

Abstract
Nitrogen dioxide is a highly toxic reactive nitrogen species (RNS) recently discovered as an inflammatory oxidant with great potential to damage tissues. We demonstrate here that cell death by RNS was caused by c-Jun N-terminal kinase (JNK). Activation of JNK by RNS was density dependent and caused mitochondrial depolarization and nuclear condensation. JNK activation by RNS was abolished in cells lacking functional Fas or following expression of a truncated version of Fas lacking the intracellular death domain. In contrast, RNS induced JNK potently in cells expressing a truncated version of tumor necrosis factor receptor 1 or cells lacking tumor necrosis factor receptor 1 (TNF-R1), illustrating a dependence of Fas but not TNF-R1 in RNS-induced signaling to JNK. Furthermore, Fas was oxidized, redistributed, and colocalized with Fas-associated death domain (FADD) in RNS-exposed cells, illustrating that RNS directly targeted Fas. JNK activation and cell death by RNS occurred in a Fas ligand- and caspase-independent manner. While the activation of JNK by RNS or FasL required FADD, the cysteine-rich domain 1 containing preligand assembly domain required for FasL signaling was not involved in JNK activation by RNS. These findings illustrate that RNS cause cell death in a Fas- and JNK-dependent manner and that this occurs through a pathway distinct from FasL. Thus, avenues aimed at preventing the interaction of RNS with Fas may attenuate tissue damage characteristic of chronic inflammatory diseases that are accompanied by high levels of RNS.
Nitrogen dioxide (NO2) is a highly reactive free-radical gas and is most commonly known as an indoor and outdoor air pollutant that causes pulmonary damage and asthma exacerbations in children (8, 47). More importantly, NO2 is also emerging as a reactive nitrogen species (RNS) that may play an important role in inflammation (see reference 1 for review). NO2 is produced by peroxidases, most notably eosinophil peroxidase (EPO) (4), following decomposition of peroxynitrite (ONOO−) or autooxidation of nitric oxide (1), and likely is a significant contributor to inflammation-associated tissue damage (18, 61). Formation of 3-nitrotyrosine residues, which occurs in many inflammatory diseases, is currently viewed as a marker of inflammation and is likely attributable to the action of NO2 (2, 30, 61).
Despite the great potential of causing damage, NO2 is a poorly studied oxidant and the mechanisms by which it evokes cell death remain enigmatic. Inhalation studies employing NO2 in rodents have demonstrated injury to the pulmonary epithelium (46, 47), without elucidating specific targets of NO2 attack. Previously, we have shown that lung epithelial cells exposed to NO2 or ONOO− undergo cell death that is density dependent and selectively occurs at the leading edge of a wounded cell culture (51). The c-Jun N-terminal kinase (JNK) member of the family of mitogen-activated protein kinases, also known as stress-activated protein kinase (SAPK), is regulated in a density-dependent manner (34) and is known to be activated by environmental stresses (12, 29), including oxidative stress (22, 32, 50, 66).
JNK has been implicated in multiple physiological processes, including survival (35) and apoptosis (12), and the consequences of its action appear to depend upon the cell type or stimulus under investigation, the extent and duration of its activity, as well as the engagement of other signaling modules. A causal relationship between JNK activation and apoptosis was first established in neuronal cells after neurotrophic factor withdrawal (68) and was confirmed in mice with a targeted disruption of the neuronal gene, jnk3 (69) or mice containing a mutation in the c-jun gene that lacked the JNK phosphorylation sites (3). This observation initially suggested that transcriptional events were important in JNK-dependent apoptosis, and one candidate gene product was Fas ligand (20, 33). However, gene knockout studies demonstrated that caspase 8, the initiator caspase required for Fas-dependent cell death, was not required for UV-induced cell death (62), suggesting that alternative pathways regulate stress-induced JNK-dependent cell death. It is now well established that mitochondria can play an important role in JNK-dependent stress-induced apoptosis (38, 60, 62), via JNK-induced phosphorylation of the BH3-only proteins Bim and Bmf, and the subsequent release from dynein and myosin V motor complexes, thereby engaging the mitochondrial apoptotic pathway (37).
Activation of death receptors is known to mediate JNK activation (7, 57, 58, 70), which in some cases contributes to the apoptotic process (57, 58), although many controversies exist (7, 10, 26, 39, 59, 64, 70). Fas and tumor necrosis factor receptor 1 (TNF-R1) receptors share characteristic cysteine-rich repeats in their extracellular domains, and cytoplasmic death domains that are critical for signaling to apoptosis (9, 65). Upon Fas stimulation, a death-inducing signaling complex (DISC) is formed by the recruitment of Fas-associated death domain protein (FADD) and caspase 8 or 10 to the intracellular death domain, which results in autoproteolytic processing of the caspases that trigger the apoptotic cascade (65). FADD also interacts with TNFR-1 via an adapter protein, TNF receptor-activated death domain (TRADD), and is required for the induction of caspase 8-dependent apoptosis (9).
The goal of the present study was to investigate whether the activation of JNK also plays a role in cell death caused by the RNS, NO2, or ONOO−. Since membrane-localized death receptors represent likely targets for interaction with RNS, which are highly membrane reactive (23), we investigated the involvement of Fas and TNF-R1 in the molecular responses to these highly reactive species.
MATERIALS AND METHODS
Cell culture and reagents.
A murine alveolar type II epithelial cell line (C10) (41) was employed. C10 cells were propagated in CMRL 1066 supplemented with l-glutamine, penicillin-streptomycin, and 10% fetal bovine serum (GIBCO BRL). A line of spontaneously transformed rat alveolar type II epithelial cells (RLE) (16) was propagated in Dulbecco's modified Eagle's medium-F12 medium (DMEM/F12) containing penicillin-streptomycin, supplemented with 7% newborn bovine serum (GIBCO BRL). Fibroblasts were isolated from lungs of C57BL/6, TNF-R1−/− mice, or LPR mice, which lack functional Fas (Jackson Laboratories, Bar Harbor, Maine), by mincing lungs and propagation of explants as described previously (49). In selected experiments, cells were plated at various densities 16 h prior to exposure. One hour prior to exposure to RNS, growth medium was switched to phenol red-free DMEM/F12 containing 0.5% serum. Flag-JNK1-APF and hemagglutinin (HA)-tagged JNK1 were kindly provided by Roger Davis (University of Massachusetts, Worcester), and HA-Fas 1-210, HA-Fas 67-210, and TNF-R1 lacking the intracellular death domain (HA-P60ΔCD) were provided by Richard Siegel and Michael Lenardo (National Institutes of Health). FADD (80-205) was provided by Peter Vandenabeele (University of Ghent, Ghent, Belgium). As a positive control, cells were treated with FasL + M2 (recombinant Flag-tagged human FasL [Alexis Biochemicals; 100 ng/ml] plus anti-Flag antibody M2 [Sigma; 1 μg/ml]) in order to cross-link recombinant FasL. TNF-α was obtained from GIBCO, the generic caspase inhibitor zVAD-FMK was from Promega, Fas:Fc was from Alexis, anti-FasL antibody (MFL-4) was from Pharmingen, and a phospho-specific MKK4 antibody was purchased from Cell Signaling Technologies.
