

Abstract
The proline-rich Akt substrate of 40 kDa (PRAS40) protein is not only a substrate of the protein kinase Akt but also a component of the mTOR complex 1 (mTORC1), thus it links the Akt and the mTOR pathways. We investigated the potential protective role of PRAS40 in cerebral ischemia and its underlying mechanisms by using rats with lentiviral over-expression of PRAS40 and mice with PRAS40 gene knockout (PRAS40 KO). Our results show that gene transfer of PRAS40 reduced infarction size in rats by promoting phosphorylation of Akt, FKHR (FOXO1), PRAS40, and mTOR. In contrast, PRAS40 KO increased infarction size. Although the PRAS40 KO under normal condition did not alter baseline levels of phosphorylated proteins in the Akt and mTOR pathways, PRAS40 KO that underwent stroke exhibited reduced protein levels of p-S6K and p-S6 in the mTOR pathway but not p-Akt, or p-PTEN in the Akt pathway. Furthermore, co-immunoprecipitation suggests that there were less interactive effects between Akt and mTOR in the PRAS40 KO. In conclusion, PRAS40 appears to reduce brain injury by converting cell signaling from Akt to mTOR.
Keywords: Focal cerebral ischemia, Akt, Stroke, mTOR, PRAS40, PTEN
Introduction
The proline-rich Akt substrate of 40-kDa (PRAS40) protein was first identified as a 14-3-3 binding protein and substrate of Akt in the Roth laboratory 10 years ago (Kovacina et al., 2003). Later it was found that PRAS40 is also an important component and a substrate of the mammalian target of rapamycin complex 1 (mTORC1) (Wang et al., 2007). Thus, PRAS40 is an interactive linker between the Akt and mTOR pathways.
Both the Akt (Zhao et al., 2006) and mTOR pathways (Guertin and Sabatini, 2005, 2007) play protective roles against brain injury induced by stroke. Akt has long been known to contribute to neuronal survival after stroke (Gao et al., 2008; Noshita et al., 2003; Ouyang et al., 1999; Zhao et al., 2005). Recently, the involvement of the mTOR pathway has also been reported, as several neuroprotectants have been shown to inhibit infarction by upregulating mTOR activity (Koh, 2008; Koh et al., 2008; Wu et al., 2012). In alignment with the roles of Akt and mTOR, PRAS40 also appears to attenuate cerebral ischemic injury after stroke (Saito et al., 2004, 2006). Previous studies from the Chan laboratory have found that stroke reduces phosphorylated PRAS40 (p-PRAS40), and decreases binding of p-PRAS40 to phosphorylated Akt (p-Akt) and the 14-3-3 protein (Saito et al., 2004, 2006), suggesting that PRAS40 is involved in brain injury induced by stroke. More importantly, liposome-mediated p-PRAS40 cDNA transfection inhibits apoptotic neuronal cell death, suggesting that PRAS40 overexpression is neuroprotective (Saito et al., 2004).
Most recently, we reported that lentiviral vector gene transfer of the Akt isoforms Akt1 and Akt3 attenuated brain injury by promoting both p-PRAS40 and p-mTOR protein levels, and mTOR inhibition by rapamycin abolished the protective effects Akt1 and Akt3 gene transfer (Xie et al., 2013a). Our results suggest that a close relationship exists between Akt and mTOR in their protection against stroke, and that PRAS40 may serve a pivotal role (Xie et al., 2013a). Nevertheless, many questions remain about how PRAS40 functions in cerebral ischemia. First, the underlying protective mechanisms of PRAS40 against stroke remain elusive, such as how PRAS40 affects Akt and mTOR activity and how they interact. Second, previously reported protective effects of PRAS40 against stroke were demonstrated using liposome-mediated PRAS40 gene transfer, which is considered to have insufficient gene transfer efficacy for neurons in vivo, and this protective effect has not been confirmed with more efficient gene transfer techniques, such as lentiviral vector systems. Third, previous studies have suggested that overexpression of PRAS40 inhibits, while knockdown of PRAS40 promotes, mTOR activity (Vander Haar et al., 2007; Wang et al., 2008). As mTOR is neuroprotective, these previous studies suggest that overexpression of PRAS40 might promote neuronal death, which contradicts with the finding that PRAS40 overexpression attenuates infarction after stroke. Whether PRAS40 is truly neuroprotective or not requires further investigation. Fourth, previous studies on the cellular functions of PRAS40 were mostly performed in cell lines; the function of PRAS40 has not been studied in rodent gene knockout animal models.
