

Abstract
The analysis of next-generation sequencing data is computationally and statistically challenging because of the massive volume of data and imperfect data quality. We present GotCloud, a pipeline for efficiently detecting and genotyping high-quality variants from large-scale sequencing data. GotCloud automates sequence alignment, sample-level quality control, variant calling, filtering of likely artifacts using machine-learning techniques, and genotype refinement using haplotype information. The pipeline can process thousands of samples in parallel and requires less computational resources than current alternatives. Experiments with whole-genome and exome-targeted sequence data generated by the 1000 Genomes Project show that the pipeline provides effective filtering against false positive variants and high power to detect true variants. Our pipeline has already contributed to variant detection and genotyping in several large-scale sequencing projects, including the 1000 Genomes Project and the NHLBI Exome Sequencing Project. We hope it will now prove useful to many medical sequencing studies.
The cost of human genome sequencing has declined rapidly, powered by advances in massively parallel sequencing technologies. This has made possible the collection of genomic information on an unprecedented scale and made large-scale sequencing a practical strategy for biological and medical studies. An initial step for nearly all sequencing studies is to detect variant sites among sampled individuals and genotype them. This analysis is challenging because errors in high-throughput sequence data are much more common than true genomic variation. There are diverse sources of trouble (base-calling errors, alignment artifacts, contaminant reads derived from other samples), and the resulting errors are often correlated. The analysis is also computationally and statistically challenging because of the volume of data involved. Using standard formats, raw sequence reads for a single deeply (30×) sequenced human genome require >100 gigabytes (GB) of storage.
Several tools are now available to process next-generation sequencing data. For example, the Genome Analysis Toolkit (GATK) (DePristo et al. 2011), SAMtools (Li 2011), and SNPTools (Wang et al. 2013) are used for variant discovery and genotyping from small to moderate numbers of sequenced samples. However, as the number of sequenced genomes grows, analysis becomes increasingly challenging, requiring complex data processing steps, division of sequence data into many small regions, management and scheduling of analysis jobs, and often, prohibitive demands on computing resources. A tempting approach to alleviate computational burden is to process samples in small batches, but this can lead to reduced power for rare variant discovery and systematic differences between samples processed in different batches.
There is a pressing need for software pipelines that support large-scale medical sequencing studies that will be made possible by decreased sequencing costs. Desirable features for such pipelines include (1) scalability to tens of thousands of samples; (2) the ability to easily stop and resume analyses; (3) the option to carry out incremental analyses as new samples are sequenced; (4) flexibility to accommodate different study designs: shallow and deep sequencing, whole-genome, whole-exome, or small targeted experiments; and, of course, (5) high-quality genotyping and variant discovery.
Here, we describe and evaluate our flexible and efficient sequence analysis software pipeline, Genomes on the Cloud (GotCloud). We show that GotCloud delivers high-quality variant sites and accurate genotypes across thousands of samples. We describe the strategies to systematically divide processing of very large data sets into manageable pieces. We also demonstrate novel automated frameworks for filtering sequencing and alignment artifacts from variant calls as well as for accurate genotyping using haplotype information.
Results
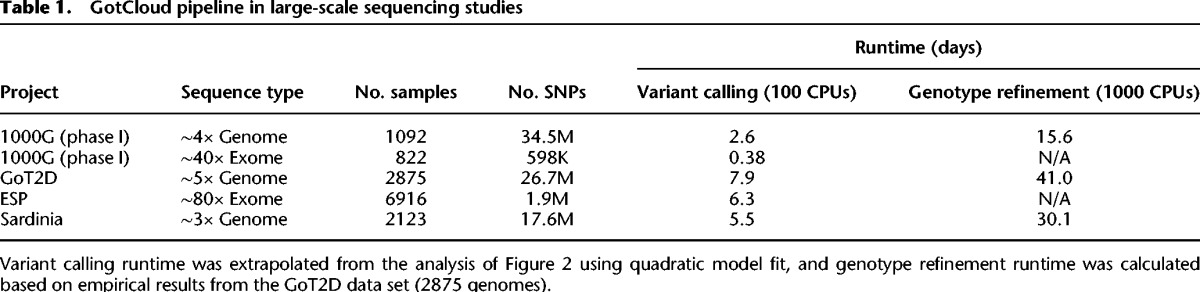
GotCloud offers a comprehensive pipeline including sequence alignment, post-alignment processing and quality control, variant calling, variant filtering, and haplotype-aware genotype refinement, as described in the Methods section (Fig. 1). In this section we highlight and evaluate key features of GotCloud, including the computational efficiency and the robustness of variant calling and filtering, compared with GATK UnifiedGenotyper. Our GotCloud variant calling pipeline has been used in many large genome and exome sequencing studies, each with thousands of samples (Table 1).
Figure 1.
