

Abstract
A diastereoselective process for the formation of intermediates suitable for the preparation of C1 substituted carbapenems was developed. The process is readily scalable and does not involve organometallics or strong bases such as LDA.
Keywords: Antibiotic, Carbapenem, β-Lactam, silyl enol ether, Lewis acid
The carbapenem class of β-lactam antibiotics represent the most potent and generally broad spectrum of all β-lactams1 and these drugs are usually reserved for use in a nosocomial setting as agents of last resort. Unfortunately, in the past two decades, potent carbapenemases, both serine and metallo, have emerged in highly resistant Gram-negative pathogens, including carbapenem resistant Enterobacteriaceae (CRE),2 Pseudomonas aeruginosa,3 and Acinetobacter baumannii.4 The SAR of the carbapenem class was optimized during the 1970s and 1980s before these highly resistant pathogens had emerged. Thus we decided to take a closer look at whether atypical (i.e. substituted at positions other than C2) carbapenem analogs might have improved biological profiles.
Carbapenems are totally synthetic antibiotics, the synthesis of which is facilitated by the commercial availability of (3S,4R)-4-acetoxy-3-[(R)-1-(tert-butyldimethylsiloxy)ethyl]azetidine-2-one (1) which can react with a wide array of nucleophiles, including some silyl enol ethers, such as 2,5 to generate the key C1-C5 C-C bond with the appropriate C5 configuration, presumably via an intermediate acyliminium ion as shown in Scheme 1.
Scheme 1.

Reported synthetic methodology leading to 3.
We desired to find a general, easily scalable, methodology for forming this crucial C-C bond that did not involve organolithium reagents. Ideally, the methodology would facilitate manipulation of the C1 substituent, would provide opportunity for stereochemical control at this position, and would omit potentially sensitive functionality, such as the diazo moiety of 2, which might interfere with subsequent synthetic manipulations prior to closure of the pyrrolidine ring. A two carbon fragment which terminated in a functional group suitable for further elaboration of the pyrrolidine ring would be ideal. Thus our target was a simple intermediate such as 4 (Scheme 2). Inspection of the literature revealed that this problem has been solved by utilization of silyl ketene acetals (5),6 silyl enol ethers of chiral carboximides (6),7 and organozinc reagents (7).8 The acetate 1 can also be allylated under indium-mediated conditions.9
Scheme 2.

Reported synthetic methodology leading to 4.
We proceeded to explore these options. Formation of silyl ketene acetals, such as 5, and silyl enol ethers of carboximides, 6, employ LDA as the base. This was deemed unsuitable for large scale synthesis, as generation of LDA itself involves use of n-butyllithium, and, in our experience commercial LDA solutions are unstable to long term storage. Our attempts to convert esters to silyl ketene acetals utilizing weaker bases, such as triethylamine, together with more potent silylating agents, such as silyl triflates were unsuccessful.10 Utilization of organozinc reagents, 7, initially seemed promising, and small scale (i.e. < 5 mmol) syntheses were successful. However, these reagents posed substantial hazards on scale-up. Reaction of activated zinc with bromoesters efficiently occurs only at slightly elevated temperatures ( > 50 °C), and is, itself, exothermic. Thus large scale reactions (> 200 mmol) involving this methodology proved difficult to control and these reagents were deemed unsuitable.
By contrast, silyl enol ethers of aldehydes and ketones are readily available through reaction of the carbonyl compound with an appropriate silylating reagent in the presence of triethylamine, and, as exemplified by the precedent in Scheme 1, should react with 1 under mild conditions. Initially, we envisioned that silyl enol ethers of pyruvic acid esters might make suitable precursors, after subsequent deprotection and oxidation to the desired substituted acetates (Scheme 3). However, while the TBS enol ether of benzyl pyruvate (9) readily formed, it refused to react with 1, even at elevated temperature (higher temperatures led to substantial decomposition of 1). Presumably, the electron withdrawing effect of the adjacent ester functionality renders the double bond less electron rich, thereby interfering with this process.
Scheme 3.

