

Abstract
Here we describe a rhodium-catalyzed intramolecular decarbonylative coupling between 3-aminocyclobutenones and alkenes for synthesis of substituted [3.1.0] bicycles. This transformation represents a formal cyclopropanation reaction, in which the cyclobutenones serve as a one-carbon-unit synthon.
Keywords: C–C cleavage, Cyclobutenone, Rhodium catalysis, Decarbonylation, Cyclopropanation
1. Introduction
Cyclobutenones are a unique class of compounds, and often serve as important synthetic intermediates due to their high reactivity driven by strain release.1 Conventionally, cyclobutenones are often employed as a vinyl-ketene equivalent through a retro-4π cyclization.2a During the past two decades, novel transformations of cyclobutenones have been realized through transition metal-catalyzed carbon-carbon (C–C) bond cleavage followed by C–C bond formation.2 These reactions often lead to complex scaffolds that are challenging to prepare using traditional approaches. For example, using Ni, Co, Ru and Rh as catalysts, intermolecular or intramolecular cyclization of cyclobutenones with alkenes, alkynes and other strained rings can provide five to eight-membered ring systems efficiently.2
Our group has been interested in applying catalytic C–C bond activation methods in complex molecule synthesis. Recently, we have demonstrated fused-ring and spirocycle synthesis through Rh-catalyzed C–C activation of benzocyclobutenones (Scheme 1a).3 An intriguing question is whether non-benzo-type cyclobutenones will behave similarly in the catalytic intramolecular coupling reactions, which, to our knowledge, has not been explored previously. One potential challenge for this study is that regioselective synthesis of poly-substituted cyclobutenone substrates is nontrivial.4,5 We were particularly inspired by an efficient method reported by Danheiser and coworkers for preparing multi-substituted 3-aminocyclobutenones5 from readily available of ynamides6 and ketenes.7 Here, as an exploratory study, we describe our development of a Rh-catalyzed decarbonylative coupling of 3-aminocyclobutenones with alkenes leading to [3.1.0] bicycles (Scheme 1b). Compared to the corresponding benzocyclobutenones, a distinct reactivity with 3-aminocyclobutenones was discovered during this study.
Scheme 1.

Rh1-catalyzed intramolecular cyclization between cyclobutenones and alkenes.
2. Results and discussions
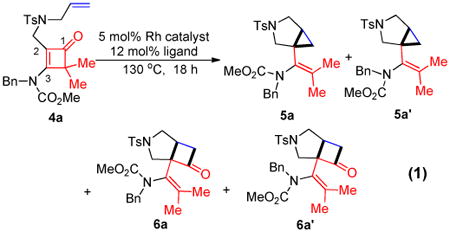
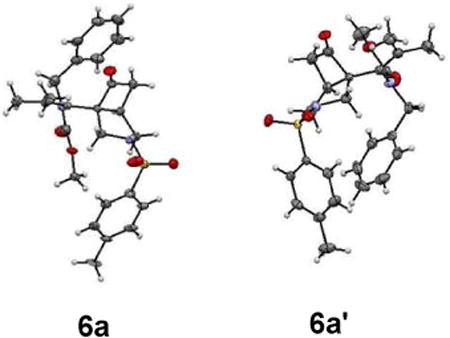
As illustrated in Scheme 2, the tethered alkene-cyclobutenone substrates can be efficiently synthesized in three steps from readily available enyne substrates.5,8 Our initial study of the intramolecular cyclization reaction employed cyclobutenone 4a9 as the model substrate. When directly heating 4a in 1,4-dioxane at 130 °C, vinyl cyclobutanones 6a and 6a′ were slowly formed (their structures were unambiguously confirmed by X-ray crystallography)10 (entry 1, Table 1). The reaction likely proceeds through a retro-[2+2] of the cyclobutenone to give a vinyl ketene and then a [2+2] cycloaddition with the alkene (vide infra, Scheme 3).11 It is interesting to note that compounds 6a and 6a′ represent a pair of conformational isomers and can be separated using regular silica-gel chromatography. The C3-N bond cannot freely rotate likely caused by the significant steric hindrance between the tetra-substituted olefin and the bulky cyclobutanone. [Rh(cod)Cl]2 did not influence the reaction, because almost the same results were obtained when using [Rh(cod)Cl]2 in combination with bidentate phosphine ligands (dppp, dppf and dppb) as well as monodentate P(C6F5)3 ligand (entry 2). However, when the more electron-deficient [Rh(CO)2Cl]2 was employed as the catalyst, an interesting bicycle[3.1.0]-hexane compound 5 was observed as the dominating product in 68% yield (entry 3). The yield can be further improved to 84% by adding P(C6F5)3 as an additional ligand, in which cyclobutanones 6a and 6a′ (the major byproducts) only formed in 10% yield (entry 4); in contrast, using more electron-rich ligands gave diminished yields (entries 5-8). The solvent effect was also investigated (entries 9-12) and 1,4-dioxane remained to be optimal.
Scheme 2.

General approach for synthesizing the 3-aminocyclobutenone substrates.
Table 1. Selected reaction optimization conditions.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Rh catalyst | Ligand | Solvent | Yield [%] a |
| 1 | none | None | 1,4-Dioxane | 6a15%, 6a′15% |
| 2 | [Rh(cod)Cl]2 | P(C6F5)3 | 1,4-Dioxane | 6a15%, 6a′15% |
| 3 | [Rh(CO)2Cl]2 | None | 1,4-Dioxane | 5c 68% |
| 4 | [Rh(CO)2Cl]2 | P(C6F5)3 | 1,4-Dioxane | 5 84% |
| 5 | [Rh(CO)2Cl]2 | dpppb | 1,4-Dioxane | 5 60% |
| 6 | [Rh(CO)2Cl]2 | dppmb | 1,4-Dioxane | 5 35% |
| 7 | [Rh(CO)2Cl]2 | dppbb | 1,4-Dioxane | 5 50% |
| 8 | [Rh(CO)2Cl]2 | PPh3 | 1,4-Dioxane | 5 43% |
| 9 | [Rh(CO)2Cl]2 | P(C6F5)3 | Toluene | 5 78% |
| 10 | [Rh(CO)2Cl]2 | P(C6F5)3 | m-Xylene | 5 80% |
| 11 | [Rh(CO)2Cl]2 | P(C6F5)3 | THF | 5 65% |
| 12 | [Rh(CO)2Cl]2 | P(C6F5)3 | PhCl | 5 81% |
Determined by 1H NMR spectroscopy using mesitylene as the internal standard.