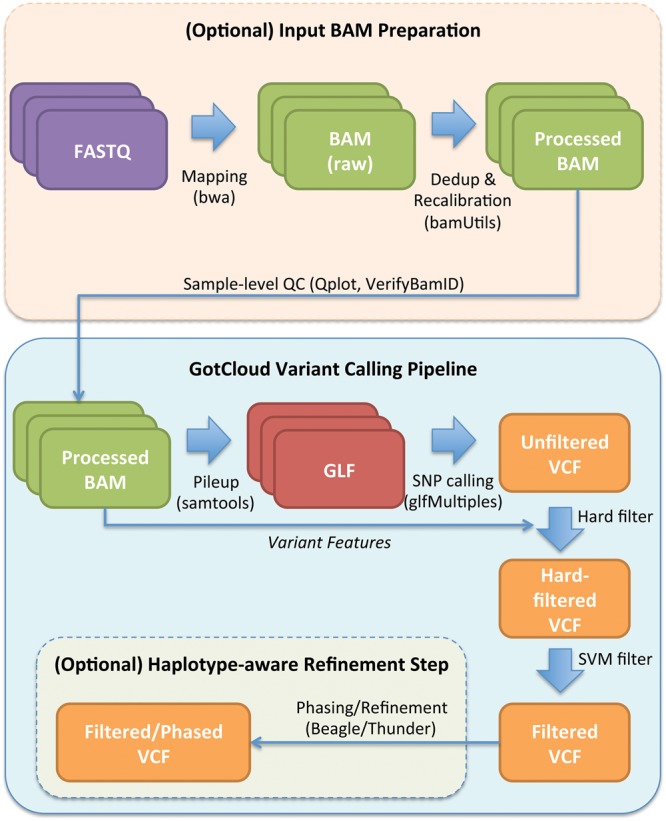
Outline of GotCloud variant calling pipeline.
Table 1.
GotCloud pipeline in large-scale sequencing studies
Evaluation of computational efficiency
Using low-coverage genome and exome data sets from the 1000 Genomes Project, we evaluate the computational efficiency of GotCloud using a minicluster with four dedicated computing nodes, where each node has 48 physical CPU cores and 64 GB of main memory. Figure 2A summarizes the total computational costs as a function of sample size, comparing the GotCloud variant calling pipeline and the GATK UnifiedGenotyper.
Figure 2.
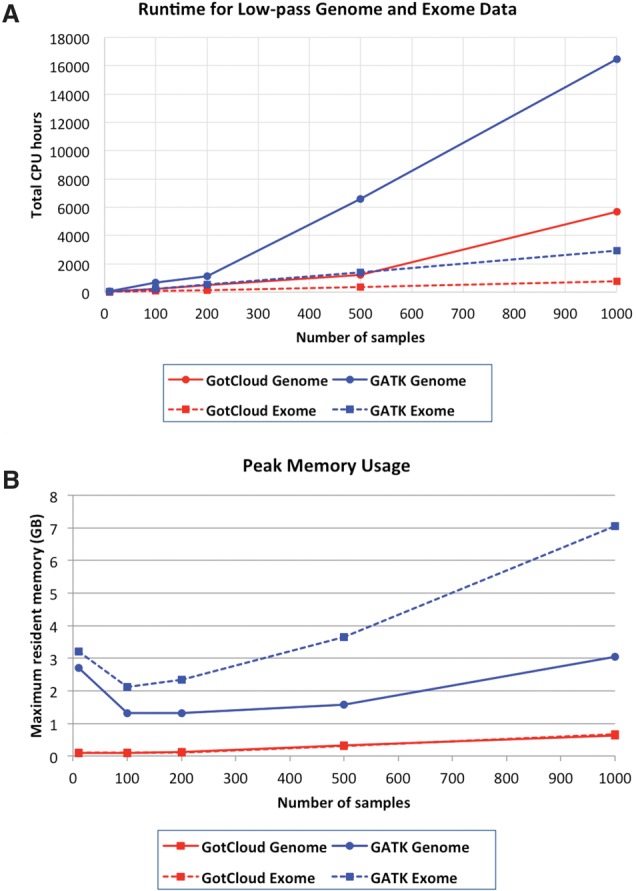
Computational costs of GATK UnifiedGenotyper and GotCloud pipelines. (A) Runtime estimated for whole-genome (6×) and whole-exome (60×) sequence data running with 40 parallel sessions on a four-node minicluster with 48 physical CPU cores. For GATK, runtimes for 1000 samples are extrapolated from analyses of a single 5-Mb block of Chromosome 20. For all other analyses, no extrapolation was used. (B) Peak memory usage estimates averaged over Chromosome 20 chunks.
For low-coverage genomes, total runtimes for both GotCloud and GATK increase at a faster than linear rate with sample size, because increased sample size increases not only the number of samples (and the amount of sequence data) to process but also results in more discovered variant sites, which must be inspected in each sample for genotyping and variant filtering. For both low-coverage genomes and deep exomes, GotCloud ran faster than GATK, most noticeably for analyses of >500 samples. This speed advantage increases gradually. For analysis of 1000 low-coverage samples, GotCloud took ∼5700 CPU hours, whereas GATK took ∼16,500 CPU hours. Similarly, for 1000 exomes, GotCloud took ∼750 CPU hours and GATK took ∼2930 CPU hours.
GotCloud also maintains an efficient memory footprint (Fig. 2B). While GATK required >7 GB of memory to analyze a thousand exomes, GotCloud on average required <1 GB of memory. In memory-bound computing environments with a large number of concurrent processes, which is a common practice for large-scale sequencing studies, GotCloud can host approximately 10 times more concurrent processes than GATK.
Unlike low-coverage genomes, the runtime for deep exomes grows almost linearly with sample size, because the majority of computational effort is spent on the “pileup” step that summarizes deep sequence data, whereas little time is spent on the “glfMultiples” variant calling processes due to the relatively small target size (Supplemental Fig. 1).
Evaluation of variant detection sensitivity
We next assessed the variant detection sensitivity of GotCloud and GATK with increasing sample sizes for low-coverage genomes. For both GATK and GotCloud, the number of detected variants per sample increased as more samples were analyzed together (Tables 2, 3; Supplemental Table S3), particularly when the coverage was low. This is consistent with our expectation because variants shared between samples are more likely to be detected when the information across samples is combined (Li et al. 2010).
Table 2.
