

Abstract
Nucleosome composition actively contributes to chromatin structure and accessibility. Cells have developed mechanisms to remove or recycle histones, generating a landscape of differentially aged nucleosomes. This study aimed to create a high-resolution, genome-wide map of nucleosome turnover in Schizosaccharomyces pombe. The recombination-induced tag exchange (RITE) method was used to study replication-independent nucleosome turnover through the appearance of new histone H3 and the disappearance or preservation of old histone H3. The genome-wide location of histones was determined by chromatin immunoprecipitation–exonuclease methodology (ChIP-exo). The findings were compared with diverse chromatin marks, including histone variant H2A.Z, post-translational histone modifications, and Pol II binding. Finally, genome-wide mapping of the methylation states of H4K20 was performed to determine the relationship between methylation (mono, di, and tri) of this residue and nucleosome turnover. Our analysis showed that histone recycling resulted in low nucleosome turnover in the coding regions of active genes, stably expressed at intermediate levels. High levels of transcription resulted in the incorporation of new histones primarily at the end of transcribed units. H4K20 was methylated in low-turnover nucleosomes in euchromatic regions, notably in the coding regions of long genes that were expressed at low levels. This transcription-dependent accumulation of histone methylation was dependent on the histone chaperone complex FACT. Our data showed that nucleosome turnover is highly dynamic in the genome and that several mechanisms are at play to either maintain or suppress stability. In particular, we found that FACT-associated transcription conserves histones by recycling them and is required for progressive H4K20 methylation.
For cellular homeostasis, it is important to preserve epigenetic stability in dividing or resting cells and to allow the epigenetic landscape to be dynamic when conditions change or cells differentiate. Nucleosome composition contains epigenetic information in the form of post-translationally modified histones and histone variants (Jenuwein and Allis 2001), and this information is used to regulate activities at a particular locus. During DNA replication, as well as during other processes such as transcription and DNA repair, nucleosomal histones are replaced or diluted by the incorporation of new histones (for review, see Das and Tyler 2012). During replication, histone H3.1 is incorporated by the CAF1 complex, while in other phases of the cell cycle, histone H3.3 is incorporated during transcription by the chaperone complex HIRA in human cells (Tagami et al. 2004). Other chaperones, such as DAXX and ATRX, have similar specific functions, leading to a distinct histone exchange depending on the histone chaperone (Goldberg et al. 2010). In the fission yeast Schizosaccharomyces pombe, the genome encodes only one histone H3, but the role of multiple histone chaperones responsible for specific H3 deposition appears to be conserved from yeast to human (Kim et al. 2007; Rufiange et al. 2007). Newly synthesized histones have post-translational modifications that differ from their nucleosomal counterparts, notably acetyl groups at the N-terminal histone tails (Loyola et al. 2006; Alvarez et al. 2011). Following incorporation into nucleosomes, histones are modified as they mature, first by deacetylation and later by successive methylation (Scharf et al. 2009a). Some modifications require a full cell cycle to reestablish the conserved epigenetic pattern of the chromatin (Scharf et al. 2009a).
High histone turnover rates are primarily associated with nucleosomes close to promoters (Guillemette et al. 2005; Zhang et al. 2005; Jin and Felsenfeld 2007). Nucleosome-depleted regions (NDRs) are found surrounding the transcribed units, and the 5′ NDR in particular is well characterized (Lee et al. 2004; Jiang and Pugh 2009; Rando and Chang 2009; Radman-Livaja and Rando 2010). However, the following question remains: Are NDRs actually depleted of nucleosomes, or do they have very high histone turnover that makes nucleosomes difficult to detect from a technical standpoint? The first nucleosomes after the 5′ NDR are positioned just downstream from the transcription start site (TSS), or in yeast at the TSS, and they are often associated with histone modifications related to high nucleosome turnover: H4 acetylation (Scharf et al. 2009b), H3K4 methylation (Kraushaar et al. 2013), and/or the histone variant H2A.Z (Guillemette et al. 2005; Zhang et al. 2005; Jin and Felsenfeld 2007). Nucleosome turnover in promoter regions is directly affected by ATP-dependent chromatin remodelers such as Ino80, which binds to the 5′ NDR and promotes turnover of TSS nucleosomes (Yen et al. 2013). In mouse, Drosophila, and Saccharomyces cerevisiae, Chd1 has been found to promote the turnover of nucleosomes surrounding the 5′ NDR but suppresses turnover at the end of the gene (Radman-Livaja et al. 2012; Skene et al. 2014). In general, nucleosome turnover appears to correlate with gene transcription (Rufiange et al. 2007; Deal et al. 2010; Huang et al. 2013; Kraushaar et al. 2013).
Stable nucleosomes that have low histone turnover rates have been found in heterochromatin, marked by H3K9me2 in S. pombe (Aygün et al. 2013). Compared to promoter regions, gene bodies show relatively low levels of nucleosome turnover (Dion et al. 2007; Jamai et al. 2007; Rufiange et al. 2007; Deal et al. 2010; Skene et al. 2014). During transcription, nucleosomes are temporarily disassembled or displaced ahead of, and subsequently assembled behind, the transcription machinery, preferentially by reassembly of parental histones and, to a lesser extent, by the incorporation of newly synthesized histones (for review, see Owen-Hughes and Gkikopoulos 2012). Histone recycling in gene bodies depends on the protein complex facilitates chromatin transcription (FACT) (Belotserkovskaya et al. 2003; Jamai et al. 2009).
To summarize, the complex interplay between the different processes leads to a nonuniform pattern of nucleosome turnover, resulting in a mixture of histones of different ages throughout the genome (Dion et al. 2007; Jamai et al. 2007; Rufiange et al. 2007; Deal et al. 2010). The relative contribution of these processes and their dependency on chromatin context remain to be elucidated. Low nucleosome turnover results in the accumulation of post-translational histone modifications. Consecutive methylation of the H4K20 residue is dependent on nucleosome age (Scharf et al. 2009a), and different methylation levels at this residue are found at distinct nuclear loci (Sims et al. 2006). Partial genome-wide maps show that H4K20me1 is a repressive mark of transcribed genes, whereas H4K20me3 is found in repetitive elements in the human genome (Congdon et al. 2010). The overwhelming majority (>80%) of H4K20 is dimethylated in human cells (Pesavento et al. 2008; Evertts et al. 2013), and at least three different lysine methyltransferases (KMTs) that methylate H4K20 have been identified in mammalian cells (Wang and Jia 2009). In contrast, lysine demethylases (KDMs) have been discovered for H4K20me1 but not for H4K20me2/3 (Liu et al. 2010; Qi et al. 2010). Knocking out the gene encoding monomethyltransferase SETD8 in mouse or in Drosophila (PR-Set7) is lethal due to defects in cell cycle progression (Karachentsev et al. 2005; Sakaguchi and Steward 2007; Oda et al. 2009). In S. pombe, all three methylation states of H4K20 are mediated by a single methyltransferase, Set9, and no KDM has been identified. In contrast to higher organisms, deletion of the set9 gene does not affect gene transcription or heterochromatin formation (Sanders et al. 2004), and H4K20me3 is not associated with constitutive heterochromatin (Carneiro et al. 2010).
Given that no demethylase has been found to act on H4K20me2/me3 in metazoans or yeast (Wang and Jia 2009), we hypothesize that the H4K20me2/me3 marks are eliminated by nucleosome turnover. In S. pombe, given the absence of a KDM, all three H4K20 methylation marks are likely to correlate with nucleosome age, as proposed for Dot1-mediated H3K79 methylation in other organisms (De Vos et al. 2011). In addition, Set9 was recently found to regulate chronological aging in S. pombe (Sideri et al. 2014).
