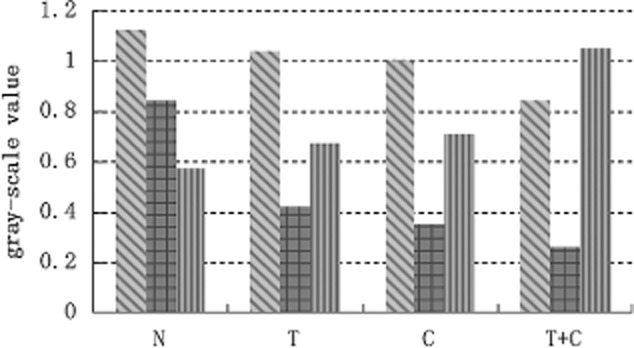
The gray-scale value of proteins. (a) The gray-scale value of Cellular Fas-associated death domain-like interleukin-1 β converting enzyme inhibitory protein (cFLIP) proteins; (b) the gray-scale value of pro-caspase-8 proteins; (c) the gray-scale value of caspase-8 proteins.  , A;
, A;  , B;
, B;  , C.
, C.
Discussion
In recent years, many studies have shown that epigenetics is closely related to the occurrence and metastasis of tumors. HDAC inhibitors can cause chromosome remodeling and histone acetylation, showing good effects in multiple myeloma animal models. Now, more and more new compounds of HDAC inhibitors are used in clinical research. They have little or no toxicity to normal cells, but can specifically kill tumor cells.
TSA is one kind of HDAC inhibitor. Its anti-tumor mechanism inhibits tumor cell proliferation, arrests the cell cycle in the G2/M phase, and induces tumor cell differentiation and apoptosis.17 TSA can increase the expression of the apoptosis protein DARP and promote the apoptosis of tumor cells.13 It can also reduce the expression of Bcl-2 protein by mitochondrial channels; therefore, promoting the apoptosis of tumor cells.18 Other studies have found that TSA can inhibit the activity of cyclooxygenase-2 in non-small cell lung cancer.19 Research has shown that TSA can not only inhibit the growth of tumor cells, but can also show a synergistic effect when combined with chemotherapy drugs as it enhances the antitumor activity of chemotherapeutic drugs. TSA combined with docetaxel or erlotinib can enhance their anti tumor effects.17
Cisplatin is one of the most commonly used drugs in combination chemotherapy. However, the side effects of cisplatin, such as digestive tract reactions, renal toxicity, bone marrow suppression, and auditory neurotoxicity, cannot be ignored. Long-term use of cisplatin can also cause drug resistance. One study has found that TSA could increase the sensitivity of cisplatin resistant lung cancer cells to cisplatin by inducing apoptosis,13 and the apoptosis rate increased with the increase of TSA concentration. TSA combined with cisplatin may enhance the anti-tumor effects of cisplatin, increasing apoptosis of tumor cells.
To further confirm our hypothesis, we designed the following experiment. We chose A549 cells from human lung adenocarcinoma as our study object. In the study, we demonstrated that cisplatin significantly inhibited cell proliferation in a concentration-dependent manner. The cell apoptosis rate increased with the increase of cisplatin concentration using MTT assay, and the results were repeatable. Notably, TSA in combination with cisplatin caused a greater inhibitory effect on A549 cells than single-drug therapy, implying that the TSA can potentiate the anti-tumor effects of cisplatin on A549 cells. In addition, A549 cells treated with the combined regimens significantly led to the appearance of chromatin condensation, nuclear fragmentation, and apoptotic bodies in co-treatment groups compared with the single drug treatment group, as evidenced by Hochst33258 staining. These morphological changes are consistent with the typical characteristics of cells undergoing apoptosis.20
cFLIP belongs to the peptidase C14A family, and is a regulatory protein of apoptosis. It can block the death receptor pathway of apoptosis by creating an inhibitory effect. Research has shown that, as cFLIP is over expressed in various tumor tissues, the expression of cFLIP in lung cancer tissues is also significantly increased. Sharp et al. used small interfering ribonucleic acid (siRNA) selective interference in the expression of cFLIP, and, thus, the pro-caspase-8 became active and the downstream molecules of caspase activation were significantly enhanced.21 cFLIP protein structure and sequence were similar to caspase-8, N containing two serial death effector domains (DED), and caspase-8. cFLIP, through its N terminal with two DED competitive FADD and (or) caspase-8, blocking the activation and pro-caspase-8 death inducing signaling complex (DISC), thereby blocking apoptosis signal transduction, play a role in the inhibition of apoptosis.22 cFLIP may be an important factor in the survival of cancer cells against chemotherapy and biological therapy. The level of cFLIP expression can be used as one of the signs of tumor cells to chemotherapy sensitivity; therefore, using this biological target as an approach may provide a new strategy for cancer treatment.
To further characterize the specific apoptotic pathway activated by these agents, cFLIP and caspase-8 expression were examined. We demonstrated that treatment of cells with TSA in combination with cisplatin significantly decreased cFLIP, decreased pro-caspase-8, and increased caspase-8 compared with a single agent, suggesting that the apoptotic process occurred in response to such stresses via a caspase-dependent pathway.
Our study does have some limitations. We used human lung adenocarcinoma A549 cells; however, there are several histological types of human lung carcinoma, such as squamous cell, small cell, large cell, and undifferentiated carcinomas. Cisplatin is used in different histological types of lung cancer. Further research is needed to determine the mechanisms and whether TSA combined with cisplatin is effective in different lung carcinomas.
