

Abstract
The recent accelerated approval for use in extensively drug-resistant and multidrug-resistant-tuberculosis (MDR-TB) of two first-in-class TB drugs, bedaquiline and delamanid, has reinvigorated the TB drug discovery and development field. However, although several promising clinical development programs are ongoing to evaluate new TB drugs and regimens, the number of novel series represented is few. The global early-development pipeline is also woefully thin. To have a chance of achieving the goal of better, shorter, safer TB drug regimens with utility against drug-sensitive and drug-resistant disease, a robust and diverse global TB drug discovery pipeline is key, including innovative approaches that make use of recently acquired knowledge on the biology of TB. Fortunately, drug discovery for TB has resurged in recent years, generating compounds with varying potential for progression into developable leads. In parallel, advances have been made in understanding TB pathogenesis. It is now possible to apply the lessons learned from recent TB hit generation efforts and newly validated TB drug targets to generate the next wave of TB drug leads. Use of currently underexploited sources of chemical matter and lead-optimization strategies may also improve the efficiency of future TB drug discovery. Novel TB drug regimens with shorter treatment durations must target all subpopulations of Mycobacterium tuberculosis existing in an infection, including those responsible for the protracted TB treatment duration. This review summarizes the current TB drug development pipeline and proposes strategies for generating improved hits and leads in the discovery phase that could help achieve this goal.
The medical community needs novel tuberculosis (TB) drug regimens that work within a shorter time period and are effective against strains that are currently drug-resistant. An improved TB drug discovery pipeline would help meet this need.
INTRODUCTION AND CURRENT DRUG DEVELOPMENT PIPELINE
The goal of tuberculosis (TB) drug discovery and development is to achieve TB drugs and drug regimens that are well understood and superior to those available today in their efficacy, speed of action, safety and tolerability, ease of use for all patient populations, and accessibility. An ideal new regimen should be rapidly bactericidal and possess potent sterilizing activity to achieve stable cure in a shorter time period than the typical 6 mo required for the current standard of care for drug-sensitive TB. To achieve a significantly shorter duration of therapy, it is expected that a regimen would need to kill all subpopulations of Mycobacterium tuberculosis (Mtb) that exist in clinical TB and are thought to vary in replication rate. In addition, new regimens should include drugs that show novel mechanisms of action (MOA) to ensure effectiveness against strains that are resistant to existing TB drugs. The regimens should also be well tolerated and possess absorption, distribution, metabolism, and excretion (ADME) properties suitable for coadministration with anti-HIV (human immunodeficiency virus) agents and appropriate for once-a-day oral dosing, optimally within a fixed-dose combination. It is also imperative that new drugs be available at relatively low cost to remain accessible to all high-burden countries.
The TB drug development pipeline can be viewed on the frequently updated website of the Stop TB Partnership’s Working Group for New TB Drugs (www.newtbdrugs.org). Strategies recently used in TB drug development include reevaluation of existing TB drugs to optimize their utility; repurposing of drugs registered for non-TB indications as components of TB drug combinations; development of improved analogs of compounds or drugs with some known but limited value for TB; and development of novel chemical entities with new modes of action against TB. TB drugs are delivered as combinations with at least three compounds to prevent resistance development and reap the efficacy benefits of different compound classes or modes of action. Evaluation of new TB drugs is therefore as part of regimens from clinical Phase II onward.
The current global TB drug development pipeline includes four compounds under evaluation for active pulmonary TB in Phase III clinical trials. Two of these four compounds, the fluoroquinolones, moxifloxacin and gatifloxacin, are being investigated as part of 4-mo regimens, combined with first-line TB drugs, for drug-sensitive TB. The other two, bedaquiline (a diarylquinoline) and delamanid (a nitroimidazole), are being evaluated as additions to background therapy for MDR-TB. Another nitroimidazole, PA-824, is poised to enter Phase III clinical trials as part of a novel drug combination with moxifloxacin and pyrazinamide, in which it will be evaluated as a 4-mo regimen against drug-sensitive TB and as a 4- or 6-month regimen against MDR-TB. Earlier in development, a variety of approaches to novel regimen development are ongoing. PA-824 and bedaquiline, both novel chemical entities, are under evaluation in Phase II as part of novel drug combinations that include the first-line TB drug pyrazinamide as well as the repurposed drugs moxifloxacin and clofazimine. A number of ongoing studies will evaluate members of the well-known anti-TB class, the rifamycins (rifampicin and rifapentine in this case), in regimens for drug-sensitive TB. Besides the nitroimidazoles and diarylquinolines, two other novel chemical classes are being developed for TB—the ethylenediamines (represented by SQ109) and the oxazolidinones (linezolid, sutezolid, and AZD5847). Notably, a gap currently exists in Phase I with no known ongoing studies of new candidate TB drugs at that stage of development. The paucity of new chemical classes represented (and hence distinct modes of action and utility against drug-resistant and drug-sensitive disease) underscores the need for diverse, well-resourced discovery efforts to increase the flow to development to account for attrition and to plug the Phase I gap.
TB Drug Discovery
Recent advances in understanding the molecular biology of Mtb have been significant, driven largely by the whole-genome sequencing of the bacterium in 1998 (Cole et al. 1998). Knowledge of the complete Mtb genome sequence enabled scientists to establish the number of essential genes both in vitro and in vivo (Sassetti and Rubin 2003; Sassetti et al. 2003), to study genome-wide DNA microarrays for patterns of gene expression under various growth conditions (Sherman et al. 2001; Schnappinger et al. 2003; Rustad et al. 2008; Zhang et al. 2012), and to more rapidly identify targets of new compounds via identification of the mutated genes of compound-resistant mutants (Abrahams et al. 2012a; La Rosa et al. 2012; Ioerger et al. 2013; Remiuinan et al. 2013). The use of elegant gene knockdown techniques has allowed in vitro and in vivo validation of potential drug targets by showing the effect of the depletion of a specific target (Wei et al. 2011; Woong et al. 2011).
There is limited commercial potential for new TB drugs, and therefore, this indication has not been an area of intense activity. In addition, the recent exodus of much of the pharmaceutical industry from antibacterial drug discovery has proven to be a loss for TB drug research and development, as the TB field no longer benefits from drug candidates being developed against other bacteria. Fortunately, some government and nongovernment organizations have taken the initiative to fill this gap, with a turning point occurring with the creation of a number of not-for-profit product-development partnerships (PDPs), such as the Global Alliance for TB Drug Development (TB Alliance) that formed in 2000. Since then, several additional consortia have formed, including the TB Drug Accelerator, the Lilly Early TB Drug Discovery Initiative, and More Medicines for TB (MM4TB) and Orchid, both funded by the European Commission. Academic and government laboratories are also involved in translational medicine of basic research to TB drug discovery, mostly funded by the National Institutes of Health (NIH) and the Bill and Melinda Gates Foundation. The TB field has recently experienced some positive developments with the approval of bedaquiline (TMC-207, Sirturo from Janssen), by the U.S. Food and Drug Administration (FDA) at the end of 2012 (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm33695.htm) (and by EMA in March of 2014), and delamanid (OPC-67683, Deltyba from Otsuka) by the European Medicines Agency (EMA) in December of 2013 (http://online.wsj.com/article/PR-CO-20131125-900844.html), both indicated for MDR-TB. However, the current drug discovery pipeline (http://www.newtbdrugs.org/pipeline-discovery.php) provides little substrate for the development of drastic treatment-shortening drug regimens that will truly change the TB treatment paradigm.
In this review, we focus on the lessons learned from screening efforts of the recent past and how these can be applied for greater future success. We then highlight the lead-generation efforts and TB drug targets that we believe to be the most promising and worthy of more attention. Specifically, we discuss advances in screening technologies that are expected to produce the next generation of progressable hits against Mtb, highlighting the potential for natural products as a source for novel hits, and mechanism- or structure-based drug discovery approaches for the generation of anti-TB leads. We then focus on a selection of novel drug targets that have been recently validated such that their inhibition should eliminate persisters (a subpopulation of organisms in an infection that is phenotypically resistant to killing by most drugs and thought to be responsible for protracted treatment durations and relapse) and contribute to treatment shortening in novel regimens.
THE CURRENT STATUS AND RECENT HISTORY IN HIT/LEAD GENERATION FOR TB
Screening Efforts
As in the antibacterial field in general, despite significant effort being expended on Mtb target-based biochemical screens (Payne et al. 2007), no TB drug or drug candidate has emerged from these efforts to date. Problems with target-based screens include their propensity to identify hits with potency against the target but that do not inhibit bacterial growth because target inhibition does not translate into bacterial killing for a variety of reasons, including poor penetration and efflux. Conversely, whole-cell screening has yielded notable successes for TB drug discovery. As a consequence, it has become the mainstay of TB hit generation. Mtb whole-cell screening is an approach in which compound libraries are screened for their ability to inhibit bacterial growth. These screens have been performed against both replicating and nonreplicating Mtb under a variety of culture conditions in an effort to identify compounds that can kill the multiple subpopulations of Mtb that exist in an infection. Whole-cell screening has the advantage that it is not target-specific, thus, it enables screening against the organism’s entire set of potential targets at once. Results of such a screen, which effectively allows the organism’s physiology to inform discovery efforts, yield accessible targets and sensitive pathways that would otherwise be unpredicted. This approach also overcomes cell-penetration and cell-efflux issues (i.e., getting and keeping compounds in cells) of target-based screens. However, whole-cell screening is not without its challenges. For example, target identification can be problematic, and if not successful, it can limit the ability to quickly improve the potency of hit compounds. A good example of whole-cell screening success is the compound TMC-207 (Andries et al. 2005); other examples include a diarylcoumarin series that targets the acyl–acyl carrier protein synthetase activity of FadD32 (Kawate et al. 2013; Stanley et al. 2013) and the indolcarboxamides NITD-304 and NITD-349, presumed to inhibit MmpL3 from the sequencing of resistant mutants (Rao et al. 2013). All of these compounds were subsequently shown to be efficacious in vivo against murine TB.
Perhaps the earliest publically accessible whole-cell screening results were from the NIH's effort to acquire and test compounds against Mtb (Tuberculosis Antimicrobial Acquisition and Coordinating Facility, TAACF) (Ananthan et al. 2009; Maddry et al. 2009). More recently, several pharmaceutical companies have performed phenotypic screens; for example, GSK recently reported a set of 177 Mtb-active compounds (Ballell et al. 2013). Some hits were further investigated in the hit-to-lead phase and have been made public (Abrahams et al. 2012b). The Novartis Institute of Tropical Diseases (NITD) has published results of a hit-to-lead program based on their selected series (Kondreddi et al. 2013; Yokokawa et al. 2013). AstraZeneca has also performed phenotypic screening and published some results (Ramanchandran et al. 2013). Similar screens against Mtb are under way by members of the TB Drug Accelerator (TBDA) consortium and MM4TB, among others.
The whole-cell screens published thus far largely used standardized aerobic culture conditions that favor replicating Mtb. More recently, specialized whole-cell screens have been performed, including those that use culture conditions considered relevant to in vivo infection Mtb inside host cells. The expectations are that these screens will identify inhibitors that will be active in vivo and against persisters. Screens against Mtb in macrophages have been successful (Christophe et al. 2010); the late preclinical compound Q203 is one example. It is an imidazo[1,2-a]pyridine-3-carboxamide derivative that targets QcrB, a component of the CytBC1 complex of the respiratory chain (Pethe et al. 2013). Interestingly, this series was also discovered through screens under standard culture conditions and for inhibitors of ATP homeostasis under nonreplicating conditions (Mak et al. 2012). Other cell-based specialized screens recently reported include those for inhibitors of ATP or pH homeostasis (Darby et al. 2013) and against engineered Mtb strains with modulated expression of pantothenate synthetase (Kumar et al. 2013). These screens are an improvement over the simple phenotypic screens because inhibitors against particular targets, pathways, or processes already proven essential for TB are likely to be discovered via a whole-cell approach, thus combining the advantages of validated targets with whole-cell screening. Because most of the whole-cell screens against Mtb were undertaken in the last 5–10 yr, it may be of some use to analyze the hits that emerged as well as the screens that identified them. Some interesting phenomena are emerging.
First, structurally identical or closely related compounds have been identified as hits many times over by various organizations; these compounds are listed in Table 1. Some examples include imidazo[1,2-a]pyridine-3-carboxamides (e.g., Q203) that were shown to kill Mtb by at least four different phenotypic screens performed by various organizations (Abrahams et al. 2012b; Mak et al. 2012; Moraski et al. 2011, 2012); tetrahydropyrazolo[1,5-a]pyrimidine-3-carboxamides were selected as hits in at least three different screens (Maddry et al. 2009); and urea analogs containing an adamantyl group or a norbonanylmethylene group were identified as hits in two different screens (Ananthan et al. 2009; Brown et al. 2011; Scherman et al. 2012). The redundant identification of these hits may reflect the similarity of both the libraries of the various organizations and the screening methods.
Table 1.
