

Abstract
Malignant glioma remains incurable despite tremendous advancement in basic research and clinical practice. The identification of the cell(s) of origin should provide deep insights into leverage points for one to halt disease progression. Here we summarize recent studies that support the notion that neural stem cell (NSC), astrocyte, and oligodendrocyte precursor cell (OPC) can all serve as the cell of origin. We also lay out important considerations on technical rigor for further exploring this subject. Finally, we share perspectives on how one could apply the knowledge of cell of origin to develop effective treatment methods. Although it will be a difficult battle, victory should be within reach as along as we continue to assimilate new information and facilitate the collaboration among basic scientists, translational researchers, and clinicians.
One or more regenerative cell types in the brain—neural stem cells, astrocytes, or oligodendrocyte precursor cells—may serve as cell(s) of origin for gliomas.
Although neurons process and relay information in the central nervous system (CNS), glial cells provide critical support for both the wiring and functions of the neural network (Barres 2008). Because of their importance, malfunctions of glial cells lead to a plethora of diseases, one of them being glioma. Malignant gliomas remain incurable because of two unique properties of the tumor cells (Holland 2001; Maher et al. 2001; Zhu and Parada 2002). The infiltrative nature of glioma cells makes complete surgical resection impossible despite great advancement in neurosurgical techniques, whereas resistance to conventional chemotherapy and radiation spares them from eradication (Stupp et al. 2005). To make matters worse, even those initially diagnosed as low grade tend to progress into malignant glioma within five to ten years. Therefore, it is imperative to gain insights into the detailed mechanisms to develop effective methods of intervention.
Years of molecular characterization of glioma, including efforts by The Cancer Genome Atlas (TCGA), revealed prevalent genetic mutations in three well-known molecular pathways in malignant gliomas: receptor tyrosine kinase (RTK) signaling, p53 signaling, and Rb-mediated G1 checkpoint machinery (Parsons et al. 2008; TCGA 2008). Recent work also showed that mutation in isocitrate dehydrogenase 1 (IDH1) is a unique signature in an identifiably separate subset of gliomas (Yan et al. 2009; Verhaak et al. 2010). Based on this knowledge, great efforts have been devoted to design molecular-targeted therapies. However, drug resistance is an anticipated problem owing to adaptive responses of the dynamic cell-signaling network (Holohan et al. 2013). Therefore, it is critical to identify the Achilles heel of glioma cells for therapeutic interventions. In this review, we will discuss current progresses on the identification of the cell of origin for gliomas and how we could turn this knowledge into clinical applications.
Although there are different ways to define “cell of origin,” the most accepted definition is the cell type that is uniquely susceptible to particular oncogenic mutation(s) (Visvader 2011). Because understanding the molecular basis of the susceptibility carries great promise for the development of effective therapy, it is of utmost importance to unequivocally identify and carefully characterize potential cell(s) of origin for glioma. We emphasize “potential” because the cell of origin refers to the identity of normal cells within a given organ that have the physiologic potential to transform into gliomas. Therefore, this definition is distinct from the “cancer stem cell” hypothesis, which focuses on a putative subset of cells in the tumor mass that serve as the renewable seed of the tumor.
To study the cell of origin of glioma, it is important to first understand the normal process of glial cell development. In the mammalian CNS, neural stem cells (NSCs) are localized in the ventricular zone of embryonic brains and the subventricular zone and subgranular zone of the dentate gyrus of adult brains, and give rise to both neurons and glial cells (Fig. 1) (Doetsch et al. 1999; Gage 2000; Alvarez-Buylla et al. 2002; Gotz and Barde 2005; Ming and Song 2011). Glial cells can be subdivided into two cell types: astrocytes and oligodendrocytes, which can be distinguished by their unique marker expressions and morphologies (Lee et al. 2000; Rowitch 2004). Although cell culture experiments initially indicated a common progenitor for all glial cells (Raff et al. 1984, 1985; Wolswijk and Noble 1989; Rao and Mayer-Proschel 1997), it now appears that in normal physiology astrocytes and oligodendrocytes develop from different subset of progenitors. Although the astrocytic progenitor cells remain elusive, the oligodendrocyte precursor cells (OPCs) have been characterized in great detail (Raff et al. 1983; Barres and Raff 1994; Woodruff et al. 2001; Dawson et al. 2003; Rowitch 2004; Dimou et al. 2008; Nishiyama et al. 2009). It is important to note that, whereas most CNS progenitor cell types terminally differentiate after embryonic development, OPCs persist into adulthood and continue to divide, accounting for up to 4% of the total adult CNS cell population (Imamoto et al. 1978; Ffrench-Constant and Raff 1986; Wolswijk and Noble 1989; Reynolds and Hardy 1997; Gensert and Goldman 2001; Dawson et al. 2003; Nunes et al. 2003). In addition to NSCs and OPCs, astrocytes may also retain some limited capacity to proliferate, especially in the context of brain injuries (Ge et al. 2012; Bardehle et al. 2013). This regenerative potential makes the NSC, OPC, and astrocyte prime suspects as the cell of origin of gliomas (Fig. 1). In the next sections, we will summarize the data that support each cell type as the cell of origin, then conclude with important clarifications of technical and conceptual issues for these types of studies, and perspective on how the identification of cell of origin could impact research approaches and treatment strategies for malignant gliomas.