In this study we constructed the lentiviral vector to express the PRAS40 gene and investigated how PRAS40 gene expression affects infarct sizes and protein expression in the Akt and mTOR pathways after stroke. We also studied the pathological outcomes of stroke in PRAS40 gene knockout (PRAS40 KO) mice and the effects of PRAS40 KO on Akt and mTOR activity, as well as their interactive effects.
Materials and methods
Animal experiments were conducted according to the protocols approved by the Stanford Institutional Animal Care and Use Committee and the NIH Guidelines for Care and Use of Laboratory Animals. Animals were housed under a 12:12 hour light:dark cycle with food and water available ad libitum.
Construction of lentiviral vectors
We constructed lentiviral vectors containing genes of constitutively-active PRAS40. Target genes were cloned from PRAS40 (pCMV5) plasmids kindly provided by Dr. Richard Roth, Department of Chemical and Systems Biology, Stanford University. Genes were cloned into the lentiviral backbone plasmid, pHR’tripCMV–IRES–eGFP, which contains a CMV promoter and an IRES sequence between its multiple cloning sites (MCS) and eGFP. The IRES sequence enables independent expression of both the target gene and eGFP simultaneously. SacII/PstI was the restriction enzyme used for cloning. The control vector was the lentiviral plasmid backbone with only eGFP inserted.
Lentiviral vector generation and titration
We used a 3 plasmid system for Lentivirus packaging: the lentiviral transfer vector (pHR′tripCMV–IRES–eGFP) that contains the coding region of various targeted genes as described above; the packaging plasmid (p-delta) that provides all vector proteins driven by the trip CMV promoter, except the envelope protein; and the envelope-encoding plasmid (p-VSVG) that encodes the heterologous vesicular stomatitis virus envelope protein (VSVG) (Hu et al., 2011). Briefly, 293T cells were grown in Dulbecco’s modified Eagle medium (DMEM, Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Gibco). A mixture of 45 μg of transfer vectors, 30 μg of packaging plasmids and 15 μg of envelope-encoding plasmids was transiently transfected into 3 separate T175 flasks containing 1.5 × 107 HEK-293T cells using the calcium phosphate precipitation (CPP) method. Supernatants were collected 72 h post-transfection and viral particles were concentrated by ultracentrifugation. Viruses were resuspended in phosphate-buffered saline (PBS) and kept at −80 °C until use. The virus particles were titered with the TCID50 method as described previously (Apolonia et al., 2007; Breckpot et al., 2003). Virus titers ranged from 1 × 108–5 × 108 TU/ml and were diluted in PBS to the final concentration of 1 × 108 TU/ml before gene transfer was conducted.
Creation of PRAS40 KO mice and genotyping
PRAS40 KO mice were kindly provided by Dr. Richard Roth, Department of Chemical and Systems Biology, Stanford University. C57BL/6 PRAS40 KO mice were generated using standard homologous recombination methods. loxP sequences were inserted between exons 1 and 2 and after exon 5. A phosphoglycerine kinase (PGK) Neo cassette flanked by FLP recombinase target sequences was used to confer resistance to C57BL/6 ES cells that had successfully integrated the targeting vector. This procedure produces ES cells with exons 2 through 5 flanked by loxP sites. ES cells were microinjected and the chimeric mice were bred to generate heterozygous F1 mice. These floxed mice were crossed with Cre-deleter C57BL/6 mice, leading to the removal of the entire coding region of the PRAS40 gene. Founder animals were backcrossed with C57BL/6 mice for more than 12 generations.
Presence and copy numbers of the transgene in the offspring were identified by polymerase chain reaction (PCR) analysis. In brief, genomic DNA was extracted from the tail of mice using a DNeasy Blood and Tissue Kit (Qiagen, Germantown, MD, USA), and genomic PCR was performed with Taq DNA Polymerase High Fidelity (Invitrogen, Carlsbad, CA, USA) under the following conditions: 94 °C for 60 s; 30 cycles at 94 °C for 60 s, 55 °C for 45 s, and 72 °C for 45 s; and 72 °C for 6 min. One primer pair (forward PCR primer: GGGGCGCTCTGAGATTAAAG, reverse PCR primer: GGTGACAGTCCTCTAGCCC) amplifies a fragment of 225 bp in mice homozygous or heterozygous for endogenous gene (no band will be generated by this oligo pair in a cre-recombinant homozygous mice). Another set of primers (forward PCR primer: GTGGTGTGCATGTGTGACTTG, reverse PCR primer: GGTGACAGTCCTCTAGCCC) generates a product of 300 bp in a cre recombined homozygous and heterozygous mice but not in non-recombined mice.
