

Abstract
The ability to provide early clinical intervention for inherited disorders is heavily dependent on knowledge of a patient’s disease-causing mutations and the resultant pathophysiologic mechanism(s). Without knowing a patient’s disease-causing gene, and how gene mutations alter the health and functionality of affected cells, it would be difficult to develop and deliver patient-specific molecular or small molecule therapies. Many believe that the field of stem cell biology holds the keys to the future development of disease-, patient-, and cell-specific therapies. In the case of the eye, which is susceptible to an extremely common late-onset degenerative disease known as age-related macular degeneration, stem cell-based therapies could increase the quality of life for millions of patients worldwide. Furthermore, autologous, patient-specific induced pluripotent stem cells could be a viable source to treat rare Mendelian retinal degenerative diseases such as retinitis pigmentosa, Stargardt disease, and Best disease, to name a few.
Induced pluripotent stem cells (iPSCs) are a promising source for cell replacement therapies. But iPSCs are also useful for studying the pathophysiology of inherited retinal disorders (e.g., retinitis pigmentosa) in vitro.
The retina is a complex tissue that is responsible for converting light energy into changes in neuronal membrane potential that are then relayed to the brain as trains of action potentials and processed to yield a picture of the surrounding visual environment. Many of the diseases that impair vision occur in the outermost layers of the retina, namely, the photoreceptor layer and the underlying supportive retinal pigment epithelium (RPE). Many of the diseases that will be discussed in this review occur within the area highlighted by the red bracket shown in Figure 1. Although the ability to identify disease-causing genetic mutations has dramatically improved in recent years, it is still challenging to investigate the pathophysiologic mechanisms associated with these mutations because of the inability to obtain samples of diseased tissue from living patients.
Figure 1.

Schematic of the human retina. The major layers of the retina are shown schematically: NFL, nerve fiber layer; GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium; BM, Bruch's membrane; and CV, choroidal vasculature. The photoreceptors, retinal pigment epithelium, and choroidal vasculature (indicated by red bracket on right of schematic) are the cell types that are most commonly affected by inherited retinal diseases.
Despite this difficulty, the retina is a good candidate for gene and cell replacement therapies for a number of reasons. Compared with other components of the central nervous system, the posterior retina is very accessible to therapeutic delivery via subretinal injection, a procedure that can be performed by most vitreoretinal surgeons. Clinical diagnosis and postintervention monitoring of the retina are also easier than other parts of the CNS because of transparency of the cornea, lens, and intraocular media.
The cells that comprise the outer retina are highly specified to perform visual processing. Cone and rod photoreceptor cells consist of a cell body and an extremely metabolically active outer segment that is responsible for converting light energy into hyperpolarization of the cell membrane. This phototransduction involves the isomerization of retinoids and the subsequent activation of other signaling molecules in an amplifying cascade (Tang et al. 2013). Hyperpolarization of the photoreceptor cells results in a graded stimulation of bipolar cells, which then cause ganglion cells to send trains of action potentials via their axons in the optic nerves and tracts to the lateral geniculate nuclei in the brain. Photoreceptor outer segments are shed in a diurnal rhythm and then phagocytized, degraded, and recycled by the RPE. The RPE is a polarized monolayer of neuroepithelial origin that is tasked with this phagocytic activity as well as the reisomerization of all-trans-retinol to the light sensitive 11-cis isomer needed for photon capture in the photoreceptors (Ramsden et al. 2013). Rod photoreceptors cannot survive long without close proximity to healthy, functional RPE. The RPE also provides some nutrients to the photoreceptors and contains pigment that limits light scattering (Ramsden et al. 2013). Both photoreceptor cells and RPE cells are highly metabolic and consume large amounts of oxygen, which is supplied by the lobular, fenestrated capillary bed, the choriocapillaris. The physiological interdependence of photoreceptors, RPE cells, and the choriocapillaris results in the injury of all three of these cell types in most inherited retinal degenerative disorders.