Figure 1.

Cell lineage in the brain. Neural stem cells (NSCs) give rise to neurons, astrocytes, and oligodendrocyte precursor cells (OPCs) that can in turn differentiate into oligodendrocytes. It should be noted that there could be transient, intermediate progenitor cell types between NSCs and progeny cell types, and that OPC is a stable cell type in the adult brain. Dotted red lines depict the cell types that could serve as the cell of origin for malignant gliomas.
NSCs AS THE CELLS OF ORIGIN
The developmental potential and regenerative plasticity of NSCs make them natural candidates to serve as cells of origin for glioma. Clinically, complex cellular composition and multilineage marker staining in the tumor mass, suggestive of ongoing differentiation, also hint the tumor origin at the NSC level, although dedifferentiation of restricted progenitors for this observation cannot be ruled out. Finally, cultured tumor cells tend to express multiple NSC markers, form renewable tumor spheres, and differentiate into multiple lineages on serum induction, all of which are characteristics of NSCs. Although these clues are tantalizing, it is naturally difficult to firmly pinpoint cell of origin with endpoint studies. Therefore, direct evidence has to come from genetically engineered animal model studies, in which introduced physiologically relevant oncogenic mutations permit analysis of the earliest tumor events.
To precisely define a cell of origin for a tumor arising in vivo, it is necessary to use highly selective methods to induce gene mutations only in specific populations of cells. Introduction of mutations by retroviral injection into brain is an effective approach to target relatively few cells and in an anatomically restricted area. This allows the distinction between tumors arising from resident cells compared with the possibility of cells escaping from the proliferative niche to colonize tumor growth in distant sites. Using this approach, several recent reports identified NSC as the likely cell of origin for gliomas in mouse models and showed that cells outside of the proliferative niche failed to form gliomas with combinations of deletion in Pten, Tp53, Nf1, and Rb1 (Holland et al. 2000; Alcantara Llaguno et al. 2009; Jacques et al. 2010). This approach is a rigorous test of the ability of tumors to initiate in specific regions, and clearly showed that the cells in the NSC niche are more readily transformed than differentiated cell types outside the niche (Fig. 2). Such studies serve to highlight the susceptibility and even propensity of NSCs to function as glioma cells of origin, but they do not rule out alternative cell types—in particular progenitor cells—as potential additional sources for originating gliomas.
Figure 2.

Supporting evidence for NSC as the cell of origin for glioma. (A) Oncogenic mutations induced by viral vector injection into the brain parenchyma failed to lead to gliomagenesis. (B) Oncogenic mutations induced by viral vector injection near NSC niche consistently led to gliomagenesis. LV, lateral ventricle.
Although viral injection is highly flexible, it does cause brain injury that could complicate the issue. The best way to get around that caveat is to use genetically engineered mouse models, which direct the expression of mutations to specific cell populations. It was found that introduction of p53 and NF1 mutations into NSCs led to consistent glioma formation (Zhu et al. 2005). Another study found that induced expression of KRASG12D alone in embryonic radial glia was able to induce gliomas, however cooperating loss of p53 was required for postnatal gliomagenesis, indicating that cells in a less differentiated more stem-like state are more readily transformed (Munoz et al. 2013a). Additionally, there are numerous examples of introducing mutations into nestin-expressing neural progenitor cells in the postnatal brain to generate high-grade gliomas, convincingly demonstrating that NSCs are susceptible to gliomagenic transformation.
Although the studies above established that the introduction of mutations into NSCs is sufficient for gliomagenesis, it would be most interesting to analyze the necessity of NSC in gliomas. Using a nestin-TK-green fluorescent protein (GFP) transgene, it was found that most dividing cells in the tumor mass after chemotherapy are GFP positive (Chen et al. 2012). More importantly, ablation of nestin+ cells by ganciclovir administration significantly arrested tumor growth, implicating the critical roles of NSCs in tumor maintenance. Interestingly, NSC-ablation did not result in complete remission of gliomas in the mouse model. This observation, although potentially attributable to technical issues, also could be the consequence of emergence of tumors derived from other pools of progenitor cells (Chen et al. 2012).
ASTROCYTE AS THE CELL OF ORIGIN
A large proportion of astrocytes in the rodent postnatal cortex are generated through local proliferation of differentiated astrocytes, which generate increasing numbers of mature astrocytes through symmetric cell division. The frequency of these proliferating astrocytes was greatest in the first 2 weeks postnatally, but they were still detectable at postnatal day 52, the latest time point evaluated in this study (Ge et al. 2012). Recent studies also found that perivascular astrocytes in adult mouse brains can enter cell cycle on injuries (Bardehle et al. 2013). The ability of mature astrocytes to undergo extensive proliferation and generate a significant cell population, lends strong support to the hypothesis that mature astrocytes could serve as cells of origin for cancer. Of course, it will be important to determine the extent and timing of this process in the human brain.