Several techniques have been used to determine nucleosome turnover (for review, see Deal and Henikoff 2010). Locus-specific nucleosome turnover is commonly measured by the incorporation of epitope-tagged H3 that is expressed using an inducible promoter (Iacovoni et al. 1999; Choi et al. 2005; Aygün et al. 2013; Kraushaar et al. 2013). However, histone expression at nonphysiological levels may be problematic in that histone transcription is normally highly regulated in a cell cycle–dependent manner, and histone expression is extremely low, except during S phase (Rustici et al. 2004). A more physiological alternative to ectoptic histone overexpression is the recombination-induced tag exchange (RITE) method, which uses the endogenous promoter of the gene of interest. This method presents an alternative to metabolic labeling of proteins (Deal et al. 2010). The RITE method measures the actual protein turnover, as it detects proteins synthesized before and after a genetic epitope tag switching during the same experiment (Terweij et al. 2013). This technique has been used in S. cerevisiae to track histone H3 during the cell cycle (Verzijlbergen et al. 2010) and to track proteins that mark intracellular compartments (Menendez-Benito et al. 2013).
In this study, we sought to understand how nucleosome age in S. pombe correlates with several chromatin marks with a focus on H4K20 methylation.
Results
Use of the RITE system to determine genome-wide, replication-independent nucleosome turnover
To adapt the RITE system for S. pombe, we targeted one of the three endogenous histone H3 loci (hht2+) using a construct containing a cassette with a hemagglutinin (HA) epitope tag flanked by two LoxP recombination sites and followed by a T7 tag (Fig. 1A). The tag switch is under the control of a hormone-dependent Cre recombinase. Using the endogenous H3 promoter and the RITE system, we can permanently swap epitope tags by adding the hormone β-estradiol to the growth medium and thereby distinguish HA-labeled H3 that is produced before Cre activation from T7-labeled H3 that is synthesized after the recombination event. To arrest and synchronize the cells at G2/M, we used cells that expressed a temperature-sensitive allele of cdc25 (cdc25-22). Cells were grown to mid-log phase at the permissive temperature and then arrested at G2/M for 3 h at the restrictive temperature. The chromatin was sampled at two time points: first, before the epitope switch was induced by addition of β-estradiol (t = 0 h); and second, after an additional 2 h at the restrictive temperature (t = 2 h). The recombination efficiency (t = 2) was 27 ± 6% (mean ± SEM, n = 3) as determined by a plating assay. The background switch rate, i.e., cells that acquired the T7 epitope without exposure to β-estradiol, was 1.6%. Chromatin immunoprecipitation using antibodies against the HA and the T7 epitopes revealed low incorporation of newly synthesized H3-T7 at the three S. pombe heterochromatin loci telomeres, mating type, and pericentric regions (Fig. 1B). In the coding regions of genes, the appearance of H3-T7 (Fig. 1C) was proportional to the transcription level (Fig. 1D). We investigated histone incorporation into the nucleosomes of four genes with different expression levels: rad50+ (encoding DNA repair protein Rad50); rad52+ (encoding DNA recombination protein Rad52, previously Rad22); spd1+ (encoding ribonucleotide reductase [RNR] inhibitor); and pyk1+ (encoding a predicted pyruvate kinase). For rad50+, two primer pairs were used to determine the differences between histone incorporation in the 5′ region and the coding sequence (CDS), and the CDS showed less incorporation of H3-T7 than did the 5′ end. The old H3-HA histones remained at 2 h both in the heterochromatin (Fig. 1F) and in the CDS of the rad52+ and rad50+ genes, which are expressed at low levels (Fig. 1G). In contrast, the level of HA-tagged H3 was drastically reduced in the spd1+ and pyk1+ genes, which are highly expressed, and in the 5′ region of rad50+.
Figure 1.

The RITE system that was used to measure genome-wide nucleosome turnover in S. pombe. (A) Model of the RITE genetic switch system. Green triangles represent LoxP sites. (B,C) T7 ChIP-qPCR analysis of heterochromatic regions (B) and genes (C). (D) RNA levels at five loci in growth-arrested (3 h at 36°C) cells with the RITE system (Hu2549 with cdc25-22 allele). (E,F) HA ChIP-qPCR of heterochromatin regions (E) and genes (F). Three independent experiments were performed, and the error bars show SEM. (G) A browser view of HA and T7 ChIP-exo results for five genomic loci before (0 h) and after (2 h) the genetic switch. Data show the average signal from two independent experiments. (H) The running average (window 500) of ChIP-exo signals of 150-bp chromatin fragments from H3-HA versus H3-T7 (RITE). (I) The running average (window 500) of ChIP-exo signals of 150-bp chromatin fragments from H3-T7 (RITE) versus pInv-H3-HA.
To investigate genome-wide nucleosome turnover, we performed ChIP followed by exonuclease digestion to trim the DNA fragments and massive parallel sequencing (ChIP-exo) (Rhee and Pugh 2011; Serandour et al. 2013; Rhee et al. 2014). This resulted in comprehensive maps of HA-tagged H3, which were synthesized before the genetic switch, and T7-tagged H3, which was produced after the genetic switch. Comparison with MNase-seq data (DeGennaro et al. 2013) revealed that ChIP-exo identified nucleosome peaks at a similar resolution (Supplemental Fig. S1A). A strand-specific analysis of just the first base in the sequence reads, i.e., the first base at which the histone H3-DNA crosslinking occurs, revealed that the histone-protected region consisted of 87 ± 5 bp (Supplemental Fig. S1B). We also observed the recently described asymmetry of the nucleosome dyads surrounding the NDR (Rhee et al. 2014; Ramachandran et al. 2015). The ChIP-exo data were used to identify locus-specific nucleosome turnover at high resolution across the genome (Fig. 1G).
To establish nucleosome turnover maps, we determined the differences between the ChIP signals from the 2- and 0-h samples for both epitope tags (ΔRPKM). A time-dependent increased T7 signal accompanied by a decreased HA signal was interpreted as a high turnover region. In contrast, old nucleosomes that were maintained in the chromatin >2 h were expected to have no increase in the T7 signal and also no decrease in the HA signal. A decrease in the HA signal was highly associated with an increase in T7-tagged H3 (Fig. 1H).
To compare the RITE method to the commonly used technique of ectopic expression of H3 under the control of an inducible promoter, we transfected cells with a plasmid encoding HA-tagged H3 under the invertase promoter (pInv) (Supplemental Fig. S1C; Iacovoni et al. 1999; Choi et al. 2005). Samples for ChIP-microarray analysis were taken 0 and 2 h after induction of H3-HA expression. To contrast the two different turnover measurement methods, we compared the normalized ChIP-exo signal for H3-T7 (RITE) with the ChIP-microarray signal from pInv-H3-HA (pInv). As expected, ectopically expressed H3-HA was also found in highly transcribed genes (Supplemental Fig. S1D). The two turnover methods showed high concordance from a genome-wide perspective (Fig. 1I). The major differences were found at the 5′ and 3′ NDRs: pInv-H3 histones were enriched, but H3 histones from the RITE system (H3-HA or H3-T7) were not (Supplemental Fig. S1E). In order to compare the two turnover methods in the same experimental detection technique, ChIP-microarray was used also in combination with the RITE method. Also with this ChIP-microarray, although with lower resolution than ChIP-exo, we detected the histones from the RITE system surrounding the NDR with H3-T7 close to promoters and H3-HA inside CDSs. Comparisons with a published data set generated using the pInv-H3 technique (Aygün et al. 2013) confirmed the overall similar results between the two histone exchange techniques (pInv-H3 and RITE) and also the incorporation of H3 from the pInv-H3 system at the NDRs (Supplemental Fig. S1F).
Replication-independent nucleosome turnover rates differ in distinct parts of the genome
We next used the RITE system to characterize the relationship between nucleosome turnover and transcription. The differences between ChIP signals before and after the epitope switch were determined as the histone H3 exchange rate and correlated with CDS and with the 5′ and 3′ untranslated regions (UTRs) of genes. Old nucleosomes were found mainly in CDS but not in 5′ and 3′ UTRs, whereas new nucleosomes were incorporated primarily at 5′ UTRs (Fig. 2A,B).
Figure 2.

