

Abstract
CHIP (c-terminal Hsp70-interacting protein) is an E3 ligase which may play different roles in different cancers. The elucidation of the VHL-HIF-1α(hypoxia inducible factor-1α)-VEGF (vascular endothelial growth factor) pathway has led to the development of targeted therapy in renal cell carcinoma (RCC). However, little is known about the role of CHIP and the relationship between CHIP and VEGF-VEGFR2 (VEGF receptor 2) pathway in RCC. In this study, we found that the expression of CHIP was downregulated and significantly correlated with pT status (P = 0.022) and TNM stage (P = 0.022) in 304 RCC and 35 normal renal tissues using tissue microarray. Moreover, low expression of CHIP is a strong and independent negative prognostic value for RCC. In vitro, CHIP negatively regulated RCC cell migration, invasion and angiogenesis. In addition, ELISA tests showed that restoration of CHIP inhibited, while knockdown promoted, the secreted level of VEGF. Furthermore, western blot indicated that the VEGFR2 protein level was reduced after CHIP overexpression. Our findings demonstrate for the first time that CHIP may be involved in RCC angiogenesis through regulating VEGF secretion and expression of VEGFR2. CHIP may serve as promising prognostic biomarker of angiogenesis and may constitute a potential therapeutic target in RCC.
RCC accounts for approximately 3% of all malignancies, and the incidence of RCC is increasing by a rate of approximately 2.5% each year1. Metastatic lesions are present in 20–30% of patients at the time of RCC diagnosis, and in 20–30% of patients with preoperative absence of metastasis, metastatic lesions appear after nephrectomy or local recurrence of disease take place2. As a result, identification and validation of appropriate biomarkers for prediction or evaluation of prognosis remain important issues.
To grow beyond 1–2 mm in diameter, the tumor requires the emergence and further development of its own blood vessels, which is known as “neoangiogenesis”. RCC is a highly vascular tumor characterized by inactivation of the VHL gene. Its gene product, pVHL, binds the hydroxylated form of HIF-1α ultimately leading to its destruction by the proteasome. In the absence of functional pVHL, HIF-1α accumulates, which causes downstream upregulation of a number of pro-angiogenic factors, including the VEGF3. VEGF exerts its biologic effect through interaction with receptors present on the cell surface. These transmembrane tyrosine kinase receptors include VEGFR1, VEGFR2 and VEGFR3. Upon binding of VEGF to the extracellular domain of the receptor, dimerization and autophosphorylation of the intracellular receptor tyrosine kinases occurs and a cascade of downstream proteins are activated. The VEGFR2 are high-affinity VEGF receptors, mediating the majority of VEGF downstream angiogenic effects4. It has been shown that blockade of VEGFR2 resulted in inhibition of tumor growth and in abrogation of tumor cell invasion5. Importantly, VEGF receptors have been identified on the surface of renal cancer cells, suggesting that VEGF may augment RCC tumor growth through an autocrine loop6.
CHIP acts as E3 ligase in combination with cytoplasmic chaperones heat shock protein 70 (HSP70) and HSP90 while mediating the ubiquitination and degradation of various chaperone-bound proteins7. CHIP has been demonstrated to regulate a number of oncogenic proteins including receptor tyrosine kinase ErbB2, HIF-1α, estrogen receptor-α (ER-α), and androgen receptor (AR)8,9,10,11. Previous studies have indicated that CHIP may play different roles in different human cancers. The phenotypes determined by CHIP should be dependent on the function of its specific targets in a specific type of cancer12. However, to date, the function of CHIP in RCC has not yet been reported. In addition, the relationship between CHIP and VEGFR2 in RCC is still unclear.
In this study, we tested the hypothesis that CHIP regulates cell migration, invasion and angiogenesis by inhibiting VEGF signaling pathway in RCC. First, we used a tissue microarray (TMA) of human RCC patients and immunohistochemistry (IHC) to evaluate the expression of CHIP in relation to clinicopathologic features. We further investigated the role of CHIP in RCC cell migration, invasion and angiogenesis. Finally, we studied the relation between CHIP expression and the secreted level of VEGF and VEGFR2 protein expression.
Results
CHIP expression is decreased in human RCC
To investigate whether CHIP expression is changed in RCC, IHC staining was performed in our established TMA slides (Fig. 1A). Positive CHIP staining was recorded in 94.1% (32/34) and 51.0% (155/304) of the biopsies in normal renal tissues (NRT) and RCC tissue, respectively (Fig. 1B).We found that CHIP staining was mainly localized in the cytoplasm and the expression of CHIP was significant lower in the carcinoma tissues than the NRT (P = 0.0001).
Figure 1. CHIP protein expression in NRT and RCC tissues.