A number of approaches have provided experimental evidence that astrocytes can serve as a cell of origin for malignant glioma. For example, overexpressing oncogenes, including EGFRvIII, PDGFR, RAS, MYC, and AKT in primary astrocyte cultures isolated from Ink4a/Arf−/− or Tp53−/− mutant mice, and then implanting the cells into host mouse brain led to malignant gliomas (Bachoo et al. 2002; Endersby et al. 2011; Liu et al. 2011b; Ghazi et al. 2012; Munoz et al. 2013a,b; Paugh et al. 2013; Radke et al. 2013). In addition to in vitro modeling, a number of genetically engineered mouse models (GEMM) also imply that astrocytes can act as a cell of origin for gliomas. However, precisely defining astrocytes as a cell of origin poses a technical challenge that is complicated by the fact that many of the markers used to identify astrocytes are also expressed in neural precursor cells (Molofsky et al. 2012). For example, the use of the GFAP promoter to drive expression of Cre recombinase results in irreversible Cre-mediated recombination in neural precursor cells and all of their progeny, in addition to mature astrocytes (Fraser et al. 2004). This developmental expression resulting in broad Cre activity can be bypassed by use of a tamoxifen–inducible GFAP-creER transgene. However, these transgenes continue to be expressed in the adult NSCs of the subventricular zone of the lateral ventrical and the subgranular zone of the dentate gyrus, the two regions of ongoing neurogenesis in the postnatal rodent brain, with expression patterns varying somewhat among different transgenic founder lines of similar transgenes.
To clarify this issue, fully penetrant mouse models of high-grade astrocytoma were induced by combined deletion of the tumor suppressors Tp53 and Pten with or without concomitant Rb deletion driven by tamoxifen induction of GFAP-CreER in mature mice (Chow et al. 2011). This approach drove widespread and unbiased deletion of these tumor suppressor genes in mature astrocytes from all regions of the central nervous system, as well as a small subpopulation of Gfap–expressing NSCs in the subventricular zone (SVZ) and subgranular layer. Seventy-eight percent of tumors arose within or contiguous with proliferative niches, although the number of these cells with Cre activity was dramatically less than that found in mature astrocytes, consistent with progenitor/stem cells having a greater propensity for malignant transformation. However, >20% of astrocytomas formed in nonproliferative zones including the cortex, brain stem, cerebellum, and spinal cord (Fig. 3). In these cases, there were no detectable other tumor foci that could have disseminated to form these distal masses, and no detectable areas of aberrant proliferation, including within the proliferative niches (Chow et al. 2011). Based on the use of GFAP-creER to induce mutations in this model, tumors may have arisen from progenitor cells that escaped from proliferative niches to populate distant sites, from locally resident progenitor cells (Laywell et al. 2000; Lee et al. 2005), or from mature astrocytes (Dufour et al. 2009; Ge et al. 2012). Clearly, GEMMs using both virally delivered Cre and mouse Cre-expressing lines have shown that gliomas can arise outside of the canonical adult neural progenitor cell niches (Hambardzumyan et al. 2009; Zhu et al. 2009; Chow et al. 2011). The nature of the tumor-initiating cell has yet to be rigorously investigated in these models.
Figure 3.
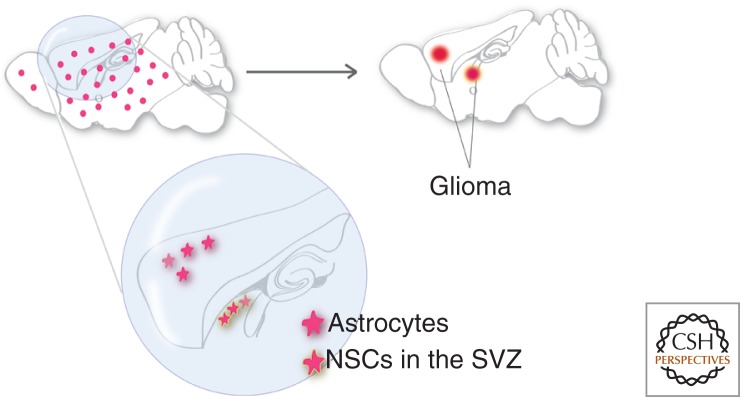
Supporting evidence for astrocyte as the cell of origin for glioma. Oncogenic mutations introduced into both astrocytes and NSCs in adult brains with GFAP-CreER led to full penetrance of glioma formation. Because >20% of tumors formed in areas clearly away from NSC niches, it suggested that adult astrocytes could be transformed into malignant gliomas. SVZ, subventricular zone.