Summary of variant calling results and the effect of filtering for low-pass sequence data
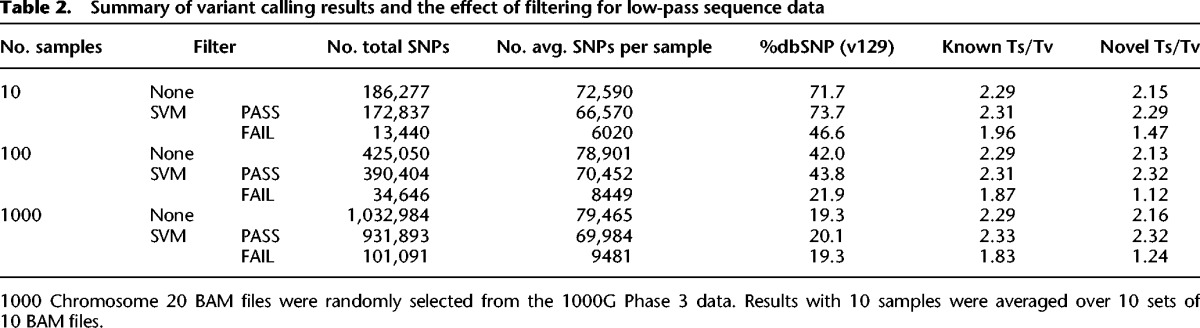
Table 3.
Comparison of SNP call sets (unfiltered and filtered) between GATK UnifiedGenotyper and GotCloud for low-coverage genome data
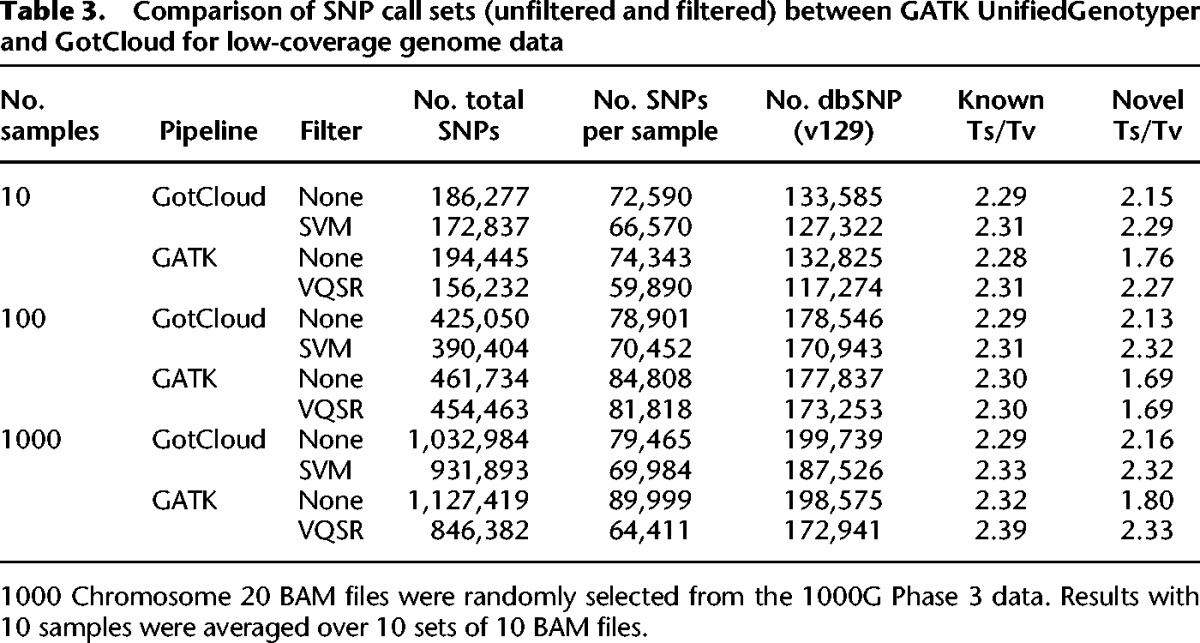
We calculated the fraction of detected variants among the polymorphic variants in HumanExome BeadChip arrays as a measure of variant detection sensitivity. We excluded the variants included in the Omni2.5 SNP genotyping array, which was used for training the SVM filters. As expected, GotCloud's HumanExome BeadChip sensitivity for low-coverage data increased from 71.4% to 75.3% as the sample size increased from 10 to 1000 (Fig. 3A). For deep exome data, the sensitivity also increased from 90.8% to 94.2% (Fig. 3B). GATK showed lower sensitivity both for low-coverage data (67.4%–71.5%), and deep exome data (78.3%–89.6%). GotCloud consistently showed higher variant detection sensitivity than GATK in every comparison.
Figure 3.