Structurally identical or closely related compounds have been identified as hits many times over by various organizations as listed below
| Series | Structures | Investigating organization(s) |
|---|---|---|
| Imidazo[1,2-a]pyridine-3-carboxamides | 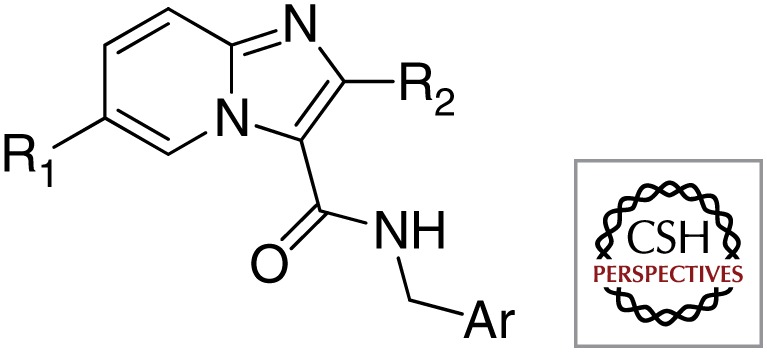 |
University of Notre Dame |
| Institut Pasteur Korea, Quro | ||
| National Institutes of Health, Novartis Institute of Tropical Diseases | ||
| GlaxoSmithKline | ||
| Tetrahydropyrazolo[1,5-a]pyrimidine-3-carboxamides | 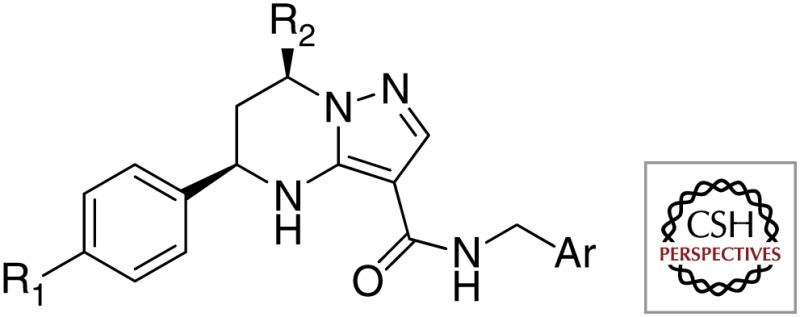 |
Tuberculosis Antimicrobial Acquisition and Coordination Facility |
| GlaxoSmithKline | ||
| Novartis Institute of Tropical Diseases | ||
| Adamantyl ureas | 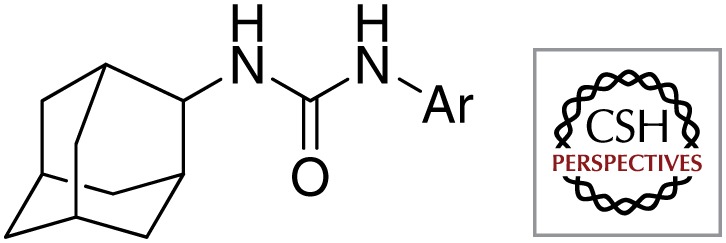 |
Colorado State University |
| Tuberculosis Antimicrobial Acquisition and Coordination Facility | ||
| Indole-2-carboxamides | 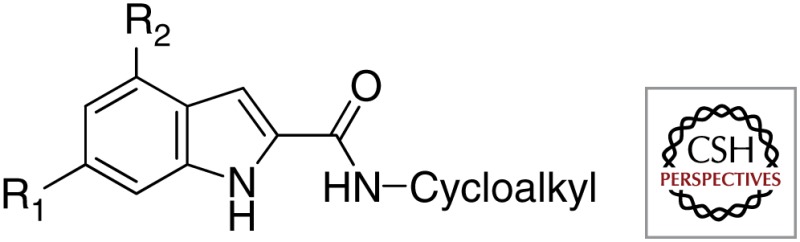 |
Novartis Institute of Tropical Diseases |
| University of Illinois at Chicago | ||
| GlaxoSmithKline |
Second, the same recurring mechanistic targets have been identified for different hits. Representative promiscuous targets are summarized in Table 2. For example, at least five structurally divergent chemical series were shown to have the same target, DprE1, an enzyme involved in isomerization of decaprenylphosphoryl-β-d-ribose to decaprenylphosphoryl-β-d-arabinose, an important cell wall component (Batt et al. 2012). Similarly, structurally divergent compounds were shown to share MmpL3 as the predominant resistant determinant; MmpL3 is a cell membrane transporter of trehalose monomycolate (Grzegorzewicsz et al. 2012a). The exclusivity of MmpL3 as a target of many of the observed hits has been challenged recently and it remains to be debated at present (Li et al. 2014).
Table 2.
Representative promiscuous targets
| Target | Compound | Structure |
|---|---|---|
| DprE1 | BTZ043 | 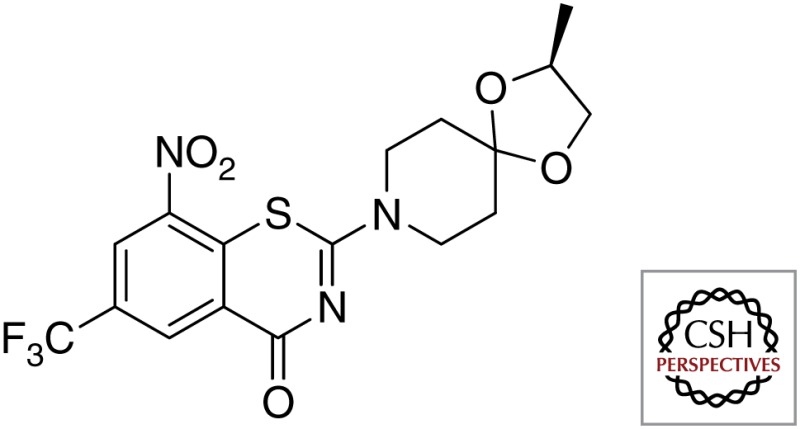 |
| DNB1 | 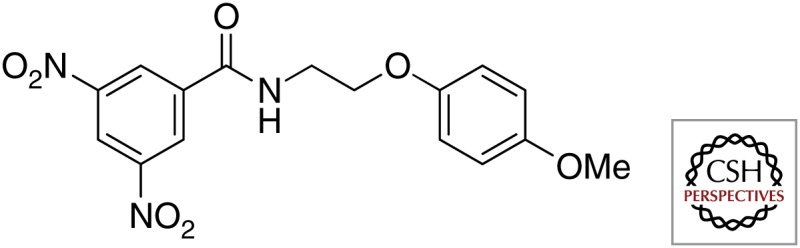 |
|
| TAACF377790 | 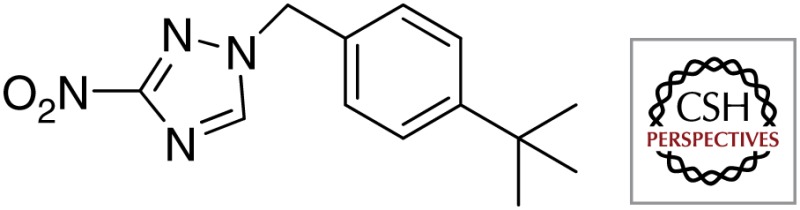 |
|
| TCA1 | 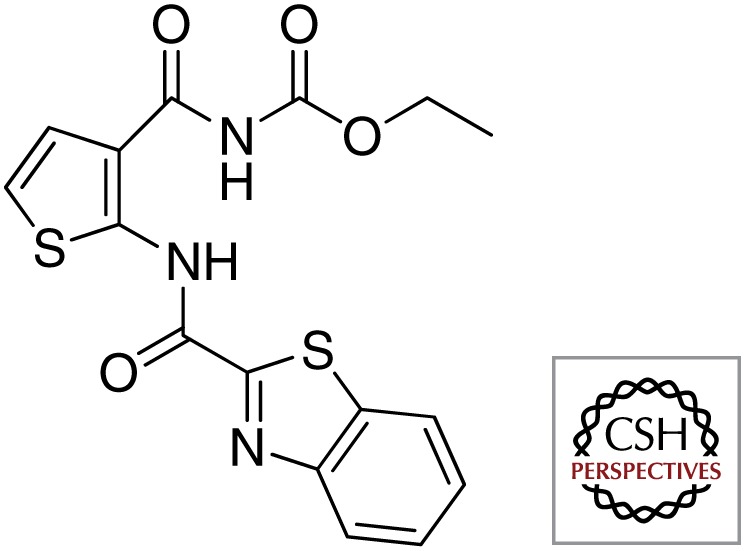 |
|
| Azaindoles | 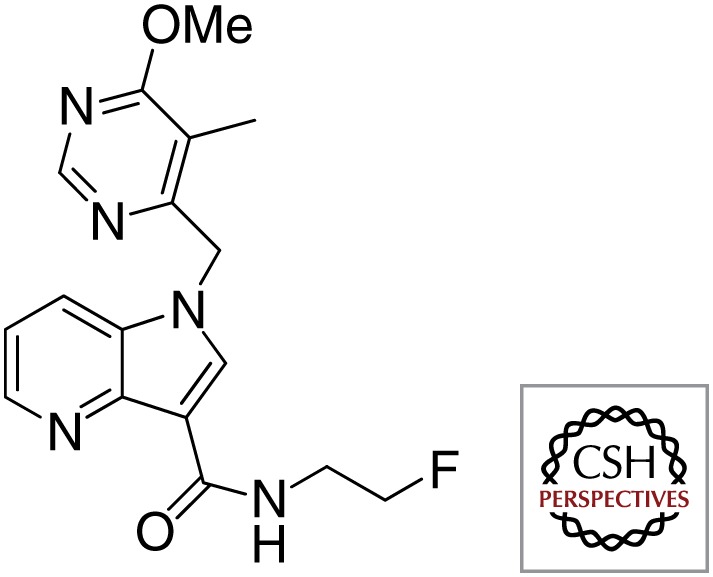 |
|
| MmpL3 | BM212 | 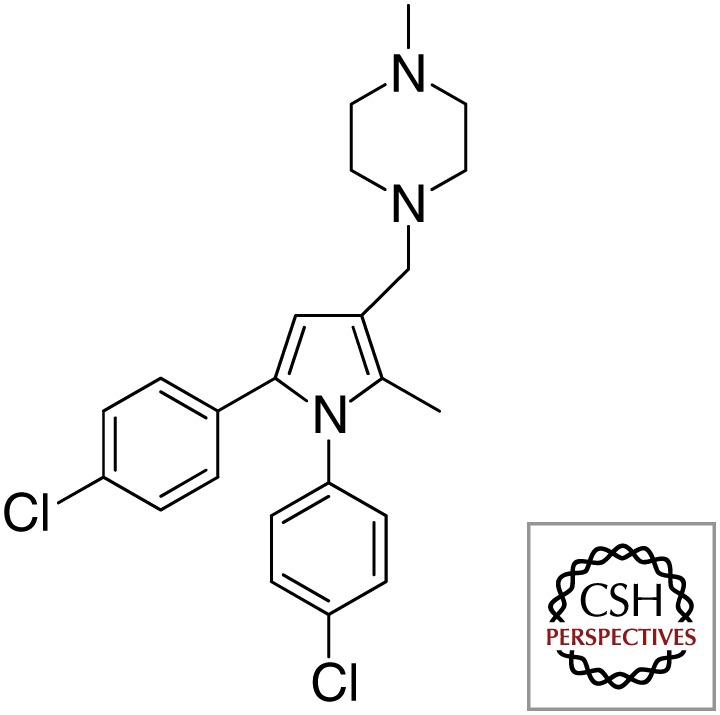 |
| AU1235 | 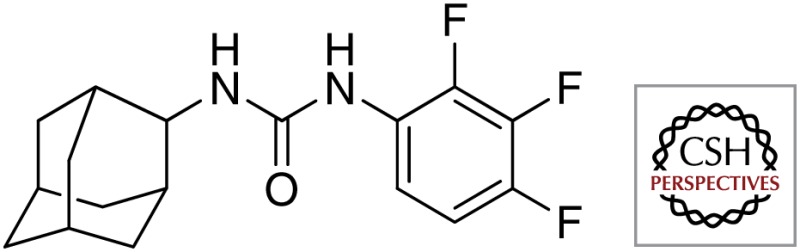 |
|
| C215 | 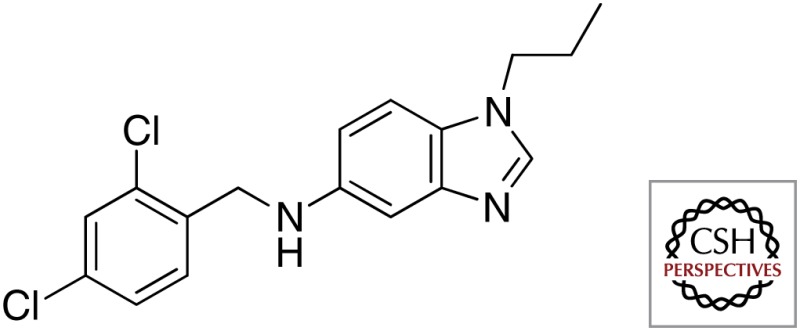 |
|
| THPP | 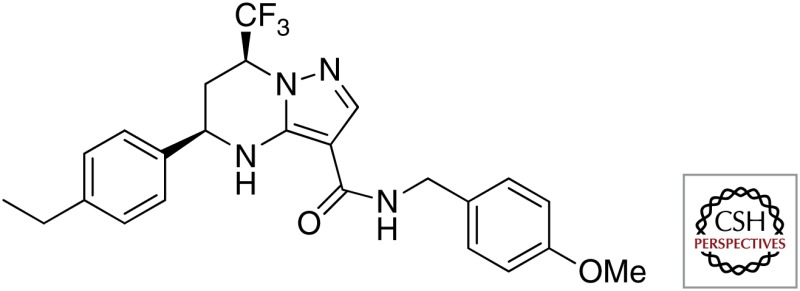 |
|
| Spiro | 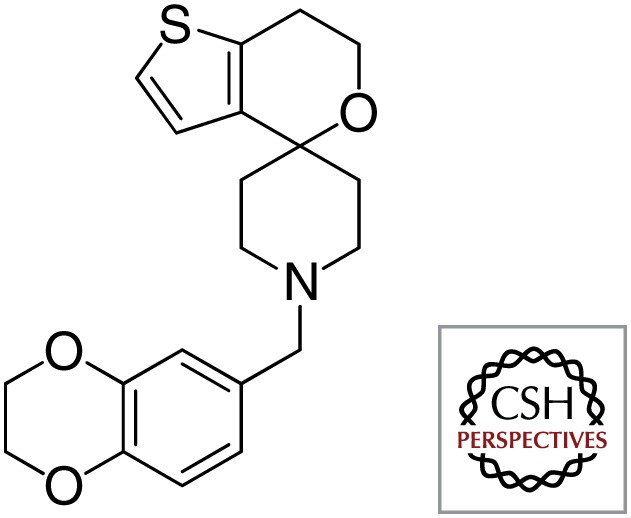 |
|
| SQ109 | 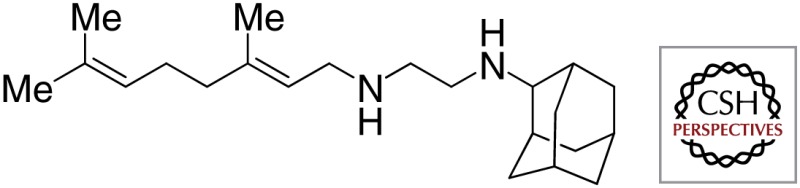 |
|
| NITD-304 | 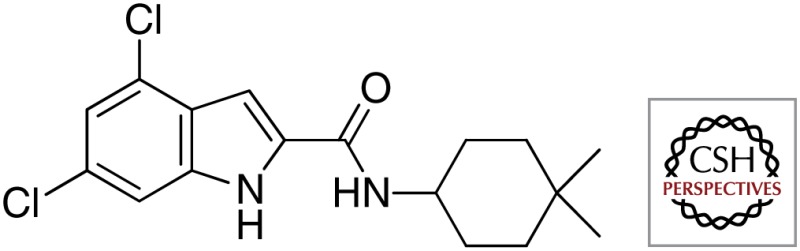 |
|
| QcrB | Imidazo[1,2-a]pyridine | 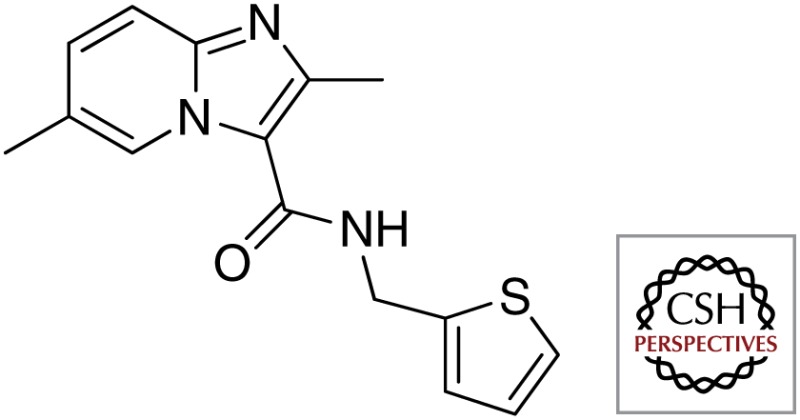 |
| Q203 | 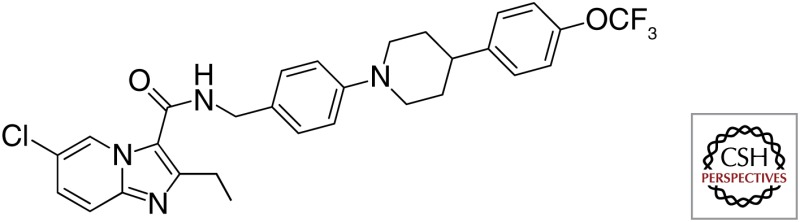 |
The reasons for the same mechanistic targets are not clear, but it may be that these are the most accessible and vulnerable targets under the screening conditions used, and they are mainly cell surface or membrane-bound proteins; the phenomenon of lipophilic compounds and membrane-localized targets has been pointed out in two recent review articles (Stanley et al. 2012; Goldman 2013). The main argument Stanley and Goldman present is that lipophilic compounds may partition and concentrate in the membranes higher than in the intracellular environment. These observations are a reminder that the methodology and conditions of a screen determine the results and the hits identified. Researchers thus may have to rethink their screens and the compound libraries they use because the lipophilic and high-molecular-weight properties of certain hits may preclude successful subsequent downstream development. It is also the case that highly lipophilic and high-molecular-weight compounds that target membrane-localized targets seem to correlate with the potential for hepatotoxicity (Chen et al. 2013), mitochondrial dysfunction (Naven et al. 2013), and nonspecific interactions (Tarcsay and Keseru 2013). They also affect pharmaceutical development owing to the lack of solubility and difficulty in formulation. The fact that bedaquiline (TMC-207), with a cLogP value of 7.3, was successfully developed amply shows that such a compound can be developed, even though the hurdles are high. Various measures can be used to correct this problem. For example, hits with preferred physicochemical properties should be prioritized over higher-potency hits with poor properties, and methods should be established to accurately determine the drug concentrations in various cellular compartments. To that point, a recently reported method for measuring the intracellular compound levels in Mycobacterium smegmatis may be useful in linking intracellular compound levels to bacterial killing (Bhat et al. 2013).
Third, instead of the completely nontarget-specific whole-cell screens, target-specific, whole-cell screens using specialized strains that focus on a specific in vivo validated target, pathway, or process should be performed. Such screens should be designed to use the current knowledge of the most sensitive targets—including for persisters—the transport functions (Sarathy et al. 2012), and the porin systems of Mtb (Siroy et al. 2008) that can be exploited to transport drugs and avoid efflux.
Fourth, smaller libraries with preferred physicochemical properties should be used. For example, NITD scientists recently screened a library of small and polar compounds, with promising results (U. Manjunatha, P. Smith, et al., pers. comm.).
Fifth, CPZN-45 (Ishizaki et al. 2013), spectinamides, and macrolides are successful chemical series in lead optimization derived from natural products. This suggests that use of natural products as a potential source for novel leads against Mtb should be revisited. New potential natural product leads are discussed below.
Underexploited Methods of Lead Generation
Natural Products
It is of interest to note among the four first-line TB drugs (rifampicin, isoniazid, pyrazinamide, and ethambutol) and six series of the second-line TB drugs (aminoglycosides, capreomycin-class polypeptides, fluoroquinolones, cycloserine, and para-aminosalicylic acid), at least four of them are derived from natural products (rifampicin, aminoglycosides, polypeptides, and cycloserine). The utility of natural products and natural product–derived compounds in antibacterial research is well-documented (Newman and Cragg 2012; Brown et al. 2014). Natural products are said to cover a wider chemical space compared with combinatorial library compounds (Feher and Schmidt 2003). This is important especially in view of the identification of frequent hits of synthetic compounds mentioned in the preceding section. Nevertheless, the drug discovery effort in the natural product area still seems to lag behind reflecting the general trend in the pharmaceutical industry, which has mostly retreated from the natural product discovery area 10–20 years ago. There are excellent recent reviews on antitubercular compounds derived from natural products (Singh et al. 2011; Garcia et al. 2012; Guzman et al. 2012; Salomon and Schmidt 2012; Kirst 2013). Here, we highlight some recent notable examples that may prove to be useful leads for TB drug discovery (Fig. 1).