The specific mutation under investigation plays a critical role in determining which cell types are most susceptible to transformation. Targeted inactivation of the retinoblastoma (RB) family of proteins by expression of a mutated form of SV40 T antigen that dominantly inactivates RB, and related proteins p107 and p130, acts as a potent initiation event in gliomagenesis. Inducing inactivation of the RB family in GFAP-expressing astrocytes of the adult brain caused widespread hyperproliferation of astrocytes similar to histopathology of grade II gliomas, even in the absence of other cooperating mutations. Expression of activated KRAS or loss of Pten failed to initiate gliomas, but cooperated with RB family loss to cause progression to high-grade gliomas (Song et al. 2013). These experiments show that some mutations are capable of initiating gliomagenesis in mature astrocytes, although others do not rapidly induce tumors in this cell background. It is worth noting that use of Kras or SV40 oncogenes to generate glioma animal models represents an artificial departure from genes that are directly implicated by mutation in human tumors. Thus, the biological conclusions from the use of such approaches must be taken with this reservation.
Taken together, we conclude that NSCs may be more readily transformed in the experimental setting, and in other circumstances, gliomas can also arise from mature astrocytes.
OPC AS THE CELL OF ORIGIN
Although the renewability of NSCs makes them an obvious candidate as the cell of origin for gliomas (Sanai et al. 2005), OPCs actually outnumber NSCs in the adult brain in both rodents and human (Dawson et al. 2003; Dimou et al. 2008; Geha et al. 2010). Equally important, OPCs continue to slowly divide in adult brains, making them the major dividing cell population of the adult CNS (Imamoto et al. 1978; Dawson et al. 2003). Because of their abundance, OPCs (also called NG2 cells or polydendrocytes) are now called a fourth cell type in the adult CNS in addition to neurons, astrocytes, and oligodendrocytes (Nishiyama 2007; Nishiyama et al. 2009). Considering its large population, proliferative capacity in adult CNS, and developmental plasticity (Kondo and Raff 2000; Nunes et al. 2003), OPCs should be as good a candidate as NSCs for the glioma cell of origin (Canoll and Goldman 2008).
There are at least three lines of evidence that support OPC as the cell of origin for gliomas. First, histopathological analysis of human glioma samples revealed prevalent expression of OPC-specific markers, such as NG2, Olig2, and O4 (Shoshan et al. 1999; Lu et al. 2001; Ligon et al. 2004; Rebetz et al. 2008). Second, the molecular signature of proneural subtype glioma based on TCGA studies matched OPC profile extensively, suggesting at least this subtype of glioma is related to OPCs (Verhaak et al. 2010). Finally, an extensive body of work with murine models convincingly showed the transforming ability of OPCs. For example, malignant gliomas were reproducibly induced when platelet-derived growth factor (PDGF), the mitogen for OPCs, is overexpressed in various populations of glial and neural progenitors in neonatal brains (Uhrbom et al. 1998; Dai et al. 2001; Lindberg et al. 2009) or introduced into the brain with retroviral vectors (Assanah et al. 2006; Hambardzumyan et al. 2009; Lei et al. 2011). Importantly, actively proliferating tumor cells in these models expressed OPC markers (PDGFRa and NG2), suggesting that OPCs are the transformed cell type (Assanah et al. 2006). In addition to the PDGF overexpression models, it was reported that S100b promoter-directed expression of v-erbB, an activated variant of EGFR, could induce glioma formation in a p53 null mouse model (Weiss et al. 2003; Persson et al. 2010). The fact that S100b does not turn on its expression in NSCs strongly suggests that cells other than NSCs can function as the glioma origin. Importantly, grafting of as few as 50 tumor cells isolated with OPC-specific surface antigen NG2 consistently generated gliomas in immunocompromised mice, whereas cells isolated with NSC marker CD15 failed to do so, suggesting that OPCs are not only the origin but also the propagating population in this model (Persson et al. 2010). An independent study also showed that Olig2, a basic helix-loop-helix (bHLH) transcription factor essential for OPC development, was functionally required for gliomagenesis in a mouse model (Ligon et al. 2007), suggesting the importance of this lineage in gliomagenesis.
Recently, a mouse model based on a genetic mosaic system called MADM (mosaic analysis with double markers) (Fig. 4A) (Zong et al. 2005; Muzumdar et al. 2007) added a new twist to our understanding of cell of origin for gliomas (Liu et al. 2011a). Because MADM generates GFP-positive mutant cells and their sibling WT cells labeled with red fluorescent protein (RFP), the aberrant expansion of each cell lineage can be examined at early stages of gliomagenesis after Tp53 and NF1 mutations were initiated in NSCs. Surprisingly, there was little expansion of NSCs, astrocytes, and neurons, suggesting that these cells were not susceptible to the loss of these two tumor suppressor genes. In stark contrast, mutant OPCs expanded over WT counterparts by more than 100-fold, a few months before malignant transformation (Fig. 4B). On tumor formation, marker staining and gene expression profiling clearly showed that OPC is the transforming cell type. Last, direct introduction of p53 and NF1 mutations into OPCs with NG2-Cre resulted in gliomas with indistinguishable signatures from those initiated from NSCs. These findings suggest that, even though the initial mutations may occur in either NSC or OPC, the latter serves as the cell of origin in this model (Liu et al. 2011a).
Figure 4.