(A) Representative photographs showed CHIP IHC staining in TMA, which were taken at different magnifications (Top panel ×100, bottom panel ×400). (B) CHIP staining was decreased in RCC tissues compared with NRT. Positive CHIP staining was recorded in 94.1% (32/34) and 51.0% (155/304) of the biopsies in NRT and RCC tissue, respectively. (P = 0.0001, χ2 test).
Expressions of CHIP correlated with clinicopathological characteristics in RCC patients
The clinicopathologic features of the 304 RCC biopsies were summarized in Table 1. Because TNM stage is an important prognostic marker for RCC patients, we studied if CHIP expression correlates with TNM stage. Two sided Fisher’s exact analysis revealed a significant difference in CHIP expression between TNM stages I and II to IV (P = 0.022). CHIP staining was dramatically decreased in TNM stages II to IV compared with stages I. Because TNM stage concludes T-depth of invasion, N-lymph node metastasis, and M-distant metastasis, we analyzed the correlation between CHIP expression and these clinicopathologic parameters. The analysis indicated a significant correlation between CHIP expression and depth of invasion when comparing pT1 versus pT2-pT4 (P = 0.022). We did not find significant correlation between CHIP expression with other clinicopathologic variables, including age, gender and tumor size.
Table 1. CHIP staining and clinicopathological characteristics of 304 renal cancer patients.
|
CHIP staining |
||||
|---|---|---|---|---|
| Variables | Negative (%) | Positive (%) | Total | P* |
| All cases | 154 (34.7) | 150 (49.3) | 304 | |
| Age | ||||
| ≤ 56 years | 69 (47.6) | 76 (52.4) | 145 | 0.182 |
| > 56 years | 85 (53.5) | 74 (46.5) | 159 | |
| Gender | ||||
| Male | 100 (49.5) | 102 (50.5) | 202 | 0.328 |
| Female | 54 (52.9) | 48 (47.1) | 102 | |
| Tumor size | ||||
| ≤ 7 cm | 114 (48.3) | 122 (51.7) | 236 | 0.082 |
| > 7 cm | 40 (58.8) | 28 (41.2) | 68 | |
| pT status | ||||
| pT1 | 92 (46.2) | 107 (53.8) | 199 | 0.022 |
| pT2- pT4 | 62 (59.0) | 43 (41.0) | 105 | |
| pN status | ||||
| pN0 | 142 (51.1) | 136 (48.9) | 278 | 0.123 |
| pN1- pN3 | 7 (35.0) | 13 (65.0) | 20 | |
| pM status | ||||
| pM0 | 132 (51.8) | 123 (48.2) | 255 | 0.332 |
| pM1 | 3 (37.5) | 5 (62.5) | 8 | |
| TNM stage | ||||
| I | 82 (45.6) | 98 (54.4) | 180 | 0.022 |
| II-IV | 54 (59.3) | 37 (40.7) | 91 | |
*Two sided Fisher's exact tests.
Decreased CHIP expression correlates with poor patient survival
To study whether reduced CHIP staining in RCC patients correlates with a worse prognosis, Kaplan-Meier survival curves were plotted using 5-year overall survival (OS) and disease-specific survival (DSS) (n = 304, follow-up time, 60 months) to compare the patients with positive CHIP staining to those with negative staining (Fig. 2). Our data revealed that positive CHIP staining correlated with both 5-year OS and DSS in RCC (P = 0.029 and P = 0.030, respectively, log-rank test). The 5-year OS dropped from 28.0% in patients with positive CHIP expression to 18.5% in those with negative expression, and the 5-year DSS dropped from 31.2% in patients with positive CHIP expression to 20.6% in those with negative expression.
Figure 2. CHIP expression associated with both 5-year OS and DSS in RCC patients.
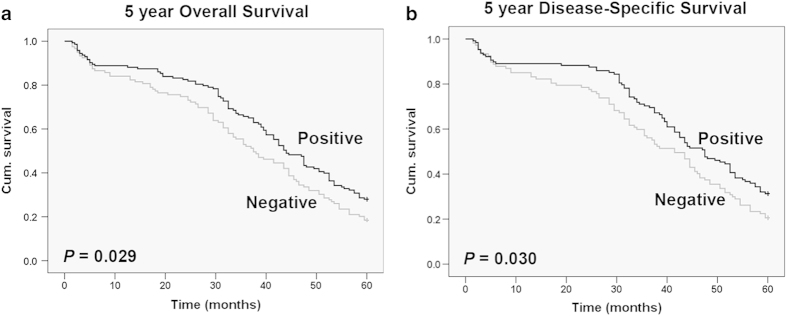
(A) Kaplan-Meier curves showed that the 5-year OS dropped from 28.0% in patients with positive CHIP expression to 18.5% in those with negative expression (P = 0.029, log-rank test). (B) Kaplan-Meier curves showed that the 5-year DSS dropped from 31.2% in patients with positive CHIP expression to 20.6% in those with negative expression (P = 0.030, log-rank test). Cum., cumulative.