Transcription-dependent nucleosome turnover is related to the position of nucleosomes within genes. (A,B) ChIP was used to measure nucleosome stability using HA epitope-tagged H3 (H3-HA; transcribed before the RITE switch) (A) or T7 epitope-tagged H3 (H3-T7; transcribed after the RITE switch) (B) in different regions of the transcript (5′ UTR, CDS, and 3′ UTR). Measurements were compared before (0 h) and after (2 h) β-estradiol addition. Bars show the average of two independent ChIP-exo experiments, and the error bars represent the range. (C,D) Heatmaps of nucleosomes at 2 h aligned at the transcription start site (TSS) for H3-HA (C) and H3-T7 (D). The top panels show genes that are expressed at intermediate levels (two to four transcripts per cell), and the bottom panels show genes with less than one transcript per cell. Short transcripts are found at the top of each panel and long transcripts are found at the bottom. The ChIP signal (RPKM for each 10 bp) is represented by color intensity. The white line marks the borders of the transcribed units. (E) Averaged ChIP signals (RPKM for each 10 bp) for epitope-tagged H3 aligned at the TSS and for all transcribed units in the genome. (F,G) The histone turnover score (normalized ΔΔRPKM) aligned at the TSS and transcription end site (TES) for all transcribed genes (F) and stratified according to transcription levels (G). (H) Average ChIP signal per transcribed unit for the H3-HA (old H3, top) and H3-T7 (new H3, bottom) ΔRPKM (2–0 h) according to the number of transcripts per cell. The running averages (window 500) are plotted against the average transcript levels within the window.
Examination of the nucleosome turnover dynamics in and around genes revealed a distinct pattern of high nucleosome turnover that peaked in regions flanking NDRs (Fig. 2C,D). The data were stratified according to the level of transcription based on previous transcription measurements (Marguerat et al. 2012). Low histone H3 exchange, evident from low signal for new histone H3 (T7-tagged), was especially pronounced in long genes (>2 kb) that were expressed at intermediate levels (Fig. 2D, top panel). In the same set of genes, the preservation of old histone H3 (HA-tagged) increased with length (Supplemental Fig. S1G). By aligning the ChIP signals from all genes at TSSs and calculating the average flanking ChIP signal (Fig. 2E), it became evident that the first two nucleosomes after the 5′ NDR showed high turnover, whereas subsequent nucleosomes were generally conserved within the 2-h window.
To summarize the different contributing signals as a single turnover value, we calculated a normalized ΔΔRPKM turnover score for each position of the genome (see Methods). This measure simplifies how turnover is represented (Fig. 2F). Stratifying the turnover scores based on transcription levels revealed that transcription played a role in nucleosome conservation (Fig. 2G). We omitted data from very highly expressed genes because low nucleosome occupancy confounds turnover measurement (note the seemingly low turnover at the NDR in Fig. 2F; Lee et al. 2004; Choi et al. 2012).
In contrast to our results, nucleosome turnover in genes has previously been reported to correlate positively with gene transcription. Accordingly, we further characterized the relationship between gene transcription and histone exchange. We determined the levels of new and old histones as a function of the transcription of genes that are expressed at similar levels in G2 and in unsynchronized cells. (Fig. 2H). These static-level G2 transcripts were defined as transcripts with levels that differed by <20% in G2 versus unsynchronized cultures (Rustici et al. 2004; Marguerat et al. 2012). The cells used in our turnover experiments were arrested in G2, as described above. The transcribed units were separated into UTR regions and CDS regions. Histone exchange of the 5′ UTR inversely correlated with gene expression. The CDS regions showed that histones were conserved in transcription except in very highly transcribed genes. At the end of the transcribed unit, i.e., in the 3′ UTR, we observed the same result as in previous studies, i.e., an increase in new histones that was proportional to the level of expression. These findings are consistent with transcription-coupled recycling of histones in CDS regions of genes with continuous expression.
The findings for genes with nonvarying G2 expression contrasted with the findings for genes that showed changed expression during the G2 stage of the cell cycle (Supplemental Fig. S2). As expected, when genes were transcriptionally regulated, they acquired new histones and lost old histones in all three regions (i.e., in the 5′ UTR, CDS, and the 3′ UTR), regardless of the absolute transcript levels.
Association of the nucleosome turnover rate with histone modifications
To systematically correlate nucleosome turnover measurements with the levels of chromatin markers that are associated with high or low turnover, we compared the data generated in this study to previous data sets. The analysis included published data sets for H2A.Z, H3K4me2, and H4K12ac, which represent histone variants and marks associated with supposedly rapid turnover of promoter nucleosomes. The analysis also included two markers of transcriptionally active regions, RNA polymerase II (Pol II) and monoubiquitinated H2B (H2Bub1). The histone modification H3K9me2 was used as a mark of heterochromatin. In addition to analyzing the levels of published marks associated with nucleosome turnover, we performed ChIP-tiling array experiments to determine the genome-wide patterns of the three H4K20 methylation marks, since the methylation state of this residue has been shown to correlate with nucleosome aging in Drosophila (Scharf et al. 2009b).
The genome was shattered in silico into nucleosome-sized fragments (87,000 DNA fragments, each 150-bp in size), and ChIP signals were calculated for all 150-bp fragments of the genome. We calculated the correlation coefficients between the different histone exchange rates, histone modifications, H2A.Z, and Pol II. The resulting correlation coefficients were subsequently hierarchically clustered (Fig. 3A). The main split in the data set clearly coincided with elements that were previously associated with different turnover rates. Within each cluster, there were high correlations (PCC > 0.5) between several elements, such as for H3K4me2, H2A.Z, and H4K12ac, for H3K9me2, H2Bub1, and for the three methylation states of H4K20. The new (T7-tagged) H3 histones did not correlate with any of these elements but rather correlated inversely with H4K20me3 (PCC: −0.2), H4K20me2 (PCC: −0.2), and to some extent with H3K9me2 (PCC: −0.1) and H2Bub1 (PCC: −0.1). H3-HA correlated very slightly with H4K20me3 (PCC: 0.1) and H4K20me2 (PCC: 0.1). Notably, slow turnover elements, as defined by inverse correlation with H3-T7, comprised features associated with condensed chromatin, such as the heterochromatin marks H3K9me2 and H4K20me2/me3. The highest correlation between chromatin features in the data set was observed between H4K20me2 and H4K20me3 (PCC: 0.83). Slightly different patterns emerged when we repeated the analysis using different functional regions of the genome, such as intergenic regions, UTRs (5′ and 3′), and CDS (Supplemental Fig. S3). Notably, at the CDS, H3-H7 correlated inversely with H4K20me2/me3 and Pol II, in addition to H3K9me2. This is consistent with Pol II–mediated transcription leading to low nucleosome turnover, and given the exclusion of H4K20 methylation at heterochromatin (Carneiro et al. 2010), H4K20me2/me3 potentially associated with the transcribed genome.
Figure 3.
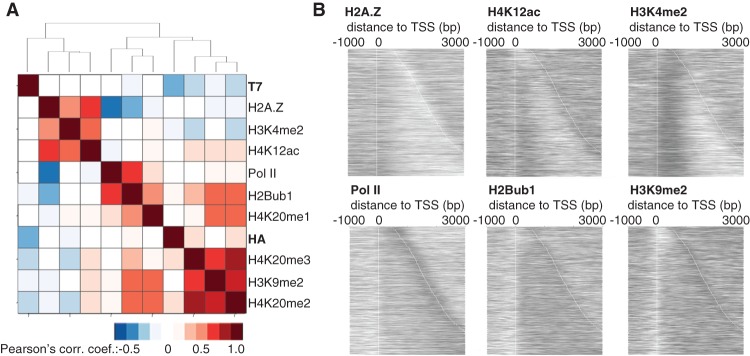
Correlation between chromatin modifications and nucleosome turnover. (A) Hierarchical clustering of the correlation coefficients from the ChIP signals for 150-bp genomic fragments. All ChIP-microarray samples were normalized to input (log2). In the ChIP-exo turnover data, i.e., H3-HA (RITE) and H3-T7 (RITE), data from the 2-h samples are relative to the 0-h time point. (B) Heatmaps of the ChIP signals (log2) from the antibody against indicated protein/protein modification at genes that were expressed at intermediate levels (two to four transcripts per cell).