In vitro oxygen glucose deprivation (OGD) model, gene transfer and cell viability assay
Primary neuronal cultures were prepared using timed-pregnant Sprague–Dawley rats (E18, Charles River Laboratories International, Wilmington, MA). Briefly, rats were anesthetized with isoflurane and the E18 embryos were removed. The cortical region of the fetal brains were dissected in warm media and pooled together. The cortices were triturated and incubated in papain for 20 min at 37 °C, then centrifuged at 1500 rpm for 5 min at room temperature (RT). Cells were resuspended in MEM (minimal essential medium) (Gibco, Grand Island, NY, USA) containing 10% fetal horse serum (Hyclone, Logan, UT, USA), 2 mmol/l glutamine (Gibco), 25 mM glucose, and 1% penicillin/streptomycin (Gibco). Cells were plated onto poly-D-lysine-coated tissue culture plates at 7.5 × 105 cells/ml. Media were completely changed after 24 h. One-half medium changes were performed at day 4. Cultures were incubated at 37 °C in a 5% CO2 incubator and experiments were performed after days 9–11.
The lentiviral vectors, diluted with PBS to 5 μl for 24-well plates and to 10 μl for 6-well plates, were directly added into the medium of 9 day primary mixed neuron cultures with the multiplicity of infection (MOI) at 5. The same amount of PBS was also added to the medium as an experimental control (Cheng et al., 2009). Then cells were incubated at 37 °C in a 5% CO2 incubator for another 2 days before OGD was conducted. Primary mixed neuron cultures were washed twice with glucose-free balanced salt solution (BSS0, pH 7.4) and the plates were transferred to a modular hypoxic chamber filled with mixed gases of 5% CO2 and 95% N2. Oxygen level was maintained at <0.02% at 37 °C. The cells were kept in the hypoxic chamber for 6 h. Cultures were then restored with glucose at a final concentration of 5.5 mM (BSS5.5, pH 7.4) and recovered at normoxic conditions (37 °C, 5% CO2) for ~18 h (OGD restoration). The control groups without OGD were washed twice with 5.5 mM glucose in balanced salt solution (BSS5.5, pH 7.4) with no oxygen deprivation.
Cell viability was quantified by measuring lactate dehydrogenase (LDH) release that is always used to evaluate both apoptosis and necrosis at 18 h after OGD restoration using a previously described colorimetric assay (Dugan et al., 1995). Briefly, 100 μl of cell-free supernatant was transferred to 96-well plates. The supernatant was incubated with 150 μl of NADH/phosphate buffer (0.15 mg/ml) for 10 min. Then 30 μl of sodium pyruvate (2.97 mg/ml) was added and the absorbance wavelength was measured at 340 nm using a microplate reader. Background absorbance was subtracted and the percentage of LDH release was calculated based on an LDH standard curve.
In vivo lentiviral vector injections in rats and mice
All viral vectors were coded, thus the surgeons who performed both virus injections and stroke models were blinded. The lentiviral vectors were injected into rat or mouse brains 5 days prior to ischemia. In rats the left distal MCA occlusion model was used, where infarction is induced only in the cortex, therefore lentiviral vectors were injected into the rat’s left cortex. In mice the left MCA was occluded by a suture insertion as described later in this article and lentiviral vectors were injected into the left striatum, which generated reproducible infarction in the left striatum, while infarction in the cortex was more variable. While under isoflurane anesthesia the animals were placed in a stereotaxic frame. For virus injection into rats, a 1 cm sagittal skin incision was made on the head and the skull was exposed. A burr hole in the left hemisphere was drilled according to the coordinate 0.96 mm posterior, 3.5 mm lateral to the bregma. A 10 μl syringe (Hamilton Company, Reno, NV) with a 26-gauge needle (Cat# 80010) was inserted to the cortex and lowered to the depth of 1.8 mm from the dura. For virus injection into mice, a burr hole in the left hemisphere was drilled according to the coordinate 0.5 mm anterior, 2.0 mm lateral to the bregma. The same syringe used for rat brains was inserted to the striatum and lowered to the depth of 2.5 mm from the dura. Each viral vector (5 μl) of GFP or PRAS40 was injected into the cortex of the rat brain or the striatum of the mouse brain using a microsyringe pump controller at 0.5 μl/min (World Precision Instruments, Inc., Sarasota, FL). For both rats and mice, the same amount of PBS was also injected as an experimental control, in addition to the GFP control vector. Following viral vector administration, the needle was left for 10 min before being withdrawn. The wound was closed and the animals were allowed to recover.