Furthermore, we examined whether CHIP expression is an independent prognostic marker for RCC. We performed univariate and multivariate Cox regression analysis including CHIP expression, age, tumor size and TNM stage to study the effects of CHIP on patient survival in RCC. The univariate Cox regression analysis showed that CHIP expression was an independent prognostic marker for RCC patients (hazard ratio, 0.738; 95% confidence interval, 0.559–0.974; P = 0.032; Supplementary Table S1). In multivariate Cox regression analysis, we found that similar to TNM stage, which has been widely accepted as an important prognostic factor for RCC patients, CHIP expression was also an independent prognostic factor for 5-year survival (hazard ratio, 0.739; 95% confidence interval, 0.595–1.074; P = 0.021; Supplementary Table S2). Because 5-year patient survival is widely used to predict outcome in RCC, our results clearly indicated that low CHIP expression is associated with poor prognosis, suggesting that CHIP may serve as a molecular prognostic marker for this aggressive disease.
CHIP regulates RCC cells migration and invasion in vitro
Because low CHIP expression is associated with poor prognosis, supporting CHIP may play important roles in one or more steps of tumor metastasis, we investigated the involvement of CHIP in RCC cells migration and invasion. To determine the effect of CHIP overexpression or knockdown on RCC cells migration and invasion, we transiently transfected 786-O and OS-RC-2 cells with Myc-control and Myc-CHIP plasmids or control siRNA and CHIP siRNA, respectively. Twenty-four hours or forty-eight after transfection, CHIP protein was considerably overexpressed or knockdown in cancer cells, respectively (Fig. 3A). In cell migration assay, we found that the ability of cell migration was drastically decreased after CHIP overexpression in both 786-O and OS-RC-2 RCC cell line. In contrast, knockdown CHIP promoted cell migration (Fig. 3B). Meanwhile, the results of the cell invasion assay corresponded with the cell migration assay (Fig. 3C).
Figure 3. CHIP negatively regulated cell migration and invasion in 786-O and OS-RC-2 cell lines.

(A) WB showed that twenty-four hours or forty-eight after transfection, CHIP protein was significantly overexpressed or knockdown in cancer cells, respectively. Actin was used as an internal control. (B) Cell migration assay was performed after reintroduction or knockdown of CHIP in 786-O and OS-RC-2 cells. The ability of cell migration was decreased after CHIP overexpression. In contrast, CHIP knockdown promoted cell migration. (C) Matrigel cell invasion assay was performed after reintroduction or knockdown of CHIP in 786-O and OS-RC-2 cells. The ability of cell invasion was decreased after CHIP overexpression while CHIP knockdown promoted cell invasion. The data are presented as mean ± s.e.m. for triplicate determinations. *** P < 0.001.
Expression of CHIP inhibits angiogenesis in vitro
Mutations of VHL gene are present in 60% of RCC patients. The VHL gene inhibits hypoxia inducible genes including proteins involved in angiogenesis, which plays important roles in the progression of RCC2. To test the effect of CHIP expression in RCC angiogenesis, we overexpressed or knocked down CHIP in 786-O and OS-RC-2 cells, respectively. The condition medium containing secreted cytokines was collected from these either overexpressing or knockdown RCC cells and used for HUVECs growth and tube formation assays. In the HUVECs growth assay, we found that conditioned medium from 786-O and OS-RC-2 cells overexpressing CHIP significantly inhibited proliferation of endothelial cells (P = 0.017 and P = 0.019, respectively). Nevertheless, conditioned medium with CHIP knockdown substantially promoted proliferation of endothelial cells (Fig. 4A). In addition, as shown in Fig. 4B, the average number of complete tubular structures formed by HUVECs in conditioned medium from CHIP overexpression led to 90% and 68% decrease, respectively; whereas increased 3.08- and 5.87-fold with knockdown of CHIP, respectively.
Figure 4. CHIP negatively regulated angiogenesis in 786-O and OS-RC-2 cell lines.

(A) HUVEC growth assay was performed to detect the HUVECs proliferation. CHIP overexpression significantly inhibited proliferation of HUVECs. Nevertheless, CHIP knockdown significantly promoted proliferation of HUVECs. (B) Representative pictures were taken in situ for tube formation in the supernatant of 786-O and OS-RC-2 cells (×100). The degree of tube formation was assessed as the number of tube. The average number of complete tubular structures formed by HUVECs from CHIP overexpression in the 786-O and OS-RC-2 cells led to 90% and 68% decrease, respectively; whereas increased 3.08- and 5.87-fold with knockdown of CHIP, respectively. All experiments were carried out in triplicate. Data are shown as mean ± s.e.m. ***P < 0.001, **P < 0.01, *P < 0.05.