Supporting evidence for OPC as the cell of origin for glioma. (A) Scheme of the mosaic analysis with double markers (MADM) system. From a heterozygous, colorless mouse, Cre-induced somatic recombination generates sparse “twin spots”: a green mutant cell and a red wild-type (WT) cell. The ratio between green and red cells in a given cell lineage reveals its transforming potential. (B) Mutations (p53 and NF1) introduced into NSCs led to massive overexpansion of OPCs but not other cell types, suggesting that OPC is the cell origin for gliomas in this model. RFP, red fluorescent protein; GFP, green fluorescent protein. (From Liu and Zong 2012; adapted, with permission, from the authors in conjunction with Elsevier © 2012.)
In summary, this set of evidence clearly shows that OPC is a cell of origin for malignant gliomas, and is susceptible to transformation by a wide range of mutations often found in human gliomas, such as p53, PTEN, v-erbB, Nf1, and Ras (Persson et al. 2010; Lei et al. 2011; Liu et al. 2011a; Barrett et al. 2012).
CONCLUDING REMARKS
From the discussion above, it is obvious that the question about which cells are the origin for glioma does not have a simple answer. It is not surprising because malignant glioma is likely a collection of diseases and, thus, may have multiple origins. Nevertheless, all three candidate cell types, NSCs, astrocytes, and OPCs, are all regenerative cell types in the brain, implying that the transformation of these cells depends on the synergy of intrinsic regenerative properties of these cells and the oncogenic mutations. Obviously, there are still many unanswered questions. For example, can molecular subtype of gliomas defined by the TCGA effort (Verhaak et al. 2010) be explained by distinct cell of origins? If that is the case, then it is puzzling why gene expression profile of proneural subtype of gliomas matches exceptionally well with OPCs, whereas profiles of other subtypes do not match as well with NSCs or astrocytes. Furthermore, mutations in RTK signaling and p53 pathways can transform multiple cells of origin, although they have distinct developmental potential and signaling context (Liu and Zong 2012). It would be of great interest to carefully investigate the transforming process in each cell type with superb temporal and spatial resolution to tease apart common and unique features that are targetable for therapeutic purpose.
To continuously advance our understanding on this subject, we would like to point out the importance of technical rigor for future studies.
The obvious advantage of patient sample analysis, either marker staining or gene expression profiling, is the direct relevance to human disease. However, the presence of niche cells in late-stage tumor mass and abnormal gene expression patterns of malignant cells could confound our findings. A few recent findings could help resolve this issue. First, the recent finding of IDH1 mutation in gliomas allows one to use the antibody-recognizing mutant IDH1 to reveal bona fide tumor cells. Costaining tumor sections with mutant IDH1 and cell-specific markers should help reveal their identities with little ambiguity. Second, single-cell RNA sequencing (Patel et al. 2014) is another approach to help understand not only the cell of origin but also the heterogeneity in the tumor mass.
We raise two cautionary notes for the reliance of marker genes to claim cell identity. One problem is that many broadly used markers are not as specific as previously thought. The other is the potential misexpression of marker genes in late-stage tumor cells. These problems could be alleviated by verifying marker choices based on recent cell-type-specific gene expression profiling analysis (Cahoy et al. 2008) and by using whole transcriptomic analysis rather than a few marker genes (Verhaak et al. 2010).
Ongoing transformative advances in genomic technology have accelerated the unraveling of molecular landscapes of tumors including gliomas. This rapidly moving field must, however, continually be tempered with the biological and physiological imperatives of organ systems and how tumors naturally occur and grow within. A complementary application of these two fields will sustain the natural tension between possibility and plausibility in the search for the cause and the cure of glioma.
Mouse genetic models are highly valuable because they allow the study of cell of origin from the initial transforming process, thus circumventing issues associated with late stage analysis. These highly relevant mouse models can also be enlisted for preclinical drug testing before clinical trials. However, we must characterize these models carefully to avoid confusion in the field. First, we must ensure that transgene expression is restricted within a specific cell type. For example, although nestin has been widely considered as an NSC marker, it was also found to express in reactive astrocytes and perinatal OPCs (Zhu et al. 2008). Second, it would be ideal to initiate gliomagenesis at clonal level during defined time window to closely mimic human physiology. Previous findings derived from transgenic mice that introduce genetic alterations to a large number of cells at early stages of brain development might need to be revisited with more advanced tools. Third, whenever we introduce mutations into NSCs or multipotent progenitor cells, we need to analyze lineage-specific expansion of progeny cell types before claiming the cell of origin, as illustrated by the MADM study that revealed transforming ability of OPCs (Liu et al. 2011a).
Finally, we must keep in mind that progenitor populations in humans could be different from those in mice. Findings from mouse models should be carefully confirmed with human samples or primary cell culture before the attempt to translate them into clinics.