Focal cerebral ischemia in rats and mice
According to the Stroke Treatment Academic Industry Roundtable (STAIR) recommendations, multiple species and stroke models are desired to test the efficacy of a neuroprotective agent (Fisher et al., 2009). In this study, to test the protective effect of PRAS40 lentiviral vectors in rats, the distal MCA occlusion model was used, which generated a well-defined infarction in the cortex only (Zhao et al., 2005). In this model, focal ischemia was generated by permanent distal MCA occlusion combined with transient bilateral CCA occlusion in male Sprague–Dawley rats (250–280 g, Charles River Laboratories International, Wilmington, MA, USA) (Zhao et al., 2005). Core body temperatures were monitored with a rectal probe maintained at 37 °C throughout the experiment under anesthesia. Rats were anesthetized by 5% isoflurane and maintained by 2%–3% isoflurane. A ventral midline incision was made and the 2 CCAs were isolated. Then a 2 cm vertical scalp incision was made midway between the left eye and ear. The temporalis muscle was bisected and a 2 mm burr hole was made at the junction of the zygomatic arch and squamous bone. The distal MCA was exposed and cauterized above the rhinal fissure at the cross of the lateral vein and MCA. The bilateral common carotid arteries (CCAs) were occluded for 60 min with suture tightening. Blood pressure, heart rate, respiratory rate, and temperature were monitored throughout the surgery and kept within physiological ranges. To study the protective effect of PRAS40 lentiviral vectors in both wild-type (WT) and PRAS40 KO mice, the transient MCA suture occlusion model was used. Transient focal ischemia in both male PRAS40 KO and WT mice (25 to 30 g) was induced by middle cerebral artery occlusion (MCAO) for 60 min, which generates infarction in both cortex and striatum, as previously described (Gu et al., 2012). Anesthesia was induced with 4% isoflurane and maintained by 1.5%–2% isoflurane in 70% air and balanced oxygen by a facemask. Rectal temperature was maintained at 37 ± 0.5°C with a heating pad (Harvard Apparatus, Holliston, MA). We introduced a silicone-coated 6–0 monofilament into the left external carotid artery and advanced it from the carotid bifurcation to occlude the MCA. Anesthesia was discontinued after suture insertion. Mice were re-anesthetized 60 min later and the filament was withdrawn. Sham-operated mice underwent an identical procedure, except that the suture was not inserted.
To study the effect of PRAS40 KO on brain injury, two stroke models were used, including the distal MCA occlusion model and transient MCA suture occlusion model. The distal MCA occlusion model was generated similarly with that described above in rats with modifications that left both the distal MCA and CCA permanently occluded. The MCA suture occlusion model was created as described above. Neurological score was evaluated 48 h after MCA suture occlusion according to a neurological grading score, (Xiong et al., 2011) from 0 (no observable neurological deficit) to 4 (unable to walk spontaneously and a depressed level of consciousness). The evaluator was blinded to genotypes and experimental treatment.
CBF measurement in PRAS40 KO mice
To examine the effect of PRAS40 KO on CBF changes in PRAS40 KO mice, relative regional cerebral blood flow (rCBF) was monitored by using a laser Doppler probe (PeriFlux 5000; Perimed AB, Stockholm, Sweden) in separate animals as described before (Xiong et al., 2013). Briefly, a small incision was made in the ischemic hemisphere to expose the skull, and the laser Doppler probe with a diameter of 0.5 mm was attached to the exposed skull (1 mm posterior to the bregma, 3.3 mm lateral to the midline) to detect CBF values at 15 min before ischemia onset (baseline), during ischemia (every 15 min after stroke onset), and immediately, 15 and 30 min after reperfusion. CBF values were expressed as percentages relative to the baseline (100%). CBF values in both PRAS40 KO mice and WT mice were compared.