Retinal degenerative disorders are the leading cause of irreversible blindness worldwide (McClements and MacLaren 2013). Some well-studied diseases within this large and heterogenous group are age-related macular degeneration (AMD), retinitis pigmentosa, Stargardt disease, Leber congenital amaurosis, and Best disease, to name just a few. AMD is a complex disorder of older individuals that results from the interaction among a number of genetic loci and the environment (Swaroop et al. 2007; Ryu et al. 2010; Priya et al. 2012). In contrast, retinitis pigmentosa and the other degenerative disorders listed above are Mendelian retinal dystrophies, each caused primarily by the absence or dysfunction of a single gene product. The commonality between AMD and the single gene disorders is that they each are associated with damage to the photoreceptors, RPE, and underlying choroidal vessels. An additional complexity to the single gene disorders is that some of them selectively injure the macula (the cone photoreceptor-rich central area of retina that is responsible for visual acuity), whereas others selectively injure the rod photoreceptor-rich mid-peripheral retina.
For the development of some types of disease-specific therapies, it is first necessary to understand the underlying disease mechanisms and the natural history of the disease in some detail. There are large clinical differences among the diseases listed above, but in addition, there are often significant differences among individuals affected with the same disease that are traceable to differences in severity of their mutations and the presence or absence of modifying alleles. These factors will cause the ideal therapeutic options to vary from patient to patient, as will the point in the patient's disease course that therapy is being contemplated.
Figure 2 depicts the relationships between various treatment options and the time course and mechanistic understanding of a given patient’s disease. When knowledge of disease mechanism is abundant, such as knowing that a person harbors a well-characterized disease allele of high penetrance, preventative measures such as preimplantation genetic testing can be taken. If the disease is already clinically manifest and there is some knowledge of the mechanism of cell loss, drug therapy specifically targeted against mediators of a specific pathway may be applied. Similarly, when specific disease-causing mutations have been identified and there are still viable retinal cells remaining to rescue, gene therapy can be used to replace the mutated gene and restore normal gene function thereby arresting or slowing the progress of the disease. When the disease has progressed to the point that extensive cellular loss has occurred, cell replacement therapy using stem cell technology will be needed to restore useful vision. If autologous induced pluripotent stem cells (iPSCs) were used for such transplantation, the patient’s disease-causing mutations would still be present and would be expected to injure the transplanted cells at the same or a faster rate as they did before treatment. However, if the disease-causing gene is first corrected with viral-mediated gene replacement or genome editing, one can take advantage of the near-perfect immunologic match of an autologous transplant without suffering the disadvantage of the disease-causing genotype. Recent advances in genome editing using RNA-mediated clustered regularly interspaced short palindromic repeats (CRISPRs) allow the delivery of corrected, fully functioning cells into diseased tissues (Cong et al. 2013; Joung and Sander 2013). Last, when knowledge of disease mechanism is sparse or in the cases of individuals that have had disease for an extended period of time and undergone near or total loss of photoreceptor cells, RPE, and/or choriocapillaris, optogenetic strategies, or a retinal prosthesis could be viable therapeutic options to restore some visual function. Optogenetics uses light to trigger responses in genetically targeted groups of cells. In contrast to traditional gene therapy approaches whose goal is to correct a mutated gene and restore normal protein function, optogenetics introduces novel light sensitive proteins into neurons like bipolar cells and ganglion cells to cause them to function as photoreceptors to restore some visual function (Garg and Federman 2013; Packer et al. 2013; Yonehara et al. 2013). Retinal prostheses typically consist of arrays of photodiodes that are surgically implanted beneath or above the retina to electrically stimulate the remaining inner retinal cells (Loudin et al. 2007; Zrenner et al. 2011). Another strategy that is being pursued for patients with very advanced disease is a three-dimensional transplants consisting of stem-cell-derived sheets of photoreceptors and RPE cells grown on a biocompatible polymer scaffold (Zhang et al. 2007; Yao et al. 2011; Tucker et al. 2011a; Shepherd et al. 2013).
Figure 2.

Relationship between knowledge of disease mechanism and disease progression on therapeutic options. Some therapeutic options are more suitable for earlier stages of disease than others. Similarly, some require a precise understanding of the cause and/or mechanism of the disease, whereas others are more generally usable.
USING STEM CELLS TO INTERROGATE PATHOPHYSIOLOGY OF DISEASE
Stem cells, which can be isolated from various tissues during different points in development, have the ability to continually self-propagate and differentiate into multiple cell types. Embryonic stem cells (ESCs), harvested from the inner cell mass of the blastocyst of preimplantation embryos, are classified as pluripotent. That is, they are capable of forming tissues of all three embryonic germ layers: endoderm, mesoderm, and ectoderm. Fetal stem cells are isolated from fetal tissue and are multipotent. They can differentiate into a limited number of cell types depending on their individual degree of differentiation at the time of isolation. Adult stem cells (ASCs) are present in small numbers in most major organs and also display multipotency. For example, ASCs can be found in bone marrow, intestinal crypts, and the eye, near the limbal region posterior to the cornea (Davanger and Evensen 1971; Jiang et al. 2002; Snippert et al. 2010).