Figure 1.

Recent notable examples of natural and natural-derived products that provide leads for TB drug discovery.
Thuggacin A, isolated from the fermentation broth of the myxobacterium Sorangium cellulosum, is active against Mtb with an minimum inhibitory concentration (MIC) of 8.0 µg/mL and an MOA believed to be inhibition of the Mtb respiratory chain—a target of great interest at present (Steinmetz et al. 2007; Irschik et al. 2007). Ergosterol peroxide, isolated from the leaves of Radermachera boniana, has an MIC of 1.5 µg/mL; although it has an unusual peroxide moiety, it appears to have a reasonable selectivity, because its cytotoxicity against Vero cells is greater than 86 µg/mL (Truong et al. 2011). Trichodermin A, isolated from a fungus from marine sponges, has an Mtb MIC of 0.12 µg/mL under both aerobic and hypoxic conditions (Pruksakorn et al. 2010); it has been shown to be active against nonreplicating mycobacteria and its MOA to be inhibition of ATP synthesis (Pruksakorn et al. 2011). Another peptide natural product, lariatin A, shows a potent inhibitory activity against Mtb, with an MIC of 0.39 µg/mL (Iwatsuki et al. 2007). Isolated from the soil bacterium Rhodococcus jostii, lariatin A forms an unusual “lasso” structure in which its tail passes through a ring structure; its MOA is speculated to be inhibition of cell wall biosynthesis (Iwatsuki et al. 2006). A thiostrepton-class compound isolated from Nocardia pseudobrasiliensis, nocardithiocin is highly potent against Mtb, with an MIC ranging from 0.025 to 1.56 µg/mL. A known natural product, pyridomycin was recently shown to have the same MOA as isoniazid (i.e., inhibition of Mtb InhA, the enoyl-ACP reductase involved in mycolic acid biosynthesis) (Hartkoorn et al. 2012); structural studies indicate that this compound blocks both the cofactor- and substrate-binding pockets (Hartkoorn et al. 2013). As discussed below, all of the inhibitors of Mtb ClpP proteases to date are natural products or natural product–derived analogs.
These examples indicate natural products; although mostly neglected by current efforts caused by cost and other factors, they remain a relevant source of leads for TB drug development. As mentioned above, natural products occupy a chemical space that is very different from that covered by the various small molecules currently being screened within most pharmaceutical company libraries. Although medicinal chemistry to improve ADME and toxicity properties of an initial natural product hit may be challenging, such compounds can provide novel templates and indicate vulnerable targets for simplified, smaller, more developable analogs. Natural products discovered for non-Mtb bacteria may also be a source of future anti-TB leads. Because of the recent accumulation of knowledge in biosynthetic genes of secondary metabolites, information on culture conditions to generate secondary metabolites, and isolation of underexplored organisms, the science of natural products is poised to make a great contribution to TB drug discovery when it is combined with advances in analytical chemistry and chemoinformatics (Li and Vederas 2009; Wright 2014).
Mechanism- and Structure-Based Drug Design
There are numerous examples of mechanism- or structure-based drug designs (Finn 2012) in TB research, only some of which will be mentioned here. An interesting case of structure-guided design is the recently reported malate synthase (GlcB) inhibitors that are analogs of phenyl-dikitoacids, designed based on the enzyme X-ray crystal structure. One analog showed efficacy against murine TB, as discussed in the section on targets below (Krieger et al. 2012). FASII enoyl-ACP reductase (InhA) is the well-validated target of the TB drug isoniazid and has been a target of rational drug design (Pan and Tonge 2012). Based on the methylthiazole compounds discovered at GSK (Ballell Pages et al. 2010; Castro Pichel et al. 2012), scientists at AstraZeneca have published their investigation of the binding mode of these agents to InhA (Shirude et al. 2013). This is part of an effort to discover direct InhA inhibitors that, unlike isoniazid, do not require activation by KatG (Encinas et al. 2014). Defective KatG activation is the dominant resistance determinant in isoniazid-resistant Mtb clinical isolates.
Another approach to modern drug design, fragment-based drug discovery, has recently succeeded in generating inhibitors of Mtb pantothenate synthetase (Pts) (Silvestre et al. 2013). In this study, 1250 fragments were initially screened by a thermal-shift method and nuclear magnetic resonance (NMR) experiments. The initial 39 hits were further characterized by isothermal titration calorimetry and X-ray crystallography to further categorize them into fragments binding at the adenine pocket, those binding at the pantoate pocket, and a fragment binding deep in the pantoate pocket. One fragment has been elaborated to provide potent inhibitors of Pts (Hung et al. 2009).
EMERGING TARGETS
Targeting Mtb Iron Acquisition and Storage
Iron is an essential nutrient for all living organisms, including pathogenic bacteria in an infection, but the mammalian immune system uses various methods to restrict a pathogen’s access to iron (Doherty 2007). Various mammalian proteins are involved in iron homeostasis, and their effect on the immune system is discussed in an excellent review (Johnson and Wessling-Resnick 2012); they include hepcidin, lactoferrin, siderocalin, haptoglobin, hemopexin, Nramp1, ferroportin, and the transferrin receptor, which underscores the importance of this nutrient in the host–pathogen relationship. Iron is sequestered in the human body by intracellular ferritin and extracellular transferrin (Weinberg 1999). During infection, Mtb localizes inside host macrophages (Kauffmann 2004), where it has access to transferrin-bound iron. Mtb secretes two classes of siderophores, mycobactins (Rodriguez 2006) and carboxymycobactins (Ratledge and Dover 2000), to bind iron, obstructing it from the mammalian iron-binding proteins, and then internalizes the iron-laden siderophores through its receptors.
Iron Acquisition
It was recently shown that mbtE deletion mutants are unable to synthesize mycobactins and are attenuated for growth in vitro, macrophages, and guinea pigs, highlighting the importance of mycobactin biosynthesis for the growth and virulence of Mtb and establishing this pathway as a potential target for the TB drug development (Reddy et al. 2013). Mycobactin analogs that inhibited MbtA, another mycobactin biosynthesis enzyme, have shown in vitro Mtb activity superior to first-line TB drugs (Neres et al. 2008).
Deletion mutants of Mtb lacking irtA and irtB are attenuated in human macrophages and mouse lungs (Rodriguez and Smith 2006). IrtAB genes encode the ABC transporter IrtAB involved in the efficient transport and usage of iron from Fe-carboxymycobactin in Mtb. The irtAB genes are located in a chromosomal region previously shown to contain genes regulated by iron and the major iron regulator IdeR.
