

Abstract
A computational study using DFT methods was performed for an array of mono and disubstituted benzynes and indolynes. The inherent distortion present in the geometry-optimized structures predicts the regioselectivity of aryne trapping by nucleophiles or cycloaddition partners. These studies will serve to enable the further use of unsymmetrical arynes in organic synthesis.
Keywords: heterocycles, arynes, computations, regioselectivities, hetarynes
Over the past 10–15 years there has been resurgence in the field of aryne chemistry.1 Arynes were once avoided because of their high reactivity, but chemists have now demonstrated that arynes can be strategically employed in a host of synthetic applications. Our laboratories have been interested in harnessing substituted arynes and heterocyclic arynes to build complex scaffolds,2 especially those seen in drugs and natural products. These efforts have led to the aryne distortion/interaction model,2c,2d,3,4 which explains aryne regioselectivities and can also be used to make reliable regioselectivity predictions. Following our recent regioselectivity studies of 3-substituted benzynes2p and substituted indolynes,2d,2e,2m we now report regioselectivity predictions for a number of disubstituted benzynes and substituted indolynes. We expect our findings will help propel the further exploitation of unsymmetrical arynes in synthesis.
A brief summary of the predictive powers of the aryne distortion model, as applied to various 3-substituted benzynes, is provided in Table 1. First, the geometry-optimized structure of a given unsymmetrical aryne is obtained using DFT calculations.5,6,7 These calculations provide the internal angles of each alkyne terminus. The site with the larger internal angle is the preferred site of attack by nucleophiles.8 Additionally, the degree of distortion (as measured by the difference in angles) can be used to provide an estimate of regioselectivity. Even a mild degree of distortion (e.g., 4° or greater), typically corresponds to synthetically useful levels of selectivity. As shown for benzynes substituted at C3 with an inductively electron-withdrawing group (entries 1–5), nucleophilic addition is predicted to occur with a preference for attack at C1. Generally speaking, distortion decreases in moving from the most inductively withdrawing groups (entries 1 and 2) to the least withdrawing group (entry 5), which has been validated experimentally.2p
Table 1.
Distortion analysis of 3-substituted benzynes due to the presence of inductively withdrawing groups.
| Entry | Aryne | Geometry-optimized structurea | Site of attack (angle difference) | Regioselectivity (N-Me-aniline)b |
|---|---|---|---|---|
| 1 |
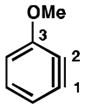
|
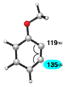
|
C1 (16°) | C1 addition exclusively |
| 2 |
|
|
C1 (17°) | C1 addition exclusively |
| 3 |
|
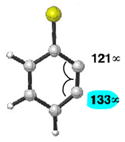
|
C1 (12°) | C1 addition favored (>20:1) |
| 4 |
|
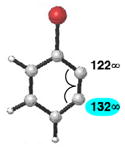
|
C1 (10°) | C1 addition favored (13:1) |
| 5 |
|

|
C1 (4°) | C1 addition favored (9:1) |
| Aryne Distortion Model -Predictive Capabilities | ||||
| ||||
Geometry optimizations were performed using DFT methods (B3LYP/6-31G*; B3LYP/LACVP was used for 3-iodobenzyne (entry 5)).
Known regioselectivities for the addition of N-Me-aniline to each aryne (ref 2p).
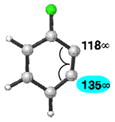
We studied benzynes bearing two substituents adjacent to the triple bond, as these have not been assessed previously using the aryne distortion model. An analysis of several 6-substituted 3-fluorobenzynes is shown in Table 2. Fluoride dominates regioselectivity in every case. Nucleophilic addition is predicted to occur at C1 due to the distortion introduced by the electronegative fluoride substituent. Selectivity increases as the C6-substituent becomes less electron-withdrawing.
Table 2.
Distortion analysis of 3-fluorobenzynes bearing a C6 inductively-withdrawing substituent.
| Entry | Aryne | Geometry-optimized structurea | Site of attack (angle difference) |
|---|---|---|---|
| 1 |
|

|
C1 (3°) |
| 2 |
|
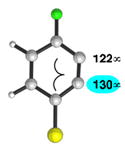
|
C1 (8°) |
| 3 |
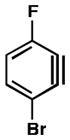
|
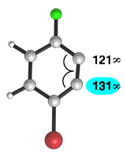
|
C1 (10°) |
| 4 |
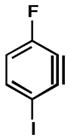
|
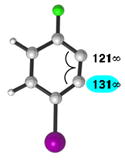
|
C1 (10°) |
Geometry optimizations were performed using DFT methods (B3LYP/6-31G*; B3LYP/LACVP was used for 3-fluoro-6-iodobenzyne (entry 4)).