6 mol% of the ligand was used.
5 is a mixture of inseparable isomers: 5a and 5a′ (1:1).
Scheme 3.

Proposed catalytic cycle.
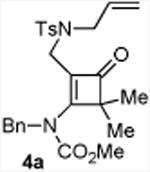
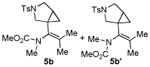
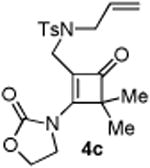
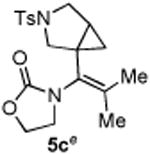
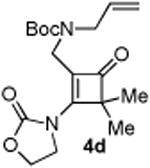
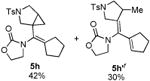
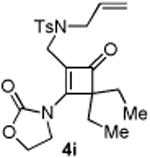
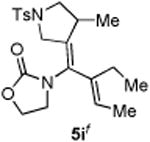
With the optimized conditions in hand, we next examined the scope of this decarbonylative cyclization reaction (Table 2).12 First, change of the substituents on the nitrogen of the aminocyclobutenones did not significantly affect the reactivity (entries 1-3). While the methyl formate substrates (4a and 4b) all gave a pair of conformational isomers, the less hindered oxazolidone substrates afforded a single isomer (entries 3-10). Besides the nitrogen-based linker (entries 3-4), oxygen and carbon-based linkers are also effective (entries 5-7). Different substituents on the cyclobutenone C2 position were also examined. When replacing the gem-dimethyls to a cyclopentane group, the desired vinyl cyclopropane 5h was isolated in 42% yield and a ring-opening diene (5h′) was found as the major byproduct (entry 8). The exact reason of forming diene 5h′ remains unclear. It is likely that 5h′ did not come from the decomposition of 5h as treatment of 5h under the reaction conditions did not yield any 5h′. It was surprising to note that the diethyl analogue (4i) gave the ring-opening diene 5i as the sole product (entry 9), which suggests the steric hindrance at the C2 position of the cyclobutenone plays an important role in the selectivity of the reaction.
Table 2.
Substrate scope.a
| Entry | Substrate | Product | Yield [%]b |
|---|---|---|---|
| 1 |
|
|
80 5a:5a′= 1:1d |
| 2 |
|
|
75 5b:5b′= 1:1d |
| 3 |
|
|
72 |
| 4 |
|
|
45 |
| 5 |
|
|
65 |
| 6 |
|
|
72 (86)c |
| 7 |
|
|
76 (90) |
| 8 |
|
|
|
| 9 |
|
|
45 |
| 10 |
|
|
84 |
| 11 |
|
0 | |
| 12 |
|
0 |
Each reaction was ran on a 0.1 mmol scale in a sealed 4 mL vial, using 5 mol% [Rh(CO)2Cl]2 and 12 mol% P(C6F5)3, in 1,4-dioxane (1.5 mL), at 130 °C, for 18 h.
Yields of isolated products.
The number in parentheses represents the yield based on recovered starting material (brsm).
The ratio was determined by 1H NMR.
The structure of 5c was confirmed by a series of 2D NMRs, DEPT, and HRMS.
5h′ and 5i were isolated as a single olefin-geometric isomer; the exact olefin geometry for each was not determined.
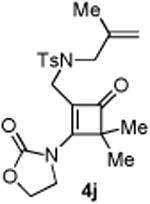
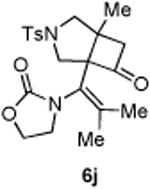
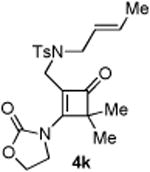
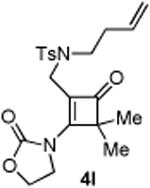
An interesting observation is that the substrate tethered with a 1,1-disubstituted olefin gave a complete selectivity for the [2+2] cyclobutanone product in 84% yield (entry 10).13 In contrast, use of a 1,2-disubstituted olefin and elongation of the linkage led to a complex unidentifiable mixture (entries 11 and 12).
It is well known that decarbonylation of cyclobutanones to cyclopropanes can be catalyzed by transition metals, such as Rh(I) complexes.14 Thus, it is natural to concern whether the [3.1.0] bicycle (e.g. 5a/5a′) comes from the direct decarbonylation of a cyclobutanone intermediate (e.g. 6a/6a′). Hence, the following experiment was conducted. When treating a mixture of 6a and 6a′ under the standard reaction conditions, no decarbonylation product 5a/5a′ was observed (eq 2), suggesting that the [3.1.0] bicycle should come from a different reaction pathway.
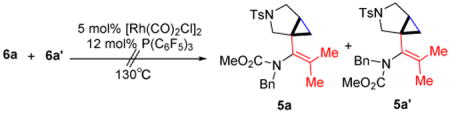 |
(2) |
The proposed catalytic cycle is depicted in Scheme 3.15 First, a retro-4π cyclization of cyclobutenones should give a reactive vinyl ketene intermediate (7), which can undergo either a [2+2] cycloaddition with the tethered alkene to give cyclobutanone 6 (background reaction) or a cyclometalation reaction mediated by Rh to give a five-membered metallacycle 8 (catalytic reaction). Subsequent CO de-insertion from complex 8 followed by reductive elimination should provide cyclopropane 5.
