

Abstract
In epithelial tissues, cells are linked to their neighbors through specialized cell-cell adhesion proteins. E-cadherin is one of the most important membrane proteins for the establishment of intimate cell-cell contacts, but the molecular mechanism by which it is recruited to contact sites is largely unknown. We report here that the cytoplasmic domain of E-cadherin interacts with C3G, a guanine nucleotide exchange factor for Rap1. In epithelial cell cultures, ligation of the extracellular domain of E-cadherin enhances Rap1 activity, which in turn is necessary for the proper targeting of E-cadherin molecules to maturing cell-cell contacts. Furthermore, our data suggest that Cdc42 functions downstream of Rap1 in this process. We conclude that Rap1 plays a vital role in the establishment of E-cadherin-based cell-cell adhesion.
An epithelial cell is connected to its neighbors through a variety of cell-cell adhesive structures to form a precisely aligned epithelial cell sheet. The structural components of these adhesion complexes include several membrane proteins. Among them, E-cadherin has been shown to be the most essential protein (1, 29, 36). The extracellular domain of E-cadherin forms Ca2+-dependent homophilic trans-dimers, providing specific interaction with adjacent cells, while the cytoplasmic domain is connected to the actin cytoskeleton via anchor proteins called catenins. The elimination of E-cadherin's adhesive function by low-calcium treatment or addition of inhibitory antibodies strongly inhibits the ability of epithelial cells to form stable cell-cell contacts.
When epithelial cells form cell-cell contacts, E-cadherin is recruited exclusively to the lateral membrane domain, the site of cell-cell contact. The process begins with the engagement of opposing E-cadherin molecules at the tips of filopodial or lamellopodial projections. Following the formation of this initial cluster of E-cadherin molecules, additional adjacent puncta assemble, generating a zipper-like structure, which then develops into a mature, linear cell-cell contact (2). During this process, E-cadherin is transported from a cytoplasmic pool (or elsewhere on the plasma membrane) to the initial cluster. However, the molecular mechanism by which E-cadherin is directionally targeted to cell-cell contact sites is still not fully understood, though it is likely that this involves the interaction of its cytoplasmic domain with a binding protein(s). Several proteins are known to interact with E-cadherin, including β-catenin, p120ctn, and Hakai (11, 29, 30), but none of these has been clearly shown to be implicated in the targeted recruitment of E-cadherin to nascent cell-cell contact sites. We assumed that there might be other E-cadherin binding proteins that are involved in this process.
Rap1 is a Ras-like small GTP-binding protein which has various roles in several cellular processes, such as proliferation, secretion, and integrin-mediated cell adhesion (4). Rap1 binds either GDP or GTP, and the change between the two states represents a molecular switch, an inactive GDP-bound and an active GTP-bound form. The conversions between the two states are controlled by two types of regulators, guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). GEFs act as activators by facilitating conversion from the GDP- to the GTP-bound form, whereas GAPs act as inactivators by inducing hydrolysis of the bound GTP to convert it into the GDP form. In this study, we investigated the functional role of Rap1 in E-cadherin-based cell-cell contacts.
MATERIALS AND METHODS
Antibodies, plasmids, and materials.
Antibodies to the cytoplasmic portion of E-cadherin and to the extracellular portion of E-cadherin (ECCD-2) were from Transduction Laboratories (San Diego, Calif.) and Zymed (South San Francisco, Calif.), respectively. The former was used for immunoprecipitation and Western blotting, and the latter was used for immunofluorescence. Anti-N-cadherin and anti-CD29 (integrin-β1) antibodies were from Transduction Laboratories. Anti-Flag and antihemagglutinin (HA) antibodies were from Sigma (St. Louis, Mo.) and Roche (Mannheim, Germany), respectively. Anti-Myc antibody was from Upstate (Charlottesville, Va.). Anti-C3G and anti-Rap1 antibodies were from Santa Cruz (Santa Cruz, Calif.), and anti-green fluorescent protein (GFP) antibody was from Invitrogen (Paisley, United Kingdom). All antibodies were used at a dilution of 1:1,000 for Western blotting and 1:100 for immunofluorescence.
The cDNAs of C3G(N) (amino acids 1 to 357) and C3G(C) (amino acids 351 to 1078) were amplified from pBS-C3G (22) by PCR with primers 5′-GGAATTCGCGGCCGCCCATGGACACAGACTCTCAG-3′ and 5′-GGAATTCGCGGCCGCTCGAGCTTGTCTATGCTGCTGCAGGGGGAG-3′ and primers 5′-GGAATTCGCGGCCGCCCTGCAGCAGCATAGACAAGCTCAGC-3′ and 5′-GGAATTCGCGGCCGCCTAGGTCTTCTCTTCCCGGTC-3′, respectively, and cloned into a NotI site of the pcDNA-Flag vector.
To construct pcDNA-Flag-C3G (full length), the cDNA of C3G was excised from pBS-C3G (NcoI and BamHI) and, after blunting the ends, inserted into an EcoRV site of the pcDNA-Flag vector. To construct pEGFP-C3G (full length), the cDNA of C3G was excised from pcDNA-Flag-C3G (full length) (BamHI and XhoI) and inserted into pEGFP-C1 (BglII/SalI). pRK5-Myc-Rap1V12, pRK5-Myc-Rap1N17, pMT2-HA-RapGAP, pMT2-HA-PDZ-GEF, and pGEX-RalGDS were kindly provided by J. L. Bos (University Medical Center, Utrecht, The Netherlands). pRK5-Myc-Cdc42 (wild type), pRK5-Myc-Cdc42L61, and pMT2-HA-Rlf were kindly provided by A. Hall (Medical Research Council Laboratory for Molecular Cell Biology, University College London, London, United Kingdom). The cDNA of p120ctn was amplified by PCR from pBS-p120ctn with the primers 5′-GGAATTCGCGGCCGCGACTGGACGACTCAGAGGTGGAGTCG-3′ and 5′-GGAATTCGCGGCCGCTAAATCTTCTGCATCAAGGGTG-3′ and cloned into a NotI site of the pcDNA-HA vector. The cDNA of β-catenin was excised from the KpnI and SalI sites of pBAT-Myc-β-catenin (17) and inserted into the KpnI and XhoI sites of pcDNA3.1 to produce pcDNA-Myc-β-catenin. Lipofectamine Plus reagent was obtained from Invitrogen.
Yeast two-hybrid screens.
pBTM-Tpr-Met-E-cadherin was constructed as described previously (11, 34). A cDNA encoding the cytoplasmic region of E-cadherin was fused to the Met sequence of pBTM-Tpr-Met lacking sequences encoding the substrate binding sites of Met. Yeast two-hybrid screens were performed as described previously (11). It should be noted that the high transactivating property of the cytoplasmic region of E-cadherin was dramatically reduced by the insertion of Tpr-Met.
Cell culture, immunoprecipitation, GST-E-cadherin pulldown assay, overlay-blotting assay, and Western blotting.