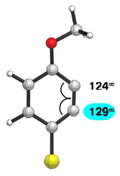
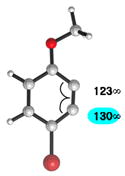
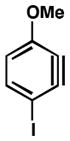
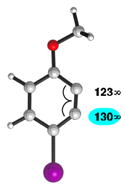
We also examined the distortion present in 3-substituted 6-methoxybenzynes (Table 3). The inductively withdrawing fluoride group governs regioselectivity in the case of entry 1. However, for the less electronegative halides, Cl, Br, and I, the methoxy group controls aryne distortion (entries 2–4). Accordingly, nucleophilic addition is predicted to occur at C2 in these three cases.
Table 3.
Distortion analysis of 6-methoxybenzynes bearing a C3 inductively withdrawing substituent.
| Entry | Aryne | Geometry-optimized structurea | Site of attack (angle difference) |
|---|---|---|---|
| 1 |

|
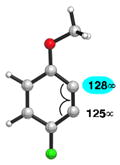
|
C1 (3°) |
| 2 |
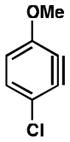
|
|
C2 (5°) |
| 3 |
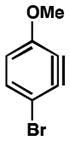
|
|
C2 (7°) |
| 4 |
|
|
C2 (7°) |
Geometry optimizations were performed using DFT methods (B3LYP/6-31G*; B3LYP/LACVP was used for 6-methoxy-3-iodobenzyne (entry 4)).
Indolynes are an important class of arynes that have gained recent attention.9 In addition to serving as building blocks for medicinally-privileged indoles, indolynes and close relatives have been used as intermediates in the total syntheses of several complex alkaloids.2i–o Although the effect of N-substituents on indolyne distortion has been previously examined computationally and experimentally,2d arene substituent effects on indolyne distortion have been largely neglected.10
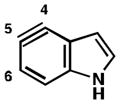
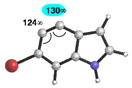
Table 4 provides a distortion analysis for the 4,5-indolyne and several C6-substituted derivatives. As we have shown previously, the unsubstituted 4,5-indolyne11 is distorted such that nucleophilic addition occurs at C5 (entry 1). Interestingly, the presence of a 6-methoxy group overturns this distortion, such that nucleophilic addition is expected to occur at C4 (entry 2). A similar prediction is seen for F, Cl, and Br substituents (entries 3–5, respectively). Finally, in the case of the 6-iodo-4,5-indolyne, the aryne distortion model predicts little unsymmetrical distortion and, consequently, low regioselectivities.12
Table 4.
Distortion analysis of 4,5-indolynes.
| Entry | Aryne | Geometry-optimized structurea | Site of attack (angle difference) |
|---|---|---|---|
| 1 |
|
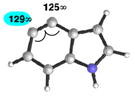
|
C5 (4°) |
| 2 |
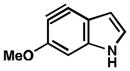
|
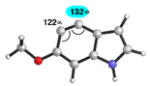
|
C4 (10°) |
| 3 |
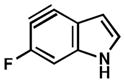
|
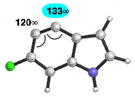
|
C4 (13°) |
| 4 |
|
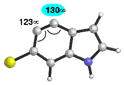
|
C4 (7°) |
| 5 |
|
|
C4 (6°) |
| 6 |
|
|
N/A (0°) |
Geometry optimizations were performed using DFT methods (B3LYP/6-31G*; B3LYP/LACVP was used for 6-iodo-4,5-indolyne (entry 6)).
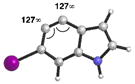
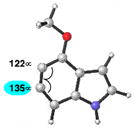
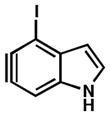
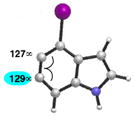
As shown in Table 5, we have also studied substituent effects for 5,6-indolynes. The parent 5,6-indolyne shows minor distortion and predicted regioselectivities that favor nucleophilic addition occurring at C5 (entry 1).2d The influence of C4 and C7 substituents were examined. C7 substituents generally lead to an increase in distortion and predicted regioselectivities; these results are given in the Supplementary Material. The presence of C4 inductively withdrawing substituents, however, leads to an overturning of the predicted regioselectivity such that C6 attack is expected to be favored (entries 2–6). Distortion is greatest in the case of the most electron-withdrawing substituents (entries 2 and 3) and becomes less significant in the cases of the Cl, Br, and I substituted analogs (entries 4–6, respectively).