Induced iPSCs were originally generated via retroviral transduction of mouse dermal fibroblasts with the transcription factors Oct4, Sox2, Klf4, and c-myc (Yamanaka and Takahashi 2006). They have since been derived from a variety of different tissues and species. Like ESCs, iPSCs have both an unlimited capacity for self-renewal and are pluripotent. They are therefore suitable for tissue-specific cell generation. Our laboratory and others have become proficient in generating retinal progenitor cells (RPCs) from iPSCs and further differentiating them into RPE and photoreceptor cells (Wang et al. 2008; Meyer et al. 2011; Tucker et al. 2011b, 2013a,b). Using these protocols we have recently been able to show that following transplantation into the subretinal space of retinal degenerative hosts, iPSC-derived photoreceptor precursor cells aid in the restoration of retinal structure and function (Tucker et al. 2011b).
Apart from being a promising cell source for autologous transplantation, a major advantage of the iPSC technology is that it provides the ability to model and study human disease in vitro. In particular, iPSCs are a useful tool for confirmation of newly identified disease-causing mutations and interrogation of disease pathophysiology in relatively inaccessible tissues such as the retina that cannot be routinely subjected to molecular analysis in living patients. As shown in Figure 2, the ability to develop an in-depth understanding of disease mechanism will increase the number of therapeutic options available to a physician. The following paragraphs provide an overview of the ways in which iPSCs have been used to model Mendelian diseases such as retinitis pigmentosa and Best disease, as well as complex disorders such as AMD.
MODELING OF MENDELIAN RETINAL DEGENERATIONS
Most inherited retinal diseases are caused by mutations in a single gene. For instance, mutations in the gene ABCA4 (ATP-binding cassette, subfamily A, member 4), cause a wide spectrum of recessive photoreceptor cell-specific retinal phenotypes including Stargardt disease, cone–rod dystrophy, and even retinitis pigmentosa (RP). ABCA4 is a flippase that facilitates the movement of the visual cycle intermediate, N-retinylidene-phosphatidylethanolamine (NR-PE), from the intradiscal leaflet of photoreceptor disc membranes to the cytoplasmic leaflet (Quazi et al. 2012). Once translocated, NR-PE is reduced to vitamin A, transferred to the RPE and reisomerized into 11-cis-retinal. When ABCA4 is mutated, the flippase function of the protein is lost, leading to the formation and accumulation of the insoluble bisretinoid N-retinylidene-N-retinyl-ethanolamine (A2E). A2E acts similarly to a detergent and is toxic to the RPE and photoreceptors (Schindler et al. 2010; Sheffield and Stone 2011; Braun et al. 2013). Stargardt disease typically presents with reduced visual acuity and a gradually enlarging central scotoma that is associated with the progressive accumulation of autofluorescent deposits of A2E-containing lipofuscin. The age of onset for Stargardt disease can vary from early childhood to adulthood (Schindler et al. 2010). Within the phenotypic spectrum of Stargardt disease, some patients have very severe degeneration, including near complete loss of RPE and cone and rod photoreceptors (resembling retinitis pigmentosa), whereas other individuals have much milder disease in which the foveal photoreceptors are relatively spared and the visual acuity remains quite good for many years. A comprehensive understanding of the function of ABCA4 and its gene product have facilitated current gene therapy trials designed to deliver wild-type ABCA4 to patients with this disease (Al-Saikhan 2013; Boye et al. 2013). As indicated above, for patients with extensive photoreceptor cell loss, cell replacement therapy to provide new functional photoreceptor and RPE cells will likely be required. For Mendelian disorders like Stargardt disease, an autologous iPSC-based approach would likely require correction of the disease-causing genetic mutation before cellular transplantation. If left uncorrected, the deleterious effect of the genetic mutation would likely reduce the longevity of the newly transplanted cells. This would likely be especially true for early onset diseases. Although undesirable for cell replacement purposes, as indicated above, one could take advantage of the presence of the mutant alleles in the iPSCs to interrogate disease pathophysiology and develop new treatment strategies.
