

Abstract
The molecular basis for the toxicity of mutant huntingtin containing long polyglutamine stretches has been unclear. We now learn that mutant huntingtin binds to a complex that imports constituent proteins across the mitochondrial inner membrane, halting bioenergetics in synaptic mitochondria and predisposing to neuronal dysfunction and death.
Mitochondria are the wellspring of life, increasingly heralded as necessary for the prevention of degeneration of vulnerable populations of neurons Mitochondrial metabolism is crucial to neuronal function1, and compromised mitochondria have been found in neurodegenerating cells2, with mitochondrial dysfunction implicated in every major neurodegenerative disorder, including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis and Huntington's disease 3 Whether mitochondrial dysfunction is the sole cause of the disorder or secondary to other deficits has continued to be debated, but understanding the implications of this question is important to determining the underpinnings of neurodegeneration. In this issue of Nature Neuroscience, Yano et al. (Yano et al., 2014) describe a breakthough in understanding the link between mitochondrial dysfunction and neuronal death in Huntington's disease. They find that mutant huntingtin protein causes a specific blockade of the mechanism that provides mitochondria with almost all of the proteins they need to run metabolism on a daily basis.
A classic hallmark of neurodegenerative disease is abnormal accumulation of distorted proteins. In Huntington's disease, patients may dominantly inherit an aberrant form of the gene huntingtin (HTT) that contains an expanded sequence of CAG repeats resulting in an extra-long string of glutamines in the huntingtin protein4 The function of normal huntingtin is unknown, but it is clear that the abnormal protein aggregates inside degenerating striatal neurons close to mitochondria5. Although human tissue and mouse models have demonstrated that aggregated huntingtin proteins do not directly cause death of striatal neurons6, death may be caused by interference of mutant huntingtin with mitochondrial calcium handling 7 or metabolic functions 8. Indeed, in addition to the severe motor and cognitive deficits found in Huntingto's disease, patients also display weight loss leading to early death9.
How could mutant huntingtin protein cause mitochondrial dysfunction? Recent work on brain samples from patients with Huntington's disease and in mouse models has revealed reduced enzymatic activity of members of the inner mitochondrial membrane electron transport chain, the main enzyme system that produces the cell's energy supply8. This finding is correlated with reduced levels of ATP in the mutant neurons and reduced uptake of substrates such as glucose by mitochondria and by the cells affected by the mutant protein. Mutant mitochondria also display abnormal morphology and calcium handling, decreased movement and a tendency to fragment 10. All of these defects are consistent with the notion that mitochondria are starved for building blocks necessary for ongoing maintenance and repair. These repair processes include replacement of damaged proteins.
How do mitochondria normally replace proteins? Mitochondria were, in ancient times, separate prokaryotic cells that incorporated themselves into eukaryotic cells and became symbiotes. They gave up most of their DNA to the more efficient host cell nuclear genome. Mitochondria now contain DNA only for 37 genes, mostly encoding subunit members of the electron transport chain, mitochondrial ribosomal proteins and tRNA11. This means that to carry out their vital tasks, mitochondria continually import over 1,200 proteins from the cytosol12. After a mitochondrial preprotein is made in the cytosol, it must translocate through two mitochondrial membranes if it is to reach the mitochondrial matrix. Each preprotein has a specific signal or targeting sequence that allows it to bind to a group of mitochondrial proteins known as translocators. There is a general translocator in the outer mitochondrial membrane (TOM complex) and at least two types of translocator in the inner mitochondrial membrane (TIM complexes) (Figure 1). These translocators work together to take up new proteins from the cytosol and sort them so they end up at their appropriate locations in the mitochondrion13.
Figure 1.