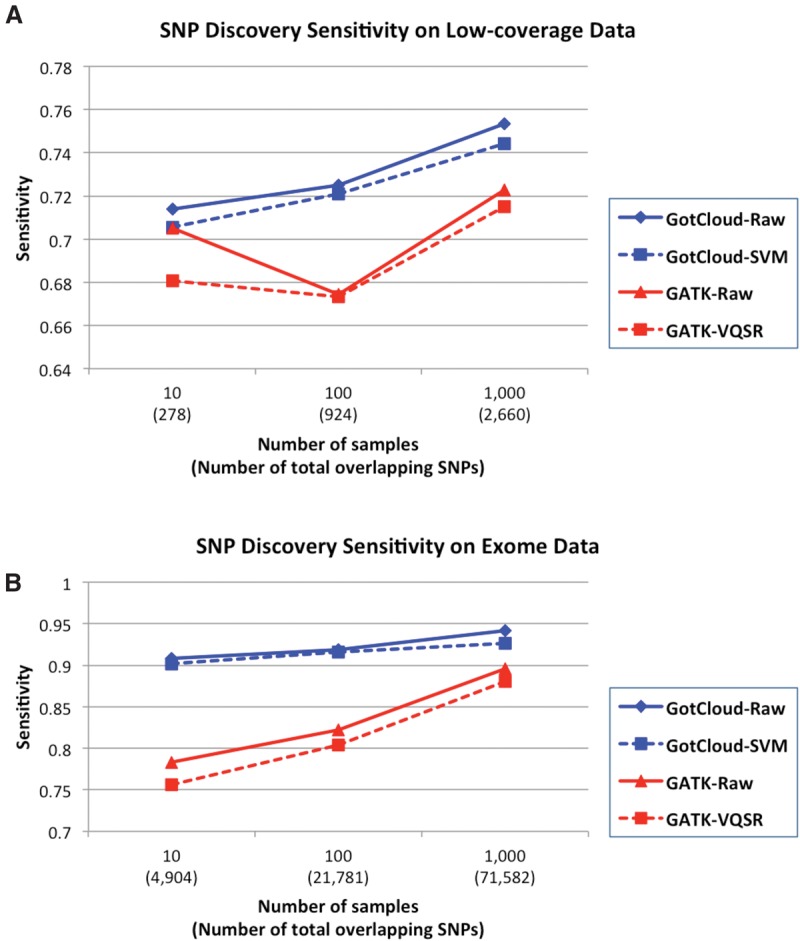
Variant discovery sensitivity comparison of GotCloud and GATK using HumanExome BeadChip excluding the SNPs contained in Omni2.5 array, because Omni2.5 variants are used to train variant filters in GotCloud and GATK. GotCloud results are shown for unfiltered (raw) and SVM-filtered sets, and GATK results are shown for unfiltered and VQSR-filtered sets, across low-coverage genome (A) and exome data (B).
Evaluation of variant filtering
We evaluated the quality of filtered variant calls by looking at the transition to transversion ratio (Ts/Tv). To complement the sensitivity analysis, we evaluated the quality of the filtered calls using the HumanExome BeadChip sensitivity described above.
Previous studies report whole-genome Ts/Tv between 2.1 and 2.3 (DePristo et al. 2011), but the exact value is affected by allele frequency, GC content, proportion of CpG sites, natural selection, and other factors. Instead of setting a specific target Ts/Tv value, we compared the Ts/Tv between “known” SNPs (those in dbSNP) and “novel” SNPs (those not in dbSNP). We used dbSNP version 129, which is the latest release without variants from next-generation sequencing. Large differences in Ts/Tv between known and novel SNPs suggest that the “novel” SNP list likely includes variants with unusual properties, indicating potential false positives.
In GotCloud's two-step filtering process (Fig. 1), variants with lower quality are first identified by applying individual hard filters (Supplemental Table S1), and variants failing multiple hard filters are used as negative examples to train a support vector machine (SVM) classifier (See Methods for details). In our analysis of 1000 low-coverage samples, the difference of Ts/Tv between known (2.29) and novel (2.16) SNPs was high before SVM filtering, suggesting that novel SNPs have a lower quality than known SNPs. After SVM filtering, known (2.33) and novel (2.32) SNPs have similar Ts/Tv, suggesting that many false positive SNPs were filtered out. The trend was similar for all other sample sizes.
Our SVM filter reduces the variant detection sensitivity by only a small amount. The HumanExome BeadChip sensitivity was reduced by only 0.5%–0.9% after removing 8%–13% of the unfiltered calls, suggesting that the vast majority of SNPs filtered out are likely false positives (Fig. 3). The variant quality score recalibration (VQSR) filter from GATK reduced sensitivity by 0.1%–2.4% after removing 1.6%–25% of the unfiltered calls, across different sample sizes.
In exomes, we expect higher Ts/Tv than in other regions because degeneracy of the genetic code means that selection against variants that alter protein sequence preferentially removes transversion alleles from the population, as reported in previous studies on population-scale exome sequencing (Tennessen et al. 2012; Fu et al. 2013). Differences between known and novel SNPs are also expected as a result of natural selection since protein-coding variants (which are more often transversions) tend to be rarer than variants that do not alter protein sequence (which are more often transitions). To facilitate interpretation, we stratify the analysis of exome samples and examine nonsynonymous variants that alter protein sequence separately from synonymous variants that do not.
In exome sequencing, we again observed that (within each functional category) Ts/Tv for known and novel variants became much more similar after filtering (Supplemental Table S2). With 1000 exomes, Ts/Tv at nonsynonymous SNPs were 2.24 and 1.61 for known and novel variants before filtering, which became 2.33 and 2.31, respectively, after filtering. For synonymous SNPs, Ts/Tv of 5.40 and 4.05 for known and novel variants before filtering became 5.55 and 5.49 after filtering. We expect that filtering becomes progressively more effective with larger sample sizes because the SVM classifier can better learn how to use diagnostics such as allele balance in heterozygotes and strand preference for the reference when more data are available. This expectation is confirmed by inspection of Table 2 and Supplemental Table S2, where differences between known and novel variants after filtering become progressively smaller as sample size increases.