CHIP suppresses secreted level of VEGF and protein expression of VEGFR2 in RCC cells
RCC is characterized by the VHL loss or its inactivation, and the process of angiogenesis is controlled by the VHL-HIF(1α)-VEGF-VEGFR2 signal pathway13. Here we investigated whether the role of CHIP on angiogenesis was mediated by regulating VEGF secretion and VEGFR2 expression. Using ELISA test, we first observed if VEGF secretion level was regulated by CHIP in RCC cells. As shown in Fig. 5A, VEGF secretion level was negatively regulated by CHIP in both 786-O and OS-RC-2 cells, respectively. Moreover, we performed western blot (WB) to measure the VEGFR2 expression in RCC cells. Our data showed that the VEGFR2 protein level was dramatically reduced in 786-O and OS-RC-2 cells after being transfected with Myc-CHIP (Fig. 5B).
Figure 5. CHIP suppresses the secreted level of VEGF and protein expression of VEGFR2 in RCC cells.
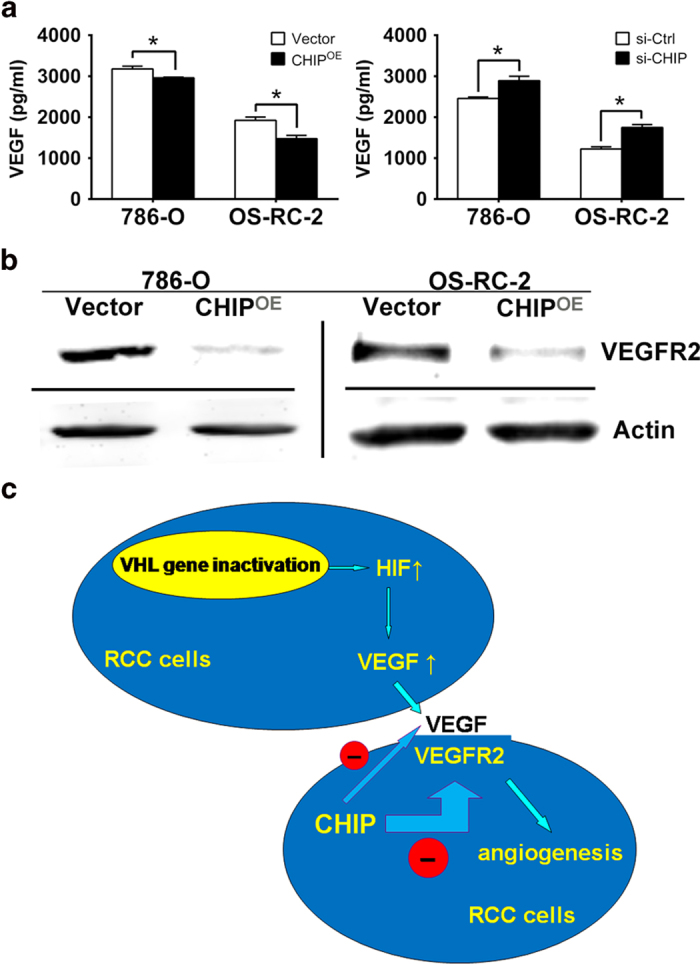
(A) The secreted level of VEGF was determined by ELISA assay. VEGF secretion level was negatively regulated by CHIP in both 786-O and OS-RC-2 cells, respectively. All experiments were carried out in triplicate. (B) Representative WB analysis of the relative protein levels of VEGFR2 in CHIP restoration and control group for both 786-O and OS-RC-2 cell lines. VEGFR2 protein level was dramatically reduced after CHIP restoration. (C) Model of mechanism of CHIP inhibiting RCC angiogenesis. CHIP decreased the secreted level of VEGF, meanwhile increased the degradation of VEGFR2 and downregulated the protein level of VEGFR2 on cell membrane. Data are shown as mean ± s.e.m. *P < 0.05.
Discussion
As a member of the E3 ubiquitin ligases, CHIP associates with molecular chaperones (Hsp70 or Hsp90), ubiquitinating the client proteins, and is demonstrated to be involved in tumorigenesis in several malignancies12. For example, CHIP regulates oncogenic proteins such as steroid receptor coactivator 3 in breast cancer14, and AR in prostate cancer8. However, there has been no attempt to evaluate the expression and function of CHIP in human RCC to date.