Attempted synthesis of intermediates 10 and 11.
We then envisioned a more direct process involving silyl enol ethers of acetaldehyde, which could react with 1 to form the aldehyde 13, and then be subsequently oxidized to the acid or reduced to the corresponding alcohol. Surprisingly, the reaction of 1 with aldehyde silyl enol ethers has not been reported. Vinyloxytrimethysilane (12a) is commercially available, but extremely expensive (5 g/$101 USD, Sigma-Aldrich, bp 74 °C), with the high cost due to its challenging synthesis11 including difficult separation from solvents due to its low boiling point. Gratifyingly, as shown in Equation 1, the reaction of 1 with 12a proceeded smoothly, yielding a mixture of 13, 14a, and unreacted 1. To circumvent the difficult preparation and high cost of 12a, we decided to prepare and employ the TBS enol ether 12b. Unfortunately, the TBS enol ether was substantially less reactive than 12a, and produced only a poor yield of 13, together with 14b. An optimal compromise of ease of preparation and isolation was produced by utilizing the triethylsilyl (TES) ether, 12c. Synthesis of 12c was achieved by treating freshly distilled acetaldehyde with triethylsilyl chloride and triethylamine in dry acetonitrile and purification (removal of TESOH) was accomplished using flash chromatography using pentane as eluent.12 Unlike trimethylsilyl ethers, triethylsilyl ethers are stable to column chromatography.13
Equation 1.

Initial results of reaction of selected vinyloxytrialkylsilanes with 1.
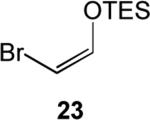
Likewise we were able to utilize this methodology to generate a number of substituted silyl enol ethers as shown in Table 1. Starting aldehydes, 17 and 18, were generated from the monoprotected diols, followed by Swern oxidation,14 as shown in Equation 2. Lastly, we were able to form the triethylsilyl bromoenol ether 23, by utilizing a two-step bromination-elimination sequence15 as shown in Equation 3.
Table 1.

| |||
|---|---|---|---|
| R | TES enol ether | Temperature | Yield |
| H |

|
rt to 35 °C | 62% |
| CH3 |
|
65 °C | 54% |
| OTES (17) |

|
65 °C | 35% |
| CH2OTES (18) |

|
65 °C | 49% |
Equation 2.

Swern oxidation of monotriethylsililylated diols 15 and 16.
Equation 3.

Bromination of 12c.
As shown in Table 2, zinc-promoted reaction of the individual stereoisomers with 1 provided the corresponding aldehydes, with, in the case of 19, a surprisingly high degree of diastereocontrol. Since diastereomeric aldehydes 24a and 24b are inseparable by column chromatography, and to confirm stereochemical identity the aldehyde was selectively reduced using NaBH3CN as shown below and the resultant, chromatographically separable, alcohols, 27a and 27b, and the major isomer, 27a, proven identical by 1H NMR with the reported spectra for this epimer.16 These alcohols were individually further converted to bicyclic β-lactams 28a and 28b as indicated in Scheme 4, with the major isomer again proving identical with previously reported spectra.17 A functionalized aldehyde (i.e. R = OR’), potentially useful in C1-substituted carbapenem synthesis, was formed by using (Z)-1,2-bis(triethylsilyloxy)ethylene, 21, as the silyl enol ether, but reaction occurred only at elevated temperature (85 °C) in the absence of solvent (with the diethyl ether solvent of the ZnCl2 allowed to distill from the reaction vessel).
Table 2.
Reaction of TES enol ethers with 1.

| ||||
|---|---|---|---|---|
| Enol Ether | Product(s) | Temperature | Yield | |

|

|
40 °C | 32% | |
|
|
||||

|
6 | 1 | 40 °C | 48% |

|
3 | 2 | 40 °C | 56% |

|

|
|||
| 4 | 1 | 85 °C | 84% | |

|
|
85 °C | 64% | |
|
No reaction | 85 °C | - | |
Scheme 4.