HEK293 cells were cultured and transiently transfected as previously described (34). MCF7 cells were transfected with Lipofectamine Plus reagent, and stably transfected cells were selected in medium containing 800 μg of G418 ml−1. For immunoprecipitation experiments with HEK293 or MCF7 cells, cells cultured in 9-cm dishes were lysed for 30 min in 1 ml of 1% Triton X-100 lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 1% Triton X-100) containing 5-μg ml−1 leupeptin, 50 mM phenylmethylsulfonyl fluoride, and 7.2 trypsin inhibitor units of aprotinin. After centrifugation at 14,000 × g for 10 min, the supernatants (about 0.5 mg of protein) were subjected to immunoprecipitation for 1 h with 5 μl of the indicated antibody bound to 10 μl of protein G-Sepharose 4B beads (Pharmacia), followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting with the indicated antibodies.
For the glutathione S-transferase (GST)-E-cadherin pulldown assay, 10 μl of glutathione-Sepharose beads (Pharmacia) attached to 20 μg of GST or GST-E-cadherin protein were incubated with cell lysates, followed by the procedures described above. pcDNA-Flag C3G (amino acids 1 to 357) and pcDNA E-cadherin (the cytoplasmic domain; amino acids 734 to 884) were used to express exogenous C3G and E-cadherin in HEK293 cells. For Western blotting, SDS-PAGE gels were transferred onto polyvinylidene difluoride membranes. After incubation with primary and secondary antibodies, proteins were visualized by enhanced chemiluminescence (ECL+; Perkin-Elmer Life Sciences, Boston, Mass.). To detect Flag-tagged and HA-tagged proteins, peroxidase-conjugated anti-Flag (Sigma) and anti-HA antibodies (Roche) were used, respectively.
Overlay-blotting assays were performed as described previously (12). A membrane was incubated with 200 nM GST-E-cadherin, and the E-cadherin protein bound to C3G on the membrane was detected by anti-E-cadherin antibody.
Immunofluorescence, microinjection, and time-lapse imaging.
Immunofluorescence was performed as follows. Cells on coverslips were washed twice in phosphate-buffered saline (PBS) and incubated in 3% paraformaldehyde-PBS for 15 min. After washing twice in PBS, cells were permeabilized in 0.5% Triton X-100-PBS for 15 min, followed by blocking in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum for 1 h. They were further incubated in primary antibody for 1 h, washed four times in PBS, incubated in Cychrome 2 (Cy2) or Cy3-conjugated secondary antibody solution for 1 h, and washed four times in PBS. The coverslip was then mounted onto Mowiol on a glass slide.
Microinjection was performed as described previously (5). Each DNA construct (0.1 μg per μl of PBS) was microinjected into the nucleus of MCF7 cells. For Rap1V12, Rap1N17, and C3G(N) constructs, 20 ng of DNA μl−1 was microinjected. For microinjection of pcDNA-Flag, a dye (Amaxa Fluor 488-conjugated dextran; Molecular Probes) was coinjected. After microinjection, cells were incubated in a low-calcium medium for 3 to 4 h until cell-cell contacts were disrupted, followed by incubation in normal-calcium medium for 30 min. It should be noted that the effect on tight junction protein ZO-1 was examined after 30, 60, and 90 min, as tight junction molecules are recruited to cell-cell contact sites more slowly than E-cadherin. The effect of RapGAP on the recruitment of ZO-1 was not clearly seen at any time point.
The effect of microinjection on E-cadherin localization at cell-cell contacts was quantified as follows. Cell-cell contacts between microinjected cells were first examined in the image showing the expression of microinjected molecules, and the E-cadherin staining at the cell-cell contact sites was evaluated. When less than 50% of the contact site was stained for E-cadherin, it was counted as a cell-cell contact with incomplete E-cadherin localization. Cells that made no contacts or adhered at only one point were not evaluated. A 2-by-2 chi square test was used to calculate the P value.
Calcium was depleted from fetal calf serum, and the low-calcium medium was reconstituted as described previously (5). Data were analyzed by confocal microscopy if not otherwise indicated. To obtain epifluorescence and confocal images, we used a Zeiss Axioskop 1 with a Roper Scientific Coolsnap camera and a Bio-Rad MRC1024 mounted on a Nikon Optiphot 2 microscope, respectively. To obtain time-lapse images, we used an Axiovert 135TV with a Ludl Electronic Products Biopoint Controller. Images were captured and analyzed with Openlab software (Improvision).
hE/Fc-coated beads.
CHO cells stably expressing human E-cadherin ectodomain-Fc chimeric protein (hE/Fc) were obtained as a gift from C. Niessen (Cologne, Germany). The hE/Fc protein was purified by protein A affinity chromatography from conditioned medium of CHO cells as described previously (9). Preparation of polystyrene beads coated with hE/Fc protein was carried out as previously described (9). Briefly, polystyrene beads (Polysciences Inc., Warrington, Pa.; 15-μm diameter) were coated with bovine serum albumin (BSA), hE/Fc protein, or anti-CD29 antibody (1 mg ml−1) in the minimum volume of coating buffer (10 mM HEPES-NaOH [pH 7.4], 50 mM NaCl, and 1 mM CaCl2) for 90 min at 4°C on an Eppendorf shaker (1,400 rpm). The beads were blocked with 10 mg of heat-denatured BSA ml−1 for 1 h at 4°C, followed by two washes with coating buffer. Immediately before use, the beads were resuspended in culture medium and kept at 37°C.
For analysis of Rap1 activation as determined by the GST-RalGDS pulldown assay, confluent MCF7 cells were incubated with BSA-, hE/Fc-, or anti-CD29 antibody-coated beads (107 beads/9-cm dish) for 10 min before being lysed. To assess the recruitment of GFP-Rap1 to cadherin complexes, semiconfluent stable MCF7 cell lines expressing GFP-Rap1 were incubated with BSA- or hE/Fc-coated beads (105 beads/coverslip) for 10 min and then fixed. The level of GFP-Rap1 recruited to hE/Fc-coated beads compared to BSA-coated beads was determined with Metamorph 6.0 digital analysis software (Universal Imaging). Briefly, a defined region was created to outline the perimeter of the bead, and a second, larger region was created to encompass the bead and the immediate region surrounding the bead. The total pixel intensity within the area (14,026 pixels) between the two created regions was determined for both BSA- and hE/Fc-coated beads. A total of 30 to 45 beads were counted for each population. Student t tests assuming unequal variance were performed for statistical analysis.
Rap1 activation assay.
The level of Rap1-GTP was determined from HEK293 and MCF7 cell lysates with a GST fusion protein containing the Rap1 binding domain of RalGDS, adapted as previously described (10). GST-RalGDS was purified as described previously (31). Prior to each experiment, MCF7 cells and HEK293 cells were seeded at 2 × 106 and 1 × 106 cells, respectively, onto a 9-cm dish. GST-RalGDS (30 μg ml−1) was precoupled to glutathione-Sepharose beads for 1 h at 4°C with rotation. Immediately after stimulation, MCF7 cells were washed with 2 ml of ice-cold PBS and lysed in 1 ml of cold lysis buffer (50 mM Tris-Cl [pH 7.5], 200 mM NaCl, 2 mM MgCl2, 10% glycerol, 1% NP-40, 2 mM sodium orthovanadate, 10 mM NaF, 10 μg ml−1 each leupeptin and aprotinin, 1 mM phenylmethylsulfonyl fluoride). Lysates were briefly precleared by centrifugation at 14,000 rpm for 3 min at 4°C. The supernatant (0.85 ml) was incubated with 30 μg of GST-RalGDS protein precoupled to glutathione-Sepharose beads, with rotation for 45 min at 4°C. The beads were then washed twice with 1 ml of lysis buffer. For HEK293 cells, 36 to 48 h following calcium phosphate transfection, cells were lysed in 1 ml of cold lysis buffer with rotation at 4°C for 5 min. Lysates were then precleared by centrifugation at 14,000 rpm for 10 min at 4°C, and the supernatant was incubated with GST-RalGDS protein precoupled to glutathione-Sepharose beads as described for MCF7 cells. Densitometric analysis was performed with the Bio-Rad image acquisition system (Gel Doc 170), and paired Student t tests were performed for statistical analysis.