3. Conclusion
In summary, a Rh-catalyzed intramolecular coupling of 3-aminocyclobutenones with alkenes has been studied. In this transformation, the cyclobutenone serves as a one-carbon synthon leading to an unprecedented cyclopropanation reaction. This reaction offers a unique approach for synthesizing heavily substituted vinyl cyclopropanes and [3.1.0]-bicycle skeletons. Stimulated by this work, efforts on investigating the coupling of less substituted cyclobutenones with alkenes, alkynes and allenes are ongoing in this laboratory.
4. Experimental
4.1. General
THF, 1,4-dioxane and toluene were purchased from Fischer Scientific, distilled freshly over sodium and freeze-pump-thawed. Other commercially available chemicals were purchased and used without additional purification unless noted otherwise. All reactions were carried out under nitrogen with stirring bar in a rubber septum sealed flask. Reaction temperatures were reported as the temperatures of the bath surrounding the flasks or vials. Sensitive reagents and solvents were transferred under nitrogen into a nitrogen-filled glove-box with standard techniques. Analytical thin-layer chromatography (TLC) was carried out using 0.2 mm commercial silica gel plates (silica gel 60, F254, EMD chemical). Vials (15 × 45 mm 1 dram (4 mL)/27.75 × 95 mm 10 dram (40 mL) with PTFE lined cap attached) were purchased from Qorpak and flame-dried or put in an oven overnight and cooled in a dessicator. High-resolution mass spectra (HRMS) were obtained on a Karatos MS9 and are reported as m/z (relative intensity). Accurate masses are reported for the molecular ion [M+Na]+, [M+H]+, [M-H]- or [M]+. Infrared spectra were recorded on a Nicolet 380 FTIR using neat thin film technique. Nuclear magnetic resonance spectra (1H NMR and 13C NMR) were recorded with a Varian Gemini (400 MHz, 1H at 400 MHz, 13C at 100 MHz). Unless otherwise noted, all spectra were acquired in CDCl3. Chemical shifts are reported in parts per million (ppm, δ), downfield from tetramethylsilane (TMS, δ = 0.00 ppm) and are referenced to residual solvent (CDCl3, δ=7.26 ppm (1H) and 77.00 ppm (13C)). Couplingconstants were reported in Hertz (Hz). Data for 1H NMR spectra were reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, dd = doublet of doublets, td = triplet of doublets, ddd = doublet of doublet of doublets, m = multiplet, coupling constant (Hz), and integration.
4.2 General procedure for synthesis of cyclobutenone compounds through [2+2] cyclization
Acyl chloride (3.0 mmol) and Et3N (0.70 ml, 5.0 mmol) were added drop wise to a solution of ynamide (1.0 mmol) in anhydrous DCM (10 mL) at 0 °C under N2. The resulting mixture was stirred for 48 h at 40°C, and then quenched with saturated aqueous NaHCO3 solution. The layers were separated, and the aqueous layers were extracted with EtOAc (3 × 20 mL). The organic layers were combined and washed with brine (20 mL), which were then dried over MgSO4 and concentrated. Compounds 4a-4l were purified via silica gel flash chromatography. (For details, see supporting information).
4.2.1. Compound 4a
Compound 4a was obtained in 85% yield as a white solid. Rf = 0.60, hexans/EtOAc = 2:1, 1H NMR (400 MHz, CDCl3) δ 7.54-7.52 (m, 2H), 7.40-7.39 (m, 2H), 7.32-7.30 (m, 1H), 7.30-7.21 (m, 4H), 5.66-5.56 (m, 1H), 5.28 (s, 2H), 5.26-5.21 (m, 1H), 5.06-5.03 (m, 1H), 3.86 (s, 3H), 3.71 (d, J = 6.0 Hz, 2H), 3.49 (s, 2H), 2.39 (s, 3H), 1.33 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 195.8, 173.0, 152.3, 143.6, 136.1, 135.1, 132.9, 129.6, 128.8, 127.5, 127.3, 125.2, 118.5, 115.9, 63.1, 54.4, 51.0, 39.7, 21.4, 20.6. IR: ν 1752, 1595, 1437, 1395, 1346, 1305, 1262, 1185, 1160, 1090, 1044, 743 cm-1. HRMS calcd for C26H30N2NaO5S+ [M+Na]+: 505.1773, found: 505.1765. Mp (°C): 100-102.
4.2.2. Compound 4b
Compound 4b was obtained in 55% yield (90% brsm yield) as a white solid. Rf = 0.30, hexans/EtOAc = 5:1, 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 5.69-5.59 (m, 1H), 5.21-5.15 (m, 1H), 5.07-5.04 (m, 1H), 3.87 (s, 3H), 3.81 (s, 2H), 3.73 (dt, J = 1.2, 6.0 Hz, 2H), 3.55 (s, 3H), 2.39 (s, 3H), 1.22 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 195.7, 173.4, 152.2, 143.6, 135.3, 133.0, 129.7, 127.3, 118.5, 115.6, 62.8, 54.2, 50.9, 39.8, 36.3, 21.4, 20.5. IR: ν 2959, 2927, 1494, 1428, 1263, 1090, 1043, 955, 916 cm-1. HRMS calcd for C20H26N2NaO5S+ [M+Na]+: 429.1460, found: 429.1456. Mp (°C): 83-85.
4.2.3. Compound 4c
Compound 4c was obtained in 60% yield (86% brsm yield) as a colorless oil. Rf = 0.55, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 7.62-7.60 (m, 2H), 7.31-7.26 (m, 2H), 5.65-5.56 (m, 1H), 5.25-5.23 (m, 1H), 5.15-5.12 (m, 1H), 4.62-4.60 (m, 2H), 4.47-4.45 (m, 2H), 3.77-3.74 (m, 4H), 2.42 (s, 3H), 1.32 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 195.1, 168.7, 151.9, 144.0, 135.2, 132.3. 129.9, 127.1, 119.5, 115.8, 63.2, 62.3, 51.0, 45.0, 38.7, 21.5, 20.3. IR: ν 1784, 1754, 1344, 1223, 1192, 1159, 1121, 1091, 913, 743 cm-1. HRMS calcd for C20H25N2O5S+ [M+H]+: 405.1484, found: 405.1482.