The identification of cell of origin will have a transformative impact on our research approaches and conceptual framework. Previously, we tended to compare the gene expression profiles between tumor samples and normal tissues to look for clues of signaling changes. The problem with that approach is that such comparisons could simply reveal signature genes of the cell of origin that may have no roles in the malignancy at all. For example, Olig2 levels would be significantly higher when an OPC-originated glioma sample is compared with neighboring tissue, simply because of the enrichment of OPCs in the tumor mass. Once we identify the cell of origin, we must use purified normal cell of origin for the comparison in this case so we could identify true candidate genes. Another important consideration is the conditions for in vitro culture. Investigators have started shifting from serum containing to serum-free media after realizing the drastic changes of cellular properties induced by serum (Lee et al. 2006). The current practice is to culture tumor cells in NSC media that contains epidermal growth factor (EGF) and fibroblast growth factor (FGF). However, this change is only suitable for NSC-originated gliomas, although authentic culture condition for astrocytes (Foo et al. 2011) and OPCs (Dugas et al. 2006) should be used for gliomas originated from these cell types because they express a distinct set of RTKs and, thus, need different growth factors in the serum-free media.
Our ultimate goal of studying the cell of origin for glioma is to discover novel therapeutic methods that could effectively treat this devastating disease (Zong et al. 2012). Because gliomas from distinct origins likely have different prognosis and unique susceptibility to treatment, the understanding of cell of origin could facilitate patient stratification and the development of tailored treatment methods. The knowledge of the cellular origin could contribute to drug development from three aspects. First, cell-type-specific membrane proteins could be used as beacons for targeted drug delivery, which should greatly reduce general toxicity to other cells in the brain. Second, the susceptibility of cell of origin to particular genetic lesions implies the existence of cell-specific signaling nodes on which oncogenic mutations take actions. Targeting the interactions between signaling nodes and mutations could provide the most effective treatment methods to halt gliomagenesis. Of course, a thorough understanding of signaling network in the cell of origin is a prerequisite for such efforts. Third, clear identification of the cell of origin for glioma would greatly promote the collaborations between cancer and stem cell/regenerative medicine fields. On one hand, studying augmented renewability of NSCs and progenitor cells in the tumor setting could provide insights on how to propagate these cells better for regeneration purposes. On the other hand, methods developed for regenerative biologists to coerce NSCs and progenitor cells into terminally differentiated cells could be applied to glioma treatment. Such strategy carries great potential to avoid drug resistance because it should reduce the tumor evolution process that tends to be facilitated by cytotoxic drugs.
In conclusion, our studies on the cell of origin for gliomas are still in progress rather than reaching the end. The key to our success as a field is to continuously adopt advanced technologies and incorporate new concepts into our existing strength. As long as our minds could evolve faster than those tumor cells, an effective treatment to glioma should be within our reach.
ACKNOWLEDGMENTS
We thank Chong Liu for critical comments and for contributing figures for the manuscript.
Footnotes
Editors: Ben A. Barres, Marc R. Freeman, and Beth Stevens
Additional Perspectives on Glia available at www.cshperspectives.org
REFERENCES
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF. 2009. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15: 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Seri B, Doetsch F. 2002. Identification of neural stem cells in the adult vertebrate brain. Brain Res Bull 57: 751–758. [DOI] [PubMed] [Google Scholar]
- Assanah M, Lochhead R, Ogden A, Bruce J, Goldman J, Canoll P. 2006. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci 26: 6781–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, et al. 2002. Epidermal growth factor receptor and Ink4a/Arf: Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1: 269–277. [DOI] [PubMed] [Google Scholar]
- Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Snippert HJ, Theis FJ, Meyer-Luehmann M, Bechmann I, et al. 2013. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 16: 580–586. [DOI] [PubMed] [Google Scholar]
- Barres BA. 2008. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 60: 430–440. [DOI] [PubMed] [Google Scholar]
- Barres BA, Raff MC. 1994. Control of oligodendrocyte number in the developing rat optic nerve. Neuron 12: 935–942. [DOI] [PubMed] [Google Scholar]
- Barrett LE, Granot Z, Coker C, Iavarone A, Hambardzumyan D, Holland EC, Nam HS, Benezra R. 2012. Self-renewal does not predict tumor growth potential in mouse models of high-grade glioma. Cancer Cell 21: 11–24. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. 2008. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci 28: 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canoll P, Goldman JE. 2008. The interface between glial progenitors and gliomas. Acta Neuropathol 116: 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. 2012. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488: 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, Broniscer A, Ellison DW, Baker SJ. 2011. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 19: 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. 2001. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 15: 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. 2003. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci 24: 476–488. [DOI] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Gotz M. 2008. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci 28: 10434–10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. 1999. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97: 703–716. [DOI] [PubMed] [Google Scholar]