Because multiple features are combined to construct SVM classifiers, GotCloud's SVM filtering outperforms the filtering approach using any individual feature. We ordered variants based on each individual feature, and evaluated the Ts/Tv of variants after applying the filters based on a single feature (Fig. 4A) and the HumanExome BeadChip sensitivity lost by applying each filter (Fig. 4B). SVM filtering showed the largest separation of Ts/Tv between filtered-in and filtered-out variants (2.32 versus 1.20) and the smallest loss of HumanExome BeadChip sensitivity (0.5%). Some filters based on individual features, such as StrandBias or AlleleBalance, achieved similar separation of Ts/Tv to SVM filters but showed >5× larger (2.8%–3.2%) loss of HumanExome BeadChip sensitivity. Our results demonstrate that the SVM filter provides an automatic and powerful framework to distinguish likely true and false variants.
Figure 4.
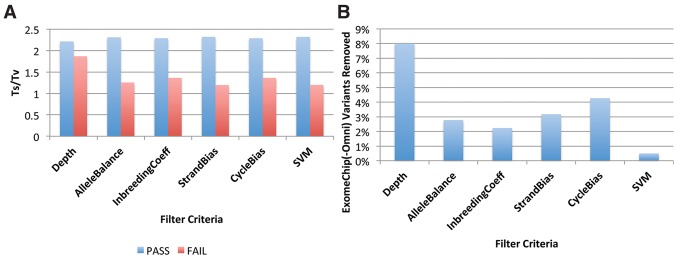
Comparison of SVM filtering with hard filtering based on a single feature. (A) Ts/Tv of filtered-in (PASS) and filtered-out (FAIL) variants using different filters. Variants are ordered by a single variant feature and a fixed fraction of variants (8%) are filtered out to match the variant counts with the default SVM filter. Absolute values are used for StrandBias correlation and CycleBias correlation. (B) Percentage of filtered-out HumanExome BeadChip (Omni2.5) variants among those that are polymorphic in the array genotypes.
Portability of SVM decision rules
GotCloud provides robust filtering for small targeted sequencing experiments in most cases, because SVM requires only a small number of positive and negative labels to find a decision boundary. In some cases, however, the number of labeled variants may be too small to develop adequate training models. GotCloud allows the transferring of SVM classification models across data sets. Our classification model is robust to the differences in feature distribution between data sets because the filtering is based on quantile-normalized feature space. We applied our model-transfer filtering trained from whole-exome sequencing data of independent samples onto small subsets of exome sequencing data, mimicking small target sequencing of 10 kb to 10 Mb. We used 500 exome samples from the 1000 Genomes Project to train the SVM classifier, and used another 500 nonoverlapping exome samples to simulate small target sequencing. Our results demonstrate that the model-transfer filtering provides higher novel Ts/Tv than the self-trained SVM filtering trained within only the target regions, especially when the target region was smaller (Fig. 5). In the experiment with the smallest target region of 10 kb, the self-trained model shows some differences between known (2.32) and novel (2.25) Ts/Tv for nonsynonymous SNPs, while the transfer model shows smaller differences between known (2.35) and novel (2.33) Ts/Tv. In our experimental setup where there is little or no systematic difference between the data sets, model-transfer filtering appears to perform as good as the self-trained SVM, even for large target regions. Based on our experiences, when there are systematic differences between the sequencing data sets, the self-trained model will likely perform better when the target region is large (e.g., >1 Mb).
Figure 5.
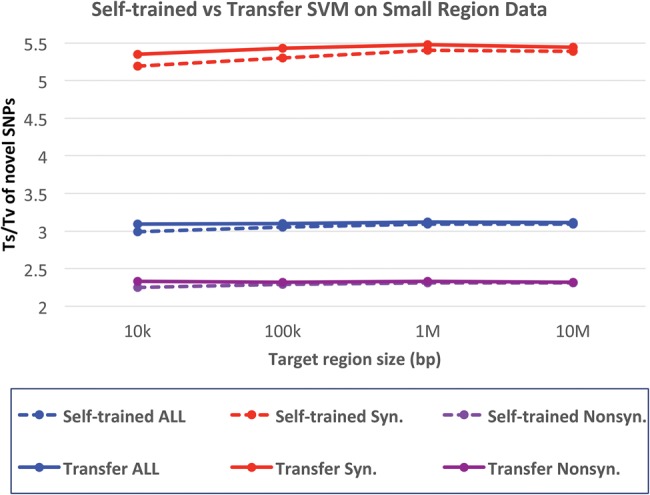
Impact of model-transfer filtering on the variant quality. The vertical axis represents Ts/Tv for novel SNPs for model-transferred SVM and self-trained SVM filters. Ts/Tv for known SNPs are ∼3.5, which is higher than novel SNPs because known SNPs contain a larger fraction of synonymous SNPs. The horizontal axis represents the size of targeted regions randomly selected from 500 whole-exome sequences. The transferred model is trained on nonoverlapping 500 exome data.
Effect of trimming overlapping fragments
One of the previously undocumented ways that GotCloud improves the quality of the variant lists is to appropriately account for overlapping fragments in paired end reads. Read pairs derived from small fragments may often overlap using our stand-alone tool bamUtil clipOverlap. If errors occur in the PCR amplification step, these overlapping fragments will carry errors forward to both paired reads. In these cases, a single-base sequencing error may appear to be two independent mismatches, resulting in false positive SNP calls. GotCloud, by default, trims the overlapping fragment with lower sequencing quality to avoid these artifacts. This artifact is more problematic for very rare variants where filtering based on multisample statistics is not as useful; hence we evaluated our approach by analyzing Ts/Tv ratios for the singletons, which are the variants with the lowest possible allele count (AC = 1). Our evaluation with low-coverage sequencing data demonstrates that the Ts/Tv of novel singletons substantially decreases from 2.21 to 1.97 if the overlapping fragments are not accounted for properly (Fig. 6).