4.2.4. Compound 4d
Compound 4d was obtained in 86% yield as a colorless oil. Rf = 0.40, DCM/Acetone = 10:1, 1H NMR (400 MHz, CDCl3) δ 5.76-5.69 (m, 1H), 5.16-5.11 (m, 2H), 4.58-4.54 (m, 2H), 4.41-4.38 (m, 2H), 3.90 (d, J = 5.6 Hz, 2H), 3.83 (s, 2H), 1.40 (s, 9H), 1.35 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 195.9, 166.7, 155.2, 152.1, 133.4, 118.4, 116.6, 80.0, 63.1, 61.9, 49.6, 44.3, 37.1, 28.3, 20.6. IR: ν 2978, 2928, 1754, 1687, 1611, 1458, 1366, 1166, 1145, 1102 cm-1. HRMS calcd for C18H26N2NaO5+ [M+Na]+: 373.1739, found: 373.1736.
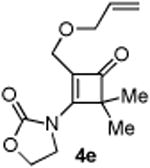
4.2.5. Compound 4e
Compound 4e was obtained in 79% yield as a colorless oil. Rf = 0.25, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 5.89-5.80 (m, 1H), 5.28-5.23 (m, 1H), 5.20-5.17 (m, 1H), 4.59-4.55 (m, 2H), 4.31-4.27 (m, 2H), 3.96-3.93 (m, 2H), 3.92 (s, 2H), 1.39 (s, 3H), 1.38 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 194.9, 168.9, 151.8, 133.9, 118.7, 117.9, 71.4, 63.0, 62.4, 58.3, 44.5, 20.7.. IR: ν 2926, 1782, 1755, 1410, 1218, 1190, 1121, 1072 cm-1. HRMS calcd for C13H18NO4+ [M+H]+: 252.1236, found: 252.1227.
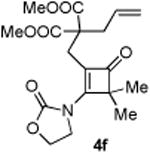
4.2.6. Compound 4f
Compound 4f was obtained in 61% yield (76% brsm yield) as a colorless oil. Rf = 0.40, DCM/Acetone = 10:1, 1H NMR (400 MHz, CDCl3) δ 5.70-5.59 (m, 1H), 5.21-5.17 (m, 1H), 5.12-5.09 (m, 1H), 4.57-4.53 (m, 2H), 4.28-4.24 (m, 2H), 3.69 (s, 6H), 2.73 (d, J = 7.2 Hz, 2H), 2.66 (s, 2H), 1.31 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 195.4, 170.9, 167.3, 152.3, 132.2, 119.8, 118.0, 63.0, 61.5, 57.5, 52.6, 44.1, 37.8, 26.5, 20.7. IR: ν 2957, 1784, 1734, 1611, 1437, 1371, 1328, 1207, 1126, 1042, 913 cm-1. HRMS calcd for C18H23NNaO7+ [M+Na]+: 388.1372, found: 388.1370.
4.2.7. Compound 4g
Compound 4g was obtained in 72% yield as a colorless oil. Rf = 0.50, DCM/Acetone = 10:1, 1H NMR (400 MHz, CDCl3) δ 5.73-5.63 (m, 1H), 5.23-5.19 (m, 1H), 5.13-5.10 (m, 1H), 4.57-4.53 (m, 2H), 4.30-4.26 (m, 2H), 4.21-4.09 (m, 4H), 2.75 (d, J = 7.6 Hz, 2H), 2.65 (s, 2H), 1.31 (s, 6H), 1.32 (t, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 195.5, 170.5, 167.3, 152.3, 132.3, 119.8. 118.2, 62.9, 61.6, 61.5, 57.4, 44.1, 37.6, 26.3, 20.8, 13.9. IR: ν 2982, 1785, 1730, 1612, 1395, 1203, 1042, 913 cm-1. HRMS calcd for C20H27NNaO7+ [M+Na]+: 416.1685, found: 416.1681.
4.2.8. Compound 4h
Compound 4h was obtained in 7% yield (70% brsm yield) as a colorless oil. Rf = 0.60, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 5.64-5.56 (m, 1H), 5.26-5.21 (m, 1H), 5.12-5.09 (m, 1H), 4.63-4.58 (m, 2H), 4.47-4.43 (m, 2H), 3.76 (s, 2H), 3.76 (d, J = 6.8 Hz, 2H), 2.41 (s, 3H), 1.99-193 (m, 2H), 1.78-1.70 (m, 2H), 1.69-1.63 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 195.1, 165.7, 152.0, 143.9, 135.4, 132.3, 129.9, 127.1, 119.5, 117.6, 70.2, 63.2, 51.0, 44.9, 38.7, 31.4, 26.5, 21.5. IR: ν 2924, 1781, 1751, 1607, 1401, 1275, 1260, 913 cm-1. HRMS calcd for C22H27N2O5S+ [M+H]+: 431.1641, found: 431.1636.
4.2.9. Compound 4i
Compound 4i was obtained in 35% yield (81% brsm yield) as a colorless oil. Rf = 0.60, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 6.4 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 5.52-5.44 (m, 1H), 5.25-5.20 (m, 1H), 5.12-5.10 (m, 1H), 4.63-4.59 (m, 2H), 4.47-4.43 (m, 2H), 3.85 (s, 2H), 3.81 (d, J = 6.4 Hz, 2H), 2.43 (s, 3H), 1.85-175 (m, 4H), 0.83 (d, J = 7.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 194.3, 166.1, 152.2, 143.9, 135.5, 131.1, 129.8, 127.3, 120.2, 119.5, 72.2, 63.3, 50.2, 45.1, 37.7, 25.5, 21.5, 10.1. IR: ν 2965, 1784, 1752, 1607, 1401, 1340, 1186, 1157, 1120, 913 cm-1. HRMS calcd for C22H29N2O5S+ [M+H]+: 433.1797, found: 433.1780.