- Dufour C, Cadusseau J, Varlet P, Surena AL, de Faria GP, Dias-Morais A, Auger N, Leonard N, Daudigeos E, Dantas-Barbosa C, et al. 2009. Astrocytes reverted to a neural progenitor-like state with transforming growth factor alpha are sensitized to cancerous transformation. Stem Cells 27: 2373–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, Barres BA. 2006. Functional genomic analysis of oligodendrocyte differentiation. J Neurosci 26: 10967–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endersby R, Zhu X, Hay N, Ellison DW, Baker SJ. 2011. Nonredundant functions for Akt isoforms in astrocyte growth and gliomagenesis in an orthotopic transplantation model. Cancer Res 71: 4106–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ffrench-Constant C, Raff MC. 1986. Proliferating bipotential glial progenitor cells in adult rat optic nerve. Nature 319: 499–502. [DOI] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, Cahoy JD, Daneman R, Zong H, Ellisman MH, et al. 2011. Development of a method for the purification and culture of rodent astrocytes. Neuron 71: 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ. 2004. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res 64: 7773–7779. [DOI] [PubMed] [Google Scholar]
- Gage FH. 2000. Mammalian neural stem cells. Science 287: 1433–1438. [DOI] [PubMed] [Google Scholar]
- Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY. 2012. Local generation of glia is a major astrocyte source in postnatal cortex. Nature 484: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geha S, Pallud J, Junier MP, Devaux B, Leonard N, Chassoux F, Chneiweiss H, Daumas-Duport C, Varlet P. 2010. NG2+/Olig2+ cells are the major cycle-related cell population of the adult human normal brain. Brain Pathol 20: 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensert JM, Goldman JE. 2001. Heterogeneity of cycling glial progenitors in the adult mammalian cortex and white matter. J Neurobiol 48: 75–86. [PubMed] [Google Scholar]
- Ghazi SO, Stark M, Zhao Z, Mobley BC, Munden A, Hover L, Abel TW. 2012. Cell of origin determines tumor phenotype in an oncogenic Ras/p53 knockout transgenic model of high-grade glioma. J Neuropathol Exp Neurol 71: 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz M, Barde YA. 2005. Radial glial cells defined and major intermediates between embryonic stem cells and CNS neurons. Neuron 46: 369–372. [DOI] [PubMed] [Google Scholar]
- Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. 2009. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol 2: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC. 2001. Gliomagenesis: Genetic alterations and mouse models. Nat Rev Genet 2: 120–129. [DOI] [PubMed] [Google Scholar]
- Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. 2000. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 25: 55–57. [DOI] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. 2013. Cancer drug resistance: An evolving paradigm. Nat Rev Cancer 13: 714–726. [DOI] [PubMed] [Google Scholar]
- Imamoto K, Paterson JA, Leblond CP. 1978. Radioautographic investigation of gliogenesis in the corpus callosum of young rats. I: Sequential changes in oligodendrocytes. J Comp Neurol 180: 115–128, 132–117. [DOI] [PubMed] [Google Scholar]
- Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh ZC OM, Naumann H, Alvarez-Buylla A, Brandner S. 2010. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J 29: 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Raff M. 2000. Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells. Science 289: 1754–1757. [DOI] [PubMed] [Google Scholar]
- Laywell ED, Rakic P, Kukekov VG, Holland EC, Steindler DA. 2000. Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proc Natl Acad Sci 97: 13883–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Mayer-Proschel M, Rao MS. 2000. Gliogenesis in the central nervous system. Glia 30: 105–121. [DOI] [PubMed] [Google Scholar]
- Lee A, Kessler JD, Read TA, Kaiser C, Corbeil D, Huttner WB, Johnson JE, Wechsler-Reya RJ. 2005. Isolation of neural stem cells from the postnatal cerebellum. Nat Neurosci 8: 723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. 2006. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9: 391–403. [DOI] [PubMed] [Google Scholar]
- Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, Bruce JN, Canoll P. 2011. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS ONE 6: e20041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, Louis DN, Stiles CD, Rowitch DH. 2004. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol 63: 499–509. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, et al. 2007. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 53: 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. 2009. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 28: 2266–2275. [DOI] [PubMed] [Google Scholar]
- Liu C, Zong H. 2012. Developmental origins of brain tumors. Curr Opin Neurobiol 22: 844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, et al. 2011a. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146: 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KW, Feng H, Bachoo R, Kazlauskas A, Smith EM, Symes K, Hamilton RL, Nagane M, Nishikawa R, Hu B, et al. 2011b. SHP-2/PTPN11 mediates gliomagenesis driven by PDGFRA and INK4A/ARF aberrations in mice and humans. J Clin Invest 121: 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Park JK, Noll E, Chan JA, Alberta J, Yuk D, Alzamora MG, Louis DN, Stiles CD, Rowitch DH, et al. 2001. Oligodendrocyte lineage genes (OLIG) as molecular markers for human glial brain tumors. Proc Natl Acad Sci 98: 10851–10856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. 2001. Malignant glioma: Genetics and biology of a grave matter. Genes Dev 15: 1311–1333. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. 2011. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 70: 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH. 2012. Astrocytes and disease: A neurodevelopmental perspective. Genes Dev 26: 891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DM, Singh S, Tung T, Agnihotri S, Nagy A, Guha A, Zadeh G, Hawkins C. 2013a. Differential transformation capacity of neuro-glial progenitors during development. Proc Natl Acad Sci 110: 14378–14383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DM, Tung T, Agnihotri S, Singh S, Guha A, Zadeh G, Hawkins C. 2013b. Loss of p53 cooperates with K-ras activation to induce glioma formation in a region-independent manner. Glia 61: 1862–1872. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Luo L, Zong H. 2007. Modeling sporadic loss of heterozygosity in mice by using mosaic analysis with double markers (MADM). Proc Natl Acad Sci 104: 4495–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A. 2007. Polydendrocytes: NG2 cells with many roles in development and repair of the CNS. Neuroscientist 13: 62–76. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X. 2009. Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nat Rev Neurosci 10: 9–22. [DOI] [PubMed] [Google Scholar]
- Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G II, Jiang L, Kang J, Nedergaard M, Goldman SA. 2003. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med 9: 439–447. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. 2008. An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. 2014. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344: 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, Bax DA, Carvalho D, Reis RM, Onar-Thomas A, et al. 2013. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res 73: 6219–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, Goldenberg DD, Vandenberg SR, Nguyen KN, Yakovenko S, et al. 2010. Non-stem cell origin for oligodendroglioma. Cancer Cell 18: 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radke J, Bortolussi G, Pagenstecher A. 2013. Akt and c-Myc induce stem-cell markers in mature primary pp53–/– astrocytes and render these cells gliomagenic in the brain of immunocompetent mice. PLoS ONE 8: e56691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Miller RH, Noble M. 1983. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature 303: 390–396. [DOI] [PubMed] [Google Scholar]
- Raff MC, Williams BP, Miller RH. 1984. The in vitro differentiation of a bipotential glial progenitor cell. EMBO J 3: 1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Abney ER, Fok-Seang J. 1985. Reconstitution of a developmental clock in vitro: A critical role for astrocytes in the timing of oligodendrocyte differentiation. Cell 42: 61–69. [DOI] [PubMed] [Google Scholar]
- Rao MS, Mayer-Proschel M. 1997. Glial-restricted precursors are derived from multipotent neuroepithelial stem cells. Dev Biol 188: 48–63. [DOI] [PubMed] [Google Scholar]
- Rebetz J, Tian D, Persson A, Widegren B, Salford LG, Englund E, Gisselsson D, Fan X. 2008. Glial progenitor-like phenotype in low-grade glioma and enhanced CD133-expression and neuronal lineage differentiation potential in high-grade glioma. PLoS ONE 3: e1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds R, Hardy R. 1997. Oligodendroglial progenitors labeled with the O4 antibody persist in the adult rat cerebral cortex in vivo. J Neurosci Res 47: 455–470. [DOI] [PubMed] [Google Scholar]
- Rowitch DH. 2004. Glial specification in the vertebrate neural tube. Nat Rev Neurosci 5: 409–419. [DOI] [PubMed] [Google Scholar]
- Sanai N, Alvarez-Buylla A, Berger MS. 2005. Neural stem cells and the origin of gliomas. N Engl J Med 353: 811–822. [DOI] [PubMed] [Google Scholar]
- Shoshan Y, Nishiyama A, Chang A, Mork S, Barnett GH, Cowell JK, Trapp BD, Staugaitis SM. 1999. Expression of oligodendrocyte progenitor cell antigens by gliomas: Implications for the histogenesis of brain tumors. Proc Natl Acad Sci 96: 10361–10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Zhang Q, Kutlu B, Difilippantonio S, Bash R, Gilbert D, Yin C, O’Sullivan TN, Yang C, Kozlov S, et al. 2013. Evolutionary etiology of high-grade astrocytomas. Proc Natl Acad Sci 110: 17933–17938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996. [DOI] [PubMed] [Google Scholar]
- TCGA. 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrbom L, Hesselager G, Nister M, Westermark B. 1998. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res 58: 5275–5279. [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE. 2011. Cells of origin in cancer. Nature 469: 314–322. [DOI] [PubMed] [Google Scholar]
- Weiss WA, Burns MJ, Hackett C, Aldape K, Hill JR, Kuriyama H, Kuriyama N, Milshteyn N, Roberts T, Wendland MF, et al. 2003. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res 63: 1589–1595. [PubMed] [Google Scholar]
- Wolswijk G, Noble M. 1989. Identification of an adult-specific glial progenitor cell. Development 105: 387–400. [DOI] [PubMed] [Google Scholar]
- Woodruff RH, Tekki-Kessaris N, Stiles CD, Rowitch DH, Richardson WD. 2001. Oligodendrocyte development in the spinal cord and telencephalon: Common themes and new perspectives. Int J Dev Neurosci 19: 379–385. [DOI] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. 2009. IDH1 and IDH2 mutations in gliomas. N Engl J Med 360: 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Parada LF. 2002. The molecular and genetic basis of neurological tumours. Nat Rev Cancer 2: 616–626. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A, Parada LF. 2005. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 8: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. 2008. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 135: 145–157. [DOI] [PubMed] [Google Scholar]
- Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, Bronson RT, Chen JW, Weissleder R, Housman DE, et al. 2009. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci 106: 2712–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. 2005. Mosaic analysis with double markers in mice. Cell 121: 479–492. [DOI] [PubMed] [Google Scholar]
- Zong H, Verhaak RG, Canoll P. 2012. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev Mol Diagn 12: 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]