Unlike ABCA4-associated disease, which is relatively well understood mechanistically, a large number of patients’ disease-causing genes are either currently unknown or the function of these genes is poorly understood. By using iPSC-derived photoreceptor precursor cells generated from patients with retinal degenerative disease, we were recently able to show that a homozygous Alu insertion, identified in exon 9 of the gene male germ cell-associated kinase (MAK), results in loss of the transcript and an inability to produce normal MAK protein (Tucker et al. 2011b). Loss of MAK was found to disrupt normal photoreceptor cell structure leading to cell death and irreversible blindness. In addition to allowing us to confirm pathogenicity of the MAK mutation, we were also able to identify and confirm the involvement of a unique MAK-specific retinal transcript in disease pathophysiology. This transcript contains an extra 75-bp exon (exon 12), which is phylogenetically conserved. It only exists in transcripts that also contain MAK exon 9. Collectively these findings have enabled the development of gene-based interventions for MAK-associated RP in which patient-specific iPSC-derived photoreceptor cells are being used to confirm therapeutic efficacy.
In a similar study, iPSC-derived photoreceptor precursor cells were used to confirm the pathogenicity of two USH2A mutations in an adult patient with autosomal recessive RP (Tucker et al. 2013b). In this study, keratinocyte-derived iPSCs were differentiated into multilayer eyecup-like structures with an outer-pigmented layer of RPE and an inner layer containing photoreceptor precursor cells. The latter cells expressed photoreceptor-specific markers and showed axonemes and basal bodies structurally characteristic of outer segments. Analysis of the identified USH2A transcripts in these cells revealed that one of the patient’s mutations causes exonification of intron 40, a translation frameshift and a premature stop codon. Up-regulation of the proteins GRP78 and GRP94 suggested that the patient’s other USH2A variant (a single point mutation resulting in an Arginine to Histidine transition at position 4192) causes disease through protein misfolding and endoplasmic reticulum (ER) stress. To further support the idea that this disease is ER stress-related rather than a developmental defect, photoreceptor precursor cells were transplanted into 4-d-old immunodeficient Crb1−/− mice. Newly integrated patient-derived cells formed morphologically and immunohistochemically recognizable photoreceptor cells, suggesting that the mutations in this patient cause degeneration of developmentally normal photoreceptors. The role of ER stress in RP-associated photoreceptor cell death is consistent with previous findings in other disease causing mutations. For instance, in a similar study, Jin and colleagues were able to show that iPSC-derived rod photoreceptor precursor cells generated from a patient with autosomal dominant rhodopsin-associated RP have elevated levels of ER stress-related proteins and subsequently succumb to apoptic cell death in vitro (Jin et al. 2011).
Unlike RP, which is usually caused by mutations in genes expressed in photoreceptor cells, Best disease results from mutations in the RPE-specific gene, BEST1. Best disease selectively affects the region of the human retina within one disk diameter of the fovea and predisposes affected individuals to vision threatening choroidal neovascular membrane formation. Late stages of Best disease are often marked by geographic loss of the RPE underlying the macula. Although BEST1 mutations have been known to cause this disease for quite some time, the mechanism by which these mutations cause Best disease remain unclear. In an elegant study recently published by Singh et al. (2013), human iPSC-derived RPE were generated from patients and unaffected siblings and used to examine the cellular and molecular processes underlying Best disease. The investigators found that RPE derived from patients with Best disease displayed disrupted fluid flux and increased accumulation of autofluorescent material after long-term feeding of photoreceptor outer segments (Singh et al. 2013). Furthermore, following chronic photoreceptor outer segment feeding, rhodopsin degradation was found to be delayed, cellular responses to calcium were up-regulated, and oxidative stress levels were altered (Abramoff et al. 2013; Singh et al. 2013). Collectively, these findings suggest that BEST1 plays an important role in photoreceptor outer segment turnover and implicates disruption of this pathway in the pathogenesis of Best disease.