Exposure to RNS.
Exposure to NO2 was performed inside a modified, stainless steel cell culture cabinet as described in detail elsewhere (51). Briefly, NO2 was generated from a tank containing 1,000 ppm of NO2 in nitrogen (Messer MG Industries). An Ionics nitric oxide analyzer (NOA) equipped with an NO2 thermal converter or an Eco-Physics analyzer was used to measure NO2 in the gas phase according to the manufacturers' instructions. Cells were exposed to 10 ppm of NO2 for 4 h on orbital rotating platforms, allowing exposure to gas-phase NO2 in 50% of the culture dish at any given time. In order to maximize the stability of gas-phase exposures, which can be problematic over a time course of 4 h, selected experiments utilized 2-h exposure regimens of 40 ppm of NO2. Both exposure schemes caused marked cell death and JNK activation (data not shown). Air-exposed mock manipulations were performed by rocking cells on an orbital rotating platform under standard culture conditions. Pure ONOO− was obtained from Calbiochem. In control experiments, ONOO− was decomposed overnight at room temperature prior to exposure to cells (31). In general, each experiment contained two independent observations per treatment group, and experiments were repeated at least two times.
Transient and stable transfections.
C10 cells were transiently transfected with 2 μg of plasmids, using Lipofectamine plus (Invitrogen) according to the manufacturer's instructions. Flag-JNK1-APF was subcloned into pcDNA3.0 (Invitrogen). Stable cell pools for either pcDNA3.0 only or pcDNA3.0-Flag-JNK1-APF were generated by selection with neomycin (G418; GIBCO-BRL).
In vitro kinase assays and Western blotting.
Cell lysates were prepared from RLE or C10 cells of lung fibroblasts, as previously described (32). JNK1 was immunoprecipitated from 200 μg of protein sample using 1 μg of rabbit polyclonal antibodies (Santa Cruz Biotechnology). In some experiments, recombinant JNK1 was immunoprecipitated by using antibody directed against HA (Roche). Immunoprecipitated JNK1 was reacted in kinase buffer containing [γ-32P]ATP and the substrate GST-Jun at 30°C for 30 min. Samples were then electrophoresed on 15% polyacrylamide gels and dried, and substrate phosphorylation was visualized by autoradiography. For phospho-specific c-Jun Western blots, whole-cell extracts were prepared by lysing cells in a hypertonic lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 5 mM EDTA, pH 8.8, 1 mM phenylmethyl sulfonyl fluoride, 1 mM sodium orthovanadate) and 20 μg of total protein from each sample was used for Western blotting with a phospho-specific c-Jun antibody (KM-1; Santa Cruz) and detection via standard conditions.
Assessment of Fas clustering and oxidative modification.
Cells grown on glass coverslips were untreated or treated with ONOO− (500 μM, 10 min) or 100-ng/ml sFasL plus 1-μg/ml M2 for 15 min. Cells were subsequently fixed in 3% para-formaldehyde in phosphate-buffered saline (PBS) for 30 min, washed, and permeabilized with 0.1% Triton X-100 for 20 min. Nonspecific binding was blocked with a mixture of 0.5% normal goat and 0.5% normal rabbit sera for 1 h, and slides were then incubated overnight at 4°C with primary antibody to FADD (Santa-Cruz FADD S18) and Fas (Santa-Cruz Fas FL-335). After three washes with PBS, slides were incubated with secondary antibodies, Alexa-568 antigoat and Alexa-488 antirabbit (Molecular Probes). Slides were subsequently washed three times in PBS and once in double-distilled water prior to mounting. Colocalization was evaluated with a Bio-Rad MRC 1240 laser-scanning confocal microscope. Control experiments included single incubations as well as incubations with nonspecific primary and secondary antibodies alone and revealed minimal cross-reactivity or background fluorescence. In order to assess whether Fas is a direct target for oxidation by ONOO−, Fas was immunoprecipitated and tyrosine nitration was assessed by Western blotting using a nitrotyrosine-specific antibody (Upstate Biotechnology). To assess the extent of cysteine oxidation of Fas, cell lysates were prepared in the presence of 0.05 mM sulfhydryl-specific biotinylating agent, N-(3-malemidylpropionyl) biocytin (MPB; Molecular Probes) for 30 min at room temperature, resulting in biotinylation of reduced cysteines. Fas was immunoprecipitated, and the extent of biotinylation was determined by using streptavidin peroxidase.
Determination of cell death.
After selected time points of exposure, cells were washed in PBS and incubated with the mitochondrial stain JC-1 (Molecular Probes) in order to visualize respiring mitochondria (red). Loss of mitochondrial respiration results in a lack of JC-1 aggregation, which causes a green staining pattern (11). The nuclear morphology was assessed with Hoechst (Sigma). The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay (Molecular Probes) was used to assess overall viability by monitoring the production of formazan at 540 nm.
RESULTS
NO2 or ONOO− induced JNK activity in log-phase cells.
Previous work from our laboratory has shown that exposure of lung epithelial cells to NO2 or ONOO− causes density-dependent cell death (51). To evaluate whether the stress-activated protein kinase JNK, known to be regulated in a density-dependent manner (34), was activated by NO2, we exposed log-phase or confluent RLE cells to 10 ppm of NO2 for 4 h. JNK activity was determined immediately after cessation of exposure and at 4 h after cessation of exposure. Figure 1A, shows that log-phase cells exposed to NO2 displayed striking increases in JNK activity compared to air-exposed controls. Increases in JNK activity were apparent immediately after cessation of the 4-h exposure period and persisted for up to 4 h. JNK activation corresponded with phosphorylation of its target, c-Jun. No increases in JNK activity or c-Jun phosphorylation were observed in confluent cultures exposed to NO2 (Fig. 1A), consistent with the lack of cell death under these conditions (51). Similarly, the highly reactive RNS, ONOO−, which generates NO2 (1), also caused a dose-dependent activation of JNK in C10 cells, which was first apparent at 15 min and persisted for at least 2 h (data not shown). When ONOO− was decomposed prior to its addition to cells, it failed to activate JNK (Fig. 1B). Analogous to our observations with NO2, ONOO−-induced cell death in C10 lung epithelial cells, which was almost completely prevented by pretreatment with the antioxidants, N-acetyl-l-cysteine, or ascorbate (Fig. 1C), confirming that RNS-induced cell death occurs though an oxidative mechanism.
FIG. 1.

RNS-induced JNK activation in lung epithelial cells. (A) Log-phase or confluent cultures of RLE cells were exposed to 10 ppm of NO2 for 4 h. Extracts prepared at different times were examined either for JNK activation via an in vitro kinase assay (upper panel) or phosphorylation of c-Jun (lower panel) by Western blotting (immunoblotting [IB]). (B) Log-phase cultures of C10 cells were exposed to different concentrations of ONOO− for 2 h, and JNK activity was evaluated. Decomposed ONOO− (Decomp) was used as a reagent control. (C) C10 cells were preincubated with N-acetyl-l-cysteine (NAC; pH 7.4) or ascorbate (Asc) at the indicated concentrations for 15 min prior to exposure to 200 μM ONOO− for 2 h. Cells were harvested for assessment of viability with the MTT assay. Results are expressed as percent viability of sham controls. *, P < 0.05 (analysis of variance) compared to ONOO−-treated cells; #, P < 0.05 (analysis of variance) compared to ONOO−-treated cells, but not different from sham controls.