Cdc42 activation assay.
The level of Cdc42 activation was determined from MCF7 cell lysates with a GST fusion protein containing the Cdc42-interacting domain of WASP (WASP-CRIB), adapted as described previously (38). The WASP-CRIB in pGEX construct (generously provided by C. Wells and G. Jones, King's College London, London, United Kingdom) was expressed in Escherichia coli strain XL1-Blue. The GST-WASP-CRIB protein was purified and stored at −80°C.
Prior to each experiment, MCF7 cells were seeded at 2 × 106 cells onto a 9-cm dish. Cells were transiently transfected with 5 μg of pcDNA-Myc-Cdc42 (wild type) with or without 5 μg of pMT2-HA-RapGAP. After 24 h of transfection, the calcium switch was performed. Immediately after the calcium switch, the pulldown assay was performed as described above for the Rap1 activation assay except that 8 μg of GST-WASP-CRIB protein was precoupled to the beads and cells were lysed in a different lysis buffer (50 mM Tris-Cl [pH 7.5], 500 mM NaCl, 10 mM MgCl2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg ml−1 each leupeptin and aprotinin, 1 mM phenylmethylsulfonyl fluoride). Membranes were probed with anti-Myc antibody. Densitometric analysis was performed with the Bio-Rad image acquisition system (Gel Doc 170), and Student t tests assuming unequal variance were performed for statistical analysis.
RESULTS
Interaction between C3G and the cytoplasmic tail of E-cadherin.
With a yeast two-hybrid screen, we identified C3G, a GEF for Rap1, as a new binding protein for the cytoplasmic domain of E-cadherin. C3G has a catalytic domain at the C terminus and a proline-rich CRK-binding region in the middle portion (15) (Fig. 1A). The E-cadherin binding site is at the N terminus (amino acids 144 to 230), which is thought to act as a regulatory domain (18). Further analyses with several truncation mutants of the cytoplasmic domain of E-cadherin revealed that the C terminus of E-cadherin (amino acids 823 to 884), comprising the CH3 domain and the C-terminal end, is responsible for the interaction with C3G (Fig. 1B). Interestingly, the cytoplasmic domain of N-cadherin did not interact with C3G (Fig. 1B).
FIG. 1.

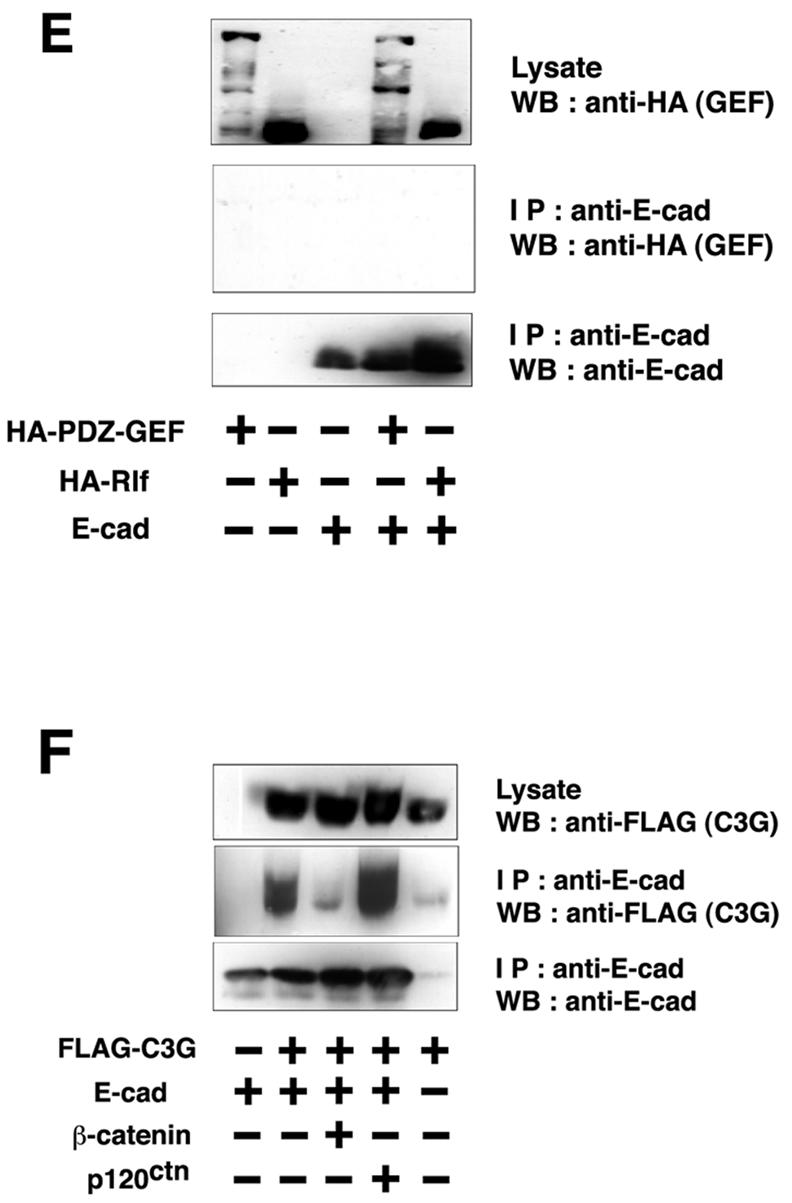
Interaction between C3G and E-cadherin. (A) Molecular domain structure of C3G. The clone isolated by the yeast two-hybrid screen encodes the N terminus of C3G (amino acids 144 to 230). (B) Summary of interactions between cadherins and C3G in yeast two-hybrid assays. The CH of CH2 and CH3 domains stands for cadherin homology (32). The CH2 and CH3 domains are conserved between classical cadherins and bind to p120ctn and β-catenin, respectively. (C) Interaction between C3G and E-cadherin by GST pulldown assay. Flag-tagged C3G (amino acids 1 to 357) expressed in HEK293 cells was pulled down by GST- or GST-E-cadherin (cytoplasmic domain)-coated beads. (D) Direct interaction between C3G and E-cadherin by overlay-blotting assay. Flag-tagged C3G (full length) expressed in HEK293 cells was immunoprecipitated, followed by SDS-PAGE and overlay-blotting assay with the purified recombinant E-cadherin protein (left panel). E-cadherin bound to C3G on the membrane was detected by anti-E-cadherin antibody. The same immunoprecipitated samples were also examined by Western blotting with anti-Flag antibody (right panel). The arrowhead indicates the position of immunoglobulin G heavy chains from anti-Flag antibody. (E) Specificity of the association between E-cadherin and other GEFs. E-cadherin and HA-PDZ-GEF or HA-Rlf were coexpressed in HEK293 cells, and the interaction was examined by immunoprecipitation with anti-E-cadherin antibody. (F) Competition between C3G and β-catenin for binding to E-cadherin. β-catenin or p120ctn was coexpressed with E-cadherin and Flag-C3G (amino acids 1 to 357), and the levels of C3G present in E-cadherin immunoprecipitates were examined.