To account for the apparent differences we found in nucleosome turnover using supposedly similar marks, we examined the relationship of the signals of the six previously published data sets for H2A.Z, H4K12ac, H3K4me2, Pol II, H2Bub1, and H3K9me2 with individual genes that were expressed at intermediate levels (Fig. 3B). H3K4me2 is a strong mark for the first ∼500 bp of all genes, but the H2A.Z signal is found both upstream of and downstream from the TSS in a subset of genes. The antibody used for Pol II ChIP largely recognized the elongating form of the polymerase, and H2Bub1 was evenly distributed throughout the gene body. These results demonstrate that neither these chromatin modifications nor Pol II alone should be used as proxies for nucleosome turnover.
Set9-mediated H4K20 methylation in protein-coding regions
H4K20me2/me3 correlated inversely with H3-T7, and H4K20me2/me3 and H3-HA were found in the same branch of the hierarchical cluster tree. This indicates that H4K20me2/me3 may be markers of old nucleosomes; accordingly, we wished to further characterize the role of H4K20 methylation in aging nucleosomes. In cells lacking the Set9 methyltransferase (set9Δ), H4K20 was only present in its unmethylated state (Fig. 4A), confirming both the specificity of the antibodies and the lack of additional methyltransferases acting on H4K20 in S. pombe. Initial ChIP results revealed that while the H4K20me1 levels were largely unaffected by transcription, the H4K20me3 levels were reduced with increasing transcription (Fig. 4B). Transcript levels in wild-type (WT) cells were obtained from the literature (Marguerat et al. 2012). The methylation states of H4K20 were mapped in a genome-wide fashion in both WT and set9Δ cells. A principal component analysis showed that the three set9Δ samples clustered with the input samples, as expected (Supplemental Fig. S4A). This confirmed previous results (Carneiro et al. 2010) and was consistent with Set9 being the only KMT acting on H4K20 in S. pombe. Thus, by normalizing the H4K20 methylation levels in the WT to the levels in set9Δ for each methylation-specific antibody, we could avoid any technical artifacts introduced by differences in antibody specificity to obtain accurate relative measurements for the different Set9-mediated methylation states of H4K20. In contrast to H4K20 methylation in mammalian cells, S. pombe heterochromatin was devoid of all three methylation states of H4K20 (Supplemental Fig. S4B). Again, this observation was consistent with previous results using marker gene insertions (Carneiro et al. 2010).
Figure 4.

H4K20 methylation is found in protein-coding regions of the genome. (A) Western blot analysis of wild-type (WT) cells (lane 1) and set9Δ cells (lanes 2,3 differ in the amount of chromatin loaded). (B) ChIP-qPCR analysis of H4K20me1 and H4K20me3 at five loci that have different transcription levels. Bars show the average and SEM of three independent experiments. (C) The average pattern of H4K20 methylation marks aligned with the TSS (left) or TES (right). (D) Patterns of H4K20me1 (left), H4K20me2 (middle), and H4K20me3 (right) aligned at the TSS, with genes sorted according to length (top, short genes; bottom, long genes). (E) Metagene analysis of the levels of H4K20me1 (top), H4K20me2 (middle), and H4K20me3 (bottom), stratified according to gene expression level.
Next we examined the H4K20 methylation status according to gene transcription level and gene length. We found that methylated H4K20 was present predominantly at the borders of transcribed units (H4K20me1) or further into and throughout the coding regions (H4K20me2/me3) (Fig. 4C). The H4K20me1 peak was downstream from, but did not overlap, the TSS and was distinct from the peaks of the higher methylation states, which were further away from the TSS and decreased less rapidly. Near the transcription end site (TES), H4K20 was preferentially monomethylated, but interestingly, the methylation degree increased with increasing distance to the TES. This appeared to depend on transcription, as the H4K20me1 signal was increasingly pronounced further from the TES in highly transcribed genes (Supplemental Fig. S4C).
To uncover more patterns in the data set, we plotted heatmaps of ChIP signals for individual genes according to gene length (Fig. 4D). In a complementary analysis, we normalized the ChIP data to nucleosome levels as measured by H4 ChIP (Supplemental Fig. S4D). With respect to gene length, H4K20me1 seemed unaffected; H4K20me2 was found throughout transcribed units >1 kb; and the H4K20me3 signal increased proportionally with transcript length, with the long genes (>2 kb) having the highest H4K20me3 levels (Supplemental Fig. S4E). To avoid bias due to different transcription levels, we only compared genes that showed similar levels of transcription (two to four transcripts per cell).
In terms of transcription activity, H4K20me1 was found at the 5′ regions of genes expressed at all levels, except for genes with the very highest level of transcription (more than 10 transcripts per cell). However, the H4K20me1 signal was found throughout the entire coding region of highly expressed genes (more than four transcripts per cell) (Fig. 4E). The strongest signals for H4K20me3 were found in the CDS regions of genes that showed low-to-intermediate expression levels (less than four transcripts per cell). In contrast, the most active genes (more than 10 transcripts per cell) were devoid of H4K20me2/me3 (Fig. 4E). These results were consistent with the ChIP-qPCR results (Fig. 4B).
H4K20 methylation at inducible genes
To determine if a subset of genes was associated with H4K20 methylated nucleosomes, we analyzed genes that had high levels of each mark (ChIP signal log2 [WT/set9Δ] > 0.5) (Supplemental Table S1) for enrichment in gene ontology (GO) categories (threshold, twofold enrichment; P-value 0.05; and at least five genes in the selection) (Supplemental Tables S2–S4). This analysis revealed some specific categories (less than 100 members) and the involvement of H4K20me1/me2-marked genes in a diverse set of functions. For example, the H4K20me1-associated genes were involved in large molecule metabolism. However, the H4K20me3-associated genes showed a more specific and focused pattern (Supplemental Table S5). The molecular functions that distinguished most of the H4K20me3-associated genes appeared to cluster around tightly regulated genes, such as those encoding proteins that regulate the cell cycle—especially meiotic division but also mitotic division—and proteins involved in the response to DNA damage. To determine if the enrichment was specific to H4K20me3 or was simply an effect on genes that were expressed at low levels, we compared this list to a GO analysis that involved the same number of genes with the same expression levels (Supplemental Table S6). This comparison confirmed that the categories related to DNA repair and cell cycle were specific for the H4K20me3-associated genes. We extracted the genes associated with the GO terms “Response to DNA Damage Stimulus” (GO:0006974, n = 217) and “Meiosis” (GO:0007126, n = 195). As expected, these categories showed an association with high H4K20me3 levels in the CDS (Fig. 5A,B). Compared to the average enrichment signal of 3.4 for all protein-coding genes (at TSS + 1000 bp), these genes had enrichment signals of 7.5 (“Responses to DNA Damage Stimulus”) and 6.6 (“Meiosis”) at the same positions relative to the TSS. As expected from H4K20 methylations accumulating in regions of low turnover nucleosomes (Fig. 1G), the H4K20 methylation pattern showed that the 5′ region of rad50+ contained H4K20me1, whereas further into the CDS, chromatin was associated with H4K20me2/me3 (Fig. 5C). In contrast, the spd1+ locus was depleted of H4K20me2/me3 (Fig. 5C).
Figure 5.

Subsets of inducible genes are associated with H4K20me3. (A) The average H4K20me ChIP signals in genes returned using the gene ontology terms “Response to DNA Damage Stimulus” and (B) “Meiosis” aligned according to the TSS. (C) ChIP data of the different H4K20 methylations (me1/me2/me3) at the spd1 and rad50 loci. Gray boxes indicate annotated features transcribed to the right (above solid line) or left (below solid line). (D) Venn diagram shows the overlap between genes associated with H4K20me3 (log2 [WT/set9Δ] > 0.5) and genes that are down-regulated in pht1Δ and swr1Δ cells. (E) A model of the relationship between transcription and FACT-mediated nucleosome recycling. High-turnover nucleosomes are marked in yellow.
To gain more information about the relationship of nucleosome turnover with genes that have particular functions, we also compared the H4K20 methylation-associated genes with a database of gene lists from other studies, with a focus on genes involved in the cell cycle and stress responses (Sadeghi et al. 2011). Two database gene lists were highly enriched with genes in the H4K20me3 gene list. These lists contained genes that require H2A.Z for their induction, i.e., genes that were down-regulated in pht1Δ (29.7% of genes in H4K20me3 list) and swr1Δ (18.7% of genes in H4K20me3 list) (Fig. 5D).