Table 5.
Distortion analysis of 5,6-indolynes.
| Entry | Aryne | Geometry-optimized structurea | Site of attack (angle difference) |
|---|---|---|---|
| 1 |
|
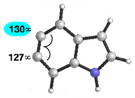
|
C5 (3°) |
| 2 |
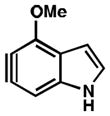
|
|
C6 (13°) |
| 3 |
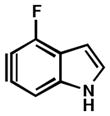
|
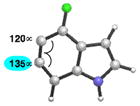
|
C6 (15°) |
| 4 |
|
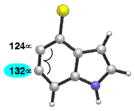
|
C6 (8°) |
| 5 |
|
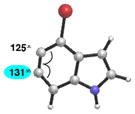
|
C6 (6°) |
| 6 |
|
|
C6 (2°) |
Geometry optimizations were performed using DFT methods (B3LYP/6- 31G*; B3LYP/LACVP was used for 4-iodo-5,6-indolyne (entry 6)).
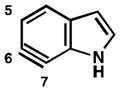
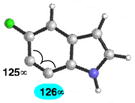
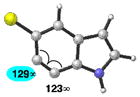
Finally, we have analyzed the distortion in several 6,7-indolynes (Table 6). The 6,7-indolyne is known to react with high regioselectivity for nucleophilic addition at C6,2d which is consistent with the significant unsymmetrical distortion seen in the geometry-optimized structure (entry 1; ca. 18°). Thus, we were curious if it would be possible to overturn this inherent selectivity using substituents. Although the presence of substituents on 6,7-indolynes partially counters the inherent selectivity, we predict that attack at C6 is still favored in nearly all cases (entries 2–6). For 5-fluoro-6,7- indolyne, selectivity is expected to be poor and may indeed favor nucleophilic attack occurring at C7.
Table 6.
Distortion analysis of 6,7-indolynes.
| Entry | Aryne | Geometry-optimized structurea | Site of attack (angle difference) |
|---|---|---|---|
| 1 |
|
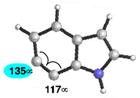
|
C6 (18°) |
| 2 |
|
|
C6 (6°) |
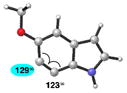
| 3 |
|
|
C7 (1°) |
| 4 |
|
|
C6 (6°) |
| 5 |
|
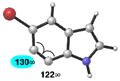
|
C6 (8°) |
| 6 |
|
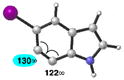
|
C6 (8°) |
Geometry optimizations were performed using DFT methods (B3LYP/6-31G*; B3LYP/LACVP was used for 5-iodo-6,7-indolyne (entry 6)).
In summary, we have applied the distortion/interaction model to a variety of mono and disubstituted benzynes and substituted indolynes. These studies give regioselectivity predictions using straightforward DFT calculations. We anticipate that our results will help encourage the use of unsymmetrical arynes in the synthesis of complex molecules and drug scaffolds.
Supplementary Material
Acknowledgments
The authors are grateful to the NIH-NIGMS (R01 GM090007 to N. K. G.), the National Science Foundation (CHE-0548209 to K. N. H.), Bristol–Myers Squibb, DuPont, the S. T. Li Foundation, the Dreyfus Foundation, the University of California, Los Angeles, and the UCLA Cota Robles Fellowship Program (E. P.) for financial support.
Footnotes
Dedicated to Professor Harry H. Wasserman
Supplementary material with this article can be found in the online version, at http:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.For reviews, see: Pellissier H, Santelli M. Tetrahedron. 2003;59:701–730.Wenk HH, Winkler M, Sander W. Angew Chem Int Ed. 2003;42:502–528. doi: 10.1002/anie.200390151.Sanz R. Org Prep Proced Int. 2008;40:215–291.Bronner SM, Goetz AE, Garg NK. Synlett. 2011;18:2599–2604.Tadross PM, Stoltz BM. Chem Rev. 2012;112:3550–3577. doi: 10.1021/cr200478h.Gampe CM, Carreira EM. Angew Chem Int Ed. 2012;51:3766–3778. doi: 10.1002/anie.201107485.Yoshida H, Takaki K. Synlett. 2012;23:1725–1732.Dubrovskiy AV, Markina NA, Larock RC. Org Biomol Chem. 2013;11:191–218. doi: 10.1039/c2ob26673c.Wu C, Shi F. Asian J Org Chem. 2013;2:116–125.Hoffmann RW, Suzuki K. Angew Chem Int Ed. 2013;52:2655–2656. doi: 10.1002/anie.201209041.Bhunia A, Biji AT. Synlett. 2014;25:608–614.Goetz AE, Garg NK. J Org Chem. 2014;79:846–851. doi: 10.1021/jo402723e.Goetz AE, Shah TK, Garg NK. Chem Commun. 2015;15:34–45. doi: 10.1039/c4cc06445c.