To confirm the interaction between C3G and the cytoplasmic domain of E-cadherin, we performed GST pulldown assays. Flag-C3G(N) (amino acids 1 to 357) was transiently expressed in HEK293 cells, and the cell lysates were incubated with GST or GST-E-cadherin beads. C3G interacted with GST-E-cadherin beads but not with GST beads (Fig. 1C). The interaction was also detected by overlay-blotting assays. Flag-C3G (full length) immobilized on a membrane interacted with the recombinant E-cadherin protein (Fig. 1D), indicating that the interaction between C3G and E-cadherin is direct. The interaction was further examined by coimmunoprecipitation in transiently transfected HEK293 cells. C3G was coimmunoprecipitated with E-cadherin (Fig. 1F), but PDZ (PSD-95, Dlg, ZO-1)-GEF (another GEF for Rap) or Rlf (a GEF for Ral) was not (Fig. 1E). Since the interaction domain of E-cadherin for C3G overlaps that for β-catenin (see Fig. 1B), we examined whether C3G competes with β-catenin for binding to E-cadherin. The overexpression of β-catenin greatly reduced the amount of C3G bound to E-cadherin, whereas overexpression of p120ctn had no effect (Fig. 1F), indicating that C3G and β-catenin compete for the interaction with E-cadherin.
To further understand the molecular dynamics of the interaction between C3G and E-cadherin in epithelial cells, we performed calcium switch experiments. In low-calcium medium, E-cadherin-based cell-cell adhesions are disrupted and cells dissociate from one another. Restoration of normal calcium levels reinitiates and reestablishes E-cadherin-based cell-cell contacts. Furthermore, in low calcium, the majority of E-cadherin is endocytosed, while residual surface E-cadherin is spread evenly over the plasma membrane (24). After the addition of calcium, small E-cadherin aggregates are initially formed between the cells. E-cadherin is then further recruited into these puncta, which develop into a continuous line of mature E-cadherin-based cell-cell adhesions (2).
We examined the interaction between endogenous E-cadherin and C3G during a calcium switch experiment in MCF7 epithelial cells (Fig. 2A). In cells containing mature, established cell-cell contacts, no interaction between E-cadherin and C3G was detected (Fig. 2A, lane 2). However, binding of C3G with E-cadherin was induced in low-calcium medium when E-cadherin-dependent cell-cell contacts were lost (Fig. 2A, lane 3). The interaction was further increased when E-cadherin-based cell-cell contacts were initiated 15 to 30 min after calcium restoration (Fig. 2A, lanes 4 and 5) and then gradually decreased as these contacts matured (Fig. 2A, lanes 6 to 8). In contrast, the interaction between E-cadherin and β-catenin was not grossly affected during calcium switch (unpublished data), indicating that the interaction between C3G and E-cadherin occurs transiently or that a small proportion of E-cadherin interacts with C3G.
FIG. 2.

Interaction and localization of C3G and E-cadherin in MCF7 cells. (A) Interaction between endogenous C3G and E-cadherin. MCF7 cells were incubated in low-calcium medium and then transferred to normal-calcium medium for the indicated times. Coimmunoprecipitation of endogenous C3G with E-cadherin was examined. Lanes: 2, no calcium switch; 1 and 3, low calcium; 4 to 8, switch from low calcium to normal calcium (lane 4, 15 min; lane 5, 30 min; lane 6, 1 h; lane 7, 2 h; lane 8, 4 h). (B) Localization of C3G during induction of cell-cell contacts. GFP-tagged C3G (full length) was transiently expressed in MCF7 cells, and the localization of C3G during calcium switch was analyzed by time-lapse images. Upper panel, phase contrast images. Lower panels, GFP-C3G. Arrows indicate the positions of transfected cells.
The localization of GFP-tagged C3G was also examined during a calcium switch in MCF7 cells. Time-lapse analyses showed that C3G is diffusely distributed in the cytoplasm and on the plasma membrane and that the overall localization of C3G was not altered during the calcium switch (Fig. 2B and video 1 [www.ucl.ac.uk/1mcb/fujita/videos]). These data suggest that the interaction between E-cadherin and C3G is regulated not by the localization of C3G but by other molecular mechanisms.
Localization and activation of Rap1 in MCF7 cells.
The identification of the interaction between E-cadherin and C3G, a GEF for Rap1, prompted us to examine the relationship between Rap1 and E-cadherin-based cell-cell adhesion. To examine the localization of Rap1 in epithelial cells, we constructed pEGFP-Rap1 (wild type) to express GFP-tagged Rap1. First, we examined whether the addition of the GFP tag affects the molecular properties of Rap1 (Fig. 3A). GFP-Rap1 was coexpressed with RapGAP or C3G in HEK293 cells, and the cell lysates were used in GST-RalGDS pulldown assays (RalGDS preferentially binds to GTP-bound Rap1). GFP-Rap1 was inactivated by RapGAP and activated by C3G, as expected. Since GFP-Rap1 interacts with its regulatory and target proteins, we assume that the addition of a GFP tag does not affect Rap1 activity.
FIG. 3.

Recruitment of Rap1 at cell-cell contact sites. (A) Characterization of GFP-Rap1 (wild type). GFP-Rap1 was coexpressed with RapGAP or C3G, and active GTP-bound GFP-Rap1 was pulled down by GST-RalGDS beads. (B and D) Colocalization of Rap1 with E-cadherin. MCF7 cells stably expressing GFP-Rap1 were incubated in a low-calcium medium and transferred to a normal-calcium medium for the indicated time, and colocalization of Rap1 and E-cadherin was examined by epifluorescent microscopy (B; 30 min and 2 h) or by confocal microscopy (D; 2 h). (C) Magnified images of GFP-Rap1 and E-cadherin from Fig. 4B (middle top and middle center panels). Arrowheads indicate zipper-like structures of newly formed cell-cell contact sites. (E) Vertical section (XZ) analysis of the localization of GFP-Rap1 in MCF7 cells. (F) Effect of overexpression of RapGAP on localization of GFP-Rap1 after the calcium switch. RapGAP cDNA was microinjected into MCF7 cells stably expressing GFP-Rap1, and the localization of Rap1 was examined 30 min after calcium restoration. Bars, 30 μm.
Next, we produced MCF7 cells stably expressing GFP-Rap1 to examine the subcellular localization of Rap1 during calcium switch experiments (Fig. 3B and video 2 [same website as that for video 1]). In the absence of cell-cell contacts, Rap1 was localized at perinuclear regions and on the plasma membrane. Soon after calcium restoration, the homogeneous plasma membrane localization was lost and Rap1 was specifically recruited to and accumulated at cell-cell contact sites (video 2). Indeed, the localization of Rap1 at contact sites overlaid with that of E-cadherin (Fig. 3B and D). However, examination of the higher-magnification images revealed that Rap1 does not colocalize with E-cadherin at nascent cell-cell contact sites, such as at zipper-like structures (Fig. 3C, arrowheads), suggesting that Rap1 is recruited into relatively matured cell-cell contact sites. It should be noted that in actively congregating cells, Rap1 was localized at cell-cell contacts but not at the free-membrane (Fig. 3D). The vertical section (XZ) analyses showed that Rap1 is enriched at the lateral membrane domain, and little Rap1 was detected at apical or basal membranes (Fig. 3E). Interestingly, overexpression of neither RapGAP nor C3G(N) (amino acids 1 to 357) affected the localization of Rap1 at cell-cell contact sites after the calcium switch (Fig. 3F and data not shown).