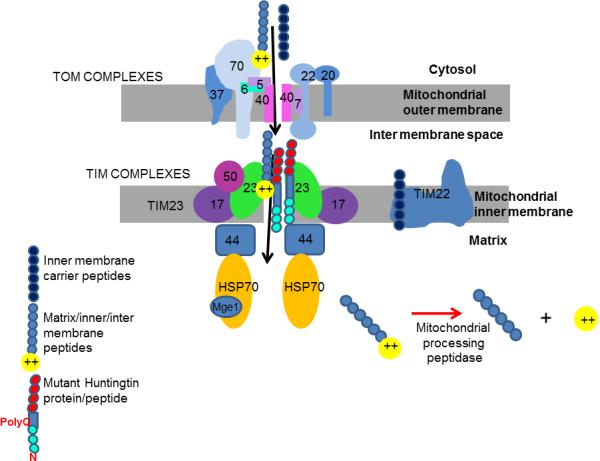
Mutant huntingtin inhibits mitochondrial protein import. Shown are cytosolic compartment, mitochondrial inner and outer membranes, intermembrane space and mitochondrial matrix. Mitochondrial preproteins translocate through the TOM complex in the outer membrane. Matrix and many inner membrane and intermembrane proteins are recognized by TIM23 subunits and translocate through the TIM23 complex. Mutant huntingtin full-length protein or mutant huntingtin N-terminal peptide, both containing long polyglutamine (polyQ) repeat sequences, bind the TIM23 complex, inhibiting protein import.
Yano et al. confirmed that mutant huntingtin protein colocalizes with mitochondria. Their surprising new finding, however, is that mutant huntingtin specifically binds to one of the inner membrane translocator complexes (Figure 1). The investigators applied recombinant tagged N-terminal fragments of mutant huntingtin or normal huntingtin to isolated brain mitochondria, then immunoprecipitated the fragments and used mass spectroscopy to find their protein binding partners. Several subunits of the inner membrane translocator complex called TIM23 were bound more tightly by the mutant protein than the wild type protein and other import complexes were not bound, suggesting specificity in the relationship between mutant huntingtin and TIM23. TIM23 is responsible for importing proteins destined for the matrix inner membrane or intermembrane space, including many members of complexes that carry out intermediary metabolism and respiration.
If mutant huntingtin is stuck to the TIM23 complex, it might disrupt protein import into the matrix, inner membrane and intermembrane space, severely hampering metabolism in cells such as neurons that arehighly energy dependent. Using an established assay, the investigators found that mutant huntingtin N-terminal peptides inhibited protein import into mitochondria isolated from mouse brain, in a manner dependent on concentration and length of the huntingtin polyglutamine sequence.
The authors then tested protein import in vivo in a mouse model of Huntington's disease. The R6/2 mouse expressesan N-terminal fragment of a human HTT gene and has become an important model because its disease progression mimics the motor abnormalities, metabolic disarray and shortened life span found in human patients9. Perhaps suspecting the importance of synaptic dysfunction in neurodegeneration, the team isolated highly purified synaptosomal mitochondria (mitochondria residing near presynaptic terminals) in addition to nonsynaptosomal mitochondria from the mutant mice. Synaptosomal mitochondria are a more select group. They are not only taken only from cells with synapses—that is, neurons—but also may also function differently from mitochondria isolated from the soma because these specialized organelles buffer calcium and provide energy for synaptic activity. Because there are many demands placed on synaptic mitochondria, the authors guessed that their function might become more easily compromised by the mutant protein. Notably, the authors found that the synaptic mitochondria exhibited strikingly compromised protein import even at presymptomatic and early disease stages (when the mice were 3 weeks old), while nonsynaptic mitochondria were protected from a decline in protein import even as late as 10–11 weeks of life. Mitochondria from liver also showed no deficit in protein import until much later in the disease, suggesting that synaptic mitochondria are particularly sensitive to these mutations. Even mitochondria isolated from embryonic neurons made from the R6/2 mouse before birth displayed the protein import defect, suggesting that this deficit is one of the earliest manifestations of disease in mutant mouse synapses.
If protein import into mitochondria is needed for normal mitochondrial respiration, then the delay in protein import should precede the onset of any respiratory chain dysfunction. The authors found that there were no deficits in respiratory function in mitochondria isolated from presymptomatic animals, even though these mitochondria showed signs of severe dysfunction in protein import, whereas respiratory function became quite compromised later in the disease. Of particular note although the deficits in respiratory chain function had been attributed previously to high levels of reactive oxygen species (ROS) causing protein and DNA damage to vulnerable mitochondria, and this may still be true, the new findings show that sublethal doses of ROS, although further damaging protein import in mitochondria from mutant huntingtin– expressing cells, do not damage the protein import pathway in normal mitochondria, ruling out ROS as the primary cause of the protein import deficit.