JNK activation mediates death by RNS.
In order to determine the causal involvement of JNK activation in cell death induced by RNS, we created pools of C10 cells that stably expressed Flag-JNK1-APF (15), a dominant-negative mutant form of JNK1. As shown in Fig. 2A, the stable expression of JNK1-APF prevented JNK activation and c-Jun phosphorylation in cells treated with ONOO−. Moreover, ONOO−-induced mitochondrial depolarization (Fig. 2B) and nuclear condensation (Fig. 2C) were markedly attenuated in JNK-APF-expressing cells compared to vector controls, illustrating the causal role for JNK in these features of cell death. The activation of peroxidases such as EPO can also generate high concentrations to NO2. We recently demonstrated that NO2 generated from EPO triggers JNK-dependent cell death (44) that involves mitochondrial depolarization, and cytochome c release. Results in Fig. 2D demonstrate that NO2 generated by catalytically active EPO causes release of apoptosis-inducing factor (AIF) from the mitochondria and promotes its nuclear localization, illustrating the pathophysiological relevance of our observations. As a complementary approach to overexpression strategies, we isolated lung fibroblasts derived from JNK1−/− mice. Results in Fig. 2E demonstrate that fibroblasts lacking JNK1 also were partially protected from ONOO−-induced cell death, compared to wild-type cells. Collectively, these data demonstrate that JNK activation plays a prominent role in death caused by RNS.
FIG. 2.
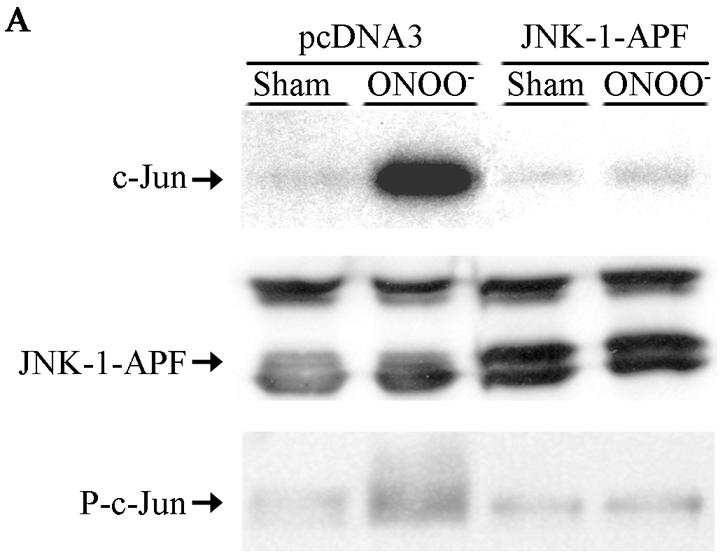




Expression of JNK1-APF inhibits RNS-induced cell death. (A) Assessment of JNK activation and c-Jun phosphorylation in pcDNA3 or JNK1-APF transfected cell pools. Cells were treated with 200 μM ONOO− for 2 h for assessment of JNK activation in an in vitro kinase assay (top) or c-Jun phosphorylation by Western blotting (bottom). (Middle panel) Western blotting for JNK1. The expression of JNK1-APF is indicated. pcDNA3 or JNK1-APF cells grown on glass coverslips were treated with 200 μM ONOO− for the indicated times for the assessment of mitochondrial membrane potential (B) or nuclear condensation (C). Mitochondrial membrane potential was determined by incubation with 5-μg/ml JC-1. Polarized mitochondria are visualized by punctate red staining, whereas depolarized mitochondria are visualized by their diffuse green staining. Cells were counted in five random fields, and data are expressed as a percentage of polarized cells. Nuclear condensation was determined after staining of cells with 5-μg/ml Hoechst. Cells were counted in five random fields, and data are expressed as a percentage of cells containing intact nuclei. Results are expressed as means ± standard error. *, P < 0.05 compared to sham controls; #, P < 0.05 compared to pcDNA3 vector controls (analysis of variance). (D) Assessment of AIF nuclear localization in cells exposed to 50 nM EPO in the presence of 15-mU/ml glucose oxidase (GO) and 100 μM sodium nitrite (NO2−) for 4 h. Nuclear presence of AIF (green) was confirmed via confocal microscopy and colocalization with propidium iodide (blue). (E) Assessment of viability of C57BL/6 or JNK1−/− lung fibroblasts exposed to 200 μM ONOO−, using the MTT assay. Data are expressed as percent survival compared to sham controls. *, P < 0.05 (analysis of variance) compared to C57BL/6-derived fibroblasts exposed to ONOO− for 1 h; #, P < 0.05 (analysis of variance) compared to C57BL/6-derived fibroblasts exposed to ONOO− for 2 h.
Fas, but not TNF-R1, is required for the activation of JNK by RNS.
ONOO− and NO2 are highly reactive oxidants that likely affect surface proteins. Since receptors of the TNF-R family, known to signal to JNK, contain multiple cysteines and tyrosines (54), which may be targets for oxidation, we questioned whether Fas or TNF-R1 could play a role in the activation of JNK by RNS. To address this, we transfected cells with the extracellular domains of Fas or TNFR-1, that lack the intracellular death domains necessary for adaptor protein recruitment (6, 55) and evaluated JNK activation. Results in Fig. 3A demonstrate that in cells expressing the extracellular domain of Fas (Fas 1-210), the ability of ONOO− to activate JNK was completely abrogated. As a control, treatment with FasL + M2, which engages Fas, caused activation of JNK, which was also prevented by the expression of the extracellular domain of Fas (Fig. 3A). Furthermore, expression of Fas 1-210 attenuated phosphorylation of MKK4 (Fig. 3B), an upstream activator of JNK. In contrast, cells expressing the extracellular domain of TNF-R1 (p60ΔCD) were fully capable of activating JNK in response to treatment with ONOO−, whereas the ability of TNF-α to activate JNK was blunted (Fig. 3C). Similarly, NO2-induced JNK activation was prevented in cells expressing extracellular domain of Fas but not TNF-R1 (Fig. 3D). In support of the observations in Fig. 3A to D, fibroblasts generated from LPR mice were completely refractory from ONOO−-induced JNK activation, whereas fibroblasts from mice lacking TNF-R1 were normally responsive to ONOO−-dependent activation of JNK (Fig. 3E). Lastly, LPR fibroblasts also were partially protected from ONOO−-induced cell death (Fig. 3F). Collectively, these data clearly demonstrate that Fas, but not TNF-R1 is an important target for JNK activation by RNS and, consequently, cell death.
FIG. 3.