4.2.10. Compound 4j
Compound 4j was obtained in 60% yield (89% brsm yield) as a colorless oil. Rf = 0.50, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 5.27 (d, J = 0.8 Hz, 1H), 4.98 (s, 1H), 4.62-4.58 (m, 2H), 4.45-4.41 (m, 2H), 3.73 (s, 2H), 3.66 (s, 2H), 2.39 (s, 3H), 1.68 (s, 3H), 1.22 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 195.1, 168.1, 151.9, 143.9, 140.3, 135.2, 129.8, 127.1, 116.0, 114.3, 63.2, 62.2, 55.0, 44.8, 39.5, 21.4, 19.9. IR: ν 2926, 1784, 1753, 1610, 1272, 1224, 1191, 1119, 1046, 913 cm-1. HRMS calcd for C21H26N2NaO5S+ [M+Na]+: 441.1460, found: 441.1456.
4.2.11. Compound 4k
Compound 4k was obtained in 52% yield (82% brsm yield) as a colorless oil. Rf = 0.50, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 5.63-5.56 (m, 1H), 5.27-5.20 (m, 1H), 4.62-4.58 (m, 2H), 4.45-4.41 (m, 2H), 3.73 (s, 2H), 3.69 (d, J = 6.4 Hz, 2H), 2.40 (s, 3H), 1.57 (dd, J = 0.8, 6.4 Hz, 3H), 1.30 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 195.1, 168.6, 152.0, 143.8, 135.4, 131.2, 129.7, 127.2, 124.5, 116.1, 63.3, 62.1, 50.4, 45.0, 38.3, 21.4, 20.4, 17.7. IR: ν 2963, 1785, 1754, 1468, 1341, 1221, 1191, 1158, 1130, 1048 cm-1. HRMS calcd for C21H26N2NaO5S+ [M+Na]+: 441.1460, found: 441.1453.
4.2.12. Compound 4l
Compound 4l was obtained in 75% yield as a colorless oil. Rf = 0.40, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 7.62-7.59 (m, 2H), 7.31-7.29 (m, 2H), 5.70-5.60 (m, 1H), 5.04-4.97 (m, 2H), 4.63-4.59 (m, 2H), 4.47-4.43 (m, 2H), 3.74 (s, 2H), 3.11-3.07 (m, 2H), 2.41 (s, 3H), 2.28-2.23 (m, 2H), 1.35 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 195.3, 169.4, 151.9, 144.0, 134.7, 134.5, 129.9, 127.2, 117.1, 115.4, 63.3, 62.5, 48.1, 45.0, 39.4, 33.4, 21.4, 20.4. IR: ν 2962, 2926, 1785, 1754, 1609, 1404, 1314, 1290, 1220, 1133, 1090 cm-1. HRMS calcd for C21H26N2NaO5S+ [M+Na]+: 441.1460, found: 441.1458.
4.3 General procedure for synthesis of Bicyclo[3.1.0]hexane and its analogues
Cyclobutenone substrates (0.1 mmol), [Rh(CO)2Cl]2 (1.95 mg, 0.05 mmol), P(C6F5)3 (6.4 mg, 0.12 mmol), and anhydrous 1,4-Dioxane (1.5 mL) were added into a 4.0 mL vial in glove box, which were then stirred at 130°C under N2 for 18h. The reaction was quenched with saturated aqueous NaHCO3 solution (10 mL). The layers were then separated, and the aqueous layers were extracted with EtOAc (3 × 10 mL). The organic layers were combined, washed with brine (20 mL), dried over MgSO4 and concentrated. The decarbonylative coupling products were purified via silica gel flash chromatography.
4.3.1. Compounds 5a and 5a′
Compounds 5a and 5a′ were obtained from 4a in 80% yield as a colorless oil, 5a and 5a′ in a 1:1 ratio (the ratio of 5a and 5a′ was determined by crude 1H NMR). Rf = 0.55, hexans/EtOAc = 2:1, 1H NMR (400 MHz, CDCl3) δ 7.63-7.60 (m, 4H), 7.33-7.30 (m, 4H), 7.27-7.20 (m, 4H), 7.12-7.07 (m, 6H), 4.70 (d, J = 14.4 Hz, 1H), 4.43 (d, J = 14.4 Hz, 1H), 4.21 (d, J = 14.4 Hz, 1H), 4.16 (d, J = 14.4 Hz, 1H), 3.70-3.62 (m, 2H), 3.62 (s, 3H), 3.40-3.34 (m, 2H), 3.32 (s, 3H), 3.03-2.99 (m, 2H), 2.85-2.79 (m, 2H), 2.42 (s, 6H), 1.69 (s, 3H), 1.62 (s, 3H), 1.43 (s, 3H), 1.38 (s, 3H), 1.29-1.23 (m, 1H), 1.12-1.08 (m, 1H), 0.73 (t, J = 4.4 Hz, 1H), 0.65 (dd, J = 4.4, 8.0 Hz, 1H), 0.48 (t, J = 4.4 Hz, 1H), 0.35 (dd, J = 4.8, 8.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 156.7, 156.6, 143.4, 143.3, 137.6, 137.5, 136.3, 134.8, 134.0, 133.5, 130.4, 130.1, 129.6, 129.5, 129.0, 128.5, 128.3, 128.2, 127.9, 127.7, 127.5, 127.3, 53.7, 53.5, 52.8, 52.4, 49.7, 49.6, 29.9, 29.8, 24.7, 23.0, 21.5, 21.5, 20.4, 20.2, 20.1, 20.1, 16.7, 14.9. IR: ν 2360, 2342, 1700, 1347, 1294, 1165, 1131, 1093, 913 cm-1. HRMS calcd for C25H30N2NaO4S+ [M+Na]+: 477.1824, found: 477.1816.