Is the change in protein import itself sufficient to produce neuronal dysfunction and death? To address this, the authors bypassed the mutant huntingtin protein to study the effect of depletion of the general import machinery directly. They first depleted the outer membrane translocator protein TOM40. They noted that cells died by apoptotic and non-apoptotic mechanisms soon after halting protein import. Next they depleted the actual target of mutant huntingtin, the inner membrane translocator component TIM23. In live neurons, they were able to watch the loss of fluorescence of a voltage-dependent mitochondrial membrane potential indicator and the accumulation of a fluorescent cell death marker as the levels of TIM23 declined. Neuronal death occurred rapidly after loss of mitochondrial membrane potential, within 1–2 days after the onset of TIM23 depletion. The authors also found that increasing TIM23 in mutant huntingtin–expressing neurons produced an improvement in metabolic health and in neuronal survival.
These striking findings—that reduced mitochondrial protein import caused by mutant huntingtin leads to metabolic dysfunction and death of neurons—raise many questions. Why do some neurons die and others survive if mutant huntingtin is expressed in all neurons? Is the amount of mutant huntingtin bound to the import machinery different in different cell populations, and if so, how does this come about? And what about the intriguing finding that synaptic mitochondrial protein import is more affected by the mutant huntingtin than that of somatic mitochondria? Is this discrepancy caused by a different level of activity of these mitochondria, increasing their vulnerability through an increase in reactive oxygen species? In turn, do these defective synaptic mitochondria cause some of the motor and cognitive symptoms of patients or animals found preceding cell death? If the protein import deficit causes neuronal death so rapidly, why do most patients develop neuronal loss and clinical disease late in life, and why is there often no neuronal loss in animal models? Even though there are still so many unanswered questions, the findings by Yano et al. that mutant huntingtin causes loss of mitochondrial protein import resulting in a decline in metabolism and respiration certainly provides an influx of ideas and a breath of fresh air to the neurodegeneration story.
References
- 1.Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 2.Petrozzi L, Ricci G, Giglioli NJ, Siciliano G, Mancuso M. Mitochondria and neurodegeneration. Bioscience reports. 2007;27:87–104. doi: 10.1007/s10540-007-9038-z. [DOI] [PubMed] [Google Scholar]
- 3.Chaturvedi RK, Beal MF. Mitochondrial approaches for neuroprotection. Ann N Y Acad Sci. 2008;1147:395–412. doi: 10.1196/annals.1427.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar A, Vaish M, Ratan RR. Transcriptional dysregulation in Huntington's disease: a failure of adaptive transcriptional homeostasis. Drug discovery today. 2014 doi: 10.1016/j.drudis.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oliveira JM. Nature and cause of mitochondrial dysfunction in Huntington's disease: focusing on huntingtin and the striatum. J Neurochem. 2010;114:1–12. doi: 10.1111/j.1471-4159.2010.06741.x. [DOI] [PubMed] [Google Scholar]
- 6.Kuemmerle S, et al. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- 7.Brustovetsky N, et al. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J Neurosci. 2003;23:4858–4867. doi: 10.1523/JNEUROSCI.23-12-04858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grunewald T, Beal MF. Bioenergetics in Huntington's disease. Ann N Y Acad Sci. 1999;893:203–213. doi: 10.1111/j.1749-6632.1999.tb07827.x. [DOI] [PubMed] [Google Scholar]
- 9.Crook ZR, Housman D. Huntington's disease: can mice lead the way to treatment? Neuron. 2011;69:423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy PH, Shirendeb UP. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington's disease. Biochim Biophys Acta. 2012;1822:101–110. doi: 10.1016/j.bbadis.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 12.Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annual review of genomics and human genetics. 2010;11:25–44. doi: 10.1146/annurev-genom-082509-141720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rehling P, Wiedemann N, Pfanner N, Truscott KN. The mitochondrial import machinery for preproteins. Critical reviews in biochemistry and molecular biology. 2001;36:291–336. doi: 10.1080/20014091074200. [DOI] [PubMed] [Google Scholar]