RNS-induced JNK activation occurs in Fas-dependent but TNF-R1-independent manner. (A) C10 cells were transfected with pcDNA3 or Fas 1-210 in the presence of HA-JNK1 (0.4 μg) and then exposed to 200 μM ONOO− for 2 h or 1-μg/ml M2 plus 100-ng/ml FasL (30 min). HA-JNK was immunoprecipitated, and JNK activity was assayed by in vitro kinase assay. Levels of HA-JNK1 (bottom) are included as a loading control. (B) Cells were transfected with pcDNA3 or Fas 1-210, treated with agents as described in panel A, prior to assessment of phospho-MKK4 (top) by Western blotting. (Bottom) β-Actin expression. (C) C10 cells were transfected with pcDNA3 or p60ΔCD (1.6 μg) in the presence of HA-JNK1 (0.4 μg) and then exposed to ONOO− as described above or to 10-ng/ml TNF-α (15 min) prior to assessment of JNK activity. Levels of HA-JNK1 are included as a loading control. (D) C10 cells were transfected with pcDNA3, Fas 1-210, or p60ΔCD (1.6 μg) in the presence of HA-JNK1 (0.4 μg) and then exposed to 40 ppm of NO2 for 2 h for assessment of JNK activity. Levels of HA-JNK1 are included as a loading control. (E) Lung fibroblasts derived from wild-type C57BL/6, LPR, or TNF-R1−/− mice were exposed to ONOO−, FasL + M2, or TNF-α as described above, and JNK activity was assessed in an in vitro kinase assay (top). The bottom panel represents a portion of a JNK1 Western blot as a loading control. (F) Assessment of viability of C57BL/6 or LPR lung fibroblasts exposed to 200 μM ONOO− for 2 h, using the MTT assay. Data are expressed as percent survival compared to sham controls. *, P < 0.05 (Student's t test) compared to C57BL/6-derived ONOO−-exposed fibroblasts.
RNS-induced activation of JNK does not require new protein synthesis and occurs independently of FasL.
We next addressed whether the Fas-dependent activation of JNK by ONOO− requires Fas Ligand. Incubation with the protein synthesis inhibitor cycloheximide did not alter the activation of JNK by ONOO−, illustrating that new protein synthesis was not required for JNK activation (Fig. 4A). Moreover, anti-FasL antibody, or Fas-Fc, did not interfere with the ability of ONOO− to activate JNK, whereas these approaches inhibited M2 + FasL-induced JNK activation, as expected (Fig. 4B and 4C). These results strongly suggest that ONOO−-induced JNK activation occurs in a Fas-dependent manner which does not require FasL.
FIG. 4.

RNS-mediated activation of JNK does not require new protein synthesis and occurs in a FasL-independent manner. C10 cells were exposed to 200 μM ONOO− for 15 min and then chased for 2 h in the presence or absence of 10 μM CHX (A) or with 10-μg/ml anti-FasL antibody (B) or 250-ng/ml Fas-Fc (C). JNK activity was assayed via an in vitro kinase assay. In control experiments, cells were treated with 1-μg/ml M2 plus 100-ng/ml FasL (30 min) in the presence of either anti-FasL antibody (B) or Fas-Fc (C).
RNS-induced activation of JNK is FADD dependent but caspase independent.
Engagement of Fas induces recruitment of the adaptor molecule FADD, which in turn recruits caspase 8 and promotes its activation, causing apoptosis. Next, we examined the role of FADD and caspases in RNS-induced JNK activation. Expression of dominant-negative FADD (80-205) lacking the death effector domain required for recruitment and activation of caspases, inhibited the ability of ONOO−, NO2, or cross-linked FasL to cause JNK activation (Fig. 5A and B). Incubation of cells with the generic caspase inhibitor zVAD or the caspase 8 inhibitor IETD inhibited JNK activation (Fig. 5C) and cell death by cross-linked FasL (Fig. 5D). In contrast, ONOO−-induced JNK activation or cell death was not attenuated in the presence of these caspase inhibitors (Fig. 5C and D). These data demonstrate that JNK activation, and the subsequent death of cells exposed to RNS, occurs in a Fas- and FADD-dependent, but caspase-independent manner.
FIG. 5.

RNS-dependent activation of JNK occurs in FADD-dependent but caspase-independent manner. C10 cells were transfected with either pcDNA3 or FADD (80-205) (1.6 μg) in the presence of HA-JNK1 (0.4 μg) and then exposed to 200 μM ONOO− for 2 h (A) or 40 ppm of NO2 for 2 h (B). HA-JNK was immunoprecipitated, and JNK activity was assayed as described before. (C) C10 cells pretreated with 10 μM zVAD or IETD (30 min) were exposed to 200 μM ONOO− (2 h) or 1-μg/ml M2 plus 100-ng/ml FasL (30 min) prior to assessment of JNK activity. (D) ONOO−-induced cell death is caspase independent. C10 cells were pretreated with 10 μM zVAD or IETD (30 min) prior to exposure to 200 μM ONOO− (2 h) or 1-μg/ml M2 plus 100-ng/ml FasL (1 h) prior to assessment of cell death by using the MTT assay.
RNS cause oxidative modification and aggregation of Fas.
Aggregation of Fas is important in the formation of the DISC and subsequent cell death (52). We next assessed whether ONOO− is capable of aggregating Fas and FADD, an essential component of the DISC. Results in Fig. 6A demonstrate that FasL + M2 or ONOO− caused a redistribution of Fas and FADD in C10 cells into punctate clusters. Merging of confocal images revealed a marked colocalization of Fas and FADD, suggestive of formation of the DISC, in cells treated with cross-linked FasL or ONOO−.
FIG. 6.

RNS cause aggregation and oxidative modification of Fas. (A) Cells grown on glass coverslips were treated with 200 μM ONOO− or 1-μg/ml M2 plus 100-ng/ml FasL for 5 min, subsequently stained with anti-Fas or anti-FADD antibodies followed by fluorophore-conjugated secondary antibodies. Fas (green), FADD (red), and their colocalization (yellow) were examined by confocal microscopy. (B) C10 cells were treated with either 200 μM ONOO− or M2 + FasL for 5 min, lysed in buffer containing 50 μM of MPB. Fas was immunoprecipitated (IP), and the extent of biotinylation was determined by using streptavidin peroxidase by Western blotting (immunoblotting [IB]). (C) C10 cells were treated with different concentrations of ONOO− for indicated times. Fas was immunoprecipitated, and its nitrotyrosine content was evaluated by Western blotting. Fas monomer is indicated by the arrow, whereas higher-molecular-weight complexes are indicated by the bracket.
The cysteine-rich repeats in the extracellular domain of Fas may be a target of oxidation by ONOO−, leading to the formation of mixed disulfides, or other oxidation intermediates. To assess the extent of cysteine oxidation of Fas, we prepared cell lysates from cells treated with ONOO− or FasL + M2 in the presence of the biotin-conjugated, sulfhydryl-specific alkylating reagent MPB. Fas was then immunoprecipitated, and the extent of its biotinylation was determined by using streptavidin peroxidase. Results in Fig. 6B demonstrate a loss of biotinylation in response to ONOO−, but not FasL + M2, suggesting that measurable cysteine oxidation of Fas occurred after treatment with ONOO−, but not FasL + M2. Finally, we assessed tyrosine nitration of Fas, which is considered a specific footprint for exposure to nitrating species (61). Fas was immunoprecipitated, and its nitrotyrosine content was assessed by Western blotting. Results in Fig. 6C demonstrate that tyrosine nitration of Fas occurred in cells exposed to ONOO−. Collectively, these results demonstrate that Fas is a direct target for oxidative modification by RNS.