Transcription-mediated histone conservation allows for H4K20 methylation
Our data showing transcription-dependent accumulation of H4K20 methylation suggested that transcription has a histone-conserving role (Fig. 5E). Others have shown that the Spt16 subunit of the FACT complex plays such a role for H3/H4 (Jamai et al. 2009). To test whether FACT-dependent recycling is required for histone conservation and H4K20 methylation, we determined the levels of H4K20me1 and H4K20me3 in cells with compromised Spt16. Using an earlier data set (Choi et al. 2012), we confirmed that in very highly transcribed genes (more than 10 transcripts per cell), H3 occupancy is relatively low throughout the transcribed unit (Fig. 6A). This was evident in WT cells but was more pronounced in spt16-18 cells grown at the restrictive temperature. In the gene body of genes that are expressed at lower levels, the H3 density was only slightly different in spt16-18 cells compared with WT cells. However, in cells depleted of functional Spt16, both NDRs were reduced or eliminated as the regions flanking the transcribed units were occupied by H3 (Fig. 6B).
Figure 6.

Transcription-mediated nucleosome conservation is required for the accumulation of H4K20 methylation. (A,B) Histone H3 density in WT cells (A) and changes in histone H3 density in spt16-18 cells relative to WT cells (B) and grown at permissive temperature (25°C) followed by 1 h at the restrictive temperature (36°). Data were stratified according to the average number of transcripts per cell during the vegetative growth phase. (C) ChIP-qPCR analysis of H4K20me1 at five genomic loci in spt16-18 cells and WT cells. (D) ChIP-qPCR analysis of H4K20me3 at five genomic loci in spt16-18 cells and WT cells. (E) Metagene analysis of Spt16 levels in WT cells. (F) ChIP-qPCR analysis of H4K20me1 at five genomic loci in htb1-K119R cells and WT cells. (C,D,F) Bars show the average of three independent experiments, and error bars show the SEM.
To determine whether the H4K20 methylation failed to appear on nucleosomes that no longer are recycled in spt16-18 cells, we determined the levels of H4K20me1/me3 at five genomic loci with different transcription levels (Fig. 6C,D). Nucleosome turnover was investigated previously at these loci using RITE (Fig. 1). As expected, the H4K20me1 mark was reduced in the transcribed genes in spt16-18 cells compared with WT, except for the rad50+ locus, which shows the lowest level of transcription (Fig. 6C). The H4K20me3 mark showed the same pattern (Fig. 6C). For pyk1+, which is highly transcribed, the low signal could be explained by nucleosome loss (Supplemental Fig. S5A). The nucleosome density at the other three loci was comparable in spt16-18 cells and WT cells and was thus not affected by the compromised Spt16 (Supplemental Fig. S5A). Differences in the transcription levels in the different strains and growth conditions did not explain the differences in H4K20 methylation (Supplemental Fig. S5E). Further, Spt16 was found at the four euchromatic loci (detected by ChIP) (Supplemental Fig. S5A–D), where it enables nucleosome recycling. The levels of Spt16 correlated with transcription level even for the most highly transcribed genes (Fig. 6E).
FACT acts in conjunction with monoubiquitinated H2B (H2Bub1) under certain conditions (Pavri et al. 2006; Tanny et al. 2007; Fleming et al. 2008). We therefore investigated whether a defect in H2B monoubiquitination affected the H4K20 methylation status of genes. At genes that are transcribed at intermediate to high levels (rad52+ and spd1+), the H4K20me1 levels were reduced in H2Bub1-deficient htb1-K119R cells but appeared unchanged in the spt16-18 cells. Notably, the H4K20me1 levels were increased at the highly transcribed gene pyk1+ in htb1-K119R cells (Fig. 6F). The H4K20me3 levels were not significantly affected by the loss of H2Bub1 (Supplemental Fig. S6). The nucleosome density appeared to be less affected at highly transcribed genes in the H2Bub1-deficient mutant than in the spt16-18 mutant. In contrast to spt16-18 cells, nucleosome occupancy in htb1-K119R cells was drastically reduced at the promoter region, relative to WT cells (Supplemental Fig. S6). As described previously (Sadeghi et al. 2014), the H2Bub1 mark increased with increasing transcription levels but was not as high as Spt16 at very highly transcribed genes but reached a plateau after intermediate levels of transcription (more than four transcripts per cell) (cf. Supplemental Fig. S6 and Fig. 6E). Taken together, these results show that H2Bub1 and FACT play different roles in H4K20 methylation and, by extension, play different roles in nucleosome turnover.
Discussion
Here we present for the first time a comprehensive high-resolution nucleosome turnover map for the fission yeast S. pombe. We utilized an epitope-tag exchange system to precisely locate endogenously expressed old and new histones at subnucleosomal resolution using ChIP-exo methodology. We also mapped the three methylation states of H4K20 throughout the genome and showed that the methylation status of H4K20 inversely correlated with nucleosome turnover in the protein-coding portion of the genome.
Transcription-mediated histone conservation
Two chromatin compartments, i.e., heterochromatin and the Pol II-transcribed genome, were associated with overall low nucleosome turnover in our data set. Turnover was particularly low in regions of the genome that translates into proteins (CDS) and was high in nucleosomes immediately flanking NDRs. The stability of nucleosomes in the heterochromatin was previously described (Aygün et al. 2013), as was the high turnover of nucleosomes at promoter regions (Guillemette et al. 2005; Zhang et al. 2005; Jin and Felsenfeld 2007; Rufiange et al. 2007). However, this study revealed several new features of nucleosome turnover in transcribed regions.
Our data clearly showed that histone turnover is determined by two opposing effects: the incorporation of new histones and the preservation of old histones. These two effects are not strictly dependent on each other given the possibility of simultaneous change in nucleosome density. Based on these data, we propose a model that explains various aspects of the intricate machinery underlying transcription-coupled nucleosome turnover (Fig. 5E).
In this model, nucleosomes are temporarily disassembled as Pol II transcribes genes in chromatin (Belotserkovskaya et al. 2003; Kaplan et al. 2003; Saunders et al. 2003). Immediate nucleosome reassembly minimizes the amount of exposed DNA, thus repressing intragenic transcription and initiation from cryptic sites (Reinberg and Sims 2006; Carvalho et al. 2013). The FACT complex disassembles the H2A/H2B dimer and reassembles the four core histones (Belotserkovskaya et al. 2003; Schwabish and Struhl 2004). In addition, Spt16 is required in S. cerevisiae for nucleosome spacing within transcribed units (Duina et al. 2007). Here we showed that transcription-coupled nucleosome stability depends on FACT as well as on the level of transcription.
A mechanism is needed to close the gaps introduced by nucleosomes that are actively or spontaneously displaced. We speculate that under normal growth conditions, there is sliding of the internal “old” nucleosomes and replenishment from the edges of the transcribed regions, creating 5′ and 3′ NDRs. As a direct consequence, the NDRs become less pronounced in cells with dysfunctional histone recycling, such as spt16-18 cells (Fig. 5). ATP-dependent chromatin remodelers regulate nucleosome dynamics at the promoter regions surrounding the TSS, but to our knowledge, no chromatin remodeler has been reported at the TES. As mentioned earlier, in mouse, Drosophila, and S. cerevisiae, the remodeling protein Chd1 primarily acts around the TSS but also appears to stimulate histone recycling in the gene body during Pol II elongation (Radman-Livaja et al. 2012; Skene et al. 2014).
The data in this study suggested that at genes that are continuously transcribed at low to intermediate levels, efficient nucleosome recycling maintains the original nucleosomes in the CDS during Pol II passage, with only occasional histone replenishment at the edges of genes. However, the nucleosome density declines at genes that have higher levels of transcription (Lee et al. 2004). Upon completion of a transcription burst (Raj et al. 2006), incorporation of newly synthesized histones result in high nucleosome turnover in the CDS of very highly transcribed genes.