- 2.(a) Bronner SM, Bahnck KB, Garg NK. Org Lett. 2009;11:1007–1010. doi: 10.1021/ol802958a. [DOI] [PubMed] [Google Scholar]; (b) Tian X, Huters AD, Douglas CJ, Garg NK. Org Lett. 2009;11:2349–2351. doi: 10.1021/ol9007684. [DOI] [PubMed] [Google Scholar]; (c) Cheong PHY, Paton RS, Bronner SM, Im GYJ, Garg NK, Houk KN. J Am Chem Soc. 2010;132:1267–1269. doi: 10.1021/ja9098643. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Im GYJ, Bronner SM, Goetz AE, Paton RS, Cheong PHY, Houk KN, Garg NK. J Am Chem Soc. 2010;132:17933–17944. doi: 10.1021/ja1086485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bronner SM, Goetz AE, Garg NK. J Am Chem Soc. 2011;133:3832–3835. doi: 10.1021/ja200437g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Bronner SM, Mackey JL, Houk KN, Garg NK. J Am Chem Soc. 2012;134:13966–13969. doi: 10.1021/ja306723r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Goetz AE, Bronner SM, Cisneros JD, Melamed JM, Paton RS, Houk KN, Garg NK. Angew Chem Int Ed. 2012;51:2758–2762. doi: 10.1002/anie.201108863. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Goetz AE, Garg NK. Nat Chem. 2013;5:54–60. doi: 10.1038/nchem.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Huters AD, Quasdorf KW, Styduhar ED, Garg NK. J Am Chem Soc. 2011;133:15797–15799. doi: 10.1021/ja206538k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Quasdorf KW, Huters AD, Lodewyk MW, Tantillo DJ, Garg NK. J Am Chem Soc. 2012;134:1396–1399. doi: 10.1021/ja210837b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Styduhar ED, Huters AD, Weires NA, Garg NK. Angew Chem Int Ed. 2013;52:12422–12425. doi: 10.1002/anie.201307464. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Goetz AE, Silberstein AL, Corsello MA, Garg NK. J Am Chem Soc. 2014;136:3036–3039. doi: 10.1021/ja501142e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Fine Nathel NF, Shah TK, Bronner SM, Garg NK. Chem Sci. 2014;5:2184–2190. doi: 10.1039/C4SC00256C. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Medina JM, McMahon TC, Jiménez-Osés G, Houk KN, Garg NK. J Am Chem Soc. 2014;136:14706–14709. doi: 10.1021/ja508635v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Weires NA, Styduhar ED, Baker EL, Garg NK. J Am Chem Soc. 2014;136:14710–14713. doi: 10.1021/ja5087672. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Medina JM, Mackey JL, Garg NK, Houk KN. J Am Chem Soc. 2014;136:15798–15805. doi: 10.1021/ja5099935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The distortion/interaction model is also referred to as the activation–strain model according to Bickelhaupt; for references, see: van Zeist WJ, Bickelhaupt FM. Org Biomol Chem. 2010;8:3118–3127. doi: 10.1039/b926828f.Fernández I, Cossío FP, Bickelhaupt FM. J Org Chem. 2011;76:2310–2314. doi: 10.1021/jo102572x.Fernández I, Bickelhaupt FM. J Comput Chem. 2012;33:509–516. doi: 10.1002/jcc.22877.Fernández I, Sola M, Bickelhaupt FM. Chem Eur J. 2013;19:7416–7422. doi: 10.1002/chem.201300648.