MODELING OF COMPLEX RETINAL DEGENERATIVE DISEASE
AMD is a genetically heterogeneous and complex disease with more than 50 different genetic risk loci identified to date (Gorin 2012; Liu et al. 2012), 19 of which meet genome wide significance (Fritsche et al. 2013). Early AMD typically presents in the sixth decade of life with the formation of lipid-like deposits, commonly referred to as drusen, which can accumulate within and beneath the RPE layer (Curcio et al. 2013). A subset of patients with early AMD later develop geographic atrophy of the retinal pigment epithelium and underlying choriocapillaris, and the loss of these tissues results in the loss of the overlying photoreceptor cells. Other patients with early AMD develop a complication sometimes referred to as “wet AMD” in which the underlying choroidal vasculature grows into the sub-RPE and/or subretinal space forming fenestrated, leaky vascular networks known as choroidal neovascular membranes (CNVs) (Jager et al. 2008). For AMD, all of the therapeutic interventions shown in Figure 2 could eventually come into play. Drug therapy using the antivascular endothelial growth factor (VEGF) agents Avastin and Lucentis have proven very efficacious in ameliorating CNVs and slowing vision loss in wet AMD (Agosta et al. 2012; Scott and Bressler 2013). Because of the fact that AMD is genetically heterogenous, traditional gene augmentation approaches seem less promising than for Mendelian disease. However, cell replacement therapy for patients with loss of photoreceptor, RPE, and choroidal (choriocapillaris) endothelial cells is being pursued by a number of groups and, if successful, would be extremely beneficial for thousands of patients. In very late stage disease, in which significant cell death has occurred, optogenetic and retinal prosthetic approaches may eventually be viable options, although the resolution of the latter devices will need to be improved if they are to offer AMD patients a significant improvement in visual acuity.
The greatest opportunity for AMD treatment lies in understanding the genetic causes and pathogenic mechanisms well enough to be able to identify those who will develop the disease and prevent it from happening through early and efficacious intervention. Although the notion that AMD has a strong genetic component was advocated as early as the 1970s (Gass 1973), our understanding of the biological mechanisms through which specific genetic loci contribute to the development and progression of AMD is still rather poorly understood. Genome wide association studies of AMD identified two loci on chromosomes 1 and 10 that are highly associated with disease risk (Edwards et al. 2005; Haines et al. 2005; Jakobsdottir et al. 2005; Klein et al. 2005; Rivera et al. 2005; Zareparsi et al. 2005; Schwartz et al. 2012). The risk associated with the chromosome 1q locus is predominantly because of a haplotype that harbors a missense mutation in the complement factor H (CFH) gene (Tyr402His) (Hageman et al. 2006; Zhang et al. 2008). The complement cascade was implicated in the pathogenesis of AMD before the association of CFH variants with the disease (Johnson et al. 2000, 2001; Mullins et al. 2000, 2001; Bora et al. 2005), and thus the involvement of this inhibitor in AMD fits into a mechanistic framework of impaired complement regulation and bystander cell injury (Mullins et al. 2011). In contrast, the mechanism by which the chromosome 10q locus increases AMD risk has been—and remains—controversial. The most common high-risk haplotype contains two genes (ARMS2 and HTRA1) with plausible disease-causing variants. The variants include a nonconservative polymorphism in the ARMS2 gene (Ala69Ser), a complex 144 bp deletion and a 54 bp insertion in the 3′ untranslated region of the ARMS2 transcript, and a promoter polymorphism in the HTRA1 gene (Fig. 3) (Fritsche et al. 2008).
Figure 3.
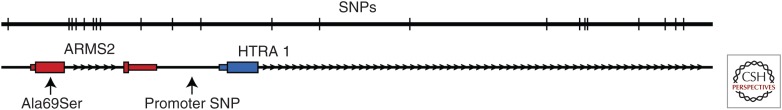
Schematic depiction of the 10q26 risk haplotype and associated mutations.
These variants are in strong linkage disequilibrium such that only a few percent of 10q alleles harboring ARMS2 Ala69Ser lack the HTRA1 promoter variant and vice versa. This, coupled with the overall genetic complexity of AMD (and the nonspecificity of the AMD phenotype recorded in written clinical records) have made it difficult to determine which, if either, of these two genes is responsible for conferring the AMD risk associated with this locus. Recently, Yang and colleagues (2014) used an unbiased proteome screen of A2E-aged patient-specific iPSC-derived RPE cell lines derived from patients with high and low risk 10q26 loci. They found that cells from patients who were homozygous for the high-risk haplotype had a reduction in superoxide dismutase 2 (SOD2)-mediated antioxidative defense. The investigators concluded that the ARMS2/HTRA1 risk alleles decrease SOD2 defense, making RPE more susceptible to oxidative damage and thereby contributing to AMD pathogenesis. By elucidating the disease mechanism(s) associated with this locus, new targets for therapeutic intervention have potentially been identified.