Conclusion
In conclusion, our study provides strong in vitro evidence that TSA, a HDAC inhibitor, inhibits cancer growth by triggering an apoptotic pathway via a caspase-dependent manner. The TSA/cisplatin combination seems to hold promise for the treatment of advanced lung cancer with limited sensitivity to cisplatin.
Acknowledgments
The study was supported by a grant from the Scientific and Technological Development Plan Projects of Shandong Province, China (2010G0020227). The authors would like to thank Prof. Jian-Feng Li of the Central Lab of Shandong Provincial Hospital, PR China for his assistance with the preparation of this manuscript.
Disclosure
No authors report any conflict of interest.
References
- Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers–a different disease. Nat Rev Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- Long N, Moore MA, Chen W, et al. Cancer epidemiology and control in north-East Asia – past, present and future. Asian Pac J Cancer Prev. 2010;11(Suppl 2):107–148. [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21:5427–5440. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- Dowdy SC, Jiang S, Zhou XC, et al. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol Cancer Ther. 2006;5:2767–2776. doi: 10.1158/1535-7163.MCT-06-0209. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kang HJ, Na H, Li MO. Trichostatin A enhances acetylation as well as protein stability of ERalpha through induction of p300 protein. Breast Cancer Res. 2010;12:R22. doi: 10.1186/bcr2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Kumar S, Kundu GC. Transcriptional regulation of human osteopontin promoter by histone deacetylase inhibitor, trichostatin A in cervical cancer cells. Mol Cancer. 2010;9:178–179. doi: 10.1186/1476-4598-9-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DD, Yang QC, Zhang ZC, Yang CX, Liu YW. Antitumor activity of histone deacetylase inhibitor trichostatin A in osteosarcoma cells. Asian Pac J Cancer Prev. 2012;13:1395–1399. doi: 10.7314/apjcp.2012.13.4.1395. [DOI] [PubMed] [Google Scholar]
- Ranganathan P, Rangnekar VM. Exploiting the TSA connections to overcome apoptosis-resistance. Cancer Biol Ther. 2005;4:391–392. doi: 10.4161/cbt.4.4.1779. [DOI] [PubMed] [Google Scholar]
- Hajji N, Wallenborg K, Vlachos P, Nyman U, Hermanson O, Joseph B. Combinatorial action of the HDAC inhibitor trichostatin A and etoposide induces caspase -mediated AIF-dependent apoptotic cell death in non-small cell lung carcinoma cells. Oncogene. 2008;27:3134–3144. doi: 10.1038/sj.onc.1210976. [DOI] [PubMed] [Google Scholar]
- Wu J, Hu CP, Gu QH, Li YP, Song M. Trichostatin A sensitizes cisplatin-resistant A549 cells to apoptosis by up-regulating death-associated protein kinase. Acta Pharmacol Sin. 2010;31:93–101. doi: 10.1038/aps.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki M, Kakinuma H, Kumazawa T, et al. Low concentrations of the histone deacetylase inhibitor, depsipeptide, enhance the effects of gemcitabine and docetaxel in hormone refractory prostate cancer cells. Oncol Rep. 2007;17:761–767. [PubMed] [Google Scholar]
- Zhang X, Yashiro M, Ren J, Hirakawa K. Histone deacetylase inhibitor, trichostatin A, increases the chemosensitivity of anticancer drugs in gastric cancer cell lines. Oncol Rep. 2006;16:563–568. [PubMed] [Google Scholar]
- Chang H, Jeung HC, Jung JJ, Kim TS, Rha SY, Chung HC. Identification of genes associated with chemo- sensitivity to SAHA/taxane combination treatment in taxane-resistant breast cancer cells. Breast Cancer Res Treat. 2011;125:55–63. doi: 10.1007/s10549-010-0825-z. [DOI] [PubMed] [Google Scholar]
- Zhang QC, Jiang SJ, Zhang S, Ma XB. Histone deacetylase inhibitor trichostatin A enhances anti-tumor effects of docetaxel or erlotinib in A549 cell line. Asian Pac J Cancer Prev. 2012;13:3471–3476. doi: 10.7314/apjcp.2012.13.7.3471. [DOI] [PubMed] [Google Scholar]
- Wu J, Hu C. Trichostatin A induced Bcl-2 protein level decrease mediated A549/CDDP cells apoptosis by mitochondria pathway. Zhongguo Fei Ai Za Zhi. 2009;12:1143–1149. doi: 10.3779/j.issn.1009-3419.2009.11.03. (In Chinese.) [DOI] [PubMed] [Google Scholar]
- Choi YH. Induction of apoptosis by trichostatin A, a histone deacetylase inhibitor, is associated with inhibition of cyclooxygenase-2 activity in human non-small cell lung cancer cells. Int J Oncol. 2005;27:473–479. [PubMed] [Google Scholar]
- Liu T, Zhang M, Zhang H, et al. Combined antitumor activity of cucurbitacin B and docetaxel in laryngeal cancer. Eur J Pharmacol. 2008;587:78–84. doi: 10.1016/j.ejphar.2008.03.032. [DOI] [PubMed] [Google Scholar]
- Sharp DA, Lawrence DA, Ashkenazi A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J Biol Chem. 2005;280:19401–19409. doi: 10.1074/jbc.M413962200. [DOI] [PubMed] [Google Scholar]
- Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46:497–510. doi: 10.1136/jmg.2009.066944. [DOI] [PubMed] [Google Scholar]