Mutants of Esx-3, a secretion system that works in concert with the ItrAB ABC transport system to take up iron-laden siderophores, have been shown to be severely impaired for growth in macrophages, indicating Esx-3 as another potential drug target in iron acquisition (Seigrist et al. 2009). A novel siderophore-export system consisting of MmpS4/MmpL4 and MmpS5/MmpL5 was recently identified in Mtb by Wells et al. (2013), and an mmpS4/S5 deletion mutant was shown to be attenuated in mouse infection models, indicating siderophore efflux as a potential drug target (Wells and Jones 2013). These investigators further showed that Mtb recycles its siderophores to enable efficient iron use and that disrupting this process causes the intracellular accumulation of deferrated siderophores, which poisons Mtb (Jones et al. 2014). This suggests that novel drugs targeting inactivation of siderophore export and recycling would deliver a one-two punch to Mtb by reducing its capacity to take up iron and by siderophore-mediated self-poisoning, making siderophore secretion a better target than siderophore biosynthesis because inhibition of siderophore biosynthesis is easily overcome by Mtb using heme (Jones and Niederweis 2011).
An Mtb heme-uptake system has been defined (Tullius et al. 2011) that consists of the secreted protein Rv0203 and the transmembrane proteins MmpL3 and MmpL11. Recent experiments showed that Rv0203 transfers heme to both MmpL3 and MmpL11 during Mtb heme uptake (Owens et al. 2013), making these proteins potential targets for TB drugs.
Iron Storage
Although iron acquisition is required for the growth and virulence of Mtb, it seems that proper iron storage within the pathogen is just as crucial. Excess free iron becomes toxic, catalyzing the production of reactive oxygen radicals that could lead to oxidative damage. Mtb has two iron storage proteins, bacterioferritin (BfrA) (Gupta et al. 2008) and a ferritin-like protein (BfrB) (McMath et al. 2010), which have been shown to be essential for Mtb protection against oxidative stress, growth in macrophages, and virulence in guinea pigs (Reddy et al. 2012). Mtb lacking ferritin suffers from iron-mediated toxicity, is unable to persist in mice, and is highly susceptible to killing by antibiotics (Pandey and Rodriguez 2012), showing that endogenous oxidative stress can enhance antibiotic killing.
Various investigators have targeted siderophores for the development of novel TB drugs. One approach has been to develop agents that directly inhibit enzymes involved in siderophore synthesis (Engelhart and Aldrich 2013). Another approach targets the iron-dependent regulator protein (IdeR) that represses siderophore synthesis genes and virulence factors when sustainable iron levels have been achieved (Mai et al. 2011); dysregulation of IdeR would lead to excess iron and oxidative damage or reduce virulence and enhance bacterial killing. The structural basis for iron activation and IdeR binding to DNA has been recently reported, and these insights have enabled the structure-based design of agents that target IdeR function. Small peptides that either enhance IdeR repression or inhibit IdeR dimerization show that IdeR activity can be rationally modulated (Monfeli and Beeson 2007).
The totality of the data reported here indicates that the biosynthesis, transport, and utilization of siderophores are potential targets for Mtb drug discovery, as Mtb survival and virulence seem to be directly related to iron availability. Indeed, the host already uses iron metabolism against pathogens: siderocalins are host proteins that sequester iron-laden siderophores (Holmes et al. 2005) as a defense mechanism.
Targeting MmpL
The Mtb genome contains 13 genes that encode 12 RND (resistance, nodulation, and cell division) proteins designated mycobacterial membrane protein large (MmpL). RND proteins transport a variety of cationic, anionic, or neutral compounds, including various drugs, heavy metals, aliphatic and aromatic solvents, bile salts, fatty acids, detergents, and dyes, across the cytoplasmic membrane (Paulsen et al. 1996; Putman et al. 2000). In a heroic attempt to decipher the role of MmpL proteins in Mtb, mutant strains inactivated in 11 mmpL genes were generated (Domenech et al. 2005). Susceptibilities to a variety of drugs were unaffected in all the MmpL-deficient strains. One gene, MmpL3, was shown to be essential for growth, and mutants in four genes, MmpL4, MmpL7, MmpL8, or MmpL11, were attenuated in mice. The role of MmpL7 in virulence is likely explained by the findings that this polypeptide is required for the transport of phthiocerol dimycocerosate (PDIM), a known virulence factor, to the cell surface (Cox et al. 1999; Jain and Cox 2005). MmpL8 was found to be involved in sulfatide biogenesis and virulence (Domenech et al. 2004), and an Mtb mutant with disruptions in mmpL4, mmpL5, mmpL7, mmpL8, mmpL10, and mmpL11 showed significant attenuation in mice (Lamichhane et al. 2005). As discussed above, Wells et al. (2013)recently showed the direct involvement of MmpL4 and MmpL5 in siderophore export; the involvement of MmpL3 and MmpL11 in heme uptake has also been shown, thus establishing the importance of these proteins in iron homeostasis and as potential targets for TB drug development. MmpL3, the only MmpL protein that has been shown to be essential, has also been shown to be the resistance determinant for a variety of unrelated drug candidates currently being evaluated (Grzegorzewicz et al. 2012a; Tahlan et al. 2012), as well as to be involved in the transport of mycolic acids across the cell membrane onto the cell surface (Grzegorzewicz et al. 2012a). Efflux of azole antibiotics in Mtb has been associated with mutations in Rv0678, which transcriptionally regulates the efflux system encoded by the mmpS5–mmpL5 efflux system (Milano et al. 2009).
The above studies clearly indicate the importance of this protein family for growth, transport of vital cell wall components with some involvement in virulence, and perhaps the efflux of small molecules that are toxic to the bacterial cell. It is therefore interesting to imagine a single molecule that might inhibit a variety of MmpL proteins and potentially affect growth, virulence, and sensitivity to other drugs. This would be reminiscent of β-lactam antibiotics that target penicillin-binding proteins and affect peptidoglycan biosynthesis and sensitivity to other drugs. A concerted search for such an inhibitor might uncover a natural product capable of such if MmpLs are as important in soil-dwelling mycobacteria. MmpLs are membrane proteins and may thus be more accessible for targeting than cytosolic enzymes, a potential explanation for the various compounds with reported associations with MmpL3. More studies are required to determine if different MmpLs are important under different in vivo conditions and their potential involvement in pathogenesis and drug efflux in the various environments of the diseased lung.
Targeting Cholesterol Metabolism
Mounting evidence suggests that Mtb uses the host’s cholesterol as a source of carbon and energy during infection. Strains defective in cholesterol transport or degradation are attenuated in activated macrophages, and Mtb requires several genes involved in cholesterol catabolism for full virulence in animal models (Pandey and Sassetti 2008; Chang et al. 2009; Yam et al. 2009; Nesbitt et al. 2010). It is unclear what carbon source for Mtb is most important during an infection, but host cholesterol use by Mtb has been shown (Van der Geize et al. 2007; Miner et al. 2009) and appears to be particularly important during the chronic phase of infection (Chang et al. 2009). Host cholesterol has been shown to be involved in a human Mtb infection (Kim et al. 2010), and high levels of cholesterol in the diet have been shown to significantly enhance the bacterial burden in the lung (Schafer et al. 2009). Specifically, cholesterol is required for phagocytosis of mycobacteria by macrophages (Gatfield and Pieters 2000; Peyron et al. 2000).
The intracellular growth operon, the igr locus, encodes enzymes of cholesterol catabolism in the Mtb genome (Chang et al. 2007) the primary function of which is the degradation of the 2′-propanoate side chain. Inactivation of the igr operon results in reduced Mtb growth on cholesterol alone or in combination with glycerol, indicating that cholesterol or its metabolites are toxic to the igr mutants. The igr locus encodes a cytochrome P450 (cyp125), two acyl-CoA dehydrogenases (fadE28 and fadE29), two conserved hypothetical proteins (Rv3541-2c), and a putative lipid-carrier protein; the locus is essential for growth in macrophages and critical in the chronic phase of infection (Thomas et al. 2011). An enzyme encoded by fadA5 has been described that catalyzes the thiolysis of acetoacetyl-CoA and is required for growth on cholesterol and virulence in the late stages of an infection (Nesbitt et al. 2010).
The other phase of cholesterol catabolism in Mtb is the degradation of the A–D rings. The 3-ketosteroid 9α-hydroxylase (KshAB) has been identified as a virulence factor involved in cholesterol ring degradation (Capyk et al. 2011). In a seminal study using bioinformatics, it was shown that 51 rhodococcal genes specifically expressed during growth on cholesterol were conserved within an 82-gene cluster in Mtb and that mycobacteria grown on cholesterol up-regulate hsaC and kshA (Van der Geize et al. 2007). Transposon mutagenesis studies suggested that the genes encoding HsaA and HsaD are essential for intracellular survival of Mtb in human macrophages (Rengarajan et al. 2005). HsaC, an iron-dependent extradiol dioxygenase, has been shown to be a virulence determinant in guinea pigs that influences dissemination, persistence, and the extent of disease pathology (Yam et al. 2009). Interestingly, cholesterol metabolism was shown to be lethal to an hsaC Mtb mutant, consistent with catechol toxicity. HsaD, the cholesterol meta-cleavage product (MCP) hydrolase (Lack et al. 2010), has been associated with the survival of Mtb in macrophages (Rengarajan et al. 2005). HsaAB, an enzyme that transforms 3-HSA to 3,4-DHSA in cholesterol catabolism, has also been identified (Dresen et al. 2010). Also, FadD3 has been shown to be a 3aα-H-4α (3′-propanoate)-7aβ-methylhexahydro-1,5-indanedione (HIP)–CoA synthetase that initiates catabolism of steroid rings C and D after side-chain degradation is complete (Casabon et al. 2013).
The rhodococcal Mce4 system is required for steroid uptake and is up-regulated on growth on cholesterol (Mohn et al. 2008). On this basis, it was predicted that all Mce4 systems are steroid transporters and that in Mtb, they transport cholesterol during an infection. Indeed, the involvement of the Mtb mce4-encoded transport system in cholesterol import has been shown, and mce4-deleted mutants have been shown to be severely attenuated in both macrophages and mouse infection models, establishing cholesterol import and metabolism as potential therapeutic targets (Senaratne et al. 2008).
From the above experimental data, it seems that although cholesterol catabolism by Mtb is important during infection, if ineffectively metabolized, cholesterol may be toxic to the pathogen. Importantly, both phenomena can be exploited for TB drug discovery, for drugs that inhibit cholesterol metabolism or those that inhibit enzymes which remove the toxic intermediates of cholesterol metabolism. In contrast to these observations, it was recently reported that cholesterol is not required as a nutritional source during infection (Yang et al. 2011).
Targeting the Mtb ClpP Protease
The ClpP proteases (cutinase-like proteins, chaperon-linked proteases, or caseinolytic proteases) have been of interest as drug targets because the antibacterial activity of an ADEP, an acyl depsipeptide that binds the Clp protease of Bacillus subtilis, was shown (Brotz-Oesterhelt et al. 2005). Clp proteases are conserved proteases involved in the degradation of damaged, poorly formed, and unfolded proteins.
Mtb encodes two ClpP homologs, ClpP1 and ClpP2, which form a self-compartmentalized protease consisting of two heptameric rings stacked on top of each other, enclosing a catalytic chamber. Within the chamber, which can be reached through two axial pores, each of the 14 identical monomers possesses a serine protease active site (Ingvarsson et al. 2007). To gain activity, the ClpP protease multimer associates with hexameric rings of Clp ATPases forming the proteolytic complex. The ATPase subunits (ClpC1 and ClpC2) are responsible for recognition, unfolding, and translocation of peptides into the ClpP degradation chamber. The resulting structure is the chamber in which the active sites are sequestered from the cytoplasm to exclude native proteins and control access to the proteolytic chamber.
It was recently shown that ClpC1 is the protein target of the natural product antibiotic cyclomarin in Mtb and that interference with the function of the ClpC1 ATPase with this noncompetitive small molecule is bactericidal in actively growing and in hypoxic nongrowing mycobacteria (Schmitt et al. 2011). The frequency of spontaneous mutations rendering Mtb resistant to cyclomarin was extremely low, and the clpC1 gene could be shown to be essential for Mtb growth, a strong indication that interference with ClpC1 is a promising approach for the development of new antitubercular therapies.
A recent Mtb global transcriptional analysis identified approximately 100 genes involved in resumption of replication on reaeration following hypoxia, which included a transcription factor, the Clp protease gene regulator ortholog ClgR (Sherrid et al. 2010). Mtb ClgR was shown to activate the transcription of at least 10 genes, including four that encode protease systems (ClpP1/C, ClpP2/C, PtrB, and HtrA-like protease Rv1043c) and three that encode chaperones (Acr2, ClpB, and the chaperonin Rv3269). clgR deletion mutants were attenuated for growth in macrophages and for controlling the phagosomal pH, compared with wild-type organisms (Estorninho et al. 2010).
Recent studies have shown that ClpP1 and ClpP2 are essential for growth and form a mixed complex that degrades missense and prematurely terminated peptides, as depletion of the protease specifically led to growth reduction in the presence of antibiotics that increase errors in translation. A recent study confirmed that ClpP1 is essential for Mtb growth in vitro and that the previously described ClpP activators (acyldepsipeptides [ADEPs], discussed below) are active against Mtb (Ollinger et al. 2012). Mtb ClpP has thus been validated as a drug target that could be exploited because of its novel mechanism—in the presence of ADEPs—of bacterial killing. It has been shown that ADEP binding leads to a change of ClpP structure, allowing access of unfolded proteins to the proteolytic chamber in the absence of the regulatory Clp ATPases (Kirstein et al. 2009), thus effectively dysregulating this protease system and leading to unregulated proteolysis and bacterial death. It was recently shown that ADEP4-activated ClpP becomes nonspecific in its proteolytic activity and kills Staphylococcus aureus persisters by degrading more than 400 proteins, causing cells to self-digest (Conlon et al. 2013). A similarly efficacious dysregulator of Mtb ClpP would represent a truly novel mechanism of sterilizing chronically infected lungs, contribute to shortening of treatment durations in novel TB drug regimens, and be effective against resistant disease.