We further examined the relationship between E-cadherin engagement and the localization of Rap1 with beads coupled with an Fc-tagged extracellular domain of E-cadherin (hE/Fc) (Fig. 4A). When the hE/Fc-coated beads made contact with MCF7 cells, β-catenin was recruited to the contact sites around the beads (Fig. 4A, lower panel, left), suggesting that the adherence of hE/Fc-coated beads to the cell surface induced the formation of new E-cadherin clustering sites (14). Similarly, Rap1 was also recruited to the cell-bead contact sites (Fig. 4A, lower panel, right), whereas integrin-β1 was not recruited (data not shown). BSA-coated beads did not induce clear recruitment of either β-catenin or Rap1 (Fig. 4A, upper panel). The level of GFP-Rap1 recruited to hE/Fc-coated beads was 1.4-fold higher (P > 0.001) than that recruited to BSA-coated beads, as determined by measuring the total pixel intensity around the bead. These data suggest that homophilic ligation of E-cadherin induces the specific recruitment of Rap1.
FIG. 4.

Effect of the establishment of E-cadherin-based cell-cell contacts on the localization and activation of Rap1. (A) Recruitment of Rap1 at E-cadherin-based cell-cell contact sites induced by the extracellular domain of E-cadherin (hE/Fc)-coated beads. BSA- or hE/Fc-coated beads were plated on MCF7 cells expressing GFP-Rap1. After 10 min, the recruitment of β-catenin or GFP-Rap1 to cell-bead contact sites was examined by confocal microscopy. The arrows indicate the positions of the beads. Bar, 30 μm. (B) Activation of Rap1 induced by hE/Fc beads. BSA-, hE/Fc-, or anti-integrin-β1 (CD29) antibody-coated beads were plated on MCF7 cells. Under the experimental conditions, approximately 70% of the cells were in contact with the beads. After 10 min, the cell lysates were used in a GST-RalGDS pulldown assay, followed by Western blotting with anti-Rap1 antibody. (C) Quantitative data for activation of Rap1 by the beads. The relative amount of active Rap1 in each sample was quantified by densitometry and corrected for the total Rap1 in the cell lysate. Results are expressed relative to the no-beads control value and represent the means ± standard deviation of five independent experimental determinations. *, significantly higher than the control (P < 0.05).
Using GST-RalGDS pulldown assays, we examined whether Rap1 is activated during the formation of E-cadherin-based cell-cell contacts induced by adherence of hE/Fc-coated beads (Fig. 4B and C). Rap1 activity was enhanced when cells were incubated with hE/Fc-coated beads (2.5-fold, P < 0.05) compared with that of nontreated cells. Incubation with BSA-coated beads did not significantly activate Rap1 (Fig. 4B and C). To exclude the possibility that increased Rap1 activation induced by hE/Fc-coated beads was merely due to firmer contacts between beads and cells, we used anti-CD29 (integrin-β1) antibody-coated beads as another control. Anti-CD29 antibody-coated beads adhered to cells as firmly as hE/Fc-coated beads did; however, this adherence did not significantly increase Rap1 activity (Fig. 4B and C). These data indicate that the formation of E-cadherin-based cell-cell contacts activates Rap1.
Involvement of active Rap1 in the establishment of E-cadherin-based cell-cell contacts.
To determine whether Rap1 is involved in the recruitment of E-cadherin to cell-cell contact sites, RapGAP, Rap1V12 (a constitutively active form), Rap1N17 (a constitutively inactive form), or the E-cadherin-binding domain of C3G [C3G(N); amino acids 1 to 357] was microinjected into MCF7 cells. Following a calcium switch (30 min after restoration of normal calcium levels), the effect of microinjection on the localization of E-cadherin was examined. As a control, an empty vector (pcDNA-Flag) was used. Microinjection of the empty vector did not affect the formation of E-cadherin-based cell-cell contacts compared with surrounding uninjected cells (data not shown) (14%: number of cell-cell contacts with incomplete E-cadherin localization/number of total contacts between microinjected cells [n = 78]; see details in Materials and Methods).
Expression of RapGAP prevented E-cadherin recruitment to newly formed cell-cell contact sites and inhibited the development of typical linear cell-cell contacts from the small E-cadherin clusters (69%, n = 104, P < 0.01) (Fig. 5A), suggesting that Rap activity is required to recruit E-cadherin into cell-cell contact sites. When RapGAP-microinjected cells were examined at later time points (e.g., 2 h after restoration of normal calcium), E-cadherin started to accumulate at cell-cell contact sites (data not shown), suggesting that there may be a Rap-independent mechanism for E-cadherin targeting. Without a calcium switch, the expression of RapGAP had little effect (data not shown). Taken together, these data indicate that Rap activity is required not for the maintenance of E-cadherin at mature cell-cell contacts but for the recruitment of E-cadherin into nascent cell-cell contact sites. Overexpression of RapGAP also had no effect on the process of endocytosis of E-cadherin during incubation in low-calcium medium (data not shown).
FIG. 5.

Role of Rap1 in E-cadherin localization. After microinjection, MCF7 cells were subjected to a calcium switch, and the effect of expression of different molecules on the localization of E-cadherin (A and B, 30 min after calcium restoration) or ZO-1 (C, 60 min after calcium restoration) was examined by immunofluorescence. The following cDNAs were microinjected: (A and C) RapGAP; (B) C3G(N) (amino acids 1 to 357). Bars, 30 μm.
Microinjection of constitutively active Rap1V12 also showed a similar but weaker inhibitory effect on E-cadherin accumulation (26%, n = 150, P < 0.05), while that of constitutively inactive Rap1N17 had no significant effect (18%, n = 100) (data not shown). Although Rap1N17 has been reported to act as a dominant negative protein in some assays, it has been shown to be incapable of inhibiting C3G-dependent exchange on Rap1 (37). The expression of the truncated mutant of C3G (amino acids 351 to 1078) lacking the E-cadherin binding site was toxic for MCF7 cells, and the effect could not be observed. However, the expression of the E-cadherin binding domain of C3G [C3G(N); amino acids 1 to 357] partially decreased E-cadherin recruitment to new cell-cell contact sites (52%, n = 87, P < 0.01) (Fig. 5B), suggesting a role for C3G in the formation of E-cadherin-based cell-cell contacts.
The recruitment of the tight junction protein ZO-1 to cell-cell contacts was not significantly affected by RapGAP (Fig. 5C) (For ZO-1, microinjection of RapGAP was 30% [n = 96] and microinjection of empty vector was 23% [n = 80]), indicating that E-cadherin and ZO-1 are recruited to cell-cell contact sites via distinct pathways.