General histology and infarct size measurement
To study the effects of PRAS40 KO on infarction in mice, we used 2,3,5-triphenyletrazolium chloride (TTC) (#T8877, Sigma-Aldrich, St. Louis, MO) staining to measure infarct sizes. At 48 h post-stroke, mice were anesthetized with isoflurane and decapitated. With a rodent brain slicer matrix we cleaved 4 coronal sections of 2 mm (Zivic Instruments, Pittsburgh, PA). After incubating these sections in 2% TTC, each infarction volume was determined by a blinded observer and corrected for edema by using the NIH ImageJ program (Image J 1.37v, Wayne Rasband, available through NIH) as described previously (Sun et al., 2012; Xiong et al., 2011). Infarct sizes were normalized to the contralateral cortex in the distal MCA occlusion model in both rats and mice, and to the contralateral hemisphere in the MCA suture occlusion model in mice, and expressed as percentages.
Since lentiviral injection could only reach a relatively small area of the brain, it was impractical to use TTC staining to measure infarction of the whole brain. Therefore, we used cresyl violet staining to measure infarct sizes in the area of the brain treated in rats and mice with lentiviral vectors. Two days after ischemia, subjects were perfused transcardially with cold 0.9% saline, followed by 4% paraformaldehyde (PFA) in PBS (pH 7.4). Brains were post-fixed in 4% PFA, and 20% sucrose for 24 h, cut into 5 coronal blocks from rostral (level 1) to caudal (level 5), and frozen at −80 °C. In each brain the block containing the needle track was sectioned into 30 μm sections using a cryostat and mounted onto glass slides. The slices with a needle track were selected for infarct measurement. Successful transfection of the lentiviral vectors in the selected slices was confirmed by observing protein expression of eGFP under fluorescent confocal microscopy (Zeiss LSM 510). Sections were stained with cresyl violet. Each infarcted area was measured by a person who was blinded to the animal’s condition, normalized to the contralateral cortex in the rat receiving distal MCA occlusion and to the contralateral hemisphere in the mouse receiving MCA suture occlusion and expressed as a percentage, as described previously (Zhao et al., 2005).
Western blot
To study the effects of PRAS40 gene transfer on protein changes in rat brains, brain tissue around the needle track was dissected at 5 and 24 h after stroke, as described (Xie et al., 2013a). Brain tissue from animals receiving sham surgery without ischemia, with or without transfection of lentiviral vectors, was also prepared for western blotting. To study the effects of PRAS40 KO on protein changes, mice were euthanized at 5, 24, and 48 h after reperfusion. The ischemic hemispheres were collected. Whole cell proteins were extracted, and western blot was performed as described in our previous study (Gu et al., 2013). Table 1 lists the primary antibodies used. In each lane, 30 μg of protein was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis using 4%–15% Ready Gel (Bio-Rad Laboratories, Hercules, CA, USA) for 1.5 h. Protein bands were transferred from the gel to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA) for 1 h. After pre-blocking the membrane with 5% nonfat dry milk (Bio-Rad Laboratories) in PBS and 0.05% Tween-20, primary antibodies were incubated overnight at 4 °C followed by incubation with horseradish-peroxidase-conjugated secondary antibody (anti-rabbit 1:2000, Cell Signaling Technology) or anti-mouse IgG for 1 h. The membrane was incubated with anti-β-actin antibodies as an even-loading control of total protein. Membranes were scanned using Typhoon trio (GE Healthcare, Waukesha, WI, USA).
Table 1.
Information of primary antibodies used in this study.
| Antibodies | Source | Dilutions | Manufacturer | Catalog# | Application |
|---|---|---|---|---|---|
| P-Akt (Ser473) | Rabbit | 1:100/1:1000 | Cell Signaling | 9271 | IF/WB |
| Akt | Rabbit | 1:1000 | Cell Signaling | 9272 | WB |
| P-FKHR (Ser256) | Rabbit | 1:1000 | Cell Signaling | 9461 | WB |
| FKHR | Rabbit | 1:50/1:1000 | Cell Signaling | 2880 | IF/WB |
| P-PRAS40 (Thr246) | Rabbit | 1:100/1:1000 | Cell Signaling | 2997 | IF/WB |
| PRAS40 | Rabbit | 1:1000 | Cell Signaling | 2691 | WB |
| P-mTOR (Ser2448) | Rabbit | 1:1000 | Cell Signaling | 2971 | WB |
| mTOR | Rabbit | 1:1000 | Cell Signaling | 2983 | WB |
| P-PTEN (Ser380) | Rabbit | 1:1000 | Cell Signaling | 9551 | WB |
| PTEN | Rabbit | 1:1000 | Cell Signaling | 9552 | WB |
| P-GSK3β (Ser 9) | Rabbit | 1:1000 | Cell Signaling | 9336 | WB |
| GSK3β | Rabbit | 1:1000 | Cell Signaling | 9332 | WB |
| P-S6K p70 (Ser371) | Rabbit | 1:500 | Cell Signaling | 9208 | WB |
| P-S6 | Rabbit | 1:100 | Cell Signaling | 2215 | WB |
| GFP | Rabbit | 1:500 | Cell Signaling | 2956 | WB |
| β-actin | Mouse | 1:300 | Sigma | A-5441 | WB |
IF = Immunofluorescence; WB = western blot.