In the present study, we used a RCC TMA containing 304 tumor biopsies and 35 NRT with robust clinical data to investigate the role of CHIP in RCC development. Our data demonstrated that CHIP expression of RCC was significantly downregulated compared with the NRT and reduced CHIP expression significantly correlated with TNM stage, depth of invasion and poorer survival of RCC patients. Furthermore, univariate and multivariate Cox proportional hazards regression analysis showed that low CHIP expression was a strong independent negative prognostic factor after adjusted by age, tumor size, pT status and TNM stages. Such information indicates that CHIP may play a significant role in the progression of RCC and testing for the presence of CHIP may help identify patient subgroups at high risk for poor disease outcome.
Reduced expression of some tumor suppressive genes may play a role in cell migration and invasion15,16. Combined with the TMA results, we supposed that CHIP may inhibit RCC cell migration and invasion abilities. Surprisingly, we found that downregulation of CHIP accelerates RCC cell migration as well as invasion through matrigel-coated chamber in vitro, whereas the cell invasion and migration abilities are dramatically suppressed when CHIP was overexpressed, respectively. As increased cell migration is a crucial step in cancer metastasis, these findings implied that CHIP may regulate the metastatic potential of RCC. Previous studies have shown that increased expression of matrix metalloproteinase-2 (MMP-2) and MMP-9 is associated with enhanced tumor invasion and metastasis17. We examined the levels of MMP-2 and MMP-9 after CHIP overexpression or knockdown by WB and gelatin zymography experiments. Unexpectedly, the results showed that the changes in CHIP levels did not affect the expression of MMP-2 and MMP-9 (see Supplementary Fig. S3 and Supplementary Methods S4). This implied that CHIP may regulate the invasive ability of RCC cells through other mechanisms, and further studies are needed to address the question.
The essential role of the formation of a new vasculature for tumor progression has been fully recognized in the last decade18,19. Meanwhile, RCC is a highly vascular tumor which originates from the proximal tubule cells of nephrons. Angiogenesis is a multi-step process, which includes endothelial cell proliferation, migration and the formation of blood vessels20. Here, we provided a novel function of CHIP in regulating angiogenesis in RCC cells. Using HUVEC growth assay and HUVEC tube formation assay, we found that the transfection of Myc-CHIP reduced, while CHIP knockdown increased, the capacity of proliferation and tube formation of HUVECs, suggesting that CHIP significantly affected angiogenic potential of RCC cells in vitro.
The majority of RCCs have clear cell histology, which is characterized by genetic and epigenetic inactivation of the VHL gene. Loss of VHL function upregulates HIF-1α, a transcription factor that leads to an overexpression of VEGF and drives tumor angiogenesis21. Otherwise, our results showed that CHIP did not affect the protein level of HIF-1α(see Supplementary Fig. S5). VEGF has been characterized with regard to multiple effects relevant to the generation and preservation of tumor vasculature. These effects include induction of endothelial cell division and migration, promotion of endothelial cell survival through protection from apoptosis, and reversal of endothelial cell senescence22,23. Of the numerous angiogenic factors discovered thus far, VEGF has been identified as a key mediator of tumor angiogenesis24. VEGFR2 is the major mediator of the mitogenic and permeability enhancing effects of VEGF4. Previous studies found the increased HIF(1α)-VEGF expression was correlated to adverse survival of RCC25,26. This led to the development of VEGF signaling pathway inhibitors which have been shown to benefit patients in randomized phase III clinical trials. Currently, there are five angiogenesis inhibitors approved by the United States Food and Drug Administration for treatment of advanced and metastatic RCC, four VEGFR2 targeted tyrosine kinase inhibitors (TKIs), including sorafenib, sunitinib, axitinib and pazopanib, and one anti-VEGF monoclonal antibody, bevacizumab27,28,29,30,31. Therefore, we hypothesized that CHIP may suppress RCC angiogenesis through inhibiting VEGF-VEGFR2 pathway. We detected the secreted level of VEGF after CHIP transfection using ELISA technology. Our data showed that VEGF secretion was increased by knockdown of CHIP, while decreased by overexpression of CHIP, respectively. These data indicated that CHIP regulates endothelial cell proliferation and blood vessel formation through controlling the VEGF secretion. Furthermore, the results of WB have shown that the expression of VEGFR2 was dramatically decreased after overexpression of CHIP, thus we speculated that CHIP may be involved in the degradation of VEGFR2.
In summary, decreased expression of CHIP in RCC speciments constitute a strong unfavorable prognostic factor for RCC patients. Downregulation of in RCC cells inhibits migration, invasion, proliferation and tube formation of cocultured endothelial cells by decreasing VEGFR2 expression and VEGF secretion (Fig. 5C). Our observations support the hypothesis that CHIP interacts with VEGF-VEGFR2 signaling pathway. We also show that decreased expression of CHIP in RCC cells mat trigger angiogenesis as well as cell migration and may be used as a promising prognostic marker for RCC patients. Although the development of TKIs targeting VEGFR2 opens a new door for advanced RCC patients, the huge costs and side effects, such as neutropenia, anemia, increased creatinine, thrombocytopenia and hand-foot syndrome may become the stumbling block of these treatments32. The search for a new and better therapy via new targets for metastatic RCC is still needed. Our results provided the first in vitro evidences that targeting CHIP might represent a new therapy to suppress RCC progression. We hope these findings might have shed light on future directions for identification of novel biomarkers for RCC and the development of targeted drugs.