CONCLUSION
For genetically heterogeneous retinal diseases like RP, a large number of disease-causing genes and mutations have yet to be identified. Each of these remaining genes is likely to be responsible for a small fraction of patients affected with the disease. The MAK gene for instance, although enriched in people of Ashkenazi Jewish ancestry, is responsible for only 1% of all cases of RP. To develop treatments for disorders like these, inexpensive model systems that faithfully recapitulate key disease phenotypes are very useful. iPSCs can provide attractive alternatives to animal models. There are now many examples in which patient-specific iPSCs have been found to accurately recapitulate a disease phenotype in a dish. One can use such systems to rapidly interrogate disease pathophysiology and test the efficacy of a variety of different drug- and gene-based therapies.
ACKNOWLEDGMENTS
The authors acknowledge grant support from National Institutes of Health (NIH) Directors New Innovator Award 1-DP2-OD007483-01, NEI EY017451, F32 EY022834, Howard Hughes Medical Institute, Foundation Fighting Blindness, Stephen A. Wynn Foundation, Grousbeck Family Foundation, Leo, Jacques & Marion Hauser Family Vision Restoration Fund.
Footnotes
Editors: Eric A. Pierce, Richard H. Masland, and Joan W. Miller
Additional Perspectives on Retinal Disorders: Genetic Approaches to Diagnosis and Treatment available at www.perspectivesinmedicine.org
REFERENCES
- Abramoff MD, Mullins RF, Lee K, Hoffmann JM, Sonka M, Critser DB, Stasheff SF, Stone EM. 2013. Human photoreceptor outer segments shorten during light adaptation. Invest Ophthalmol Vis Sci 54: 3721–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agosta E, Lazzeri S, Orlandi P, Figus M, Fioravanti A, Di Desidero T, Sartini MS, Nardi M, Danesi R, Bocci G. 2012. Pharmacogenetics of antiangiogenic and antineovascular therapies of age-related macular degeneration. Pharmacogenomics 13: 1037–1053. [DOI] [PubMed] [Google Scholar]
- Al-Saikhan FI. 2013. The gene therapy revolution in ophthalmology. Saudi J Ophthalmol 27: 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bora PS, Sohn J-H, Cruz JMC, Jha P, Nishihori H, Wang Y, Kaliappan S, Kaplan HJ, Bora NS. 2005. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J Immunol 174: 491–497. [DOI] [PubMed] [Google Scholar]
- Boye SE, Boye SL, Lewin AS, Hauswirth WW. 2013. A comprehensive review of retinal gene therapy. Mol Ther 21: 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun TA, Mullins RF, Wagner AH, Andorf JL, Johnston RM, Bakall BB, DeLuca AP, Fishman GA, Lam BL, Weleber RG, et al. 2013. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet 22: 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio CA, Messinger JD, Sloan KR, McGwin G, Medeiros NE, Spaide RF. 2013. Subretinal drusenoid deposits in non-neovascular age-related macular degeneration: Morphology, prevalence, topography, and biogenesis model. Retina 33: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davanger M, Evensen A. 1971. Role of the pericorneal papillary structure in renewal of corneal epithelium. Nature 229: 560–561. [DOI] [PubMed] [Google Scholar]
- Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. 2005. Complement factor H polymorphism and age-related macular degeneration. Science 308: 421–424. [DOI] [PubMed] [Google Scholar]
- Fritsche LG, Loenhardt T, Janssen A, Fisher SA, Rivera A, Keilhauer CN, Weber BHF. 2008. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet 40: 892–896. [DOI] [PubMed] [Google Scholar]
- Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, et al. 2013. Seven new loci associated with age-related macular degeneration. Nat Genet 45: 433-9–439e1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SJ, Federman J. 2013. Optogenetics, visual prosthesis and electrostimulation for retinal dystrophies. Curr Opin Ophthalmol 24: 407–414. [DOI] [PubMed] [Google Scholar]
- Gass JD. 1973. Drusen and disciform macular detachment and degeneration. Arch Ophthalmol 90: 206–217. [DOI] [PubMed] [Google Scholar]
- Gorin MB. 2012. Genetic insights into age-related macular degeneration: Controversies addressing risk, causality, and therapeutics. Mol Aspects Med 33: 467–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, Radeke MJ, Kavanagh D, Richards A, Atkinson J, et al. 2006. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: Characterization, ethnic distribution and evolutionary implications. Ann Med 38: 592–604. [PMC free article] [PubMed] [Google Scholar]