A recent report has described lassomycin, an antibiotic that was specifically bactericidal against both replicating and nonreplicating Mtb (Gavrish et al. 2014). Lassomycin was found to bind to the ClpC1 ATPase complex and stimulate its ATPase activity, and it was its uncoupling of ATPase from proteolytic activity that accounted for the bactericidal activity.
Alternatively, a structurally diverse series of β-lactone inhibitors has been shown to form a covalent adduct at the ClpP2 catalytic serine and inhibit Mtb growth (Compton et al. 2013). This finding provides yet another pharmacological validation of the ClpP protease as a potential drug target, but in this case, the mechanism is by inhibition of ClpP instead of by dysregulating ClpP activity as occurs with acyldepsipeptides.
Targeting Central Carbon Metabolism
Mtb adapts its metabolism to the environmental conditions to which it is exposed (Rhee et al. 2011). Several metabolic enzymes have been validated as drug targets; the multifunctionality of some of these enzymes makes them of particular interest.
The enzymes of the glyoxylate shunt, isocitrate lyase (ICL) (Honer et al. 1999) and malate synthase (GlcB) (Smith et al. 2003), have long been and remain of interest to TB drug discovery. The glyoxylate shunt serves to bypass the two CO2− generating steps of the tricarboxylic acid (TCA) cycle when carbon is limiting, including during growth on fatty acids. ICL-deficient Mtb cannot establish an infection in mice (McKinney et al. 2000; Munoz-Elias and McKinney 2005), and icl is up-regulated during infection (Timm et al. 2003). Although this suggests reliance on this pathway and subsistence on fatty acids in vivo, the importance of ICL may result from its several roles—ICL in the glyoxylate shunt, methyl-ICL in the methylcitrate cycle of propionyl-coA metabolism (Gould et al. 2006; Munoz-Elias et al. 2006), ICL in ATP homeostasis during adaptation to slow growth (Gengenbacher et al. 2010), and ICL in succinate generation for proton motive force (PMF) maintenance under hypoxia (Eoh and Rhee 2013). Perhaps owing to the small polar active site of ICL (Sharma et al. 2000), only weakly efficacious ICL inhibitors have been reported to date (Kraty and Vinsova 2012). Efforts continue to target ICL, including via target-based whole-cell screening (Abrahams et al. 2012a). Conversely, novel, efficacious phenyl-diketo acid inhibitors of Mtb GlcB were recently discovered (Krieger et al. 2012). As for ICL, GlcB appears to play unexpected metabolic roles in carbohydrate and cholesterol metabolism—another example of the metabolic flexibility of Mtb (de Carvalho et al. 2010; Beste et al. 2011; Griffin et al. 2011).
Like ICL, phosphoenolpyruvate carboxykinase (PEPCK) is required by Mtb for growth on fatty acids and survival in rodents (Liu et al. 2003; Marrero et al. 2010), and it is induced during infection (Timm et al. 2003). PEPCK catalyzes the first committed step of gluconeogenesis, by which sugars are synthesized from TCA intermediates during growth on fatty acids (Mukhopadhyay et al. 2001). As with ICL, the importance of PEPCK in vivo suggests subsistence on fatty acids. However, the glycolytic enzyme glucokinase is also required for survival of Mtb in mice (Marrero et al. 2013), suggesting a requirement for metabolism of carbohydrates during late infection. No drug discovery efforts have been reported yet against these interesting targets.
Two better explored targets are lipoamide dehydrogenase (Lpd) and dihydrolipoamide acyltransferase (DlaT). DlaT and Lpd function as components of pyruvate dehydrogenase, which supplies glycolysis-derived acetyl-CoA to the TCA cycle (Tian et al. 2005). Both are also components of the Mtb peroxynitrite reductase/peroxidase, which functions to resist host-reactive nitrogen intermediates (Bryk et al. 2000,2002). Lpd also functions within branched-chain amino acid metabolism, perhaps explaining the more profound attenuation for growth in mice on disruption of lpdC compared with dlaT (Shi and Ehrt 2006; Venogopal et al. 2011). Efforts to target both Lpd (Bryk et al. 2010) and DlaT are ongoing, the latter motivated by the demonstration that DlaT-deficient Mtb fails to establish an infection in guinea pigs (Bryk et al. 2008). Discovery efforts focused on DlaT have identified inhibitors that are selectively active against nonreplicating Mtb.
Finally, the maltosyltransferase GlgE was recently identified as a target of interest (Kalscheuer and Jacobs 2010). This enzyme participates in a pathway from trehalose to α-glucan, and GlgE-deficient Mtb dies in vitro and in mice caused by accumulation of its toxic substrate, maltose-1-phosphate. This pathway also shows a synthetic lethal interaction with the glucosyltransferase Rv3032, suggesting that synergistic TB drugs may be discovered by targeting components of these pathways.
Targeting Energy Generation: Inhibitors of the Respiratory Chain and ATP Synthesis
ATP synthesis and PMF generation are among the best-validated Mtb drug targets owing to the clinical successes of the ATP synthase inhibitor TMC207 (bedaquiline, Sirturo) (Andries et al. 2005; Diacon et al. 2009) and the cornerstone TB drug pyrazinamide, which disrupts the PMF (Zhang et al. 2003). PMF and ATP homeostasis are required by replicating and nonreplicating Mtb, under a variety of conditions (Koul et al. 2008; Rao et al. 2008), and efforts to target these processes include pathway screens using membrane particles, cell-based screens for ATP homeostasis disruptors, target-based screens, and repurposing of existing drugs known to inhibit this pathway.
Mtb operates a branched respiratory chain, the components of which operate variably, depending on the environment (Kana et al. 2001; Matsoso et al. 2005; Weinstein et al. 2005; Small et al. 2013). A menaquinone pool is reduced by NADH dehydrogenase (Ndh, provided by a 14-subunit NdhI complex and single subunit NdhII) and succinate:menaquinone oxidoreductase (Sdh). Electrons flow from the menaquinone pool to a quinol oxidase (cytochrome bd oxidase) or to a cytochrome bc1–aa3 oxidoreductase supercomplex wherein a bc1 c-type cytochrome reductase transfers electrons to the terminal aa3-type oxidase. Both oxidases use molecular oxygen as the terminal electron acceptor. In the absence of respiration, Mtb maintains the PMF and ATP synthesis using secreted succinate generated by fumarate reductase (Watanabe et al. 2011) and/or produced by ICL.
TB drug discovery efforts have focused on NdhII, encoded in Mtb by ndh and ndhA. The phenothiazines (a class of central nervous system drugs) have anti-Mtb activity in vitro and in vivo (Amarai et al. 1996; Bettencourt et al. 2000), and their Mtb target is NdhII (Warman et al. 2013), pharmacologically validating this enzyme. In addition, the leprosy drug clofazimine, which is efficacious against murine TB, targets NdhII as part of its mode of action (Yano et al. 2011). The existence of transposon mutants of ndhA but not ndh (McAdam et al. 2002) suggests the relative importance of the ndh-encoded component of NdhII. However, a novel compound identified through a screen for ATP synthesis inhibitors appears to target NdhA (Ioerger et al. 2013). The importance of NdhI and Sdh for Mtb energy metabolism is not fully understood, although recent evidence suggests a crucial role for Sdh under hypoxia (Eoh and Rhee 2013).
The biosynthesis of menaquinone is expected to be critical under aerobic and hypoxic conditions (Dhiman et al. 2009; Kurosu and Crick 2009). Menaquinone is synthesized from chorismate by a series of at least eight enzymes. Of these, MenC, MenD, and MenE appear to be essential in vitro (Sassetti and Rubin 2003). A recent report of anti-Mtb MenB inhibitors cites a personal communication from C.M. Sassetti that MenB is essential (Li et al. 2011). A report on MenA inhibitors active against replicating and nonreplicating Mtb mentioned an unpublished demonstration (by D. Schnappinger) that MenA is essential for growth of Mtb in mice, providing genetic validation for MenA and menaquinone biosynthesis in general as a drug target (Debnath et al. 2012). Target-based efforts are ongoing against Mtb MenA (Kurosu et al. 2007; Kurosu and Crick 2009), MenB, and MenE (Lu et al. 2012).
The cytochrome b subunit (QcrB) of the cytochrome bc1-aa3 complex has recently emerged as a target of interest. Previous studies (Sassetti and Rubin 2003) indicated that QcrB is essential and that the aa3-type cytochrome c oxidase is important for aerobic growth, although the less bioenergetically favorable cytochrome bd oxidase is more important for microaerobic growth (Kana et al. 2001). However, disruption of Mtb cytochrome c maturation results in only impaired growth in vitro and in mice, which is partially compensated for by induction of cytochrome bd oxidase (Small et al. 2013). This suggests a role for cytochrome bd oxidase during aerobic growth and in infection and suggests limited target vulnerability of the cytochrome bc1 complex. Despite this, the QcrB inhibitor, Q203, shows potent efficacy against murine TB. Q203 and other compounds confirmed or presumed to inhibit QcrB are described in more detail above (Mak et al. 2012a,b). Although Mtb QcrB is pharmacologically validated, further efficacy profiling of QcrB inhibitors is ongoing.
Following generation of the PMF, ATP synthesis occurs via F0F1 ATP synthase. Its c subunit is the target of the diarylquinoline TMC207. Efforts to target ATP synthase are limited by its complex nature. However, cell-based (Mak et al. 2012a) or membrane particle–based screens for ATP synthesis inhibitors may identify new series targeting ATP synthase.
ROS and NOS Generation
Although somewhat controversial (Keren et al. 2013), one hypothesis states that all bactericidal antibiotics kill bacteria by generating reactive oxygen or nitrogen species (ROS/RNS) (Dwyer et al. 2009). Recently, it was shown that a relatively small change (20%) in dissolved oxygen can affect killing of bacterial persiters (Grant et al. 2012), a subpopulation of bacteria in an infection that is phenotypically resistant to killing by most antibiotics but still sensitive, however, to high quantities of radicals. This observation can be critical for killing Mtb in granulomas, which have hypoxic cores (Via et al. 2008). Drugs such as clofazimine, PA-824, and delamanid have been shown to generate ROS and/or RNS (Singh et al. 2008; Yano et al. 2011), which is presumably the mechanism for their activity against Mtb. Recently, the function of the Mtb deazaflavin-dependent nitroreductase (Ddn), an enzyme that activates PA-824, has been hypothesized to be what provides Mtb protection from oxidative stress and bactericidal agents (Gurumurthy et al. 2013); the authors of the report noted that Ddn mutants defective in the formation of deazaflavin were hypersensitive to isoniazid, moxifloxacin, and clofazimine. This suggests that any agent that can inhibit Ddn or deazaflavin biosynthesis could synergize with existing anti-TB drugs. Thus, novel mechanisms to generate ROS/NOS at sufficiently high concentrations should be investigated as potential novel therapy for TB, as long as they do not cause oxidative damage to the host cells (Domann 2013).
CONCLUDING REMARKS
A robust global TB drug discovery portfolio is crucial to achieving the goal of new, simpler, shorter, better TB drug regimens useful for all patient populations. The current status of anti-TB lead generation is much improved compared with the situation 10–15 years ago. However, it is still slow and drastically lacking in success; significant changes are needed to produce novel regimens with efficacy against drug-resistant TB and shorter treatment durations. A review of the processes that have led to the current global pipeline suggests a few solutions that could be used to improve the situation and produce better, more easily developable leads. Smaller, targeted compound libraries with favorable physicochemical properties should be used for hit generation; hits with preferred physicochemical properties should be prioritized over higher-potency hits with poor properties; hits with improved cell penetration and with activity against multiple subpopulations of Mtb should be prioritized. When possible, leads that can penetrate caseum and have efficacy within hypoxic lesions should be prioritized, and natural products should be revisited to allow the identification of currently unexplored chemical matter.
In addition, structure- and fragment-based approaches should be used wherever practicable. Instead of either target-based biochemical screening or whole-cell-based screening, whole-cell-based, target-specific screens using specialized strains that focus on in-vivo-validated targets, pathways, or processes should be performed. Such screens should be designed using what is currently known about the various bacterial populations in the infected lung; the nature and metabolic properties of the organisms in an infection, including persisters; their transport systems and porins that could be exploited for drug uptake; and the most sensitive targets at the different stages of the disease. Such vulnerable pathways and processes might include the in-vivo-validated iron acquisition and storage pathways; central carbon, cholesterol, and energy metabolism pathways; the MmpL transport systems; and the Clp protease system. Nevertheless, because both knowledge of the biology of Mtb remains incomplete (Orme 2014) and the nature of clinical TB is so complex, any current screening approach necessarily remains limited. Multiple subpopulations of bacteria, differing in their replication rate and metabolic state, exist in an infection in the multiple and changing environments of the tuberculous lung (Lin et al. 2014). Any one set of screening conditions is thus unlikely to include key features of all of these environments, and few individual targets and pathways are likely crucial to all physiological states of the organism and disease (Dartois and Barry 2013). It is unlikely that any in-vitro-or even mouse-based screen will select and identify compound series with sufficient exposure for efficacy in necrotic, hypoxic, and cavitary lesions (Kjellsson et al. 2012), or eliminate viable but nonculturable (VBNC) (Pai et al. 2000; Manina and McKinney 2013) organisms.
Yet, we believe that combining the lessons learned from recent and ongoing TB drug discovery efforts with emerging technologies and an evolving understanding of Mtb biology should provide a path toward safer, novel regimens with greater treatment-shortening potential.
ACKNOWLEDGMENTS
We acknowledge the various collaborators and funders of the TB Alliance.