RNS-induced JNK activation requires CRD2 and CRD3, but not CRD1.
FasL-Fas receptor interaction are believed to occur predominantly via the cysteine-rich domains (CRDs) in the extracellular regions of these receptors (56). In the absence of membrane-bound ligand, inactive complexes of Fas are formed via the self-association domain in CRD1, termed the preligand assembly domain (PLAD) (55), which is required for apoptosis signaling. To explore whether this domain is also required to engage ONOO− to cause JNK activation, we expressed a truncated version of Fas lacking both the PLAD and death domains (Fas 67-210; Fig. 7A), and evaluated JNK activation by ONOO−. As shown before (Fig. 3), cells expressing extracellular domain of Fas with a truncated death domain (positions 1 to 210) were completely resistant to JNK activation by ONOO− (Fig. 7C). Cells expressing Fas 67-210, were also largely refractory to JNK activation by ONOO−, illustrating that dominant interference with signaling still occurs in the absence of CRD1. In contrast, JNK activation by FasL + M2 was partially restored in cells expressing Fas 67-210, in line with the requirement of CRD1 in FasL-induced cell death (55). These data demonstrate the requirement of CRD2 and CRD3, rather than CRD1, for ONOO−-induced JNK activation and illustrate the lack of requirement of the PLAD for interaction with RNS.
FIG. 7.

ONOO− requires CRD2 and/or CRD3 but not CRD1 of Fas for signaling to JNK. (A) Schematic representation of Fas receptor. (B) C10 cells were transfected with 2 μg of the indicated constructs, and expression was confirmed by probing Western blots (immunoblots [IB]) with antibody directed against HA. Arrows represent the expected proteins. ns, nonspecific reactivity. (C) C10 cells were transfected with either pcDNA3, Fas 1-210, or Fas 67-210 in the presence of HA-JNK1 and then exposed to ONOO− or FasL + M2 for assessment of JNK activity, as described before. Levels of HA-JNK1 are included as a loading control.
DISCUSSION
Interest in the oxidant NO2 was renewed after it was determined that this highly reactive free radical gas is formed during inflammation (4). Furthermore, this species is the most likely culprit in attacking tyrosine residues to cause the stable posttranslational modification, 3-nitrotyrosine, which is present under many inflammatory conditions (1). Not surprisingly, investigations into the mechanisms by which NO2 injures cells are scant, due to the highly unstable nature of this gas, which makes cell-based exposures challenging. Previous work in our laboratory demonstrated that NO2 preferentially kills migrating or dividing cells (51). We report here that the activation of JNK is responsible for cell death and that the death receptor, Fas, is attacked by nitrating species and signals to JNK.
JNK has been shown to be important in cell death after exposure to oxidants, such as γ- or UV-C irradiation (5, 10) or H2O2 (45, 66). Here, we demonstrate that RNS are potent activators of JNK and that JNK is causally involved in cell death, measured by mitochondrial depolarization and nuclear condensation, which were almost completely prevented in cells expressing JNK1-APF and attenuated in cells lacking JNK1. Furthermore, we also demonstrate that RNS require Fas, but not TNF-R1, to activate JNK and that Fas is a direct target for RNS-induced oxidation and membrane relocalization. Surprisingly, RNS-induced Fas signaling to JNK occurred in a caspase- and FasL-independent manner, but required FADD. While engagement of TNF-R1 is well known to cause JNK activation via recruitment of TRADD and TRAF-2 (9, 28) and that JNK activation controls apoptosis via TNF-R1 (57, 58), the link between Fas ligation, JNK activation, and apoptosis remains a topic of considerable debate (7, 10, 26, 39, 59, 64, 70). Fas was reported to engage JNK via the Daxx-dependent activation of apoptosis signal-regulating kinase 1 (ASK1) (7, 70). However, this finding has been brought into question by other investigators (26, 59, 64). Furthermore, JNK activation by Fas ligation has been demonstrated to occur as a result of caspase-dependent activation of the JNK kinase kinase, mitogen-activated protein kinase/Erk kinase kinase 1 (MEKK1) (13), and consequently, caspase inhibitors attenuated FasL-induced JNK activation (26, 39, 64), suggesting that JNK activation following Fas ligation may be the consequence of activation of the apoptotic machinery, rather than the effector. Our results strongly suggest that Fas can directly signal to JNK in cells exposed to RNS, pointing to a unique FasL-independent mechanism of activation of Fas by RNS. These observations distinguish RNS from other stresses that cause JNK-dependent apoptosis, which required transcriptional activation of FasL (17, 19, 20, 40). Although we have not ruled out that transcriptional events do not contribute to RNS-induced JNK-dependent cell death, the relatively short time frame after which cell death becomes apparent (Fig. 2) (data not shown) and the JNK-dependent mitochondrial perturbations strongly suggest that a mitochondrial-dependent pathway (37, 38, 60, 62) rather than JNK-dependent activation of death effector genes is responsible for RNS-induced cell death.
The lack of caspase involvement in JNK activation or cell death by RNS is not surprising given the fact that RNS are strong cysteine oxidizers (1) (Fig. 6) and that caspases contain a critical cysteine in their catalytic domain, required for activation. Compelling data exist demonstrating that nitrosylation of caspases represses their activity (42) and that the oxidant hydrogen peroxide (H2O2) can interfere with the caspase activation process (36). Furthermore, the activation of caspase 8 in the DISC is dependent on glutathione (25), which is depleted by RNS (44). A mixed mode of cell death with features of necrosis has also been reported in cells exposed to hyperoxia, although this was protracted and associated with recruitment of caspase 8 to Fas (67). Studies demonstrating that Fas ligation can lead to necrotic cell death when caspases are inhibited, the requirement of FADD herein, the loss of mitochondrial membrane potential, and the production of reactive oxygen species (14, 21, 43, 63) are in line with our present observations. Although we demonstrated a requirement of FADD in the Fas-dependent JNK activation by RNS, the subsequent biochemical events leading to JNK activation remain elusive, but may involve MKK4 (Fig. 3B). It is tempting to speculate that the kinase activity of the serine/threonine kinase, receptor-interacting protein (RIP), which binds to the death domain of Fas and is required for necrotic signaling by Fas (24, 27), also could be required for signaling by RNS. The molecular mechanisms that connect FADD and RIP to the execution of necrotic cell death and the involvement of JNK herein remain to be elucidated. Nonetheless, our present observations demonstrating that JNK activation by RNS was inhibited by a FADD mutant lacking the death effector domain and was caspase independent strongly suggest a similar mode of non-caspase-dependent cell death through Fas, although additional experiments will be required to test this possibility formally.