4.3.2. Compounds 5b and 5b′
Compound 5b and 5b′ were obtained in 75% yield as a colorless oil from 4b, 5b and 5b′ in a 1:1 ratio determined by crude 1H NMR. Rf = 0.50, hexans/EtOAc = 2:1, 1H NMR (400 MHz, CDCl3) δ 7.66-7.63 (m, 4H), 7.32-7.29 (m, 4H), 3.65-3.62 (m, 2H), 3.59 (s, 3H), 3.53-3.50 (m, 2H), 3.25 (s, 3H), 3.21-3.16 (m, 1H), 3.06-3.02 (m, 3H), 2.83 (s, 3H), 2.77 (s, 3H), 2.42 (s, 3H), 2.41 (s, 3H), 1.69 (s, 3H), 1.68 (s, 3H), 1.54 (s, 3H), 1.53 (s, 3H), 1.51-1.46 (m, 1H), 1.43-1.39 (m, 1H), 0.92-0.90 (m, 1H), 0.82-0.79 (m, 1H), 0.74-0.68 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 156.2. 143.5, 134.9, 134.2, 133.1, 132.9, 131.8, 131.4, 129.6, 129.5, 127.6, 127.4, 53.9, 53.4, 52.6, 52.2, 50.0, 49.7, 36.5, 36.4, 30.1, 30.0, 23.5, 23.1, 21.5, 21.4, 19.9, 19.8, 19.7, 15.9, 15.6. IR: ν 1773, 1418, 1346, 1275, 1260, 1161, 1113, 913 cm-1. HRMS calcd for C19H27N2O4S + [M+H]+: 379.1692, found: 379.1693.
4.3.3. Compounds 5c
Compound 5c was obtained in 72% yield as a white solid from 4c. Rf = 0.50, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 4.33 (d, J = 8.4 Hz, 1H), 4.31 (d, J = 7.6 Hz, 1H), 3.63-3.46 (m, 4H), 3.23 (dd, J = 3.6, 9.6 Hz, 1H), 3.04 (d, J = 9.2 Hz, 1H), 2.42 (s, 3H), 1.68-1.58 (m, 7H), 0.86-0.80 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 156.5, 143.6, 137.5, 133.2, 129.6, 127.4, 125.6, 61.9, 53.2, 49.7, 45.9, 28.5, 24.3, 21.5, 20.2, 20.0, 14.9. IR: ν 2918, 2361, 1748, 1410, 1344, 1165, 1107, 1033, 913 cm-1. HRMS calcd for C19H24N2NaO4S+ [M+Na]+: 399.1354, found: 399.1349. Mp (°C): 103-105.
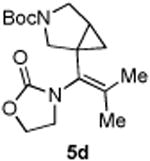
4.3.4. Compounds 5d
Compound 5d was obtained in 45% yield as a colorless oil from 4d. Rf = 0.30, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 4.41-4.37 (m, 2H), 3.77-3.74 (m, 2H), 3.65-3.55 (m, 2H), 3.49-3.46 (m, 1H), 3.27-3.24 (m, 1H), 1.86 (d, J = 4.0 Hz, 3H), 1.69 (s, 3H), 1.69-1.67 (m, 1H), 1.42 (s, 9H), 0.99 (dd, J = 4.4, 8.0 Hz, 1H), 0.66-0.64 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 156.6, 154.8, 136.8, 126.3, 79.6, 61.9, 52.1, 48.2, 47.9, 46.1, 45.9, 28.4, 24.0, 20.2, 16.5. IR: ν 2360, 2342, 1750, 1690, 1406, 1259, 1171, 913 cm-1. HRMS calcd for C17H26N2NaO4+ [M+Na]+: 345.1790, found: 345.1786.
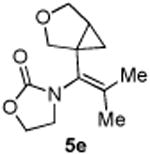
4.3.5. Compounds 5e
Compound 5e was obtained in 65% yield as a colorless oil. Rf = 0.20, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 4.42-4.34 (m, 2H), 3.87-3.84 (m, 3H), 3.80-3.77 (m, 1H), 3.64-3.58 (m, 2H), 1.87 (s, 3H), 1.82-1.76 (m, 1H), 1.70 (s, 3H), 0.89 (dd, J = 4.4, 8.0 Hz, 1H), 0.82 (t, J = 4.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 156.6, 126.4, 125.2, 72.4, 69.6, 61.9, 46.1, 29.7, 25.1, 20.2, 14.0. IR: ν 2919, 1746, 1410, 1297, 1275, 1105, 1039, 913 cm-1. HRMS calcd for C12H17NNaO3+ [M+Na]+: 246.1106, found: 246.1101.
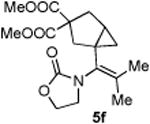
4.3.6. Compounds 5f
Compound 5f was obtained in 72% yield as a colorless oil from 4f. Rf = 0.30, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 4.40 (t, J = 8.0 Hz, 2H), 3.74-3.68 (m, 2H), 3.72 (s, 3H), 3.69 (s, 3H), 2.83 (d, J = 13.6 Hz, 1H). 2.64-2.58 (m, 2H), 2.41 (dd, J = 1.6, 13.6 Hz, 1H), 1.84 (s, 3H), 1.66 (s, 3H), 1.60-1.55 (m, 1H), 0.77-0.73 (m, 1H), 0.44-0.42 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 173.0, 172.1, 156.5, 128.2, 61.9, 59.4, 53.0, 52.9, 45.9, 41.3, 36.1, 29.8, 20.2, 20.0. IR: ν 2953, 1734, 1436, 1256, 1203, 1098, 913 cm-1. HRMS calcd for C17H23NNaO6+ [M+Na]+: 360.1423, found: 360.1420.
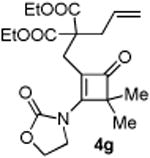
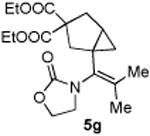
4.3.7. Compounds 5g
Compound 5g was obtained in 76% yield as a colorless oil from 4g. Rf = 0.45, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 4.37 (t, J = 7.6 Hz, 2H), 4.21-4.10 (m, 4H), 3.79-3.82 (m, 1H), 3.72-3.65 (m, 1H), 2.82 (d, J = 14.0 Hz, 1H), 2.64-2.56 (m, 2H), 2.38 (dd, J = 1.2, 13.6 Hz, 1H), 1.84 (s, 3H), 1.66 (s, 3H), 1.63-1.55 (m, 1H), 1.26 (t, J = 7.2 Hz, 3H), 1.20 (t, J = 7.2 Hz, 3H), 0.75-0.72 (m, 1H), 0.47-0.45 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 172.6, 171.7, 156.5, 128.3, 61.9, 61.8, 61.6, 59.6, 45.8, 41.2, 36.0, 29.7, 20.2, 20.0, 13.9. IR: ν 2983, 1729, 1444, 1410, 1251, 1197, 1096, 1069, 913 cm-1. HRMS calcd for C19H27NNaO6+ [M+Na]+: 388.1736, found: 388.1732.