Footnotes
Editors: Stefan H.E. Kaufmann, Eric J. Rubin, and Alimuddin Zumla
Additional Perspectives on Tuberculosis available at www.perspectivesinmedicine.org
REFERENCES
- Abrahams GL, Kumar A, Savvi S, Hung AW, Wen S, Abell C, Barry CE III, Sherman DR, Boshoff HI, Mizrahi V. 2012a. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem Biol 19: 844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams KA, Cox JA, Spivey VL, Loman NJ, Pallen MJ, Constantinidou C, Fernández R, Alemparte C, Remuiñán MJ, Barros D, et al. 2012b. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS ONE 7: e52951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral L, Kristiansen JE, Abebe LS, Millett W. 1996. Inhibition of the respiration of multi-drug resistant clinical isolates of Mycobacterium tuberculosis by thioridazine: Potential use for initial therapy of freshly diagnosed tuberculosis. J Antimicrob Chemother 38: 1049–1053. [DOI] [PubMed] [Google Scholar]
- Ananthan S, Faaleolea ER, Goldman RC, Hobrath JV, Kwong CD, Laughon BE, Maddry JA, Mehta A, Rasmussen L, Reynolds RC, et al. 2009. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinb) 89: 334–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andries K, Verhasselt P, Guillemont J, Göhlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, et al. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307: 223–227. [DOI] [PubMed] [Google Scholar]
- Ballell L, Bates RH, Young RJ, Alvarez-Gomez D, Alvarez-Ruiz E, Barroso V, Blanco D, Crespo B, Escribano J, González R, et al. 2013. Fueling open-source drug discovery: 177 Small-molecule leads against tuberculosis. ChemMedChem 8: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballell Pages L, Castro Pichel J, Fernandez Menendez R, Fernandez Velando EP, Gonzalez Del Valle S, Mendoza Losana A, Wolfendale MJ. 2010. (Pyrazol-3-yl)-1,3,4-thiadiazole-2-amine and(Pyrazol-3-yl)-1,3,4-thiazole-2-amine compounds. PCT publication No. WO 2010/118852 A1.
- Batt SM, Jabeen T, Bhowruth V, Quill L, Lund PA, Eggeling L, Alderwick LJ, Fütterer K, Besra GS. 2012. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc Natl Acad Sci 109: 11354–11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beste DJ, Bonde B, Hawkins N, Ward JL, Beale MH, Noack S, Nöh K, Kruger NJ, Ratcliffe RG, McFadden J. 2011. 13C Metabolic flux analysis identifies an unusual route for pyruvate dissimilation in mycobacteria which requires isocitrate lyase and carbon dioxide fixation. PLoS Pathog 7: e1002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt MV, Bosne-David S, Amaral L. 2000. Comparative in vitro activity of phenothiazines against multidrug-resistant Mycobacterium tuberculosis. Int J Antimicrob Agents 16: 69–71. [DOI] [PubMed] [Google Scholar]
- Bhat J, Narayan A, Venkatraman J, Chatterji M. 2013. LC-MS based assay to measure intracellular compound levels in Mycobacterium smegmatis: Linking compound levels to cellular potency. J Microbiol Methods 94: 152–158. [DOI] [PubMed] [Google Scholar]
- Brötz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, et al. 2005. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med 11: 1082–1087. [DOI] [PubMed] [Google Scholar]
- Brown JR, North EJ, Hurdle JG, Morisseau C, Scarborough JS, Sun D, Korduláková J, Scherman MS, Jones V, Grzegorzewicz A, et al. 2011. The structure-activity relationship of urea derivatives as anti-tuberculosis agents. Bioorg Med Chem 19: 5585–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DG, Lister T, May-Dracka TL. 2014. New natural products as new leads forantibacterial drug discovery. Bioorg Med Chem Lett 24: 413–418. [DOI] [PubMed] [Google Scholar]
- Bryk R, Griffin P, Nathan C. 2000. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 407: 211–215. [DOI] [PubMed] [Google Scholar]
- Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. 2002. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science 295: 1073–1077. [DOI] [PubMed] [Google Scholar]
- Bryk R, Gold B, Venugopal A, Singh J, Samy R, Pupek K, Cao H, Popescu C, Gurney M, Hotha S, et al. 2008. Selective killing of nonreplicating mycobacteria. Cell Host Microbe 3: 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk R, Arango N, Venugopal A, Warren JD, Park YH, Patel MS, Lima CD, Nathan C. 2010. Triazaspirodimethoxybenzoyls as selective inhibitors of mycobacterial lipoamide dehydrogenase. Biochemistry 49: 1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capyk JK, Casabon I, Gruninger R, Strynadka NC, Eltis LD. 2011. Activity of 3-ketosteroid 9α-hydroxylase (KshAB) indicates cholesterol side chain and ring degradation occur simultaneously in Mycobacterium tuberculosis. J Biol Chem 286: 40717–40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casabon I, Crowe AM, Liu J, Eltis LD. 2013. FadD3 is an acyl-CoA synthetase that initiates catabolism of cholesterol rings C and D in actinobacteria. Mol Microbiol 87: 269–283. [DOI] [PubMed] [Google Scholar]
- Castro Pichel J, Fernandez Menendez R, Fernandez Velando EP, Gonzalez Del Valle S, Mallo-Rubio A. 2012. 3-Amino-pyrazole derivatives useful against tuberculosis. PCT publication No. WO2012/049161.
- Chang JC, Harik NS, Liao RP, Sherman DR. 2007. Identification of mycobacterial genes that alter growth and pathology in macrophages and in mice. J Infect Dis 196: 788–795. [DOI] [PubMed] [Google Scholar]
- Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, Sherman DR. 2009. igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol 191: 5232–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Borlak J, Tong W. 2013. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 58: 388–396. [DOI] [PubMed] [Google Scholar]
- Christophe T, Ewann F, Jeon HK, Cechetto J, Brodin P. 2010. High-content imaging of Mycobacterium tuberculosis-infected macrophages: An in vitro model for tuberculosis drug discovery. Future Med Chem 2: 1283–1293. [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544. [DOI] [PubMed] [Google Scholar]
- Compton CL, Schmitz KR, Sauer RT, Sello JK. 2013. Antibacterial activity of and resistance to small molecule inhibitors of the ClpP peptidase. ACS Chem Biol 8: 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. 2013. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503: 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JS, Chen B, McNeil M, Jacobs WR Jr. 1999. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402: 79–83. [DOI] [PubMed] [Google Scholar]
- Darby CM, Ingólfsson HI, Jiang X, Shen C, Sun M, Zhao N, Burns K, Liu G, Ehrt S, Warren JD, et al. 2013. Whole cell screen for inhibitors of pH homeostasis in Mycobacterium tuberculosis. PLoS ONE 8: e68942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartois V, Barry CE III. 2013. A medicinal chemists’ guide to the unique difficulties of lead optimization for tuberculosis. Bioorg Med Chem Lett 23: 4741–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Siricilla S, Wan B, Crick DC, Lenaerts AJ, Franzblau SG, Kurosu M. 2012. Discovery of selective menaquinone biosynthesis inhibitors against Mycobacterium tuberculosis. J Med Chem 55: 3739–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. 2010. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol 17: 1122–1131. [DOI] [PubMed] [Google Scholar]
- Dhiman RK, Mahapatra S, Slayden RA, Boyne ME, Lenaerts A, Hinshaw JC, Angala SK, Chatterjee D, Biswas K, Narayanasamy P, et al. 2009. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol Microbiol 72: 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diacon AH, Pym A, Grobusch M, Patientia R, Rustomjee R, Page-Shipp L, Pistorius C, Krause R, Bogoshi M, Churchyard G, et al. 2009. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med 360: 2397–2405. [DOI] [PubMed] [Google Scholar]
- Doherty CP. 2007. Host-pathogen interactions: The role of iron. J Nutr 137: 1341–1344. [DOI] [PubMed] [Google Scholar]
- Domann FE. 2013. Aberrant free radical biology is a unifying theme in the etiology and pathogenesis of major human diseases. Int J Mol Sci 14: 8491–8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech P, Reed MB, Dowd CS, Manca C, Kaplan G, Barry CE III. 2004. The role of MmpL8 in sulfatide biogenesis and virulence of Mycobacterium tuberculosis. J Biol Chem 279: 21257–21265. [DOI] [PubMed] [Google Scholar]
- Domenech P, Reed MB, Barry CE III. 2005. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect Immun 73: 3492–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresen C, Lin LY, D’Angelo I, Tocheva EI, Strynadka N, Eltis LD. 2010. A flavin-dependent monooxygenase from Mycobacterium tuberculosis involved in cholesterol catabolism. J Biol Chem 285: 22264–22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DJ, Kohanski MA, Collins JJ. 2009. Role of reactive oxygen species in antibiotic action and resistance. Curr Opin Microbiol 12: 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas L, O’Keefe H, Neu M, Remuiñán MJ, Patel AM, Guardia A, Davie CP, Pérez-Macías N, Yang H, Convery MA, et al. 2014. Encoded library technology as a source of hits for the discovery and lead optimization of a potent and selective class of bactericidal direct inhibitors of Mycobacterium tuberculosis InhA. J Med Chem 57: 1276–1288. [DOI] [PubMed] [Google Scholar]
- Engelhart CA, Aldrich CC. 2013. Synthesis of chromone, quinolone, and benzoxazinone sulfonamide nucleosides as conformationally constrained inhibitors of adenylating enzymes required for siderophore biosynthesis. J Org Chem 78: 7470–7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eoh H, Rhee KY. 2013. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc Natl Acad Sci 110: 6554–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estorninho M, Smith H, Thole J, Harders-Westerveen J, Kierzek A, Butler RE, Neyrolles O, Stewart GR. 2010. ClgR regulation of chaperone and protease systems is essential for Mycobacterium tuberculosis parasitism of the macrophage. Microbiology 156: 3445–3455. [DOI] [PubMed] [Google Scholar]
- Feher M, Schmidt JM. 2003. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J Chem Inf Comput Sci 43: 218–227. [DOI] [PubMed] [Google Scholar]
- Finn J. 2012. Application of SBDD to the discovery of new antibacterial drugs. Methods Mol Biol 841: 291–319. [DOI] [PubMed] [Google Scholar]
- García A, Bocanegra-García V, Palma-Nicolás JP, Rivera G. 2012. Recent advances in antitubercular natural products. Eur J Med Chem 49: 1–23. [DOI] [PubMed] [Google Scholar]
- Gatfield J, Pieters J. 2000. Essential role for cholesterol in entry of mycobacteria into macrophages. Science 288: 1647–1650. [DOI] [PubMed] [Google Scholar]
- Gavrish E, Sit CS, Cao S, Kandror O, Spoering A, Peoples A, Ling L, Fetterman A, Hughes D, Bissell A, et al. 2014. Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem Biol 21: 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gengenbacher M, Rao SP, Pethe K, Dick T. 2010. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology 156: 81–87. [DOI] [PubMed] [Google Scholar]
- Goldman RC. 2013. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis? Tuberculosis (Edinb) 93: 569–588. [DOI] [PubMed] [Google Scholar]
- Gould TA, van de Langemheen H, Muñoz-Elías EJ, McKinney JD, Sacchettini JC. 2006. Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol Microbiol 61: 940–947. [DOI] [PubMed] [Google Scholar]
- Grant SS, Kaufmann BB, Chand NS, Haseley N, Hung DT. 2012. Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc Natl Acad Sci 109: 12147–12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7: e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzegorzewicz AE, Pham H, Gundi VA, Scherman MS, North EJ, Hess T, Jones V, Gruppo V, Born SE, Korduláková J, et al. 2012a. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat Chem Biol 8: 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzegorzewicz AE, Korduláková J, Jones V, Born SE, Belardinelli JM, Vaquié A, Gundi VA, Madacki J, Slama N, Laval F, et al. 2012b. A common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J Biol Chem 287: 38434–38441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Gupta RK, Khare G, Salunke DM, Tyagi AK. 2008. Cloning, expression, purification, crystallization and preliminary X-ray crystallographic analysis of bacterioferritin A from Mycobacterium tuberculosis. Acta Crystallogr Sect F Struct Biol Cryst Commun 64: 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurumurthy M, Rao M, Mukherjee T, Rao SP, Boshoff HI, Dick T, Barry CE III, Manjunatha UH. 2013. A novel F(420)-dependent anti-oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol Microbiol 87: 744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JD, Gupta A, Bucar F, Gibbons S, Bhakta S. 2012. Antimycobacterials from natural sources: Ancient times, antibiotic era and novel scaffolds. Front Biosci (Landmark Ed) 17: 1861–1881. [DOI] [PubMed] [Google Scholar]
- Hartkoorn RC, Sala C, Neres J, Pojer F, Magnet S, Mukherjee R, Uplekar S, Boy-Röttger S, Altmann KH, Cole ST. 2012. Towards a new tuberculosis drug: Pyridomycin—Nature’s isoniazid. EMBO Mol Med 4: 1032–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartkoorn RC, Pojer F, Read JA, Gingell H, Neres J, Horlacher OP, Altmann KH, Cole ST. 2013. Pyridomycin bridges the NADH- and substrate-binding pockets of the enoyl reductase InhA. Nat Chem Biol 10: 96–98. [DOI] [PubMed] [Google Scholar]
- Holmes MA, Paulsene W, Jide X, Ratledge C, Strong RK. 2005. Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure 13: 29–41. [DOI] [PubMed] [Google Scholar]
- Höner Zu Bentrup K, Miczak A, Swenson DL, Russell DG. 1999. Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis. J Bacteriol 181: 7161–7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung AW, Silvestre HL, Wen S, Ciulli A, Blundell TL, Abell C. 2009. Application of fragment growing and fragment linking to the discovery of inhibitors of Mycobacterium tuberculosis pantothenate synthetase. Angew Chem Int Ed Engl 48: 8452–8456. [DOI] [PubMed] [Google Scholar]
- Ingvarsson H, Maté MJ, Högbom M, Portnoï D, Benaroudj N, Alzari PM, Ortiz-Lombardía M, Unge T. 2007. Insights into the inter-ring plasticity of caseinolytic proteases from the X-ray structure of Mycobacterium tuberculosis ClpP1. Acta Crystallogr D Biol Crystallogr 63: 249–259. [DOI] [PubMed] [Google Scholar]