Co-immunoprecipitation
To study protein-to-protein interactions between mTOR and Akt and GSK3β in PRAS40 KO and WT mice, we performed co-immunoprecipitation using anti-mTOR antibodies. Protein concentrations were determined as described previously (Saito et al., 2004). For co-immunoprecipitation, 1 mg of lysate from the whole cell protein was used (Pierce® Classic IP Kit, Pierce Biotechnology, USA) and combined with 10 μg of rabbit polyclonal anti-mTOR antibody in order to capture the immune complex with protein A/G plus agarose. Whole-brain extract was included as a positive control. The negative control was prepared with protein A/G plus agarose without an antibody. The immune complex was then eluted and a western blot was performed. Primary antibodies were a 1:1000 dilution of anti-Akt or a 1:1000 dilution of anti-GSK3β.
Statistical analyses
Data are expressed as mean ± SEM. Differences were considered statistically significance for P < 0.05. Student’s t-tests were used when 2 groups were compared. Two-way ANOVAs were used when both genotype and treatment were taken into account, followed by the Bonferroni post-tests using Prism 5 (GraphPAD Software for Science, San Diego, CA). All assessments were by blinded observers.
Results
Construction and validation of lentiviral vectors expressing PRAS40
We first confirmed if the PRAS40 lentiviral vector was successfully constructed, transfected and overexpressed in vitro. Lentiviral vectors that express PRAS40 (Fig. 1A) were transfected into primary mixed neuronal cultures. Western blot results showed that PRAS40 gene transfer into these cells increased both p-PRAS40 and total PRAS40 protein levels (Fig. 1B). Note that we constructed and confirmed PRAS40 lentiviral vectors at the same time with cAkt1, cAkt3 and AktDN, as described previously (Xie et al., 2013a), and protein bands of PRAS40 were cut from an original gel (Supplementary Fig. 1). Confocal microscopy confirmed lentiviral-mediated PRAS40 expression, as indicated by GFP and PRAS40 expression (Fig. 1C).
Fig. 1.

Construction of lentiviral vectors of PRAS40 and its expression in primary neuronal cultures. A. Schematic backbone of lentiviral vector of pHR’tripCMV–IRES–eGFP. B. Western blot confirmation of the effects of gene transfer in primary neuronal cultures on protein expression of p-PRAS40 and PRAS40. Protein levels of both PRAS40 and p-PRAS40 were increased in cells transfected with PRAS40. All protein bands were cut from the same gel (see Supplementary Fig. 1 for the original gel). C. Triple staining of GFP, PRAS40 and DAPI in a mixed primary neuronal culture transfected with PRAS40 confirms the successful transfection of neurons. Scale bar, 50 μm.
PRAS40 gene transfer inhibited neuronal death both in vitro and in vivo stroke
We next studied the effect of PRAS40 gene transfer on infarct size in the in vivo rat stroke model. Lentivirus-expressing PRAS40 were stereotaxically delivered into the cortex 5 days before stroke and infarct size was examined using cresyl-violet staining. The injection areas and transfected cells were confirmed by observing GFP protein expression (Figs. 2A,B). A lower power GFP image to show gene transfer is available in the Supplementary materials (Supplementary Fig. 2). Infarct size measured at the needle track was significantly reduced by PRAS40 injection (Fig. 2C). Note that this distal MCA occlusion model produced infarction in the cortex only, thus infarct size was normalized to the contralateral cortex and expressed as a percentage. This differs from that in the mouse stroke model as depicted in Fig. 6, in which infarct size was normalized to the contralateral hemisphere, as the MCA suture occlusion resulted in infarction in both the cortex and striatum.