Figure 6.
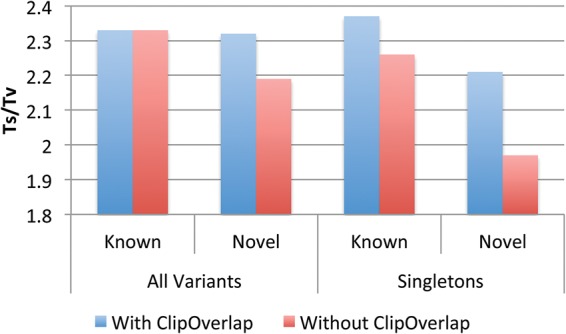
Comparison of known and novel Ts/Tv with and without trimming overlapping reads for 1000 low-coverage Chromosome 20 sequences from the 1000 Genomes Project. Overlapping fragments lowers novel Ts/Tv and the effect is more eminent in the singletons (with allele count of one).
Haplotype-aware genotype refinement
GotCloud also provides an automated pipeline to parallelize haplotype-aware genotype refinement. We evaluated the benefits of haplotype-aware refinement for low-coverage whole genomes. SVM-filtered VCF files were supplied to Beagle (50 rounds), and Beagle haplotypes were used to seed ThunderVCF (20 rounds). We measured nonreference genotype concordances using Omni2.5 array genotypes (Fig. 7). For 10 samples, nonreference discordance was reduced from 10.0% before refinement to 6.56% after Beagle refinement, and then further reduced to 5.36% after ThunderVCF. Since haplotype-aware refinement depends on the number of available haplotypes, the improvements were greater with more samples. After refinement, the nonreference discordance for the 100 sample experiment was reduced from 10.38% to 2.37%, and for 1000 samples it was reduced from 10.21% to 1.48%, consistent with our previous experiments (Li et al. 2011; The 1000 Genomes Project Consortium 2012).
Figure 7.
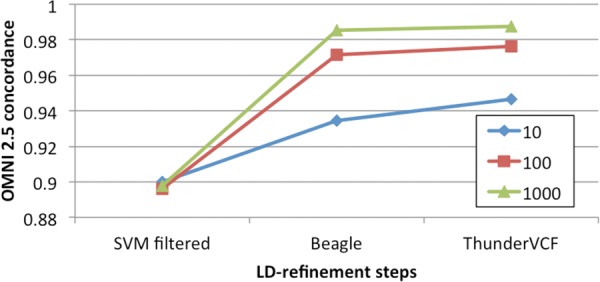
Nonreference genotype concordance for low-pass genome data calculated using Omni2.5 array genotypes. The haplotype-aware refinement steps significantly improve genotype accuracy, especially with larger sample sizes.
Discussion
The GotCloud pipeline provides an efficient and flexible framework for analyses of large-scale sequence data. Experimental results show that GotCloud discovers and genotypes high-quality SNPs by combining population-based multisample calling and machine-learning-based filtering. It also requires less computational time and has a smaller memory footprint than the popular GATK pipeline. GotCloud provides a complete end-to-end pipeline from raw sequence data to variant calls, to haplotype-aware genotype refinement that substantially improves accuracy for low-pass whole-genome sequencing.
GotCloud achieves a small memory footprint even with many deeply sequenced samples because the “pileup” step summarizes one sample at a time into a compact likelihood representation that is largely independent of sequencing depth. Once data for each sample has been summarized, variant calling reviews “pileup” results for many samples, one region at a time, using much less computing resources than would be required for accessing BAM files directly. The advantages of this approach will become more marked as sequencing depth increases. As a result, GotCloud can process larger sample sizes with the same memory or (by accommodating more parallel processes) achieve an even greater speed advantage.
Another advantage of separating the “pileup” and the “variant calling” steps is the possibility of incremental processing. Large-scale DNA sequencing studies can take many years of time and effort, and it is common to produce sequence data in multiple batches and generate multiple iterations of the variant calls for intermediate analyses and quality assurance. When a new batch of samples is added for a new round of variant calling, GotCloud needs to run the time-consuming pileup step only on the new samples, while one-pass pipelines need to reprocess all BAM files at each iteration of the analysis. For example, consider a scenario where an additional 200 exome sequencing samples are added to the 1000 existing samples (Fig. 2A; Supplemental Fig. 1B). To generate a new SNP set over all 1200 samples, GotCloud requires ∼150 CPU hours for pileup of the 200 new samples and <50 CPU hours for glfMultiples to regenerate variant lists, achieving ∼75% of reduction in computing time compared with the case of doing everything from scratch.
The GotCloud pipeline has customizable options such as the size of genomic chunks to be processed in parallel, the regions for targeted sequencing, and filtering parameters. Default parameter settings are provided for common study designs, but these parameters can be changed to leverage expert knowledge based on sequencing protocol, study design, objectives, and computing environments.
For filtering, default parameters (Supplemental Table S1) should be adequate for most scenarios, because the final SVM-filtered results from GotCloud are not very sensitive to any single threshold. When generating a variant ranking strategy, the SVM classifier combines information across the many variants that fail multiple hard filters, unlike the hard-filtering approach where one inadequate threshold directly affects the filtering results. For unusual scenarios such as drastic changes in sequencing technologies used, we provide detailed guidelines for parameter tuning in the user's manual (http://www.gotcloud.org).
In summary, GotCloud is an efficient, flexible, scalable, and integrated framework that can transform raw sequence data into high-quality variants calls and genotypes. GotCloud has already proven useful in several large-scale sequencing studies. With unprecedented growth of the sequencing throughput now enabling us to produce tens of thousands of deep genomes, we expect that GotCloud will continue to contribute to our common goal of completing the map of human genetic variation and its consequences. GotCloud is under active development with several key improvements expected in the near future and will continue to include updates to cutting edge open source methods.