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, et al. 2005. Complement factor H variant increases the risk of age-related macular degeneration. Science 308: 419–421. [DOI] [PubMed] [Google Scholar]
- Jager RD, Mieler WF, Miller JW. 2008. Age-related macular degeneration. N Engl J Med 358: 2606–2617. [DOI] [PubMed] [Google Scholar]
- Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB. 2005. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet 77: 389–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, et al. 2002. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418: 41–49. [DOI] [PubMed] [Google Scholar]
- Jin Z-B, Okamoto S, Osakada F, Homma K, Assawachananont J, Hirami Y, Iwata T, Takahashi M. 2011. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PLoS ONE 6: e17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LV, Ozaki S, Staples MK, Erickson PA, Anderson DH. 2000. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res 70: 441–449. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Staples MK, Anderson DH. 2001. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res 73: 887–896. [DOI] [PubMed] [Google Scholar]
- Joung JK, Sander JD. 2013. TALENs: A widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, et al. 2005. Complement factor H polymorphism in age-related macular degeneration. Science 308: 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MM, Chan C-C, Tuo J. 2012. Genetic mechanisms and age-related macular degeneration: Common variants, rare variants, copy number variations, epigenetics, and mitochondrial genetics. Hum Genomics 6: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loudin JD, Simanovskii DM, Vijayraghavan K, Sramek CK, Butterwick AF, Huie P, McLean GY, Palanker DV. 2007. Optoelectronic retinal prosthesis: System design and performance. J Neural Eng 4: S72–S84. [DOI] [PubMed] [Google Scholar]
- McClements ME, MacLaren RE. 2013. Gene therapy for retinal disease. Transl Res 161: 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JS, Howden SE, Wallace KA, Verhoeven AD, Wright LS, Capowski EE, Pinilla I, Martin JM, Tian S, Stewart R, et al. 2011. Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 29: 1206–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RF, Russell SR, Anderson DH, Hageman GS. 2000. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J 14: 835–846. [PubMed] [Google Scholar]
- Mullins RF, Aptsiauri N, Hageman GS. 2001. Structure and composition of drusen associated with glomerulonephritis: Implications for the role of complement activation in drusen biogenesis. Eye (Lond) 15: 390–395. [DOI] [PubMed] [Google Scholar]
- Mullins RF, Dewald AD, Streb LM, Wang K, Kuehn MH, Stone EM. 2011. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp Eye Res 93: 565–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer AM, Roska B, Häusser M. 2013. Targeting neurons and photons for optogenetics. Nat Neurosci 16: 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya RR, Chew EY, Swaroop A. 2012. Genetic studies of age-related macular degeneration: Lessons, challenges, and opportunities for disease management. Ophthalmology 119: 2526–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quazi F, Lenevich S, Molday RS. 2012. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun 3: 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden CM, Powner MB, Carr A-JF, Smart MJK, da Cruz L, Coffey PJ. 2013. Stem cells in retinal regeneration: Past, present and future. Development 140: 2576–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera A, Fisher SA, Fritsche LG, Keilhauer CN, Lichtner P, Meitinger T, Weber BHF. 2005. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet 14: 3227–3236. [DOI] [PubMed] [Google Scholar]
- Ryu E, Fridley BL, Tosakulwong N, Bailey KR, Edwards AO. 2010. Genome-wide association analyses of genetic, phenotypic, and environmental risks in the age-related eye disease study. Mol Vis 16: 2811–2821. [PMC free article] [PubMed] [Google Scholar]
- Schindler EI, Nylen EL, Ko AC, Affatigato LM, Heggen AC, Wang K, Sheffield VC, Stone EM. 2010. Deducing the pathogenic contribution of recessive ABCA4 alleles in an outbred population. Hum Mol Genet 19: 3693–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz SG, Agarwal A, Kovach JL, Gallins PJ, Cade W, Postel EA, Wang G, Ayala-Haedo J, Spencer KM, Haines JL, et al. 2012. The ARMS2 A69S variant and bilateral advanced age-related macular degeneration. Retina 32: 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AW, Bressler SB. 2013. Long-term follow-up of vascular endothelial growth factor inhibitor therapy for neovascular age-related macular degeneration. Curr Opin Ophthalmol 24: 190–196. [DOI] [PubMed] [Google Scholar]