- 4.For non-aryne applications of the distortion/interaction model, see: Ess DH, Houk KN. J Am Chem Soc. 2007;129:10646–10647. doi: 10.1021/ja0734086.Legault CY, Garcia Y, Merlic CA, Houk KN. J Am Chem Soc. 2007;129:12664–12665. doi: 10.1021/ja075785o.Ess DH, Houk KN. J Am Chem Soc. 2008;130:10187–10198. doi: 10.1021/ja800009z.Lam Y-h, Cheong PH-Y, Blasco Mata JM, Stanway SJ, Gouverneur V, Houk KN. J Am Chem Soc. 2009;131:1947–1957. doi: 10.1021/ja8079548.Hayden AE, Houk KN. J Am Chem Soc. 2009;131:4084–4089. doi: 10.1021/ja809142x.Garcia Y, Schoenebeck F, Legault CY, Merlic CA, Houk KN. J Am Chem Soc. 2009;131:6632–6639. doi: 10.1021/ja9004927.Schoenebeck F, Ess DH, Jones GO, Houk KN. J Am Chem Soc. 2009;131:8121–8133. doi: 10.1021/ja9003624.Osuna F, Houk KN. Chem Eur J. 2009;15:13219–13231. doi: 10.1002/chem.200901761.Kolakowski RV, Williams LJ. Nat Chem. 2010;2:303–307. doi: 10.1038/nchem.577.Paton RS, Kim S, Ross AG, Danishefsky SJ, Houk KN. Angew Chem Int Ed. 2011;50:10366–10368. doi: 10.1002/anie.201103998.Lan Y, Wheeler SE, Houk KN. J Chem Theory Comput. 2011;7:2104–2111. doi: 10.1021/ct200293w.Liang Y, Mackey JL, Lopez SA, Liu F, Houk KN. J Am Chem Soc. 2012;134:17904–17907. doi: 10.1021/ja309241e.Gordon CG, Mackey JL, Jewett JC, Sletten EM, Houk KN, Bertozzi CR. J Am Chem Soc. 2012;134:9199–9208. doi: 10.1021/ja3000936.Lopez SA, Munk ME, Houk KN. J Org Chem. 2013;78:1576–1582. doi: 10.1021/jo302695n.Lopez SA, Houk KN. J Org Chem. 2013;78:1778–1783. doi: 10.1021/jo301267b.Zou L, Paton RS, Eschenmoser A, Newhouse TR, Baran PS, Houk KN. J Org Chem. 2013;78:4037–4048. doi: 10.1021/jo400350v.Kamber DN, Nazarova LA, Liang Y, Lopez SA, Patterson DM, Shih HW, Houk KN, Prescher JA. J Am Chem Soc. 2013;135:13680–13683. doi: 10.1021/ja407737d.Liu F, Paton RS, Kim S, Liang Y, Houk KN. J Am Chem Soc. 2013;135:15642–15649. doi: 10.1021/ja408437u.Yang J, Liang Y, Šekut J, Houk KN, Devaraj NK. Chem Eur J. 2014;20:3365–3375. doi: 10.1002/chem.201304225.Hong X, Liang Y, Griffith AK, Lambert TH, Houk KN. Chem Sci. 2014;5:471–475.Green AG, Liu P, Merlic CA, Houk KN. J Am Chem Soc. 2014;136:4575–4583. doi: 10.1021/ja411699u.Liu F, Liang Y, Houk KN. J Am Chem Soc. 2014;136:11483–11493. doi: 10.1021/ja505569a.
- 5.Spartan ’10. Wavefunction, Inc; Irvine, CA: 2010. Geometry-optimizations were carried out using B3LYP/6-31G* (or B3LYP/LACVP in the case of iodide-containing arynes) with MacSpartan software. [Google Scholar]
- 6.CYLview, 1.0b. Université de Sherbrooke; Québec, Montreal, Canada: 2009. Images of geometry-optimized structures were prepared using CYLview: Legault, C. Y. http://www.cylview.org. [Google Scholar]
- 7.The distortion present in each unsymmetrical aryne is caused by the proximal inductively withdrawing groups, which deform the triple bond as a result of Bent’s rule; Bent H. Chem Rev. 1961;61:275–311.
- 8.This pathway is favored because it requires minimal distortion to reach the transition state. For more information regarding the aryne distortion model, see references 2c,d.
- 9.For a recent review, see reference 1m.
- 10.We have previously studied the 6-bromo-4,5-indolyne and employed this species in the total synthesis of several indolactam alkaloids; see references 2e and 2m.
- 11.A stable silyltriflate precursor to the parent 4,5-indolyne is commercially available from Aldrich Chemical Co., Inc. (product #L511625).
- 12.The predictive capabilities of the aryne distortion model strictly using geometry-optimization does not take into account steric factors. As such, one might expect nucleophilic addition in the case of 6-iodo-4,5-indolyne (Table 4, entry 6) to occur with some selectivity for nucleophilic addition at C4.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.