Rap1 may function upstream of Cdc42 in the formation of E-cadherin-based cell-cell contacts.
Cdc42, a member of the Rho small GTPase family, has been reported to be involved in the formation of cadherin-based cell-cell contacts (6, 13). We examined whether Cdc42 is involved in the initial formation of E-cadherin-based cell-cell contacts and its possible relationship to Rap1. Expression of constitutively active Cdc42L61 by microinjection enhanced the recruitment of E-cadherin into newly formed cell-cell contacts (Fig. 6A). Furthermore, coexpression of Cdc42L61 completely blocked the inhibitory effect of RapGAP, allowing E-cadherin to be recruited to cell-cell contact sites (incomplete E-cadherin localization, 5%, n = 37, P < 0.01, significantly lower than that by RapGAP alone) (Fig. 6B). The expression of Cdc42L61 did not affect the RapGAP expression level and vice versa (data not shown). These data suggest that Cdc42L61 can rescue the inhibitory effect of RapGAP on E-cadherin targeting.
FIG. 6.

Role of Rap and Cdc42 in E-cadherin localization. (A and B) Constitutively active Cdc42 enhanced the recruitment of E-cadherin into newly formed cell-cell contacts. After microinjection, MCF7 cells were subjected to a calcium switch, and the effect of expression of different molecules on the localization of E-cadherin was examined 30 min after calcium restoration by immunofluorescence. The following cDNAs were microinjected: (A) constitutively active Cdc42 (Cdc42L61); (B) Cdc42L61 plus RapGAP. Bar, 30 μm. (C) Expression of RapGAP inhibited the activation of Cdc42 during calcium switch. Wild-type Cdc42 (Myc tagged) was transiently expressed with or without RapGAP in MCF7 cells. After the calcium switch, the cell lysates were used in a GST-WASP-CRIB pulldown assay, followed by Western blotting with anti-Myc antibody. The time indicates the time after the calcium switch. (D) Quantitative data for activation of Cdc42 during the calcium switch. The relative amount of active Myc-Cdc42 in each sample was quantified by densitometry and corrected for the total Myc-Cdc42 in the cell lysate. Results are expressed relative to the time zero value and represent the means ± standard deviation of several independent experimental determinations (n = 7 for 0, 30, and 60 min; n = 4 for 120 min). * and **, significantly higher without RapGAP than with RapGAP (*, P < 0.002; **, P < 0.0005).
We further studied the possibility that Cdc42 may act downstream of Rap1 in the formation of E-cadherin-based cell-cell contacts. Kim et al. reported that initiation of E-cadherin-based cell-cell contacts after the calcium switch increased the amount of active Cdc42 in MCF7 cells (20). They also showed that incubation with anti-E-cadherin antibody diminished this activation of Cdc42, indicating that the formation of E-cadherin-based contacts itself is involved in the activation of Cdc42 (20). With this system, we examined whether expression of RapGAP affects the activation of Cdc42. Wild-type Cdc42 (Myc tagged) was transiently expressed with or without RapGAP in MCF7 cells, followed by calcium switch treatment. The amount of active Myc-Cdc42 was examined by pulldown assays with the Cdc42-binding domain (CRIB) of WASP that preferentially binds to GTP-bound Cdc42. When Myc-Cdc42 was expressed in the absence of RapGAP, the amount of active Cdc42 increased significantly after 30 min of calcium restoration and reached a maximum after 60 min (Fig. 6C and D). In contrast, when Myc-Cdc42 was coexpressed with RapGAP, the increase in Cdc42 activity was strongly inhibited (P < 0.002 for 30 min, P < 0.0005 for 60 min; significantly higher without RapGAP than with RapGAP) (Fig. 6C and D). Thus, overexpression of RapGAP inhibits the activation of Cdc42 during the formation of E-cadherin-based cell-cell contacts, suggesting that Cdc42 functions downstream of Rap1.
Rap1 is not involved in the formation of N-cadherin-based cell-cell contacts.
Finally, we examined whether Rap1 is involved in the establishment of N-cadherin-based cell-cell adhesions. We used HEK293 cells, in which N-cadherin but not E-cadherin is highly expressed (Fig. 7A). The N-cadherin-based cell-cell contacts in HEK293 cells are not as compact as the E-cadherin-based cell-cell contacts in MCF7 cells, and HEK293 cells require a longer time (60 to 90 min) to reestablish cell-cell contacts after a calcium switch (data not shown). After the calcium switch, transiently expressed GFP-Rap1 was localized at perinuclear regions and cell-cell contact sites, and GFP-Rap1 and N-cadherin were colocalized at cell-cell contact sites (Fig. 7B). Interestingly, the expression of RapGAP did not affect the formation of N-cadherin-based cell-cell contacts after the calcium switch (Fig. 7C). C3G localized in the cytoplasm of HEK293 cells, and the expression of the N terminus of C3G (amino acids 1 to 357) did not affect N-cadherin-based cell-cell contacts (data not shown).
FIG. 7.

Localization and role of Rap1 in N-cadherin-based cell-cell contacts in HEK293 cells. (A) N-cadherin but not E-cadherin is expressed in HEK293 cells. Cell lysates from HEK293 and MCF7 cells were examined for expression of E-cadherin and N-cadherin by Western blotting. (B) GFP-Rap1 is colocalized with N-cadherin at cell-cell contact sites in HEK293 cells after a calcium switch. GFP-Rap1 was transiently expressed in HEK293 cells, and the localization of GFP-Rap1 and N-cadherin was examined 1 h after calcium restoration. (C) The expression of RapGAP does not affect the localization of N-cadherin at cell-cell contact sites after a calcium switch. RapGAP was transiently expressed in HEK293 cells, and the localization of N-cadherin was examined 1 h after calcium restoration. Bar, 30 μm.
DISCUSSION
Recently, the involvement of Rap1 in E-cadherin-based cell-cell contacts was genetically demonstrated in Drosophila melanogaster (21), although the molecular basis of this involvement was not revealed. In this study, we show that ligation of the extracellular domain of E-cadherin enhances Rap1 activity and that active Rap1 is necessary for the subsequent accumulation of E-cadherin at the newly formed cell-cell contact sites. We also found that C3G, a Rap GEF, interacts with E-cadherin during cell-cell contact formation and that overexpression of a truncated mutant form of C3G, C3G(N), partially inhibits the recruitment of E-cadherin to cell-cell contact sites. Based on these data, we propose the following model. When epithelial cells first contact one another, small clusters of E-cadherin appear at the nascent cell-cell contact sites. This homophilic ligation of E-cadherin induces the binding of C3G to the cytoplasmic tail of E-cadherin, which may in turn induce the activation of Rap1. Activation of Rap1 mediates the further recruitment of E-cadherin from the cytoplasmic or plasma membrane pool, facilitating the development of mature E-cadherin-based cell-cell contacts.