The role of H4K20 methylation
As for methylation of H4K20, given the progressive nature of Set9 and the lack of a demethylase, this modification was expected to accumulate on histones over time (Wang and Jia 2009; De Vos et al. 2011). In agreement with this prediction, H4K20me3 was detected inside the CDS of genes transcribed at low levels, particularly in long genes (>2 kb). Our data showed a correlative but not necessarily causal relationship between H4K20 methylation and nucleosome age in euchromatic regions. Interestingly, Set9 was not recruited to stable heterochromatin nucleosomes in S. pombe. Instead, the highest levels of H4K20me3 were found in repressed genes that were linked to essential cellular functions and that were regulated via H2A.Z and rapidly inducible upon changes in growth conditions. The cellular function of H4K20 methylation may not regulate nucleosome stability but instead be a mere consequence of nucleosome stability. Indeed, H4K20 methylation is involved in the selection of the DSB repair pathway by stabilizing the interaction between Crb2 and gamma-H2A (Sanders et al. 2004; Wang et al. 2009; Carneiro et al. 2010). Crb2 acts by inhibiting homologous recombination repair and interacts with H4K20me1/me2, but not with H4K20me3. However, as a proxy of nucleosome age, measuring the methylation of H4K20 can extend the time window to determine nucleosome turnover for more than one cell cycle, which otherwise poses natural limitations on studies of replication-independent turnover.
The RITE technique
The RITE technique is a highly versatile and reliable tool for studying protein replacement (Verzijlbergen et al. 2010; Menendez-Benito et al. 2013; Terweij et al. 2013). Here we adapted it to study histone turnover, but it can be used to study any protein with a relatively short mRNA half-life. mRNA instability makes the genetic switch rapid, and histones are ideal in this respect because of their nonpolyadenylated transcripts (Marzluff et al. 2008). There are a few limitations to keep in mind about our turnover calculations. Calculations of H3-HA expression after the genetic switch showed low levels of transcription, as would be expected in G2 cells (data not shown). More importantly, the turnover measurements underestimated the actual turnover rate, as the genetic switch at 2 h was incomplete (∼30%), resulting in continued H3-HA expression at 2 h in many of the cells. Further, we tagged just one of the three identical H3 genes. Nevertheless, even with just one tagged H3 gene and incomplete suppression of H3-HA expression on a population basis, the technique was sensitive enough, especially when used in combination with ChIP-exo, to unambiguously determine nucleosome turnover. One intrinsic limitation of tagging H3 is that we do not get any information about nucleosome turnover in regions where H3 is, to a large extent, replaced by the centromeric H3 variant Cnp1, such as in the central domain of the centromere. Despite these limitations, the RITE tool is a more physiologically relevant tool than ectopic protein expression, which has been used previously by others and by us to estimate nucleosome turnover (Aygün et al. 2013; Kraushaar et al. 2013; Sadeghi et al. 2014).
The major difference in the results we obtained using the RITE tool versus ectopic protein expression was that ectopically expressed histones bound to NDRs, probably as a consequence of histones having an affinity for DNA. That is, when histones are present in excess, mechanisms that deplete them from supposedly open chromatin structures become exhausted. This potential artifact can be reduced by MNase digestion of the ChIP samples, as reported recently (Aygün et al. 2013). In contrast to pInv-H3-HA, the RITE H3-T7 nucleosomes did not become enriched at the NDRs. The RITE and pInv systems showed concordant results at heterochromatic regions and within transcribed genes. By combining the RITE technology, which we used to examine endogenous levels of proteins, with the subnucleosome resolution of ChIP-exo, we were able to determine histone turnover with unprecedented precision. By further linking the actual turnover measurements to the chromatin modification of H4K20, we extended the window in which we can study the aging of nucleosomes.
To summarize, our data showed that nucleosome turnover is highly dynamic in the genome and that there are several mechanisms at play that either maintain or suppress stability, depending on the chromatin function. In particular, we found that FACT-associated transcription conserves histones by recycling them and is required for progressive H4K20 methylation.
Methods
Strains and plasmids
Strains are listed in Supplemental Table S7. H3.2 RITE strain was constructed by PCR amplification of the HA/T7 RITE cassette (Verzijlbergen et al. 2010) followed by integration at the endogenous locus by homologous recombination. The Cre-EBD open reading frame was PCR amplified from pTW040 (Verzijlbergen et al. 2010) and recloned in pRAD15 vector (gift from Y. Watanabe) under an attenuated adh1 promoter. The plasmid was integrated at the ala1 locus at the MluI site. Strains were grown in YES or PMG media with the appropriate supplements.
RITE
Cells were cultured in YES media to mid-log phase (∼5 × 106 cells/mL) at 25°C. Cells were arrested in G2/M by incubation for 3 h at 36°C. Samples were taken for t = 0 (G2/M-arrested cells before switch). The genetic switch was induced by 1 µM β-estradiol for 2 h at 36°C. Samples were taken for t = 2 h (G2/M-arrested cells, after switch).
pInv-H3-HA
Cells transfected with the pInv-H3HA plasmid were grown to mid-log phase at 30°C, after which they were glucose shocked (8%) for 4 h, and then the growth medium was replaced to a high-sucrose (4%) medium to induce expression from the invertase promoter. Cells were spun down and harvested (t = 0 h), or cultures were continued in fresh sucrose containing media and grown for an additional 2 h, after which cells were harvested (t = 2 h).
ChIP
Samples were immediately subjected to formaldehyde crosslinking (final concentration, 1%), and chromatin was isolated according to a method previously described (Durand-Dubief and Ekwall 2009). Antibodies against H4K20me1 (Abcam ab9051), H4K20me2 (Abcam ab9052), H4K20me3 (Abcam ab9053), HA (Abcam ab9110), T7 (EMD chemicals 69522), H3cter (Abcam ab1791), and H4 Pan (Millipore 05-858, clone 62-141-13) were used.
ChIP-qPCR or ChIP-microarrays
ChIP-qPCR and ChIP-microarray was performed as previously described (Durand-Dubief and Ekwall 2009). Briefly, ChIP-recovered DNA was fragmented, labeled, and hybridized to microarrays (Affymetrix GeneChip S. pombe tiling 1.0FR) at the BEA facility using the manufacturer's instructions. Raw data were initially processed and normalized by TAS (Affymetrix). Normalized (scaled to intensity 100, band width 100, PM only) values are given in log2. The microarray sequences were realigned to the ASM294v2 genome (using Bowtie 2) to map the signals to genome positions. For ChIP-qPCR, the following primers were used: Q-MAT2 F, TAGTATTCTGTCGAAATTATCGAAAGCTA; Q-MAT2 R, GAAACTGAAGCAGGGAAAAATGTAG; Q-702(tel2R) F, GTTACTCGCCTGCCTCTACCAT; and Q-702(tel2R) R, GTCAGTCAAGTTAATGAGTCATGAAGAA.
ChIP-exo
S. pombe strains were grown in rich media and subjected to formaldehyde crosslinking, then processed through the ChIP-exo assay (Rhee and Pugh 2011; Serandour et al. 2013). Briefly, cells were disrupted using glass beads, and chromatin was fragmented by sonication (Bioruptor Pico, 20 cycles of 30 sec on/30 sec off). Fragmented chromatin was immunoprecipitated using antibodies directed either against HA or T7. Protein A–coated magnetic beads (NEB) were used to bind the antibodies. Beads were washed, and with the immunoprecipitate still on the beads, the DNA was polished, A-tailed, and ligated to an Illumina sequencing library adaptor. Digestion lambda exonuclease (final concentration, 2.5U/reaction) removed nucleotides from 5′ ends of double-stranded DNA until blocked by the formaldehyde-induced protein–DNA crosslink. The crosslinks were reversed (4 h at 65°C) and DNA was eluted from the beads. The single-stranded DNA was subsequently made double-stranded by primer annealing and extension. A second sequencing adaptor was ligated. Using indexing primers, the fragments were PCR amplified and gel purified (Qiagen MinElute). Samples were quantified using Qubit (HS dsDNA) and sequenced on Illumina HiSeq 2000 (36 or 50 cycles, single end sequencing) at the BEA facility following the manufacturer's instructions.