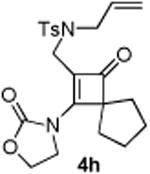
4.3.8. Compounds 5h and 5h′
Compound 5h was obtained in 42% yield as a colorless oil from 4h. Rf = 0.30, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 6.8 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 4.33 (t, J = 8.0 Hz, 2H), 3.66-3.61 (m, 2H), 3.56-3.52 (m, 2H), 3.21 (dd, J = 3.6, 9.6 Hz, 1H), 3.06 (d, J = 9.2 Hz, 1H), 2.43 (s, 3H), 2.23-2.15 (m, 3H), 2.04-1.99 (m, 1H), 1.65-1.59 (m, 5H), 0.87 (dd, J = 4.4, 8.0 Hz, 1H), 0.80 (t, J = 4.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 155.9, 148.2, 143.5, 133.3, 129.6, 127.4, 122.3, 62.0, 52.5, 49.7, 45.4, 30.9, 30.5, 29.2, 26.4, 25.7, 23.9, 21.5, 14.6. IR: ν 2954, 2869, 1747, 1410, 1343, 1164, 1095, 1036 cm-1. HRMS calcd for C21H26N2NaO4S+ [M+Na]+: 425.1511, found: 425.1508. Compound 5h′ was obtained in 30% yield as colorless oil: Rf = 0.45, hexans/EtOAc = 1:1, 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 6.4 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 5.76-5.75 (m, 1H), 4.42 (t, J = 8.0 Hz, 2H), 3.96 (d, J = 15.2 Hz, 1H), 3.83 (d, J = 15.2 Hz, 2H), 3.66-3.57 (m, 2H), 3.32 (dd, J = 6.4, 9.2 Hz, 1H), 3.14-3.11 (m, 1H), 3.04 (dd, J = 3.2, 9.2 Hz, 1H), 2.44-2.39 (m, 7H), 1.95-1.89 (m, 2H), 1.08 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 155.5, 143.8, 140.8, 137.0, 132.6, 131.9, 129.7, 127.8, 124.9. 62.1, 55.2, 50.5, 44.8, 35.3, 33.7, 32.3, 23.5, 21.5, 19.6. IR: ν 2923, 2342, 1750, 1409, 1343, 1161, 1092, 913 cm-1. HRMS calcd for C21H26N2NaO4S+ [M+Na]+: 425.1511, found: 425.1505.
4.3.9. Compounds 5i
Compound 5i was obtained in 45% yield as a colorless oil from 4i. Rf = 0.45, hexans/EtOAc = 1:1, H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 7.6 Hz, 2H), 7.33 (d, J = 7.6 Hz, 2H), 5.54 (t, J = 6.8 Hz, 1H), 4.39-4.30 (m, 2H), 3.92 (d, J = 14.4 Hz, 1H), 3.77 (dd, J = 1.6, 14.4 Hz, 1H), 3.54-3.49 (m, 2H), 3.44-3.40 (m, 1H), 3.09-3.03 (m, 1H), 2.79 (dd, J = 5.6, 9.6 Hz, 1H), 2.43 (s, 3H), 2.23-2.16 (m, 1H), 1.88-1.79 (m, 1H), 1.69 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H), 0.92 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 155.0, 143.7, 137.6, 135.6, 132.2, 129.7, 129.5, 128.1, 127.8, 62.0, 55.2, 50.6, 44.5, 35.4, 21.5, 21.4, 18.4, 13.4, 12.5. IR: ν 2968, 2924, 1750, 1407, 1343, 1161, 1092, 913 cm-1. HRMS calcd for C21H29N2O4S+ [M+H]+: 405.1848, found: 405.1839.
4.3.10. Compounds 6j
Compound 6j was obtained in 84% yield as a colorless oil from 4j. Rf = 0.2, hexans/EtOAc = 1:1, H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 4.44-4.39 (m, 1H), 4.35-4.28 (m, 1H), 3.87-3.80 (m, 2H), 3.68 (d, J = 9.6 Hz, 1H), 3.52-3.46 (m, 1H), 3.19 (d, J = 18.4 Hz, 1H), 3.05 (d, J = 6.4 Hz, 1H), 2.98-2.92 (m, 2H), 2.44 (s, 3H), 1.67 (s, 3H), 1.48 (s, 3H), 1.25 (s, 3H). C NMR (100 MHz, CDCl3) δ 208.3, 157.4, 144.2, 139.1, 131.4, 129.7, 127.9, 123.1, 76.1, 62.5, 59.7, 57.6, 57.2, 46.5, 44.1, 21.5, 21.2, 20.2, 19.4. IR: ν 1780, 1742, 1419, 1347, 1252, 1167, 1091, 1037, 913 cm -1. HRMS calcd for C21H26N2NaO5S+ [M+Na]+: 441.1460, found: 441.1456.
Supplementary Material
Acknowledgments
We thank UT Austin and CPRIT for a startup fund, NIGMS (R01GM109054-01) and the Welch Foundation (F 1781) for research grants. GD is a Searle Scholar. We are also grateful to Johnson Matthey for a generous donation of Rh salts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.For reviews of cyclobutenones, see: Bellus D, Ernst B. Angew Chem Int Ed. 1988;27:797–827.Seiser T, Saget T, Tran DN, Cramer N. Angew Chem Int Ed. 2011;50:7740–7752. doi: 10.1002/anie.201101053.