The inhibitory effect of C3G(N) overexpression on E-cadherin recruitment was weaker than that of RapGAP, suggesting that not only C3G-E-cadherin interaction but also other molecular mechanisms might be involved in the Rap1-mediated E-cadherin recruitment process. Recently, DOCK4, another Rap GEF, was shown to be involved in the formation of adherens junctions in osteosarcoma cells (39). Osteosarcoma cells with mutations in the DOCK4 gene fail to form adherens junctions and subsequently become invasive. However, it is not known whether DOCK4 is also involved in the formation of E-cadherin-based adherens junctions in epithelial cells. In addition, it has been shown that β-catenin interacts with novel proteins KIAA0313 and KIAA0705, which are homologous to Rap1 GEFs, Epac and S-SCAM, respectively (19). It is possible that not only C3G but also other Rap1 GEFs may be involved in the regulation of Rap1 activity during the formation of cell-cell contacts. Although activation of Rap1 is necessary for the formation of E-cadherin-based cell-cell contacts, overexpression of constitutively active Rap1 partially inhibits the formation of E-cadherin-based cell-cell contacts. This suggests that the activity of Rap1 should be temporally and spatially regulated and/or that cycling between GDP- and GTP-Rap1 is necessary.
In this study, we show that interaction between C3G and E-cadherin is increased during formation of new cell-cell contacts and decreased as cell-cell contacts mature. How is this interaction regulated? It is unlikely that the interaction is regulated by the localization of C3G, since time-lapse image analyses showed that the subcellular localization of C3G is not dramatically altered during a calcium switch (Fig. 2B and video 1). A more likely possibility is that the interaction is modulated by the competition between C3G and β-catenin for E-cadherin. It has been reported that the interaction between β-catenin and E-cadherin is regulated by phosphorylation of E-cadherin (16, 25). Phosphorylation of E-cadherin and/or of other molecules may affect the interaction between C3G and E-cadherin. E-cadherin interacts with the N terminus of C3G, which acts as an inhibitory domain suppressing the catalytic activity of the C terminus (18). Interaction with E-cadherin may enhance the activity of C3G by relieving the inhibitory function of the N terminus.
In epithelial cells, Rap1 is recruited at matured cell-cell contact sites but is absent at nascent contact sites. Importantly, overexpression of RapGAP inhibited the localization of E-cadherin but not of Rap1 at newly formed cell-cell contact sites. These data suggest that it is not the localization of Rap1 per se but the activity of Rap1 that is necessary for the formation of E-cadherin-based cell-cell contacts. However, the presence of Rap1 at cell-cell contacts may be involved in other unknown processes. Which molecules are involved in anchoring Rap1 to cell-cell contacts? One candidate is afadin/AF6, which colocalizes with E-cadherin at cell-cell contact sites and was shown to interact with GTP-Rap1 (35). However, an additional molecule(s) may also bind GDP-Rap1 at cell-cell contact sites. A very important question is where Rap1 is activated during the formation of cell-cell contacts. Live-cell imaging techniques, such as the fluorescent resonance energy transfer system, may help answer this question in the future.
How does active Rap1 target E-cadherin to cell-cell contact sites? We have shown that constitutively active Cdc42 can rescue the inhibitory effect of RapGAP on the formation of E-cadherin-based cell-cell contacts and that overexpression of RapGAP suppresses the activation of Cdc42 during cell-cell contact formation. These data indicate that Cdc42 may function downstream of Rap1 in this process, but the possibility cannot be ruled out that other pathways may also function downstream of Rap1. Cdc42 has been reported to regulate the trafficking of basolateral membrane proteins (23) as well as the formation of filopodia (27). Activation of Rap1 may induce activation of Cdc42, which in turn may facilitate the directional vesicle transport of E-cadherin and/or induce filopodium formation, enabling neighboring cells to contact one another.
Interestingly, a homologous system has been reported in Saccharomyces cerevisiae (7), where the determination of a new budding site is crucial for establishment of cell polarity. Initially, assembly of cortical patch proteins, including the transmembrane protein Axl2, marks the budding site. The cytoplasmic domain of Axl2 binds to Bud5, a GEF for Rsr1, the likely orthologue of Rap1 (26). Either deletion of the Rsr1 gene or introduction of a constitutively active Rsr1 causes randomization of the bud site, indicating its crucial role in bud site determination (3, 33). Indeed, it has been shown that Cdc42 is involved in bud formation and acts downstream of Rsr1 (8, 28). It is intriguing that the molecular mechanism regulating these early events in cell polarity may be at least partially conserved between S. cerevisiae and mammals.
Acknowledgments
We thank Alan Hall and Martin Raff (London) for critical reading of the manuscript. Sabine Wilhelm (Berlin) is acknowledged for technical help. We also thank Andrew Vaughan (London) for help with microscopic analysis, Julien Cau (London) for help with Metamorph software, Johannes L. Bos (Utrecht, The Netherlands) for providing various Rap constructs, Albert B. Reynolds (Nashville, Tenn.) for the pBS-p120ctn construct, Claire Wells and Gareth Jones (London) for the pGEX-WASP-CRIB construct, and Carien Niessen (Cologne, Germany) for providing stably transfected CHO cells expressing hE/Fc.
V.M.M.B. is an MRC Senior Research Fellow.
REFERENCES
- 1.Adams, C. L., and W. J. Nelson. 1998. Cytomechanics of cadherin-mediated cell-cell adhesion. Curr. Opin. Cell Biol. 10:572-577. [DOI] [PubMed] [Google Scholar]
- 2.Adams, C. L., W. J. Nelson, and S. J. Smith. 1996. Quantitative analysis of cadherin-catenin-actin reorganization during development of cell-cell adhesion. J. Cell Biol. 135:1899-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bender, A., and J. R. Pringle. 1989. Multicopy suppression of the cdc24 budding defect in yeast by CDC42 and three newly identified genes including the ras-related gene RSR1. Proc. Natl. Acad. Sci. USA 86:9976-9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bos, J. L., J. de Rooij, and K. A. Reedquist. 2001. Rap1 signalling: adhering to new models. Nat. Rev. Mol. Cell. Biol. 2:369-377. [DOI] [PubMed] [Google Scholar]
- 5.Braga, V. M. M. 2002. Cadherin adhesion regulation in keratinocytes, p. 1-36. In T. P. Fleming (ed.), Cell-cell interactions: a practical approach, 2nd ed. Oxford University Press, Oxford, England.