Methods
An overview of our GotCloud pipeline is given in Figure 1. GotCloud combines several components, including: alignment, variant calling, variant filtering, and genotype refinement. The alignment step takes raw sequence reads stored in FASTQ files as input and generates sample level sequence quality summaries and binary sequence aligned/mapped (BAM) files as output. Subsequent steps take these BAM files as input and generate progressively more refined variant call format (VCF) files as output. The variant calling, filtering, and genotyping steps consist of four major tasks: building a pileup summary of variation per individual, identifying an initial set of variant sites, filtering poor quality variants and, finally, an optional genotype refinement step which is recommended for whole-genome sequence data and improves initial genotype calls by leveraging haplotype information.
Each processing step is divided into a series of small tasks and file dependencies are managed by the GNU make utility. GNU make handles scheduling of the different tasks and deploys tasks in highly parallel environments, such as high-performance computing clusters. Dividing work into thousands of small jobs increases memory efficiency, avoiding monolithic steps that must process many terabytes of data directly. In the remainder of the Methods section, we provide additional details on each step.
Automated sequence alignment, post-processing, and quality assessment
The first step of analysis with GotCloud is to align raw sequence reads (in FASTQ format) to the reference genome and post-process the aligned reads (in BAM format) to be ready for variant calling. GotCloud uses widely available alignment software, such as BWA (Li and Durbin 2009) and MOSAiK (Zhao et al. 2013), to generate initial BAM files. After the initial alignment, each BAM file is sorted by genomic coordinates and post-processed to remove duplicated reads and recalibrate base quality scores in a computationally and memory efficient manner using our bamUtil tool included in GotCloud. After these steps, several quality control metrics (such as the number of mapped reads, base-quality distribution, insert size distribution, GC bias profile, sample identity checks, and estimated DNA sample contamination) are produced and stored into summary files (Jun et al. 2012; Li et al. 2013). These quality assessment steps provide a snapshot of data quality and help identify problems such as low library complexity, insufficient read depth, DNA sample swaps, and sample contamination. Removal of poor performing samples at early steps of the analysis chain helps improve the overall quality of study results.
Parallelized and incremental variant discovery
GotCloud's variant discovery step generates an initial unfiltered VCF from a set of BAM files, based on two major tasks–pileup and glfMultiples. The first task, “pileup,” summarizes overlapping bases for each position one sample at a time and produces genotype likelihood files by calculating the probability of observed bases given hypothetical true genotypes at each genomic position. GotCloud uses a modified version of SAMtools to generate genotype likelihoods for all 10 possible diploid SNP genotypes and store them in GLF format (Li 2011). This “pileup” task processes one sample at time and the resulting genotype likelihood files are divided into short chromosomal segments to facilitate downstream analyses. When a read pair has overlapping fragments due to short insert size, the fragment with lower average base quality is trimmed to avoid false positive variants due to PCR artifacts.
The second task, “glfMultiples,” reviews the “pileup” results of the same chromosomal segment across all samples to identify variant sites. The computational complexity of the glfMultiples step is largely insensitive to sequencing depth and increases almost linearly with the number of samples and the length of the genome targeted for analysis. As a result, GotCloud can efficiently handle thousands of deeply sequenced samples together, which is challenging for most variant callers. Our multisample SNP caller, glfMultiples, uses a naïve Bayes model to compute the probability that an alternative allele is present given observed data and a population-based prior. It has high power to detect shared variants among individuals, especially with large numbers of sequenced samples. A more detailed description of glfMultiples algorithm is provided by Li et al. (2011).
Variant filtering by leveraging machine-learning methods
High-throughput sequencing reads are prone to sequencing errors and alignment artifacts. As a result, initial variant site lists typically contain many false positives. To improve the quality of variant lists, we apply a filtering step that evaluates a series of features at each potential site. Using machine-learning techniques, these features are then used to identify the highest quality variants and reduce the number of false positive variants.
Features for each potential site are extracted from BAM files one sample at a time in a highly parallelized manner, and the results are organized into small files each representing a short stretch of the genome. We calculate features reflecting site-specific sequencing quality, such as sequencing depth and the fraction of bases with low-quality scores, and features reflecting the quality of the evidence for a variant, such as the fraction of bases with the reference allele in heterozygous samples (allele balance) and the correlation between observed alleles and the read direction (strand bias). The complete list of features is provided in Supplemental Table 1. Most of these become progressively more informative as they are cumulated across many samples. For example, observing that 75% of bases match the reference allele in a single heterozygous sample is not strong evidence of an artifact, but the same observation averaged across many heterozygotes can suggest systematic biases due to mapping artifacts or the existence of nearby complex variants.
One possible approach for combining many features together for variant filtering is to set thresholds for each feature based on expert knowledge (“hard filtering”). This is extremely laborious to calibrate and hard to replicate across different data sets. We utilize a machine-learning-based approach, based on support vector machines (SVM) that combine all available features into a variant quality score (Cortes and Vapnik 1995). GotCloud uses an open-source implementation of SVM, libSVM (Chang and Lin 2011).