Raw data from the HiSeq (FASTQ files) were aligned to ASM294v2 using Bowtie 2 with default parameters (Langmead and Salzberg 2012). Sequencing statistics are available in Supplemental Table S8. The ASM294v2.24 annotation (including UTR and CDS information) was downloaded from pombase.org and used in Podbat. The aligned data (SAM files) were imported (MAPQ > 30) and normalized to million reads and original quantity based on Qubit measurements for nonsaturated samples (T7). Data from independent biological duplicates were averaged.
Data analysis
RPKM (reads per kilobase per million) was calculated for all features of the ASM294v2 annotation and also all fragments of 10 and 150 bp. The ΔΔRPKM score was calculated based on the RPKM (reads per kilobase per million) for each 10 bases in the genome. ΔΔRPKM was defined as the gain in T7 signal after 2 h when subtracting the gain in HA signal in the same window (i.e., the addition of the loss of HA): in short, (RPKMT7,2 h – RPKMT7,0 h) – (RPKMHA,2 h − RPKMHA,0 h). The ΔΔRPKM values were then normalized by setting the genome average to zero.
Data visualization and analysis were performed in R version 3.1.1 (R Core Team 2014) and Podbat (Sadeghi et al. 2011). Running averages were calculated by ordering signals from fragments or genes and determining the average within a 500-element window. Metagene and heatmap analyses were performed with window size 50 and step 10, with the exception of heatmaps from the data generated by the ChIP-array protocol where a window size of five was used. Averages over fragments, genes, and gene fragments were calculated using average signal (ChIP-array) or RPKM (ChIP-exo). GO and gene list enrichment analysis was performed in Podbat. Running averages were calculated in R with a window size of 500. Hierarchical clustering was done using the distance measure complete in the function hclust. For PCA analysis, CEL files were read and normalized (“quantile”) using the Starr package in R (Zacher et al. 2009).
Accession numbers to previously published data sets
For GEO the numbers are as follows: GSE49874 (Sadeghi et al. 2014) H2Bub1, H3K9me2, H3; GSE66106 H3 in WT and spt16-18; GSM1103380 (Aygün et al. 2013) pInvH3HA, MNase; GSM1201968 (DeGennaro et al. 2013) MNase-seq. For ArrayExpress the numbers are as follows: E-MEXP-1850 (Durand-Dubief et al. 2010) Pol II; E-MEXP-3044 (Stralfors et al. 2011) H2A.Z, H4K12ac.
Data access
The sequencing and microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE66866.
Supplementary Material
Acknowledgments
We thank S.L. Sanders and Y. Watanabe for providing biological material and Elisabet Jemt, Michelle Rönnerblad, and Patrick Müller for technical assistance. This study was supported by grants from Vetenskapsrådet (VR-M2579 and VR-NT4448) and Cancerfonden (CAN2012/238) to K.E. and by grants from Åke Wibergs Stiftelse (M14-0205), Vetenskapsrådet, and KI Fonder to J.P.S.
Author contributions: J.P.S. conceived and performed the ChIP-exo and ChIP-qPCR experiments, analyzed data, and wrote the paper. M.S. constructed the RITE strains and set up the RITE system. V.M-B. analyzed RITE data, performed experiments using the RITE method, and wrote the paper. U.N.-A. performed ChIP-microarray experiments and analyzed data. P.A. performed HA and T7 ChIP experiments. I.S. performed the H4K20me ChIP-microarray experiments. J.C.T. provided the Spt16-myc data. R.C.A. initiated the RITE S. pombe project. K.E. conceived the project, supervised I.S., interpreted data, and wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.188870.114.
References
- Alvarez F, Munoz F, Schilcher P, Imhof A, Almouzni G, Loyola A. 2011. Sequential establishment of marks on soluble histones H3 and H4. J Biol Chem 286: 17714–17721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygün O, Mehta S, Grewal SI. 2013. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat Struct Mol Biol 20: 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. 2003. FACT facilitates transcription-dependent nucleosome alteration. Science 301: 1090–1093. [DOI] [PubMed] [Google Scholar]
- Carneiro T, Khair L, Reis CC, Borges V, Moser BA, Nakamura TM, Ferreira MG. 2010. Telomeres avoid end detection by severing the checkpoint signal transduction pathway. Nature 467: 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, Carmo-Fonseca M, de Almeida SF. 2013. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res 41: 2881–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi ES, Shin JA, Kim HS, Jang YK. 2005. Dynamic regulation of replication independent deposition of histone H3 in fission yeast. Nucleic Acids Res 33: 7102–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi ES, Stralfors A, Catania S, Castillo AG, Svensson JP, Pidoux AL, Ekwall K, Allshire RC. 2012. Factors that promote H3 chromatin integrity during transcription prevent promiscuous deposition of CENP-A(Cnp1) in fission yeast. PLoS Genet 8: e1002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon LM, Houston SI, Veerappan CS, Spektor TM, Rice JC. 2010. PR-Set7-mediated monomethylation of histone H4 lysine 20 at specific genomic regions induces transcriptional repression. J Cell Biochem 110: 609–619. [DOI] [PubMed] [Google Scholar]
- Das C, Tyler JK. 2012. Histone exchange and histone modifications during transcription and aging. Biochim Biophys Acta 1819: 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos D, Frederiks F, Terweij M, van Welsem T, Verzijlbergen KF, Iachina E, de Graaf EL, Altelaar AFM, Oudgenoeg G, Heck AJR, et al. 2011. Progressive methylation of ageing histones by Dot1 functions as a timer. EMBO Rep 12: 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, Henikoff S. 2010. Capturing the dynamic epigenome. Genome Biol 11: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, Henikoff JG, Henikoff S. 2010. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328: 1161–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGennaro CM, Alver BH, Marguerat S, Stepanova E, Davis CP, Bahler J, Park PJ, Winston F. 2013. Spt6 regulates intragenic and antisense transcription, nucleosome positioning, and histone modifications genome-wide in fission yeast. Mol Cell Biol 33: 4779–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. 2007. Dynamics of replication-independent histone turnover in budding yeast. Science 315: 1405–1408. [DOI] [PubMed] [Google Scholar]
- Duina AA, Rufiange A, Bracey J, Hall J, Nourani A, Winston F. 2007. Evidence that the localization of the elongation factor Spt16 across transcribed genes is dependent upon histone H3 integrity in Saccharomyces cerevisiae. Genetics 177: 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand-Dubief M, Ekwall K. 2009. Chromatin immunoprecipitation using microarrays. Methods Mol Biol 529: 279. [DOI] [PubMed] [Google Scholar]
- Durand-Dubief M, Persson J, Norman U, Hartsuiker E, Ekwall K. 2010. Topoisomerase I regulates open chromatin and controls gene expression in vivo. EMBO J 29: 2126–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evertts AG, Manning AL, Wang X, Dyson NJ, Garcia BA, Coller HA. 2013. H4K20 methylation regulates quiescence and chromatin compaction. Mol Biol Cell 24: 3025–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming AB, Kao CF, Hillyer C, Pikaart M, Osley MA. 2008. H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol Cell 31: 57–66. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo XY, Li X, et al. 2010. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette B, Bataille AR, Gevry N, Adam M, Blanchette M, Robert F, Gaudreau L. 2005. Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol 3: 2100–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Zhang ZQ, Xu M, Li YF, Li Z, Ma YT, Cai T, Zhu B. 2013. H3.3-H4 tetramer splitting events feature cell-type specific enhancers. PLoS Genet 9: e1003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovoni JS, Russell P, Gaits F. 1999. A new inducible protein expression system in fission yeast based on the glucose-repressed inv1 promoter. Gene 232: 53–58. [DOI] [PubMed] [Google Scholar]
- Jamai A, Imoberdorf RM, Strubin M. 2007. Continuous histone H2B and transcription-dependent histone H3 exchange in yeast cells outside of replication. Mol Cell 25: 345–355. [DOI] [PubMed] [Google Scholar]
- Jamai A, Puglisi A, Strubin M. 2009. Histone chaperone Spt16 promotes redeposition of the original H3-H4 histories evicted by elongating RNA polymerase. Mol Cell 35: 377–383. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293: 1074–1080. [DOI] [PubMed] [Google Scholar]
- Jiang CZ, Pugh BF. 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin CY, Felsenfeld G. 2007. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev 21: 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CD, Laprade L, Winston F. 2003. Transcription elongation factors repress transcription initiation from cryptic sites. Science 301: 1096–1099. [DOI] [PubMed] [Google Scholar]