- 2.For a recent review, see: Xu T, Dermenci A, Dong G. Top Curr Chem. 2014;346:233–258. doi: 10.1007/128_2014_545. For selected examples, see: Huffman MA, Liebeskind LS. Organometallics. 1990;9:2194–2196.Huffman MA, Liebeskind LS. Organometallics. 1992;11:255–256.Huffman MA, Liebeskind LS. J Am Chem Soc. 1990;112:8617–8618.Huffman MA, Liebeskind LS. J Am Chem Soc. 1991;113:2771–2772.Huffman MA, Liebeskind LS. J Am Chem Soc. 1993;115:4895–4896.Kondo T, Tagushi Y, Kaneko Y, Niimi M, Mitsudo T. Angew Chem Int Ed. 2004;43:5369–5372. doi: 10.1002/anie.200461002.Kondo T, Miimi M, Nomura M, Wada K, Mitsudo T. Tetrahedron Lett. 2007;48:2837–2839.Auvinet AL, Harrity JPA. Angew Chem Int Ed. 2011;50:2769–2772. doi: 10.1002/anie.201007598.
- 3.(a) Xu T, Dong G. Angew Chem Int Ed. 2012;51:7567–7571. doi: 10.1002/anie.201202771. [DOI] [PubMed] [Google Scholar]; (b) Xu T, Ko HM, Savage NA, Dong G. J Am Chem Soc. 2012;134:20005–20008. doi: 10.1021/ja309978c. [DOI] [PubMed] [Google Scholar]; (c) Chen PH, Xu T, Dong G. Angew Chem Int Ed. 2014;53:1674–1678. doi: 10.1002/anie.201310100. [DOI] [PubMed] [Google Scholar]; (d) Xu T, Savage NA, Dong G. Angew Chem Int Ed. 2014;53:1891–1895. doi: 10.1002/anie.201310149. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xu T, Dong G. Angew Chem Int Ed. 2014;53:10733–10736. doi: 10.1002/anie.201404802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Danheiser RL, Sard H. Tetrahedron Lett. 1983;24:23. [Google Scholar]; (b) Danheiser RL, Savariar S, Cha DD. Organic Syntheses. VIII. Wiley; New York: 1993. pp. 82–86. Collect. [Google Scholar]; (c) Wasserman HH, Piper JU, Dehmlow EV. J Org Chem. 1973;38:1451. [Google Scholar]; (d) Danheiser RL, Gee SK. J Org Chem. 1984;49:1672. [Google Scholar]; (e) Danheiser RL, Nishida A, Savariar S, Trova MP. Tetrahedron Lett. 1988;29:4917. [Google Scholar]; (f) Kowalski CJ, Lal GS. J Am Chem Soc. 1988;110:3693. [Google Scholar]
- 5.Kohnen AL, Mak XY, Lam TY, Dunetz JR, Danheiser RL. Tetrahedron. 2006;62:3815–3822. doi: 10.1016/j.tet.2005.11.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For recent reviews on the chemistry of ynamides, see: DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem Rev. 2010;110:5064–5106. doi: 10.1021/cr100003s.Evano G, Coste A, Jouvin K. Angew Chem, Int Ed. 2010;49:2840–2859. doi: 10.1002/anie.200905817.
- 7.(a) Danheiser RL, editor. Science of Synthesis (Houben–Weyl) Vol. 23. Thieme; Stuttgart, Germany: 2006. [Google Scholar]; (b) Tidwell TT. Ketenes. 2nd. Wiley Interscience; Hoboken, NJ: 2006. [Google Scholar]; (c) Tidwell TT. Eur J Org Chem. 2006:563–576. [Google Scholar]; (d) Fu N, Tidwell TT. Tetrahedron. 2008;64:10465–10496. [Google Scholar]; (e) Arrieta A, Lecea B, Cossio FP. Top Heterocycl Chem. 2010;22:313–347. [Google Scholar]
- 8.Cyclobutenone was synthesized from known compound in three steps, detail please see support information and references below: Lam TY, Wang YP, Danheiser RL. J Org Chem. 2013;78:9396–9414. doi: 10.1021/jo401635c.Song H, Liu Y, Wang Q. Org Lett. 2013;15:3274–3277. doi: 10.1021/ol401303f.
- 9.The corresponding di-hydrogen and mono-methyl substituted cyclobutenones are much more challenging to prepare compared to the gem-dialkyl substituted ones. For details, see refs 1, 4 and 5.
-
10.X-ray structures of 6a and 6a′ (For details, see SI). CCDC 1045429 and 1045428 contain the supplementary crystallographic data for compounds 6a and 6a′ respectively. These data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
- 11.For a related transformation, see: Xu SL, Moore HW. J Org Chem. 1989;54:6018–6021.
- 12.For the substrate preparation, see SI and references below: Lam TY, Wang YP, Danheiser RL. J Org Chem. 2013;78:9396–9414. doi: 10.1021/jo401635c.Song H, Liu Y, Wang Q. Org Lett. 2013;15:3274–3277. doi: 10.1021/ol401303f.Kohnen AL, Mak XY, Lam TY, Dunetz JR, Danheise RL. Tetrahedron. 2006;54:3815–3822. doi: 10.1016/j.tet.2005.11.088.Miura K, Saito H, Fujisawa N, Hosomi A. J Org Chem. 2000;65:8119–8122. doi: 10.1021/jo005567c.Sylvester KT, Chirik PJ. J Am Chem Soc. 2009;131:8772–8774. doi: 10.1021/ja902478p.Hamada T, Ye X, Stahl SS. J Am Chem Soc. 2008;130:833–835. doi: 10.1021/ja077406x.
- 13.Almost the same yield of cyclobutanone 6j can be obtained in the absence of the Rh catalyst, suggesting that 6j comes from a background reaction.
- 14.For seminal examples, see: Murakami M, Amii H, Ito Y. Nature. 1994;370:540–541.Murakami M, Amii H, Shigeto K, Ito Y. J Am Chem Soc. 1996;118:8285–8290.
- 15.Another possible mechanism was suggested by one reviewer, which can be found in the supporting information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.