Our observations demonstrating that RNS require Fas, but not TNF-R1, to signal to JNK are intriguing, given the high degree of homology between these proteins. We recently demonstrated that the oxidant H2O2 utilizes TNF-R1 to signal to JNK (49). Similarly, JNK activation by H2O2 was also attenuated in cells expressing Fas 1-210 (data not shown), illustrating that JNK activation by H2O2 requires Fas and TNF-R1, whereas RNS preferentially target Fas. This difference may stem from the unique reactivity of RNS, compared to H2O2, and/or the location of the oxidative attack. We demonstrate here that Fas is indeed directly oxidized by RNS, illustrating that this receptor is a direct target. The extracellular domains of death receptors, including Fas and TNF-R1, contain multiple CRDs. Fas contains three CRDs, whereas TNFR-1 has four CRDs. The amino acid residues in CRD2 and CRD3 of Fas are conserved in murine and human Fas, but not in TNF-R1 (56). Furthermore, CRD1 but not CRD2 or CRD3 of Fas can be exchanged for the homologous domain found in TNFR-1 for Fas ligand binding (48). These observations, coupled with our findings that Fas but not TNFR-1 is required for RNS-induced activation of JNK and that CRD2 and CRD3 of the extracellular domain of Fas receptor are the required domain to sustain JNK activation by RNS, may explain the specificity of RNS for its interaction with Fas rather than TNFR-1. Finally, our observation demonstrating that CRD1 of Fas, which is required for formation of preassembled Fas complexes, interaction with FasL, and downstream signaling (55), is not required for JNK activation by RNS suggests that RNS interacts with Fas in a manner distinct from FasL. It is plausible that cysteine or other oxidation events in CRD2 and/or CRD3 are responsible for RNS-induced aggregation of Fas (Fig. 6), although the exact sites and modes of oxidation are unknown at this time. Tyrosine nitration of Fas was recently described in rat livers following administration of lipopolysaccharide, illustrating that RNS directly target Fas under inflammatory conditions in vivo. However, the same report demonstrated that although ONOO− directly targeted Fas, it prevented FasL-induced DISC formation and subsequent apoptosis independently of JNK (53). The discrepancy between these findings and our work is puzzling and may be related to the requirement of epidermal growth factor receptor association with Fas in hepatocytes and its subsequent tyrosine phosphorylation, which was prevented by ONOO− (53). While that study demonstrated that ONOO− also caused apoptosis in hepatocytes, the contribution of Fas therein was not conclusively elucidated. Nonetheless, their findings and the findings from our study demonstrate that Fas is an important target of RNS.
In summary, we have demonstrated here that RNS directly attack Fas and consequently trigger JNK-dependent cell death. Further investigation into the precise location of oxidative attack by RNS and confirmation that this occurs in intact tissues will be important in designing strategies to limit death signaling by RNS, which might be beneficial in combating the tissue-damaging effects of RNS that accompany chronic inflammatory diseases.
Acknowledgments
We thank Michael Lenardo, Brian Seed, Roger Davis, and Peter Vandenabeele for providing the various plasmid constructs and Stan Hazen (Cleveland Clinic Foundation) for providing eosinophil peroxidase. We also thank Albert van der Vliet, Charles Irvin, Brooke Mossman, and Nicholas Heintz for scientific suggestions; Pamela Vacek for statistical analyses; and Beth Langford-Corrigan for editorial assistance.
This work was supported by the Public Health Service: P20 RL15557 (NCRR COBRE), NIH RO1 HL60014, and PO1 HL67004.
REFERENCES
- 1.Augusto, O., M. G. Bonini, A. M. Amanso, E. Linares, C. C. Santos, and S. L. De Menezes. 2002. Nitrogen dioxide and carbonate radical anion: two emerging radicals in biology. Free Radic. Biol. Med. 32:841-859. [DOI] [PubMed] [Google Scholar]
- 2.Aulak, K. S., M. Miyagi, L. Yan, K. A. West, D. Massillon, J. W. Crabb, and D. J. Stuehr. 2001. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc. Natl. Acad. Sci. USA 98:12056-12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behrens, A., M. Sibilia, and E. F. Wagner. 1999. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 21:326-329. [DOI] [PubMed] [Google Scholar]
- 4.Brennan, M. L., W. Wu, X. Fu, Z. Shen, W. Song, H. Frost, C. Vadseth, L. Narine, E. Lenkiewicz, M. T. Borchers, A. J. Lusis, J. J. Lee, N. A. Lee, H. M. Abu-Soud, H. Ischiropoulos, and S. L. Hazen. 2002. A Tale of Two Controversies. Defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J. Biol. Chem. 277:17415-17427. [DOI] [PubMed] [Google Scholar]
- 5.Butterfield, L., B. Storey, L. Maas, and L. E. Heasley. 1997. c-Jun NH2-terminal kinase regulation of the apoptotic response of small cell lung cancer cells to ultraviolet radiation. J. Biol. Chem. 272:10110-10116. [DOI] [PubMed] [Google Scholar]
- 6.Chan, F. K., H. J. Chun, L. Zheng, R. M. Siegel, K. L. Bui, and M. J. Lenardo. 2000. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288:2351-2354. [DOI] [PubMed] [Google Scholar]
- 7.Chang, H. Y., H. Nishitoh, X. Yang, H. Ichijo, and D. Baltimore. 1998. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 281:1860-1863. [DOI] [PubMed] [Google Scholar]
- 8.Chauhan, A. J., M. T. Krishna, A. J. Frew, and S. T. Holgate. 1998. Exposure to nitrogen dioxide (NO2) and respiratory disease risk. Rev. Environ. Health 13:73-90. [PubMed] [Google Scholar]
- 9.Chen, G., and D. V. Goeddel. 2002. TNF-R1 signaling: a beautiful pathway. Science 296:1634-1635. [DOI] [PubMed] [Google Scholar]
- 10.Chen, Y. R., X. Wang, D. Templeton, R. J. Davis, and T. H. Tan. 1996. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J. Biol. Chem. 271:31929-31936. [DOI] [PubMed] [Google Scholar]
- 11.Cossarizza, A., M. Baccarani-Contri, G. Kalashnikova, and C. Franceschi. 1993. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem. Biophys. Res. Commun. 197:40-45. [DOI] [PubMed] [Google Scholar]
- 12.Davis, R. J. 2000. Signal transduction by the JNK group of MAP kinases. Cell 103:239-252. [DOI] [PubMed] [Google Scholar]
- 13.Deak, J. C., J. V. Cross, M. Lewis, Y. Qian, L. A. Parrott, C. W. Distelhorst, and D. J. Templeton. 1998. Fas-induced proteolytic activation and intracellular redistribution of the stress-signaling kinase MEKK1. Proc. Natl. Acad. Sci. USA 95:5595-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denecker, G., D. Vercammen, M. Steemans, T. Vanden Berghe, G. Brouckaert, G. Van Loo, B. Zhivotovsky, W. Fiers, J. Grooten, W. Declercq, and P. Vandenabeele. 2001. Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ. 8:829-840. [DOI] [PubMed] [Google Scholar]