Conversion of 24 to reported compounds for stereochemical verification
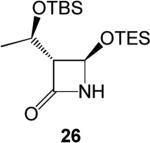
Unfortunately, (Z)-1,3-bis(triethylsilyloxy)-1-propene, 22, produced the unexpected substitution product, 26, presumably due to ZnCl2 promoted decomposition of the [.gamma]-substituted silyl enol ether, and (Z)-1-bromo-2-triethylsilyoxyethylene, 23, refused to react, even under forcing conditions.
Lastly, since we were unable to couple triethylsilyl bromoenol ether 22, we decided to try to brominate aldehyde 13, to generate a C1-substituted carbapenem synthon. As shown in Scheme 5, this was successfully achieved by converting 13 to the trisilylated enol ether 29, and then treating with NBS to produce an inseparable 4:1 mixture of diastereomers 30a and 30b.18
Scheme 5.

Bromination of 12
Discussion
Silyl enol ethers of aldehydes are less frequently utilized in Mukaiyama aldol reactions since the resultant product aldehyde can itself undergo reaction with the enol ether resulting in lowered yields19 and forming oligomers.20 The Mukaiyama-like reaction of aldehyde triethylsilyl enol ethers and 1 presented herein has advantages in that the iminium ion intermediate (Scheme 1) may be more reactive than the product aldehyde, thus allowing for selectivity and improved yields. Secondly, the use of triethysilyl enol ethers has advantages, since TES enol ethers are readily prepared, chromatographically stable, and have enhanced reactivity relative to TBS enol ethers.
In conclusion, we have developed a new method for forming the carbapenem C1-C5 bond using silyl enol ethers which, in the case of C2 substituted silyl enol ethers, is capable of producing the product with high diastereoselectivity. We have discovered that vinyloxytriethylsilane (8c) is an easily prepared and cost effective substitute for vinyloxytrimethylsilane, 8a, an acetaldehyde equivalent that has been extensively employed in organic synthesis, despite its challenging preparation and high cost. Lastly we have generated a number of highly functional carbapenem precursors that we anticipate will be useful in the preparation of C1 modified carbapenem antibiotics.
Supplementary Material
Figure 1.