Methods
Ethics Statement
This study was performed under a protocol approved by the Institutional Review Boards of Affiliated Hospital of Xuzhou Medical College and all examinations were performed after obtaining written informed consents. All experiments involving human subjects were performed in accordance with relevant guidelines and regulations.
Patients and samples
The study material consists of 304 consecutive cases of RCC and 35 cases of NRT, from Affiliated Hospital of Xuzhou Medical College, between 2005 and 2008. All these patients were treated with surgery only or with postoperative adjuvant therapy. The patients’ clinicopathologic information was obtained from the archive of the pathology department and confirmed by the medical record of the hospital. Five-year clinical follow-up results were available for 262 patients.
IHC of TMA
IHC was performed as described before33. According to the streptavidin-peroxidase (Sp) method using a standard Sp Kit (Zhongshan biotech, Beijing, China). The TMA slides were incubated with rabbit anti-CHIP antibody (1:100) (Bethyl Laboratories, Montgomery, USA) overnight at 4 °C, and diaminobenzidine (DAB; Zhongshan Biotech, Beijing, China) was used to produce a brown precipitate. The immunoreactivity was assessed blindly by two independent observers using light microscopy (Olympus BX-51 light microscope), and the image was collected by Camedia Master C-3040 digital camera. The expression of CHIP was graded as positive when 5% of tumor cells showed immunopositivity. Biopsies with < 5% tumor cells showing immunostaining were considered negative.
Cell lines and transfection
Human RCC cell lines 786-O and OS-RC-2 were purchased from the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Human umbilical vascular endothelial cells (HUVECs) were obtained from Key-GEN biotech (Nanjing, China). 786-O, OS-RC-2 and HUVEC cells were cultured in RPMI1640 medium supplemented with 10% fetal calf serum (Invitrogen, Shanghai, China). Cells were in a 37 °C humidified incubator with 5% CO2. The CHIP overexpression plasmids were obtained from Dr. Shouyu Wang (Nanjing Medical University, Jiangsu, China). Non-specific control siRNA or CHIP siRNA was purchased from GenePharma biotech (Shanghai, China). Transfection of the plasmids into the renal carcinoma cells were carried out using Lipofectamine 2000 transfection reagent (Invitrogen, Shanghai, China) following the manufacturer’s protocol. Transfection of the non-specific control siRNA or CHIP siRNA were carried out using siLentFect Lipid Reagent (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions.
Migration assay
Cell migration was determined by using a modified two chamber migration assay with a pore size of 8 μm34. For migration assay, 3 × 104 786-O and OS-RC-2 cells were seeded in serum-free medium in the upper chamber. After 12 h incubation at 37 °C, cells in the upper chamber were carefully removed with a cotton swab and the cells that had traversed the membrane were fixed in methanol and stained with leucocrystal violet. The number of migration cells was determined by counting the leucocrystal violet stained cells. For quantification, cells were counted under a microscope in five fields (up, down, median, left, right. × 200).
Invasion assay
The invasion assay was performed using a modified two chamber plates with a pore size of 8 μm15. The transwell filter inserts were coated with matrigel (BD Biosciences, NJ, USA). 5 × 104 786-O and OS-RC-2 cells were seeded in serum free medium in the upper chamber. After 24 h incubation at 37 °C, no invasive cells were gently removed from the top of the matrigel with a cotton-tipped swab. Invasive cells at the bottom of the matrigel were fixed in methanol, stained with leucocrystal violet and counted.
HUVECs growth assay
The HUVECs growth was assayed using cell counting kit-8 (CCK-8) purchased from Beyotime Institute of Biotechnology (Nanjing, China)16. In brief, 6×103 HUVECs suspended in 100 μl conditioned medium from control, CHIP overexpression or CHIP siRNA cells in a 96-well culture plate, respectively. Then HUVECs were incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 24 h. Then, cell proliferation was detected according to the manufacturer’s instructions.
Endothelial cell tube formation assay
Transfected 786-O and OS-RC-2 cells (1 × 106) were cultured in 6-well plate with fresh complete medium for 24 h, and the medium was collected and centrifuged to remove any cell debris before its use as a conditioned medium. 48-well plate was coated with matrigel and kept in 37 °C for 30 min. Then, 2 × 104 HUVECs were suspended in 100 μl conditioned medium and applied to the pre-coated 48-well plate. After incubation at 37 °C for another 24 h, the number of capillary-like tubes from four randomly chosen fields was counted ( × 100).