- 15.Derijard, B., M. Hibi, I. H. Wu, T. Barrett, B. Su, T. Deng, M. Karin, and R. J. Davis. 1994. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76:1025-1037. [DOI] [PubMed] [Google Scholar]
- 16.Driscoll, K. E., J. M. Carter, P. T. Iype, H. L. Kumari, L. L. Crosby, M. J. Aardema, R. J. Isfort, D. Cody, M. H. Chestnut, J. L. Burns, et al. 1995. Establishment of immortalized alveolar type II epithelial cell lines from adult rats. In Vitro Cell Dev. Biol. Anim. 31:516-527. [DOI] [PubMed] [Google Scholar]
- 17.Eichhorst, S. T., M. Müller, M. Li-Weber, H. Schulze-Bergkamen, P. Angel, and P. H. Krammer. 2000. A novel AP-1 element in the CD95 ligand promoter is required for induction of apoptosis in hepatocellular carcinoma cells upon treatment with anticancer drugs. Mol. Cell. Biol. 20:7826-7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eiserich, J. P., M. Hristova, C. E. Cross, A. D. Jones, B. A. Freeman, B. Halliwell, and A. van der Vliet. 1998. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature 391:393-397. [DOI] [PubMed] [Google Scholar]
- 19.Faris, M., N. Kokot, K. Latinis, S. Kasibhatla, D. R. Green, G. A. Koretzky, and A. Nel. 1998. The c-Jun N-terminal kinase cascade plays a role in stress-induced apoptosis in Jurkat cells by up-regulating Fas ligand expression. J. Immunol. 160:134-144. [PubMed] [Google Scholar]
- 20.Faris, M., K. M. Latinis, S. J. Kempiak, G. A. Koretzky, and A. Nel. 1998. Stress-induced Fas ligand expression in T cells is mediated through a MEK kinase 1-regulated response element in the Fas ligand promoter. Mol. Cell. Biol. 18:5414-5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiers, W., R. Beyaert, W. Declercq, and P. Vandenabeele. 1999. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene 18:7719-7730. [DOI] [PubMed] [Google Scholar]
- 22.Go, Y. M., R. P. Patel, M. C. Maland, H. Park, J. S. Beckman, V. M. Darley-Usmar, and H. Jo. 1999. Evidence for peroxynitrite as a signaling molecule in flow-dependent activation of c-Jun NH(2)-terminal kinase. Am. J. Physiol. 277:H1647-H1653. [DOI] [PubMed] [Google Scholar]
- 23.Goss, S. P., R. J. Singh, N. Hogg, and B. Kalyanaraman. 1999. Reactions of *NO, *NO2 and peroxynitrite in membranes: physiological implications. Free Radic. Res. 31:597-606. [DOI] [PubMed] [Google Scholar]
- 24.Grimm, S., B. Z. Stanger, and P. Leder. 1996. RIP and FADD: two “death domain”-containing proteins can induce apoptosis by convergent, but dissociable, pathways. Proc. Natl. Acad. Sci. USA 93:10923-10927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hentze, H., I. Schmitz, M. Latta, A. Krueger, P. H. Krammer, and A. Wendel. 2002. Glutathione dependence of caspase-8 activation at the death-inducing signaling complex. J. Biol. Chem. 277:5588-5595. [DOI] [PubMed] [Google Scholar]
- 26.Hofmann, T. G., A. Moller, S. P. Hehner, D. Welsch, W. Droge, and M. L. Schmitz. 2001. CD95-induced JNK activation signals are transmitted by the death-inducing signaling complex (DISC), but not by Daxx. Int. J. Cancer 93:185-191. [DOI] [PubMed] [Google Scholar]
- 27.Holler, N., R. Zaru, O. Micheau, M. Thome, A. Attinger, S. Valitutti, J. L. Bodmer, P. Schneider, B. Seed, and J. Tschopp. 2000. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1:489-495. [DOI] [PubMed] [Google Scholar]
- 28.Hsu, H., H. B. Shu, M. G. Pan, and D. V. Goeddel. 1996. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84:299-308. [DOI] [PubMed] [Google Scholar]
- 29.Ip, Y. T., and R. J. Davis. 1998. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell Biol. 10:205-219. [DOI] [PubMed] [Google Scholar]
- 30.Ischiropoulos, H., and J. S. Beckman. 2003. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J. Clin. Investig. 111:163-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janssen, Y. M., S. Matalon, and B. T. Mossman. 1997. Differential induction of c-fos, c-jun, and apoptosis in lung epithelial cells exposed to ROS or RNS. Am. J. Physiol. 273:L789-L796. [DOI] [PubMed] [Google Scholar]
- 32.Janssen-Heininger, Y. M., I. Macara, and B. T. Mossman. 1999. Cooperativity between oxidants and tumor necrosis factor in the activation of nuclear factor (NF)-kappaB: requirement of Ras/mitogen-activated protein kinases in the activation of NF-kappaB by oxidants. Am. J. Respir. Cell Mol. Biol. 20:942-952. [DOI] [PubMed] [Google Scholar]
- 33.Kasibhatla, S., T. Brunner, L. Genestier, F. Echeverri, A. Mahboubi, and D. R. Green. 1998. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol. Cell 1:543-551. [DOI] [PubMed] [Google Scholar]
- 34.Lallemand, D., J. Ham, S. Garbay, L. Bakiri, F. Traincard, O. Jeannequin, C. M. Pfarr, and M. Yaniv. 1998. Stress-activated protein kinases are negatively regulated by cell density. EMBO J. 17:5615-5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamb, J. A., J. J. Ventura, P. Hess, R. A. Flavell, and R. J. Davis. 2003. JunD mediates survival signaling by the JNK signal transduction pathway. Mol. Cell 11:1479-1489. [DOI] [PubMed] [Google Scholar]
- 36.Lee, Y. J., and E. Shacter. 2000. Hydrogen peroxide inhibits activation, not activity, of cellular caspase-3 in vivo. Free Radic. Biol. Med. 29:684-692. [DOI] [PubMed] [Google Scholar]
- 37.Lei, K., and R. J. Davis. 2003. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 100:2432-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lei, K., A. Nimnual, W.-X. Zong, N. J. Kennedy, R. A. Flavell, C. B. Thompson, D. Bar-Sagi, and R. J. Davis. 2002. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH2-terminal kinase. Mol. Cell. Biol. 22:4929-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lenczowski, J. M., L. Dominguez, A. M. Eder, L. B. King, C. M. Zacharchuk, and J. D. Ashwell. 1997. Lack of a role for Jun kinase and AP-1 in Fas-induced apoptosis. Mol. Cell. Biol. 17:170-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le-Niculescu, H., E. Bonfoco, Y. Kasuya, F.-X. Claret, D. R. Green, and M. Karin. 1999. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol. Cell. Biol. 19:751-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malkinson, A. M., L. D. Dwyer-Nield, P. L. Rice, and D. Dinsdale. 1997. Mouse lung epithelial cell lines—tools for the study of differentiation and the neoplastic phenotype. Toxicology 123:53-100. [DOI] [PubMed] [Google Scholar]