- Karachentsev D, Sarma K, Reinberg D, Steward R. 2005. PR-Set7-dependent methylation of histone H4 Lys 20 functions in repression of gene expression and is essential for mitosis. Genes Dev 19: 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Seol JH, Han JW, Youn HD, Cho EJ. 2007. Histone chaperones regulate histone exchange during transcription. EMBO J 26: 4467–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar DC, Jin WF, Maunakea A, Abraham B, Ha M, Zhao KJ. 2013. Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol 14: R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. 2004. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet 36: 900–905. [DOI] [PubMed] [Google Scholar]
- Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT, Ohgi KA, Benner C, Garcia-Bassets I, Aggarwal AK, et al. 2010. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 466: 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G. 2006. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol Cell 24: 309–316. [DOI] [PubMed] [Google Scholar]
- Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, Bahler J. 2012. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151: 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff WF, Wagner EJ, Duronio RJ. 2008. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Genet 9: 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez-Benito V, van Deventer SJ, Jimenez-Garcia V, Roy-Luzarraga M, van Leeuwen F, Neefjes J. 2013. Spatiotemporal analysis of organelle and macromolecular complex inheritance. Proc Natl Acad Sci 110: 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Okamoto I, Murphy N, Chu JH, Price SM, Shen MM, Torres-Padilla ME, Heard E, Reinberg D. 2009. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol Cell Biol 29: 2278–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen-Hughes T, Gkikopoulos T. 2012. Making sense of transcribing chromatin. Curr Opin Cell Biol 24: 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Zhu B, Li GH, Trojer P, Mandal S, Shilatifard A, Reinberg D. 2006. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell 125: 703–717. [DOI] [PubMed] [Google Scholar]
- Pesavento JJ, Yang H, Kelleher NL, Mizzen CA. 2008. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol Cell Biol 28: 468–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi HH, Sarkissian M, Hu GQ, Wang ZB, Bhattacharjee A, Gordon DB, Gonzales M, Lan F, Ongusaha PP, Huarte M, et al. 2010. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 466: 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Radman-Livaja M, Rando OJ. 2010. Nucleosome positioning: how is it established, and why does it matter? Dev Biol 339: 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman-Livaja M, Quan TK, Valenzuela L, Armstrong JA, van Welsem T, Kim T, Lee LJ, Buratowski S, van Leeuwen F, Rando OJ, et al. 2012. A key role for Chd1 in histone H3 dynamics at the 3′ ends of long genes in yeast. PLoS Genet 8: e1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. 2006. Stochastic mRNA synthesis in mammalian cells. PLoS Biol 4: 1707–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Zenter GE, Henikoff S. 2015. Asymmetric nucleosomes flank promoters in the budding yeast genome. Genome Res 25: 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, Chang HY. 2009. Genome-wide views of chromatin structure. Annu Rev Biochem 78: 245–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinberg D, Sims RJ. 2006. de FACTo nucleosome dynamics. J Biol Chem 281: 23297–23301. [DOI] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2011. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147: 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Bataille AR, Zhang L, Pugh BF. 2014. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 159: 1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. 2007. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3K56 acetylation and Asf1. Mol Cell 27: 393–405. [DOI] [PubMed] [Google Scholar]
- Rustici G, Mata J, Kivinen K, Lio P, Penkett CJ, Burns G, Hayles J, Brazma A, Nurse P, Bahler J. 2004. Periodic gene expression program of the fission yeast cell cycle. Nat Genet 36: 809–817. [DOI] [PubMed] [Google Scholar]
- Sadeghi L, Bonilla C, Stralfors A, Ekwall K, Svensson JP. 2011. Podbat: a novel genomic tool reveals Swr1-independent H2A.Z incorporation at gene coding sequences through epigenetic meta-analysis. PLoS Comput Biol 7: e1002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghi L, Siggens L, Svensson JP, Ekwall K. 2014. Centromeric histone H2B monoubiquitination promotes noncoding transcription and chromatin integrity. Nat Struct Mol Biol 20: 236–243. [DOI] [PubMed] [Google Scholar]
- Sakaguchi A, Steward R. 2007. Aberrant monomethylation of histone H4 lysine 20 activates the DNA damage checkpoint in Drosophila melanogaster. J Cell Biol 176: 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. 2004. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119: 603–614. [DOI] [PubMed] [Google Scholar]
- Saunders A, Werner J, Andrulis ED, Nakayama T, Hirose S, Reinberg D, Lis JT. 2003. Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science 301: 1094–1096. [DOI] [PubMed] [Google Scholar]
- Scharf AND, Barth TK, Imhof A. 2009a. Establishment of histone modifications after chromatin assembly. Nucleic Acids Res 37: 5032–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf AND, Meier K, Seitz V, Kremmer E, Brehm A, Imhof A. 2009b. Monomethylation of lysine 20 on histone H4 facilitates chromatin maturation. Mol Cell Biol 29: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabish MA, Struhl K. 2004. Evidence for eviction and rapid deposition of histories upon transcriptional elongation by RNA polymerase II. Mol Cell Biol 24: 10111–10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serandour AA, Brown GD, Cohen JD, Carroll JS. 2013. Development of an Illumina-based ChIP-exonuclease method provides insight into FoxA1-DNA binding properties. Genome Biol 14: R147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sideri T, Rallis C, Bitton DA, Lages BM, Suo F, Rodriguez-Lopez M, Du LL, Bähler J. 2014. Parallel profiling of fission yeast deletion mutants for proliferation and for lifespan during long-term quiescence. G3 (Bethesda) 5: 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JK, Houston SI, Magazinnik T, Rice JC. 2006. A trans-tail histone code defined by monomethylated H4 Lys-20 and H3 Lys-9 demarcates distinct regions of silent chromatin. J Biol Chem 281: 12760–12766. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Hernandez AE, Groudine M, Henikoff S. 2014. The nucleosomal barrier to promoter escape by RNA polymerase II is overcome by the chromatin remodeler Chd1. Elife 3: e02042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stralfors A, Walfridsson J, Bhuiyan H, Ekwall K. 2011. The FUN30 chromatin remodeler, Fft3, protects centromeric and subtelomeric domains from euchromatin formation. PLoS Genet 7: e1001334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. 2004. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116: 51–61. [DOI] [PubMed] [Google Scholar]
- Tanny JC, Erdjument-Bromage H, Tempst P, Allis CD. 2007. Ubiquitylation of histone H2B controls RNA polymerase II transcription elongation independently of histone H3 methylation. Genes Dev 21: 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terweij M, van Welsem T, van Deventer S, Verzijlbergen KF, Menendez-Benito V, Ontoso D, San-Segundo P, Neefjes J, van Leeuwen F. 2013. Recombination-induced tag exchange (RITE) cassette series to monitor protein dynamics in Saccharomyces cerevisiae. G3 (Bethesda) 3: 1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzijlbergen KF, Menendez-Benito V, van Welsem T, van Deventer SJ, Lindstrom DL, Ovaa H, Neefjes J, Gottschling DE, van Leeuwen F. 2010. Recombination-induced tag exchange to track old and new proteins. Proc Natl Acad Sci 107: 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Jia ST. 2009. Degrees make all the difference The multifunctionality of histone H4 lysine 20 methylation. Epigenetics 4: 273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Reddy B, Thompson J, Wang HB, Noma K, Yates JR, Jia ST. 2009. Regulation of Set9-mediated H4K20 methylation by a PWWP domain protein. Mol Cell 33: 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen KY, Vinayachandran V, Pugh BF. 2013. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell 154: 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacher B, Soeding J, Siebert M, Tresch A. 2009. Starr: simple tiling array analysis of Affymetrix ChIP-chip data. R package version 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Roberts DN, Cairns BR. 2005. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell 123: 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.