Current commercial carbapenem antibiotics with conventional numbering scheme
Acknowledgments
This work was supported by National Institutes of Health grants 1R15AI109624 and 1R41AI102507 to JDB. We also thank the SMU Department of Chemistry and the SMU Undergraduate Research Assistantship (URA) Program for valuable support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.a Papp-Wallace KM, Endimiani A, Taracila MA, Bonomo RA. Antimicrob. Agents Chemother. 2011;55:4943–4960. doi: 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; b El-Gamal MI, Oh C-H. Curr. Top. Med. Chem. 2010;10:1882–1897. doi: 10.2174/156802610793176639. [DOI] [PubMed] [Google Scholar]; c Breilh D, Texier-Maugein J, Allaouchiche B, Saux M-C, Boselli EJ. Chemother. 2013;25:1–17. doi: 10.1179/1973947812Y.0000000032. [DOI] [PubMed] [Google Scholar]
- 2.a van Duin D, Kaye KS, Neuner EA, Bonomo RA. Diagn. Microbiol. Infect. Dis. 2013;75:115–120. doi: 10.1016/j.diagmicrobio.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tzouvelekis LS, Markogiannakis A, Psichogiou M, Tassios PT, Daikos GL. Clin. Microbiol. Rev. 2012;25:682–707. doi: 10.1128/CMR.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Canton R, Akova M, Carmeli Y, Giske CG, Glupczynski Y, Gniadkowski M, Livermore DM, Miriagou V, Naas T, Rossolini GM, Samuelsen O, Seifert H, Woodford N, Nordmann P. Clin. Microbiol. Infect. 2012;18:413–431. doi: 10.1111/j.1469-0691.2012.03821.x. [DOI] [PubMed] [Google Scholar]; d Nordmann P, Dortet L, Poirel L. Trends Mol. Med. 2012;18:263–272. doi: 10.1016/j.molmed.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Voor In 't Holt AF, Severin JA, Lesaffre EMEH, Vos MC. Antimicrob. Agents Chemother. 2014;58:2626–2637. doi: 10.1128/AAC.01758-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans BA, Hamouda A, Amyes SG. B. Curr. Pharm. Des. 2013;19:223–238. [PubMed] [Google Scholar]
- 5.Hart DJ, Ha DC. Tetrahedron Lett. 1985;26:5493–5496. [Google Scholar]
- 6.Chiba T, Nagatsuma M, Nakai T. Chem. Lett. 1985:1343–1346. [Google Scholar]
- 7.Fuentes LM, Shinkai I, Salzmann TN. J. Am. Chem. Soc. 1986;108:4675–4676. [Google Scholar]
- 8.a Ito Y, Terashima S. Tetrahedron Lett. 1987;28:6625–6628. [Google Scholar]; b Endo M. Can. J. Chem. 1987;65:2140–2145. [Google Scholar]
- 9.Kang S-K, Baik T-G, Jiao X-H, Lee K-J, Lee CH. Synlett. 1999:447–449. [Google Scholar]
- 10.Treatment of benzyl acetate with TBSOTf and Et3N in CH2Cl2 at rt for 24 h resulted in recovered starting materials.
- 11.Olofson RA, Cuomo JJ. Org. Chem. 1980;45:2538–2541. [Google Scholar]
- 12.A solution of freshly distilled acetaldehyde (40 g, 0.908 mol) in dry acetonitrile (300 mL) was cooled to 0 °C and then treated with triethylamine (110 g, 152 mL, 1.09 mol) and triethylsilyl chloride (137 g, 0.908 mol). The reaction was stirred at rt for 5 h, then heated to 35 °C overnight. The product was isolated by pouring the reaction mixture into 600 mL of a 1:1 solution of diethyl ether and pentanes, filtering the precipitated triethylammonium hydrochloride, and washing the combined organic layers with water (2 × 300 mL), sat. aq. ammonium chloride (1 × 300 mL), then with water (300 mL) and brine. The organic layer was dried over Na2SO4, filtered, carefully evaporated on the rotovap (predicted product bp 147 °C at 760 mm), and the product purified by flash chromatography using pentanes as eluent to produce 89 g (62%) of 11c.
- 13.Although 12c requires chromatography to separate it from TESOH, due to the similarity of boiling points (predicted bp of 12c is 147 °C, and TESOH is 158 °C at 760 mm), higher mw TES ethers can be purified by vacuum distillation.
- 14.Loelsberg W, Werle S, Neudoerfl J-M, Schmalz H-G. Org. Lett. 2012;14:5996–5999. doi: 10.1021/ol302898h. [DOI] [PubMed] [Google Scholar]
- 15.a Zembayashi M, Tamao K, Kumada M. Synthesis. 1977:422–423. [Google Scholar]; b Ito H, Kishi Y, Nishikawa Y, Tada T, Ishida Y, Saigo K. Synlett. 2010:1811–1814. [Google Scholar]
- 16.a Tsukada N, Shimada T, Gyoung YS, Asao N, Yamamoto Y. J. Org. Chem. 1995;60:143–148. [Google Scholar]; b Wasserman HH;, Henke SL, Nakanishi E, Schulte G. J. Org. Chem. 1992;57:2541–2645. [Google Scholar]
- 17.Fuentes LM, Shinkai I, King A, Purick R, Reamer RA, Schmitt SM, Cama L, Christensen BG. J. Org. Chem. 1987;52:2563–2567. [Google Scholar]
- 18.For use of a structurally related bromoketone in carbapenem synthesis, see: Feigelson GB. Tetrahedron Lett. 1993;34:4747–50.
- 19.Denmark SE, Ghosh SK. Angew. Chem. Int. Ed. 2001;40:4759–4762. doi: 10.1002/1521-3773(20011217)40:24<4759::aid-anie4759>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 20.Brady PB, Yamamoto H. Angew. Chem. Int. Ed. 2012;51:1942–1946. doi: 10.1002/anie.201108325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.