ELISA for VEGF
786-O and OS-RC-2 cells were plated in 6-well tissue culture plates at a density of 1 × 106 cells per well. Then, cells were transfected with CHIP overexpression or CHIP siRNA with serum starvation. The supernatants were collected 24 h after transfection. VEGF concentration was determined using Quantikine ELISA kits according to the manufacturer’s instructions (Westang biotech, Shanghai, China).
WB analysis
For cancer cells, twenty-four hours (overexpression) or forty-eight (RNAi) after transfection, cells were harvested from the plates. Then aliquots of cell extracts were separated on a 10% SDS-polyacrylamide gel. The proteins were then transferred to nitrocellulose membrane and incubated overnight at 4 °C with the following antibodies: rabbit anti-CHIP (1: 1000; Cell Signaling Technology, Beverly, MA, USA), rabbit anti-VEGFR2 (1: 500; Bioworld Technology, St. Louis Park, MN, USA) and mouse anti-β-actin (1: 1000; Boster Biotechnology, Wuhan, China). Membranes were then washed and incubated with secondary antibody (1: 2000; goat anti-rabbit and goat anti-mouse IgG) for 2 h. Membranes were then washed and scanned on the Odyssey Two-Color Infrared Imaging System (LI-COR Biotechnology, Lincoln, Nebraska, USA). Each blot was repeated three times.
Statistical analysis
For TMA, statistical analysis was performed with SPSS 20.0 software (SPSS, Chicago, IL). Data are expressed as the means ± SEM. Two-factor analysis of variance procedures and the Dunnett’s t test were used to assess differences within treatment groups. The association between CHIP staining and the clinicopathologic parameters of the RCC patients, including age, gender, tumor size, pT status and TNM stage, was evaluated by χ2 test. The Kaplan-Meier method and log-rank test were used to evaluate the correlation between CHIP expression and patient survival. Univariate or multivariate Cox proportional hazards regression models were performed to estimate the crude hazard ratios (HRs) and their 95% confidential intervals (CIs). For cell migration, invasion, HUVECs tube formation, VEGF ELISA and CCK-8 cell proliferation assays, statistical analysis was performed with GraphPad Prism 6.0 software, Student t test was used. Differences were considered significant when P < 0.05.
Additional Information
How to cite this article: Sun, C. et al. Decreased expression of CHIP leads to increased angiogenesis via VEGF-VEGFR2 pathway and poor prognosis in human renal cell carcinoma. Sci. Rep. 5, 9774; doi: 10.1038/srep09774 (2015).
Supplementary Material
Acknowledgments
This project is supported by grants from the National Natural Science Foundation of China (No.81472663, 81201636), the Science and Technology Department of Jiangsu Province (No.BK2012139), the China Postdoctoral Science Foundation (No. 2014T70549, 2012M511323), and the Priority Academic Program for the Development of Jiangsu Higher Education Institutions (Public Health and Preventive Medicine).
Footnotes
Author Contributions Conceived and designed the experiments: C.S. and J.B. Performed the experiments: C.S. and H.L. Analyzed the data: M.S. and Q.L. Wrote the manuscript: Z.P. and J.Z. Revised the manuscript: H.C. All authors reviewed the manuscript.