- 42.Mannick, J. B., A. Hausladen, L. Liu, D. T. Hess, M. Zeng, Q. X. Miao, L. S. Kane, A. J. Gow, and J. S. Stamler. 1999. Fas-induced caspase denitrosylation. Science 284:651-654. [DOI] [PubMed] [Google Scholar]
- 43.Matsumura, H., Y. Shimizu, Y. Ohsawa, A. Kawahara, Y. Uchiyama, and S. Nagata. 2000. Necrotic death pathway in Fas receptor signaling. J. Cell Biol. 151:1247-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McElhinney, B., M. E. Poynter, P. Shrivastava, S. L. Hazen, and Y. M. Janssen-Heininger. 2003. Eosinophil peroxidase catalyzes JNK mediated membrane blebbing in a Rho kinase dependent manner. J. Leukoc. Biol. 74:897-907. [DOI] [PubMed] [Google Scholar]
- 45.Minamino, T., T. Yujiri, P. J. Papst, E. D. Chan, G. L. Johnson, and N. Terada. 1999. MEKK1 suppresses oxidative stress-induced apoptosis of embryonic stem cell-derived cardiac myocytes. Proc. Natl. Acad. Sci. USA 96:15127-15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moldeus, P. 1993. Toxicity induced by nitrogen dioxide in experimental animals and isolated cell systems. Scand. J. Work Environ. Health 19:28-36. [PubMed] [Google Scholar]
- 47.Mustafa, M. G. 1994. Health effects and toxicology of ozone and nitrogen dioxide, p. 351-404. In J. O. Nrigau and M. S. Simmons (ed.), Environmental oxidants, vol. 28. John Wiley & Sons, Inc., New York, N.Y. [Google Scholar]
- 48.Orlinick, J. R., A. Vaishnaw, K. B. Elkon, and M. V. Chao. 1997. Requirement of cysteine-rich repeats of the Fas receptor for binding by the Fas ligand. J. Biol. Chem. 272:28889-28894. [DOI] [PubMed] [Google Scholar]
- 49.Pantano, C., P. Shrivastava, B. McElhinney, and Y. M. Janssen-Heininger. 2003. Hydrogen peroxide signaling through tumor necrosis factor receptor 1 leads to selective activation of C-JUN-N-terminal kinase. J. Biol. Chem. 278:44091-44096. [DOI] [PubMed] [Google Scholar]
- 50.Park, H. S., S. H. Huh, M. S. Kim, S. H. Lee, and E. J. Choi. 2000. Nitric oxide negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase by means of S-nitrosylation. Proc. Natl. Acad. Sci. USA 97:14382-14387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Persinger, R. L., W. M. Blay, N. H. Heintz, D. R. Hemenway, and Y. M. W. Janssen-Heininger. 2001. Nitrogen dioxide induces death in lung epithelial cells in a density-dependent manner. Am. J. Respir. Cell Mol. Biol. 24:1-8. [DOI] [PubMed] [Google Scholar]
- 52.Peter, M. E., and P. H. Krammer. 2003. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10:26-35. [DOI] [PubMed] [Google Scholar]
- 53.Reinehr, R., B. Gorg, A. Hongen, and D. Haussinger. 2004. CD95-tyrosine nitration inhibits hyperosmotic and CD95 ligand-induced CD95 activation in rat hepatocytes. J. Biol. Chem. 279:10364-10373. [DOI] [PubMed] [Google Scholar]
- 54.Schulze-Osthoff, K., D. Ferrari, M. Los, S. Wesselborg, and M. E. Peter. 1998. Apoptosis signaling by death receptors. Eur. J. Biochem. 254:439-459. [DOI] [PubMed] [Google Scholar]
- 55.Siegel, R. M., J. K. Frederiksen, D. A. Zacharias, F. K. Chan, M. Johnson, D. Lynch, R. Y. Tsien, and M. J. Lenardo. 2000. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 288:2354-2357. [DOI] [PubMed] [Google Scholar]
- 56.Starling, G. C., J. Bajorath, J. Emswiler, J. A. Ledbetter, A. Aruffo, and P. A. Kiener. 1997. Identification of amino acid residues important for ligand binding to Fas. J. Exp. Med. 185:1487-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang, F., G. Tang, J. Xiang, Q. Dai, M. R. Rosner, and A. Lin. 2002. The absence of NF-κB-mediated inhibition of c-Jun N-terminal kinase activation contributes to tumor necrosis factor alpha-induced apoptosis. Mol. Cell. Biol. 22:8571-8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang, G., Y. Minemoto, B. Dibling, N. H. Purcell, Z. Li, M. Karin, and A. Lin. 2001. Inhibition of JNK activation through NF-kappaB target genes. Nature 414:313-317. [DOI] [PubMed] [Google Scholar]
- 59.Torii, S., D. A. Egan, R. A. Evans, and J. C. Reed. 1999. Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). EMBO J. 18:6037-6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tournier, C., P. Hess, D. D. Yang, J. Xu, T. K. Turner, A. Nimnual, D. Bar-Sagi, S. N. Jones, R. A. Flavell, and R. J. Davis. 2000. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288:870-874. [DOI] [PubMed] [Google Scholar]
- 61.van der Vliet, A., J. P. Eiserich, M. K. Shigenaga, and C. E. Cross. 1999. Reactive nitrogen species and tyrosine nitration in the respiratory tract: epiphenomena or a pathobiologic mechanism of disease? Am. J. Respir. Crit. Care Med. 160:1-9. [DOI] [PubMed] [Google Scholar]
- 62.Varfolomeev, E. E., M. Schuchmann, V. Luria, N. Chiannilkulchai, J. S. Beckmann, I. L. Mett, D. Rebrikov, V. M. Brodianski, O. C. Kemper, O. Kollet, T. Lapidot, D. Soffer, T. Sobe, K. B. Avraham, T. Goncharov, H. Holtmann, P. Lonai, and D. Wallach. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9:267-276. [DOI] [PubMed] [Google Scholar]
- 63.Vercammen, D., G. Brouckaert, G. Denecker, M. Van de Craen, W. Declercq, W. Fiers, and P. Vandenabeele. 1998. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 188:919-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Villunger, A., D. C. Huang, N. Holler, J. Tschopp, and A. Strasser. 2000. Fas ligand-induced c-Jun kinase activation in lymphoid cells requires extensive receptor aggregation but is independent of DAXX, and Fas-mediated cell death does not involve DAXX, RIP, or RAIDD. J. Immunol. 165:1337-1343. [DOI] [PubMed] [Google Scholar]
- 65.Wajant, H. 2002. The Fas signaling pathway: more than a paradigm. Science 296:1635-1636. [DOI] [PubMed] [Google Scholar]
- 66.Wang, X., J. L. Martindale, Y. Liu, and N. J. Holbrook. 1998. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem. J. 333:291-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang, X., S. W. Ryter, C. Dai, Z. L. Tang, S. C. Watkins, X. M. Yin, R. Song, and A. M. Choi. 2003. Necrotic cell death in response to oxidant stress involves the activation of the apoptogenic caspase-8/bid pathway. J. Biol. Chem. 278:29184-29191. [DOI] [PubMed] [Google Scholar]
- 68.Xia, Z., M. Dickens, J. Raingeaud, R. J. Davis, and M. E. Greenberg. 1995. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270:1326-1331. [DOI] [PubMed] [Google Scholar]
- 69.Yang, D. D., C. Y. Kuan, A. J. Whitmarsh, M. Rincon, T. S. Zheng, R. J. Davis, P. Rakic, and R. A. Flavell. 1997. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389:865-870. [DOI] [PubMed] [Google Scholar]
- 70.Yang, X., R. Khosravi-Far, H. Y. Chang, and D. Baltimore. 1997. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89:1067-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]