- Sheffield VC, Stone EM. 2011. Genomics and the eye. N Engl J Med 364: 1932–1942. [DOI] [PubMed] [Google Scholar]
- Shepherd RK, Shivdasani MN, Nayagam DAX, Williams CE, Blamey PJ. 2013. Visual prostheses for the blind. Trends Biotechnol 10: 562–571. [DOI] [PubMed] [Google Scholar]
- Singh R, Shen W, Kuai D, Martin JM, Guo X, Smith MA, Perez ET, Phillips MJ, Simonett JM, Wallace KA, et al. 2013. iPS cell modeling of Best disease: Insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet 22: 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, et al. 2010. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143: 134–144. [DOI] [PubMed] [Google Scholar]
- Swaroop A, Branham KE, Chen W, Abecasis G. 2007. Genetic susceptibility to age-related macular degeneration: A paradigm for dissecting complex disease traits. Hum Mol Genet 16: R174–R182. [DOI] [PubMed] [Google Scholar]
- Tang PH, Kono M, Koutalos Y, Ablonczy Z, Crouch RK. 2013. New insights into retinoid metabolism and cycling within the retina. Prog Retin Eye Res 32: 48–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker BA, Park I-H, Qi SD, Klassen HJ, Jiang C, Yao J, Redenti S, Daley GQ, Young MJ. 2011a. Transplantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in degenerative mice. PLoS ONE 6: e18992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker BA, Scheetz TE, Mullins RF, DeLuca AP, Hoffmann JM, Johnston RM, Jacobson SG, Sheffield VC, Stone EM. 2011b. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc Natl Acad Sci 108: E569–E576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker BA, Anfinson KR, Mullins RF, Stone EM, Young MJ. 2013a. Use of a synthetic xeno-free culture substrate for induced pluripotent stem cell induction and retinal differentiation. Stem Cells Transl Med 2: 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker BA, Mullins RF, Streb LM, Anfinson K, Eyestone ME, Kaalberg E, Riker MJ, Drack AV, Braun TA, Stone EM. 2013b. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. eLife 2: e00824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Girman S, Lu B, Bischoff N, Holmes T, Shearer R, Wright LS, Svendsen CN, Gamm DM, Lund RD. 2008. Long-term vision rescue by human neural progenitors in a rat model of photoreceptor degeneration. Invest Ophthalmol Vis Sci 49: 3201–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S, Takahashi K. 2006. Induction of pluripotent stem cells from mouse fibroblast cultures. Tanpakushitsu Kakusan Koso 51: 2346–2351. [PubMed] [Google Scholar]
- Yang J, Li Y, Chan L, Tsai Y-T, Wu W-H, Nguyen HV, Hsu C-W, Li X, Brown LM, Egli D, et al. 2014. Validation of genome-wide association study (GWAS)-identified disease risk alleles with patient-specific stem cell lines. Hum Mol Genet 23: 3445–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Tucker BA, Zhang X, Checa-Casalengua P, Herrero-Vanrell R, Young MJ. 2011. Robust cell integration from co-transplantation of biodegradable MMP2-PLGA microspheres with retinal progenitor cells. Biomaterials 32: 1041–1050. [DOI] [PubMed] [Google Scholar]
- Yonehara K, Farrow K, Ghanem A, Hillier D, Balint K, Teixeira M, Jüttner J, Noda M, Neve RL, Conzelmann K-K, et al. 2013. The first stage of cardinal direction selectivity is localized to the dendrites of retinal ganglion cells. Neuron 79: 1078–1085. [DOI] [PubMed] [Google Scholar]
- Zareparsi S, Branham KEH, Li M, Shah S, Klein RJ, Ott J, Hoh J, Abecasis GR, Swaroop A. 2005. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet 77: 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Klassen HJ, Tucker BA, Perez M-TR, Young MJ. 2007. CNS progenitor cells promote a permissive environment for neurite outgrowth via a matrix metalloproteinase-2-dependent mechanism. J Neurosci 27: 4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Morrison MA, Dewan A, Adams S, Andreoli M, Huynh N, Regan M, Brown A, Miller JW, Kim IK, et al. 2008. The NEI/NCBI dbGAP database: Genotypes and haplotypes that may specifically predispose to risk of neovascular age-related macular degeneration. BMC Med Genet 9: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zrenner E, Bartz-Schmidt KU, Benav H, Besch D, Bruckmann A, Gabel V-P, Gekeler F, Greppmaier U, Harscher A, Kibbel S, et al. 2011. Subretinal electronic chips allow blind patients to read letters and combine them to words. Proc Biol Sci 278: 1489–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]