References
- Jemal A., Siegel R., Xu J. & Ward E. Cancer statistics, 2010. CA Cancer J. Clin. 60, 277–300 (2010). [DOI] [PubMed] [Google Scholar]
- Cohen H. T. & McGovern F. J. Renal-cell carcinoma. N. Engl. J. Med. 353, 2477–2490 (2005). [DOI] [PubMed] [Google Scholar]
- Patel P. H., Chadalavada R. S., Chaganti R. S. & Motzer R. J. Targeting von Hippel-Lindau pathway in renal cell carcinoma. Clin. Cancer Res. 12, 7215–7220 (2006). [DOI] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor: Basic science and clinical progress. Endocrine Reviews 25, 581–611 (2004). [DOI] [PubMed] [Google Scholar]
- Skobe M., Rockwell P., Goldstein N., Vosseler S. & Fusenig N. E. Halting angiogenesis suppresses carcinoma cell invasion. Nat. Med. 3, 1222–1227 (1997). [DOI] [PubMed] [Google Scholar]
- Fox S. B. et al. Phosphorylated KDR is expressed in the neoplastic and stromal elements of human renal tumours and shuttles from cell membrane to nucleus. J. Pathol. 202, 313–320 (2004). [DOI] [PubMed] [Google Scholar]
- Connell P. et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3, 93–96 (2001). [DOI] [PubMed] [Google Scholar]
- Sarkar S., Brautigan D. L., Parsons S. J. & Larner J. M. Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene 33, 26–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan C. I. et al. Tid1, CHIP and ErbB2 interactions and their prognostic implications for breast cancer patients. J. Pathol. 225, 424–437 (2011). [DOI] [PubMed] [Google Scholar]
- Luo W. et al. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J. Biol. Chem. 285, 3651–3663 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X. et al. Histone deacetylase inhibitor SAHA induces ERalpha degradation in breast cancer MCF-7 cells by CHIP-mediated ubiquitin pathway and inhibits survival signaling. Biochem. Pharmacol. 75, 1697–1705 (2008). [DOI] [PubMed] [Google Scholar]
- Sun C. et al. Diverse roles of C-terminal Hsp70-interacting protein (CHIP) in tumorigenesis. J. Cancer Res. Clin. Oncol. 140, 189–197 (2014). [DOI] [PubMed] [Google Scholar]
- Brauch H. et al. VHL alterations in human clear cell renal cell carcinoma: association with advanced tumor stage and a novel hot spot mutation. Cancer Res. 60, 1942–1948 (2000). [PubMed] [Google Scholar]
- Kajiro M. et al. The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat. Cell Biol. 11, 312–319 (2009). [DOI] [PubMed] [Google Scholar]
- Bai J. et al. JWA regulates melanoma metastasis by integrin alphaVbeta3 signaling. Oncogene 29, 1227–1237 (2010). [DOI] [PubMed] [Google Scholar]
- Chen F. et al. RUNX3 suppresses migration, invasion and angiogenesis of human renal cell carcinoma. PLoS One 8, e56241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson A. R., Fingleton B., Rothenberg M. L. & Matrisian L. M. Matrix metalloproteinases: biologic activity and clinical implications. J. Clin. Oncol. 18, 1135–1149 (2000). [DOI] [PubMed] [Google Scholar]
- Carmeliet P. & Jain R. K. Angiogenesis in cancer and other diseases. Nature 407, 249–257 (2000). [DOI] [PubMed] [Google Scholar]
- Ferrara N. & Alitalo K. Clinical applications of angiogenic growth factors and their inhibitors. Nat. Med. 5, 1359–1364 (1999). [DOI] [PubMed] [Google Scholar]
- Weis S. M. & Cheresh D. A. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med. 17, 1359–1370 (2011). [DOI] [PubMed] [Google Scholar]
- Kaelin W. G. Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 8, 865–873 (2008). [DOI] [PubMed] [Google Scholar]
- Watanabe Y., Lee S. W., Detmar M., Ajioka I. & Dvorak H. F. Vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) delays and induces escape from senescence in human dermal microvascular endothelial cells. Oncogene 14, 2025–2032 (1997). [DOI] [PubMed] [Google Scholar]
- Dvorak H. F., Brown L. F., Detmar M. & Dvorak A. M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 146, 1029–1039 (1995). [PMC free article] [PubMed] [Google Scholar]
- Cao Y. et al. VEGF exerts an angiogenesis-independent function in cancer cells to promote their malignant progression. Cancer Res. 72, 3912–3918 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen J., Rasmuson T., Grankvist K. & Ljungberg B. Vascular endothelial growth factor as prognostic factor in renal cell carcinoma. J. Urol. 163, 343–347 (2000). [PubMed] [Google Scholar]
- Klatte T. et al. Hypoxia-inducible factor 1 alpha in clear cell renal cell carcinoma. Clin. Cancer Res. 13, 7388–7393 (2007). [DOI] [PubMed] [Google Scholar]
- Escudier B. et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 356, 125–134 (2007). [DOI] [PubMed] [Google Scholar]
- Motzer R. J. et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 356, 115–124 (2007). [DOI] [PubMed] [Google Scholar]
- Rixe O. et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 8, 975–984 (2007). [DOI] [PubMed] [Google Scholar]
- Hutson T. E. et al. Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 28, 475–480 (2010). [DOI] [PubMed] [Google Scholar]
- Escudier B. et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet 370, 2103–2111 (2007). [DOI] [PubMed] [Google Scholar]
- Jain R. K., Duda D. G., Clark J. W. & Loeffler J. S. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol. 3, 24–40 (2006). [DOI] [PubMed] [Google Scholar]
- Bai J. et al. BRG1 expression is increased in human glioma and controls glioma cell proliferation, migration and invasion in vitro. J. Cancer Res. Clin. Oncol. 138, 991–998 (2012). [DOI] [PubMed] [Google Scholar]
- Mei P. J. et al. RUNX3 expression is lost in glioma and its restoration causes drastic suppression of tumor invasion and migration. J. Cancer Res. Clin. Oncol. 137, 1823–1830 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.