- 6.Braga, V. M. M. 2000. Epithelial Cell Shape: cadherins and small GTPases. Exp. Cell Res. 261:83-90. [DOI] [PubMed] [Google Scholar]
- 7.Chang, F., and M. Peter. 2003. Yeasts make their mark. Nat. Cell Biol. 5:294-299. [DOI] [PubMed] [Google Scholar]
- 8.Chant, J., K. Corrado, J. R. Pringle, and I. Herskowitz. 1991. Yeast BUD5, encoding a putative GDP-GTP exchange factor, is necessary for bud site selection and interacts with bud formation gene BEM1. Cell 65:1213-1224. [DOI] [PubMed] [Google Scholar]
- 9.Chappuis-Flament, S., E. Wong, L. D. Hicks, C. M. Kay, and B. M. Gumbiner. 2001. Multiple cadherin extracellular repeats mediate homophilic binding and adhesion. J. Cell Biol. 154:231-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke, B., J. W. Akkerman, and J. L. Bos. 1997. Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 16:252-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujita, Y., G. Krause, M. Scheffner, D. Zechner, H. E. Leddy, J. Behrens, T. Sommer, and W. Birchmeier. 2002. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 4:222-231. [DOI] [PubMed] [Google Scholar]
- 12.Fujita, Y., H. Shirataki, T. Sakisaka, T. Asakura, T. Ohya, H. Kotani, S. Yokoyama, H. Nishioka, Y. Matsuura, A. Mizoguchi, R. H. Scheller, and Y. Takai. 1998. Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron 20:905-915. [DOI] [PubMed] [Google Scholar]
- 13.Fukata, M., M. Nakagawa, S. Kuroda, and K. Kaibuchi. 2002. Effects of Rho family GTPases on cell-cell adhesion. Methods Mol. Biol. 189:121-128. [DOI] [PubMed] [Google Scholar]
- 14.Goodwin, M., E. M. Kovacs, M. A. Thoreson, A. B. Reynolds, and A. S. Yap. 2003. Minimal mutation of the cytoplasmic tail inhibits the ability of E-cadherin to activate Rac but not phosphatidylinositol 3-kinase: direct evidence of a role for cadherin-activated Rac signaling in adhesion and contact formation. J. Biol. Chem. 278:20533-20539. [DOI] [PubMed] [Google Scholar]
- 15.Gotoh, T., S. Hattori, S. Nakamura, H. Kitayama, M. Noda, Y. Takai, K. Kaibuchi, H. Matsui, O. Hatase, H. Takahashi, and et al. 1995. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol. Cell. Biol. 15:6746-6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huber, A. H., and W. I. Weis. 2001. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell 105:391-402. [DOI] [PubMed] [Google Scholar]
- 17.Hulsken, J., W. Birchmeier, and J. Behrens. 1994. E-cadherin and APC compete for the interaction with beta-catenin and the cytoskeleton. J. Cell Biol. 127:2061-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichiba, T., Y. Hashimoto, M. Nakaya, Y. Kuraishi, S. Tanaka, T. Kurata, N. Mochizuki, and M. Matsuda. 1999. Activation of C3G guanine nucleotide exchange factor for Rap1 by phosphorylation of tyrosine 504. J. Biol. Chem. 274:14376-14381. [DOI] [PubMed] [Google Scholar]
- 19.Kawajiri, A., N. Itoh, M. Fukata, M. Nakagawa, M. Yamaga, A. Iwamatsu, and K. Kaibuchi. 2000. Identification of a novel beta-catenin-interacting protein. Biochem. Biophys. Res. Commun. 273:712-717. [DOI] [PubMed] [Google Scholar]
- 20.Kim, S. H., Z. Li, and D. B. Sacks. 2000. E-cadherin-mediated cell-cell attachment activates Cdc42. J. Biol. Chem. 275:36999-37005. [DOI] [PubMed] [Google Scholar]
- 21.Knox, A. L., and N. H. Brown. 2002. Rap1 GTPase regulation of adherens junction positioning and cell adhesion. Science 295:1285-1288. [DOI] [PubMed] [Google Scholar]
- 22.Knudsen, B. S., S. M. Feller, and H. Hanafusa. 1994. Four proline-rich sequences of the guanine-nucleotide exchange factor C3G bind with unique specificity to the first Src homology 3 domain of Crk. J. Biol. Chem. 269:32781-32787. [PubMed] [Google Scholar]
- 23.Kroschewski, R., A. Hall, and I. Mellman. 1999. Cdc42 controls secretory and endocytic transport to the basolateral plasma membrane of MDCK cells. Nat. Cell Biol. 1:8-13. [DOI] [PubMed] [Google Scholar]
- 24.Le, T. L., A. S. Yap, and J. L. Stow. 1999. Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J. Cell Biol. 146:219-232. [PMC free article] [PubMed] [Google Scholar]
- 25.Lickert, H., A. Bauer, R. Kemler, and J. Stappert. 2000. Casein kinase II phosphorylation of E-cadherin increases E-cadherin/beta-catenin interaction and strengthens cell-cell adhesion. J. Biol. Chem. 275:5090-5095. [DOI] [PubMed] [Google Scholar]
- 26.Marston, A. L., T. Chen, M. C. Yang, P. Belhumeur, and J. Chant. 2001. A localized GTPase exchange factor, Bud5, determines the orientation of division axes in yeast. Curr. Biol. 11:803-807. [DOI] [PubMed] [Google Scholar]
- 27.Nobes, C. D., and A. Hall. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81:53-62. [DOI] [PubMed] [Google Scholar]
- 28.Park, H. O., P. J. Kang, and A. W. Rachfal. 2002. Localization of the Rsr1/Bud1 GTPase involved in selection of a proper growth site in yeast. J. Biol. Chem. 277:26721-26724. [DOI] [PubMed] [Google Scholar]
- 29.Perez-Moreno, M., C. Jamora, and E. Fuchs. 2003. Sticky business: orchestrating cellular signals at adherens junctions. Cell 112:535-548. [DOI] [PubMed] [Google Scholar]
- 30.Pokutta, S., and W. I. Weis. 2002. The cytoplasmic face of cell contact sites. Curr. Opin. Struct. Biol. 12:255-262. [DOI] [PubMed] [Google Scholar]
- 31.Ren, X. D., and M. A. Schwartz. 2000. Determination of GTP loading on Rho. Methods Enzymol. 325:264-272. [DOI] [PubMed] [Google Scholar]
- 32.Rimm, D. L., and J. S. Morrow. 1994. Molecular cloning of human E-cadherin suggests a novel subdivision of the cadherin superfamily. Biochem. Biophys. Res. Commun. 200:1754-1761. [DOI] [PubMed] [Google Scholar]
- 33.Ruggieri, R., A. Bender, Y. Matsui, S. Powers, Y. Takai, J. R. Pringle, and K. Matsumoto. 1992. RSR1, a ras-like gene homologous to Krev-1 (smg21A/rap1A): role in the development of cell polarity and interactions with the Ras pathway in Saccharomyces cerevisiae. Mol. Cell. Biol. 12:758-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaeper, U., N. H. Gehring, K. P. Fuchs, M. Sachs, B. Kempkes, and W. Birchmeier. 2000. Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J. Cell Biol. 149:1419-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takai, Y., and H. Nakanishi. 2003. Nectin and afadin: novel organizers of intercellular junctions. J. Cell Sci. 116:17-27. [DOI] [PubMed] [Google Scholar]
- 36.Takeichi, M. 1995. Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 7:619-627. [DOI] [PubMed] [Google Scholar]
- 37.van den Berghe, N., R. H. Cool, G. Horn, and A. Wittinghofer. 1997. Biochemical characterization of C3G: an exchange factor that discriminates between Rap1 and Rap2 and is not inhibited by Rap1A(S17N). Oncogene 15:845-850. [DOI] [PubMed] [Google Scholar]
- 38.Wells, C. M., M. Walmsley, S. Ooi, V. Tybulewicz, and A. J. Ridley. 2004. Rac1-deficient macrophages exhibit defects in cell spreading and membrane ruffling but not migration. J. Cell Sci. 117:1259-1268. [DOI] [PubMed] [Google Scholar]
- 39.Yajnik, V., C. Paulding, R. Sordella, A. I. McClatchey, M. Saito, D. C. Wahrer, P. Reynolds, D. W. Bell, R. Lake, S. van den Heuvel, J. Settleman, and D. A. Haber. 2003. DOCK4, a GTPase activator, is disrupted during tumorigenesis. Cell 112:673-684. [DOI] [PubMed] [Google Scholar]