- Ioerger TR, O’Malley T, Liao R, Guinn KM, Hickey MJ, Mohaideen N, Murphy KC, Boshoff HI, Mizrahi V, Rubin EJ, et al. 2013. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS ONE 8: e75245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irschik H, Reichenbach H, Höfle G, Jansen R. 2007. The thuggacins, novel antibacterial macrolides from Sorangium cellulosum acting against selected Gram-positive bacteria. J Antibiot (Tokyo) 60: 733–738. [DOI] [PubMed] [Google Scholar]
- Ishizaki Y, Hayashi C, Inoue K, Igarashi M, Takahashi Y, Pujari V, Crick DC, Brennan PJ, Nomoto A. 2013. Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J Biol Chem 288: 30309–30319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsuki M, Tomoda H, Uchida R, Gouda H, Hirono S, Omura S. 2006. Lariatins, antimycobacterial peptides produced by Rhodococcus sp. K01-B0171, have a lasso structure. J Am Chem Soc 128: 7486–7491. [DOI] [PubMed] [Google Scholar]
- Iwatsuki M, Uchida R, Takakusagi Y, Matsumoto A, Jiang CL, Takahashi Y, Arai M, Kobayashi S, Matsumoto M, Inokoshi J, et al. 2007. Lariatins, novel anti-mycobacterial peptides with a lasso structure, produced by Rhodococcus jostii K01-B0171. J Antibiot (Tokyo) 60: 357–363. [DOI] [PubMed] [Google Scholar]
- Jain M, Cox JS. 2005. Interaction between polyketide synthase and transporter suggests coupled synthesis and export of virulence lipid in M. tuberculosis. PLoS Pathog 1: e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EE, Wessling-Resnick M. 2012. Iron metabolism and the innate immune response to infection. Microbes Infect 14: 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CM, Niederweis M. 2011. Mycobacterium tuberculosis can utilize heme as an iron source. J Bacteriol 193: 1767–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CM, Wells RM, Madduri AV, Renfrow MB, Ratledge C, Moody DB, Niederweis M. 2014. Self-poisoning of Mycobacterium tuberculosis by interrupting siderophore recycling. Proc Natl Acad Sci 111: 1945–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer R, Jacobs WR Jr. 2010. The significance of GlgE as a new target for tuberculosis. Drug News Perspect 23: 619–624. [DOI] [PubMed] [Google Scholar]
- Kana BD, Weinstein EA, Avarbock D, Dawes SS, Rubin H, Mizrahi V. 2001. Characterization of the cydAB-encoded cytochrome bd oxidase from Mycobacterium smegmatis. J Bacteriol 183: 7076–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffmann SHE. 2004. New issues in tuberculosis. Ann Rheum Dis 63: 1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawate T, Iwase N, Shimizu M, Stanley SA, Wellington S, Kazyanskaya E, Hung DT. 2013. Synthesis and structure-activity relationships of phenyl-substituted coumarins with anti-tubercular activity that target FadD32. Bioorg Med Chem Lett 23: 6052–6059. [DOI] [PubMed] [Google Scholar]
- Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. 2013. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339: 1213–1216. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, Visser A, Wang W, Hsu FF, Wiehart U, et al. 2010. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med 2: 258–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirst HA. 2013. Developing new antibacterials through natural product research. Expert Opin Drug Discov 8: 479–493. [DOI] [PubMed] [Google Scholar]
- Kirstein J, Hoffmann A, Lilie H, Schmidt R, Rübsamen-Waigmann H, Brötz-Oesterhelt H, Mogk A, Turgay K. 2009. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol Med 1: 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjellsson MC, Via LE, Goh A, Weiner D, Low KM, Kern S, Pillai G, Barry CE III, Dartois V. 2012. Pharmacokinetic evaluation of the penetration of antituberculosis agents in rabbit pulmonary lesions. Antimicrob Agents Chemother 56: 446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondreddi RR, Jiricek J, Rao SPS, Lakshminarayana SB, Camacho LR, Rao R, Herve M, Bifani P, Ma NL, Kuhen K, et al. 2013. Design, synthesis, and biological evaluation of indole-2-carboxamides: A promising class of antituberculosis agents. J Med Chem 56: 8849–8859. [DOI] [PubMed] [Google Scholar]
- Koul A, Vranckx L, Dendouga N, Balemans W, Van den Wyngaert I, Vergauwen K, Göhlmann HW, Willebrords R, Poncelet A, Guillemont J, et al. 2008. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J Biol Chem 283: 25273–25280. [DOI] [PubMed] [Google Scholar]
- Krátký M, Vinšová J. 2012. Advances in mycobacterial isocitrate lyase targeting and inhibitors. Curr Med Chem 19: 6126–6137. [DOI] [PubMed] [Google Scholar]
- Krieger IV, Freundlich JS, Gawandi VB, Roberts JP, Gawandi VB, Sun Q, Owen JL, Fraile MT, Huss SI, Lavandera JL, et al. 2012. Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem Biol 19: 1556–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Casey A, Odingo J, Kesicki EA, Abrahams G, Vieth M, Masquelin T, Mizrahi V, Hipskind PA, Sherman DR, et al. 2013. A high-throughput screen against pantothenate synthetase (PanC) identifies 3-biphenyl-4-cyanopyrrole-2-carboxylic acids as a new class of inhibitor with activity against Mycobacterium tuberculosis. PLoS ONE 8: e72786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosu M, Crick DC. 2009. MenA is a promising drug target for developing novel lead molecules to combat Mycobacterium tuberculosis. Med Chem 5: 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosu M, Narayanasamy P, Biswas K, Dhiman R, Crick DC. 2007. Discovery of 1,4-dihydroxy-2-naphthoate prenyltransferase inhibitors: New drug leads for multidrug-resistant Gram-positive pathogens. J Med Chem 50: 3973–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lack NA, Yam KC, Lowe ED, Horsman GP, Owen RL, Sim E, Eltis LD. 2010. Characterization of a carbon-carbon hydrolase from Mycobacterium tuberculosis involved in cholesterol metabolism. J Biol Chem 285: 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane G, Tyagi S, Bishai WR. 2005. Designer arrays for defined mutant analysis to detect genes essential for survival of Mycobacterium tuberculosis in mouse lungs. Infect Immun 73: 2533–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa V, Poce G, Canseco JO, Buroni S, Pasca MR, Biava M, Raju RM, Porretta GC, Alfonso S, Battilocchio C, et al. 2012. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob Agents Chemother 56: 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JW, Vederas JC. 2009. Drug discovery and natural products: End of an era or an endless frontier? Science 325: 161–165. [DOI] [PubMed] [Google Scholar]
- Li X, Liu N, Zhang H, Knudson SE, Li HJ, Lai CT, Simmerling C, Slayden RA, Tonge PJ. 2011. CoA Adducts of 4-Oxo-4-Phenylbut-2-enoates: Inhibitors of MenB from the M. tuberculosis menaquinone biosynthesis pathway. ACS Med Chem Lett 2: 818–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, Fontes FL, Yang H, Rao G, Zhu W, et al. 2014. Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem 57: 3126–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, Sacchettini J, Fortune SM, Flynn JL. 2014. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med 20: 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Yu J, Russell DG. 2003. pckA-Deficient Mycobacterium bovis BCG shows attenuated virulence in mice and in macrophages. Microbiology 149: 1829–1835. [DOI] [PubMed] [Google Scholar]
- Lu XI, Zhou R, Sharma I, Li X, Kumar G, Swaminathan S, Tonge PJ, Tan DS. 2012. Stable analogues of OSB-AMP: Potent inhibitors of MenE, the o-succinatebenzoate-CoA synthetase from bacterial menaquinone biosynthesis. Chembiochem 13: 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddry JA, Ananthan S, Goldman RC, Hobrath JV, Kwong CD, Maddox C, Rasmussen L, Reynolds RC, Secrist JA III, Sosa MI, et al. 2009. Antituberculosis activity of the molecular libraries screening center network library. Tuberculosis (Edinb) 89: 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai D, Jones J, Rodgers JW, Hartman JL IV, Kutsch O, Steyn AJ. 2011. A screen to identify small molecule inhibitors of protein-protein interactions in mycobacteria. Assay Drug Dev Technol 9: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak PA, Rao SP, Ping Tan M, Lin X, Chyba J, Tay J, Ng SH, Tan BH, Cherian J, Duraiswamy J, et al. 2012a. A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS Chem Biol 7: 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manina G, McKinney JD. 2013. A single-cell perspective on non-growing but metabolically active (NGMA) bacteria. Curr Top Microbiol Immunol 374: 135–161. [DOI] [PubMed] [Google Scholar]
- Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. 2010. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci 107: 9819–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero J, Trujillo C, Rhee KY, Ehrt S. 2013. Glucose phosphorylation is required for Mycobacterium tuberculosis persistence in mice. PLoS Pathog 9: e1003116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsoso LG, Kana BD, Crellin PK, Lea-Smith DJ, Pelosi A, Powell D, Dawes SS, Rubin H, Coppel RL, Mizrahi V. 2005. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J Bacteriol 187: 6300–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdam RA, Quan S, Smith DA, Bardarov S, Betts JC, Cook FC, Hooker EU, Lewis AP, Woollard P, Everett MJ, et al. 2002. Characterization of a Mycobacterium tuberculosis H37Rv transposon library reveals insertions in 351 ORFs and mutants with altered virulence. Microbiology 148: 2975–2986. [DOI] [PubMed] [Google Scholar]
- McKinney JD, Höner zu Bentrup K, Muñoz-Elías EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR Jr, Russell DG. 2000. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406: 735–738. [DOI] [PubMed] [Google Scholar]
- McMath LM, Habel JE, Sankaran B, Yu M, Hung LW, Goulding CW. 2010. Crystallization and preliminary X-ray crystallographic analysis of a Mycobacterium tuberculosis ferritin homolog, BfrB. Acta Crystallogr Sect F Struct Biol Cryst Commun 66: 1657–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano A, Pasca MR, Provvedi R, Lucarelli AP, Manina G, Ribeiro AL, Manganelli R, Riccardi G. 2009. Azole resistance in Mycobacterium tuberculosis is mediated by the MmpS5-MmpL5 efflux system. Tuberculosis (Edinb) 89: 84–90. [DOI] [PubMed] [Google Scholar]
- Miner MD, Chang JC, Pandey AK, Sassetti CM, Sherman DR. 2009. Role of cholesterol in Mycobacterium tuberculosis infection. Indian J Exp Biol 47: 407–411. [PubMed] [Google Scholar]
- Mohn WW, van der Geize R, Stewart GR, Okamoto S, Liu J, Dijkhuizen L, Eltis LD. 2008. The actinobacterial mce4 locus encodes a steroid transporter. J Biol Chem 283: 35368–35374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monfeli RR, Beeson C. 2007. Targeting iron acquisition by Mycobacterium tuberculosis. Infect Disord Drug Targets 7: 213–220. [DOI] [PubMed] [Google Scholar]
- Moraski GC, Markley LD, Hipskind PA, Boshoff H, Cho S, Franzblau SG, Miller MJ. 2011. Advent of Imidazo[1,2-a]pyridine-3-carboxamides with potent multi- and extended drug resistant antituberculosis activity. ACS Med Chem Lett 2: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraski GC, Markley LD, Chang M, Cho S, Franzblau SG, Hwang CH, Boshoff H, Miller MJ. 2012. Generation and exploration of new classes of antitubercular agents: The optimization of oxazolines, oxazoles, thiazolines, thiazoles to imidazo[1,2-a]pyridines and isomeric 5,6-fused scaffolds. Bioorg Med Chem 20: 2214–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Elías EJ, McKinney JD. 2005. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med 11: 638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Elías EJ, Upton AM, Cherian J, McKinney JD. 2006. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol 60: 1109–1122. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay B, Concar EM, Wolfe RS. 2001. A GTP-dependent vertebrate-type phosphoenolpyruvate carboxykinase from Mycobacterium smegmatis. J Biol Chem 276: 16137–16145. [DOI] [PubMed] [Google Scholar]
- Naven RT, Swiss R, Klug-McLeod J, Will Y, Greene N. 2013. The development of structure-activity relationships for mitochondrial dysfunction: Uncoupling of oxidative phosphorylation. Toxicol Sci 131: 271–278. [DOI] [PubMed] [Google Scholar]
- Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE III, Bennett EM, Aldrich CC. 2008. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: Structure-activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J Med Chem 51: 5349–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt NM, Yang X, Fontán P, Kolesnikova I, Smith I, Sampson NS, Dubnau E. 2010. A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect Immun 78: 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM. 2012. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75: 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollinger J, O’Malley T, Kesicki EA, Odingo J, Parish T. 2012. Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. J Bacteriol 194: 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme IM. 2014. A new unifying theory of the pathogenesis of tuberculosis. Tuberculosis (Edinb) 94: 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens CP, Chim N, Graves AB, Harmston CA, Iniguez A, Contreras H, Liptak MD, Goulding CW. 2013. The Mycobacterium tuberculosis secreted protein Rv0203 transfers heme to membrane proteins MmpL3 and MmpL11. J Biol Chem 288: 21714–21728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai SR, Actor JK, Sepulveda E, Hunter RL Jr, Jagannath C. 2000. Identification of viable and non-viable Mycobacterium tuberculosis in mouse organs by directed RT-PCR for antigen 85B mRNA. Microb Pathog 28: 335–342. [DOI] [PubMed] [Google Scholar]
- Pan P, Tonge PJ. 2012. Targeting InhA, the FASII enoyl-ACP reductase: SAR studies on novel inhibitor scaffolds. Curr Top Med Chem 12: 672–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci 105: 4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R, Rodriguez GM. 2012. A ferritin mutant of Mycobacterium tuberculosis is highly susceptible to killing by antibiotics and is unable to establish a chronic infection in mice. Infect Immun 80: 3650–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen IT, Brown MH, Skurray RA. 1996. Proton-dependent multidrug efflux systems. Microbiol Rev 60: 575–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6: 29–40. [DOI] [PubMed] [Google Scholar]
- Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, et al. 2013. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med 19: 1157–1160. [DOI] [PubMed] [Google Scholar]
- Peyron P, Bordier C, N’Diaye EN, Maridonneau-Parini I. 2000. Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol-anchored proteins. J Immunol 165: 5186–5191. [DOI] [PubMed] [Google Scholar]
- Pruksakorn P, Arai M, Kotoku N, Vilchèze C, Baughn AD, Moodley P, Jacobs WR Jr, Kobayashi M. 2010. Trichoderins, novel aminolipopeptides from a marine sponge-derived Trichoderma sp., are active against dormant mycobacteria. Bioorg Med Chem Lett 20: 3658–3663. [DOI] [PubMed] [Google Scholar]
- Pruksakorn P, Arai M, Liu L, Moodley P, Jacobs WR Jr, Kobayashi M. 2011. Action-mechanism of trichoderin A, an anti-dormant mycobacterial aminolipopeptide from marine sponge-derived Trichoderma sp. Biol Pharm Bull 34: 1287–1290. [DOI] [PubMed] [Google Scholar]
- Putman M, van Veen HW, Konings WN. 2000. Molecular properties of bacterial multidrug transporters. Microbiol Mol Biol Rev 64: 672–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Panda M, Mukherjee K, Choudhury NR, Tantry SJ, Kedari CK, Ramachandran V, Sharma S, Ramya VK, Guptha S, et al. 2013. Synthesis and structure activity relationship of imidazo[1,2-a]pyridine-8-carboxamides as a novel antimycobacterial lead series. Bioorg Med Chem Lett 23: 4996–5001. [DOI] [PubMed] [Google Scholar]
- Rao SP, Alonso S, Rand L, Dick T, Pethe K. 2008. The proton motive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci 105: 11945–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SP, Lakshminarayana SB, Kondreddi RR, Herve M, Camacho LR, Bifani P, Kalapala SK, Jiricek J, Ma NL, Tan BH, et al. 2013. Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci Transl Med 5: 214ra168. [DOI] [PubMed] [Google Scholar]
- Ratledge C, Dover LG. 2000. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol 54: 881–941. [DOI] [PubMed] [Google Scholar]
- Reddy PV, Puri RV, Khera A, Tyagi AK. 2012. Iron storage proteins are essential for the survival and pathogenesis of Mycobacterium tuberculosis in THP-1 macrophages and the guinea pig model of infection. J Bacteriol 194: 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PV, Puri RV, Chauhan P, Kar R, Rohilla A, Khera A, Tyagi AK. 2013. Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. J Infect Dis 208: 1255–1265. [DOI] [PubMed] [Google Scholar]
- Remuiñán MJ, Pérez-Herrán E, Rullás J, Alemparte C, Martínez-Hoyos M, Dow DJ, Afari J, Mehta N, Esquivias J, Jiménez E, et al. 2013. Tetrahydropyrazolo[1,5-a]pyrimidine-3-carboxamide and N-benzyl-6’,7’-dihydrospiro-[piperidine-4,4’-thieno[3,2-c]pyran] analogues with bactericidal efficacy against Mycobacterium tuberculosis targeting MmpL3. PLoS ONE 8: e60933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J, Bloom BR, Rubin EJ. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci 102: 8327–8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee KY, de Carvalho LP, Bryk R, Ehrt S, Marrero J, Park SW, Schnappinger D, Venugopal A, Nathan C. 2011. Central carbon metabolism in Mycobacterium tuberculosis: An unexpected frontier. Trends Microbiol 19: 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GM. 2006. Control of iron metabolism in Mycobacterium tuberculosis. Trends Microbiol 14: 320–327. [DOI] [PubMed] [Google Scholar]
- Rodriguez GM, Smith I. 2006. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J Bacteriol 188: 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustad TR, Harrell MI, Liao R, Sherman DR. 2008. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE 30: e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon CE, Schmidt LE. 2012. Natural products as leads for tuberculosis drug development. Curr Top Med Chem 12: 735–765. [DOI] [PubMed] [Google Scholar]
- Sarathy JP, Dartois V, Lee EJ. 2012. The role of transport mechanisms in Mycobacterium tuberculosis drug resistance and tolerance. Pharmaceuticals (Basel) 5: 1210–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci 100: 12989–12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48: 77–84. [DOI] [PubMed] [Google Scholar]
- Schafer G, Guler R, Murray G, Brombacher F, Brown GD. 2009. The role of scavenger receptor B1 in infection with Mycobacterium tuberculosis in a murine model. PLoS ONE 4: e8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherman MS, North EJ, Jones V, Hess TN, Grzegorzewicz AE, Kasagami T, Kim IH, Merzlikin O, Lenaerts AJ, Lee RE, et al. 2012. Screening a library of 1600 adamantyl ureas for anti-Mycobacterium tuberculosis activity in vitro and for better physical chemical properties for bioavailability. Bioorg Med Chem 20: 3255–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt EK, Riwanto M, Sambandamurthy V, Roggo S, Miault C, Zwingelstein C, Krastel P, Noble C, Beer D, Rao SP, et al. 2011. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew Chem Int Ed Engl 50: 5889–5891. [DOI] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, et al. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J Exp Med 198: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senaratne RH, Sidders B, Sequeira P, Saunders G, Dunphy K, Marjanovic O, Reader JR, Lima P, Chan S, Kendall S, et al. 2008. Mycobacterium tuberculosis strains disrupted in mce3 and mce4 operons are attenuated in mice. J Med Microbiol 57: 164–170. [DOI] [PubMed] [Google Scholar]
- Sharma V, Sharma S, Hoener zu Bentrup K, McKinney JD, Russell DG, Jacobs WR Jr, Sacchettini JC. 2000. Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis. Nat Struct Biol 7: 663–668. [DOI] [PubMed] [Google Scholar]
- Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK. 2001. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding α-crystallin. Proc Natl Acad Sci 98: 7534–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrid AM, Rustad TR, Cangelosi GA, Sherman DR. 2010. Characterization of a Clp protease gene regulator and the reaeration response in Mycobacterium tuberculosis. PLoS ONE 5: e11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Ehrt S. 2006. Dihydrolipoamide acyltransferase is critical for Mycobacterium tuberculosis pathogenesis. Infect Immun 74: 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirude PS, Madhavapeddi P, Naik M, Murugan K, Shinde V, Nandishaiah R, Bhat J, Kumar A, Hameed S, Holdgate G, et al. 2013. Methyl-thiazoles: A novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J Med Chem 56: 8533–8542. [DOI] [PubMed] [Google Scholar]
- Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, Cheng TY, Siddiqi N, Fortune SM, Moody DB, Rubin EJ. 2009. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci 106: 18792–18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestre HL, Blundell TL, Abell C, Ciulli A. 2013. Integrated biophysical approach to fragment screening and validation for fragment-based lead discovery. Proc Natl Acad Sci 110: 12984–12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SB, Young K, Miesel L. 2011. Screening strategies for discovery of antibacterial natural products. Expert Rev Anti Infect Ther 9: 589–613. [DOI] [PubMed] [Google Scholar]
- Singh R, Munjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, et al. 2008. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322: 1392–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siroy A, Mailaender C, Harder D, Koerber S, Wolschendorf F, Danilchanka O, Wang Y, Heinz C, Niederweis M. 2008. Rv1698 of Mycobacterium tuberculosis represents a new class of channel-forming outer membrane proteins. J Biol Chem 283: 17827–17837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small JL, Park SW, Kana BD, Ioerger TR, Sacchettini JC, Ehrt S. 2013. Perturbation of cytochrome c maturation reveals adaptability of the respiratory chain in Mycobacterium tuberculosis. MBio 4: e0047513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CV, Huang CC, Miczak A, Russell DG, Sacchettini JC, Höner zu Bentrup K. 2003. Biochemical and structural studies of malate synthase from Mycobacterium tuberculosis. J Biol Chem 278: 1735–1743. [DOI] [PubMed] [Google Scholar]
- Stanley SA, Grant SS, Kawate T, Iwase N, Shimizu M, Wivagg C, Silvis M, Kazyanskaya E, Aquadro J, Golas A, et al. 2012. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem Biol 7: 1377–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley SA, Kawate T, Iwase N, Shimizu M, Clatworthy AE, Kazyanskaya E, Sacchettini JC, Ioerger TR, Siddiqi NA, Minami S, et al. 2013. Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc Natl Acad Sci 110: 11565–11570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz H, Irschik H, Kunze B, Reichenbach H, Höfle G, Jansen R. 2007. Thuggacins, macrolide antibiotics active against Mycobacterium tuberculosis: Isolation from myxobacteria, structure elucidation, conformation analysis and biosynthesis. Chemistry 13: 5822–5832. [DOI] [PubMed] [Google Scholar]
- Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE III, et al. 2012. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother 56: 1797–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarcsay Á, Keserű GM. 2013. Contributions of molecular properties to drug promiscuity. J Med Chem 56: 1789–1795. [DOI] [PubMed] [Google Scholar]
- Thomas ST, VanderVen BC, Sherman DR, Russell DG, Sampson NS. 2011. Pathway profiling in Mycobacterium tuberculosis: Elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism. J Biol Chem 286: 43668–43678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Bryk R, Shi S, Erdjument-Bromage H, Tempst P, Nathan C. 2005. Mycobacterium tuberculosis appears to lack alpha-ketoglutarate dehydrogenase and encodes pyruvate dehydrogenase in widely separated genes. Mol Microbiol 57: 859–868. [DOI] [PubMed] [Google Scholar]
- Timm J, Post FA, Bekker LG, Walther GB, Wainwright HC, Manganelli R, Chan WT, Tsenova L, Gold B, Smith I, et al. 2003. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc Natl Acad Sci 100: 14321–14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong NB, Pham CV, Doan HT, Nguyen HV, Nguyen CM, Nguyen HT, Zhang HJ, Fong HH, Franzblau SG, Soejarto DD, et al. 2011. Antituberculosis cycloartane triterpenoids from Radermachera boniana. J Nat Prod 74: 1318–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullius MV, Harmston CA, Owens CP, Chim N, Morse RP, McMath LM, Iniguez A, Kimmey JM, Sawaya MR, Whitelegge JP, et al. 2011. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc Natl Acad Sci 108: 5051–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, et al. 2007. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci 104: 1947–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal A, Bryk R, Shi S, Rhee K, Rath P, Schnappinger D, Ehrt S, Nathan C. 2011. Virulence of Mycobacterium tuberculosis depends on lipoamide dehydrogenase, a member of three multienzyme complexes. Cell Host Microbe 9: 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, Taylor K, Klein E, Manjunatha U, Gonzales J, et al. 2008. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun 76: 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warman AJ, Rito TS, Fisher NE, Moss DM, Berry NG, O’Neill PM, Ward SA, Biagini GA. 2013. Antitubercular pharmacodynamics of phenothiazines. J Antimicrob Chemother 68: 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Zimmermann M, Goodwin MB, Sauer U, Barry CE III, Boshoff HI. 2011. Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS Pathog 7: e1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei JR, Krishnamoorthy V, Murphy K, Kim JH, Schnappinger D, Alber T, Sassetti CM, Rhee KY, Rubin EJ. 2011. Depletion of antibiotic targets has widely varying effects on growth. Proc Natl Acad Sci 108: 4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg ED. 1999. Iron loading and disease surveillance. Emerg Infect Dis 5: 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein EA, Yano T, Li LS, Avarbock D, Avarbock A, Helm D, McColm AA, Duncan K, Lonsdale JT, Rubin H. 2005. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc Natl Acad Sci 102: 4548–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RM, Jones CM, Xi Z, Speer A, Danilchanka O, Doornbos KS, Sun P, Wu F, Tian C, Niederweis M. 2013. Discovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog 9: e1003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woong Park S, Klotzsche M, Wilson DJ, Boshoff HI, Eoh H, Manjunatha U, Blumenthal A, Rhee K, Barry CE III, Aldrich CC, et al. 2011. Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog 7: e1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GD. 2014. Something old, something new: Revisiting natural products in antibiotic drug discovery. Can J Microbiol 60: 147–154. [DOI] [PubMed] [Google Scholar]
- Xuequan L, Zhou R, Sharma I, Li X, Kumar G, Swaminathan S, Tonge PF, Tan DS. 2012. Stable analogues of OSB-AMP: Potent inhibitors of MenE, the o-succinylbenzoate-CoA synthetase from bacterial menaquinone. Biosynthesis. Chembiochem 13: 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam KC, D’Angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V, Ly LH, Converse PJ, Jacobs WR Jr, Strynadka N, et al. 2009. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog 5: e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Gao J, Smith I, Dubnau E, Sampson NS. 2011. Cholesterol is not an essential source of nutrition for Mycobacterium tuberculosis during infection. J Bacteriol 193: 1473–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T, Kassovska-Bratinova S, Teh JS, Winkler J, Sullivan K, Isaacs A, Schechter NM, Rubin H. 2011. Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: A pathway for the generation of bactericidal levels of reactive oxygen species. J Biol Chem 286: 10276–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokokawa F, Wang G, Chan WL, Ang SH, Wong J, Ma I, Rao SPS, Manjunatha U, Lakshminarayana SB, Herve M, et al. 2013. Discovery of tetrahydropyrazolopyrimidine carboxamide derivatives as potent and orally active novel anti-tubercular agents. ACS Chem Lett 4: 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wade MM, Scorpio A, Zhang H, Sun Z. 2003. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J Antimicrob Chemother 52: 790–795. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, Sacchettini JC, Rubin EJ. 2012. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog 8: e1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]