To train the classifier, we first generate a list of positive and negative examples. We utilize external information to generate a list of likely true positives and use an initial set of hard filters to generate a list of likely false positives. By default, the list of likely true positives is the union of array-based polymorphic sites identified from the HapMap Project (The International HapMap Consortium 2007) and the 1000 Genomes Project (Li et al. 2011; The 1000 Genomes Project Consortium 2012). Lists of likely false positives are seeded with sites that fail multiple stringent hard-filters, set as shown in Supplemental Table 1. The SVM classifier defines a decision boundary in the high-dimensional coordinate space defined by all available features, maximizing the distance between the decision boundary and the likely true false positives. We utilize the commonly used radial basis function (RBF) kernel (Amari and Wu 1999).
GotCloud also offers the ability to transfer a SVM model to another data set. This is especially useful for small targeted sequencing experiments where sufficient positive and negative examples might not be available for training (because of limited overlap with HapMap or 1000 Genome site lists, for example). Once a SVM classifier is trained on a large data set, the model can be stored and reused for filtering of other smaller-sized sequencing studies. In our experience, model-transfer SVM will likely perform better than self-trained SVM when the target region is <1 Mb.
Haplotype-aware genotype refinement
The final step of the GotCloud pipeline is genotype refinement. In this step, genotype calls are refined using haplotype information. This step is based on the observation that genotypes at any site are likely to be similar for individuals that share a stretch of sequence (or haplotype) surrounding that site. In the 1000 Genomes Project Consortium analyses, haplotype-based genotype refinement improved genotype accuracy of low-coverage whole-genome data, resulting in genotype accuracies for low-coverage data that were similar to those for deeply sequenced exomes (The 1000 Genomes Project Consortium 2012), in sites shared by multiple individuals. Haplotype-based genotype refinement is especially useful for improving genotype accuracy for low-coverage whole-genome sequences and also for phasing whole-genome sequences for any coverage, although at the expense of additional computational cost (Table 1). The procedure is less useful for targeted exome sequencing, because identifying shared haplotypes is challenging without long contiguous stretches of sequence.
GotCloud uses two tools for genotype refinement: Beagle (Browning and Yu 2009) and ThunderVCF (Li et al. 2011). Beagle is computationally efficient, but the resulting haplotypes can be made more accurate by additionally running ThunderVCF, which is based on a model used by MaCH (Li et al. 2010). As shown in the 1000 Genomes Project (The 1000 Genomes Project Consortium 2012), initializing ThunderVCF with Beagle-phased haplotypes further improves genotype accuracies of Beagle-phased haplotypes, and is much faster than running ThunderVCF alone from random haplotypes. This two-step approach is implemented in GotCloud's genotype refinement pipeline.
Experimental setup
To evaluate the performance of the GotCloud pipeline, we analyzed Chromosome 20 across 1000 low-coverage (∼6x) genomes and 1000 deep exomes. Low-coverage genomes were randomly selected from Phase 3 data of the 1000 Genomes Project and have an average sequencing depth of 5.9× (standard deviation: 2.7). Exomes were randomly selected from Phase 1 data of the 1000 Genomes Project and have an average on target depth of 86× (standard deviation: 35). For the targeted sequencing experiment with a 1 Mb or smaller region, we randomly selected from the Phase 3 exomes, by randomly selecting up to 1 Mb of regions within the exome target region. For comparison, we also ran analyses using the GATK UnifiedGenotyper (DePristo et al. 2011) with default options. Overall runtimes were estimated using a four-node cluster with 48 physical CPU cores, running 40 parallel sessions. For the 1000 sample GATK experiment, we used Chromosome 20 data and extrapolated the number into the whole genome due to larger memory footprints. Peak memory usages are measured by averaging over Chromosome 20 using 5-Mb chunks. When evaluating the sensitivity of variant discovery, we used HumanExome BeadChip variants polymorphic in the sequenced samples, excluding variants included in the Omni2.5 arrays that are also used for positive labels in SVM filtering.
Software availability
The GotCloud pipeline is available for public download (http://www.gotcloud.org) and is prepared for several different cloud computing environments, including the Amazon Web Services (AWS) Elastic Computer Cloud (EC2). It is also possible to run GotCloud on single machines for small projects.
Supplementary Material
Acknowledgments
We thank the 1000 Genomes Project Consortium for making the sequence data publicly available. We thank Michael Boehnke for valuable feedback on the manuscript. This research was funded through grants from the National Institutes of Health (National Human Genome Research Institute; U01 HG006513).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.176552.114.
References
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amari S, Wu S. 1999. Improving support vector machine classifiers by modifying kernel functions. Neural Netw 12: 783–789. [DOI] [PubMed] [Google Scholar]
- Browning BL, Yu Z. 2009. Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. Am J Hum Genet 85: 847–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Lin CJ. 2011. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol 2: 27:1–27:27. [Google Scholar]
- Cortes C, Vapnik V. 1995. Support-vector networks. Mach Learn 20: 273–297. [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, et al. 2013. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493: 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G, Filckinger M, Hetrick KN, Romm JM, Doheny KF, Abecasis GR, Boehnke M, Kang HM. 2012. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am J Hum Genet 91: 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27: 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. 2010. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 34: 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sidore C, Kang HM, Boehnke M, Abecasis GR. 2011. Low-coverage sequencing: implications for design of complex trait association studies. Genome Res 21: 940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Zhan X, Wing M, Anderson P, Kang HM, Abecasis GR. 2013. QPLOT: a quality assessment tool for next generation sequencing data. Biomed Res Int 2013: 865181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lu J, Yu J, Gibbs RA, Yu F. 2013. An integrative variant analysis pipeline for accurate genotype/haplotype inference in population NGS data. Genome Res 23: 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Lee W, Garrison EP, Marth GT. 2013. SSW library: an SIMD Smith-Waterman C/C++ library for use in genomic applications. PLoS One 8: e82138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.