Fig. 2.

Neuroprotective effects of PRAS40 gene transfer in vitro and in vivo. A. Representative coronal sections of brains show infarction areas and an injection site receiving PRAS40 virus. The injection site is marked by a square in which a needle track is visible. Gene transfer of PRAS40 reduced brain infarction after focal ischemia. B. Representative immunostaining of GFP protein in brains transfected with control GFP and PRAS40 lentiviral vectors. Both GFP and PRAS40 are represented by green color; DAPI, blue. Normal, a brain section without stroke; post-ischemia, 48 h after stroke. C. Average infarct size is determined from brain sections with needle tracts. Infarct area was measured and normalized to the non-ischemic contralateral cortex, and expressed as a percentage. *, vs control vector of GFP, P < 0.05. D. Effects of gene transfer of PRAS40 on neuronal death as measured by LDH release in primary neuronal cultures. Cultured mixed neurons were transfected with vectors containing PRAS40; controls were cultures transfected with GFP vectors or treated with vehicle solution. Cultures were subjected to 6 h of OGD 2 days after transfection. LDH release was measured 18 h post-OGD. All data after OGD was normalized to the values of the control (vehicle), non-OGD samples transfected with the corresponding vectors. n = 14/group. Φ, ΦΦP < 0.05, and 0.01, respectively, between the two indicated groups.
Fig. 6.
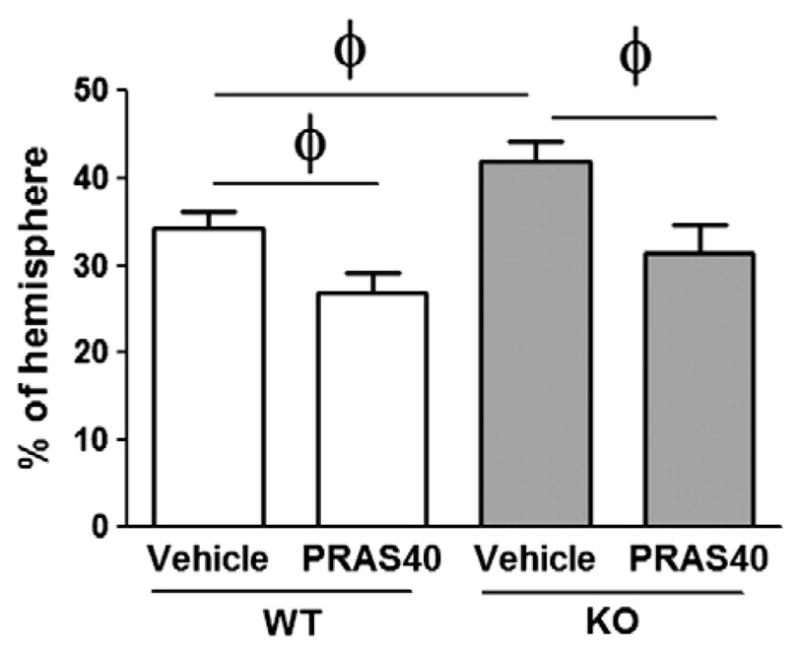
PRAS40 gene transfer recovered infarct sizes in KO mice comparable to those in WT mice. Infarct area was measured and normalized to the non-ischemic contralateral hemisphere, and expressed as a percentage. Administration of PRAS40 lentiviral vector before middle cerebral artery occlusion (MCAO) protects both wild-type (WT) and PRAS40 KO mice, n= 9–11/group. ϕ,P < 0.05, between the two indicated groups.
We then investigated the effects of PRAS40 gene transfer on neuronal death induced by OGD in vitro. We measured LDH release 18 h after OGD by using primary neuronal cultures that were transferred with Lentivirus-expressing PRAS40. The results showed that compared with control vector, PRAS40 gene transfer reduced LDH release and ameliorated neuronal death (Fig. 2D).
We have previously shown that these lentiviral vectors transfected neurons in vivo by using immunofluorescent staining of neuronal marker MAP-2 with GFP (Xie et al., 2013a). As GFP protein levels expressed by the Lentivirus are indicators of tissue survival after stroke, we examined GFP protein levels by western blot. Our results showed that, compared to control vectors, PRAS40 gene transfer enhanced GFP protein levels in the ischemic hemisphere (Supplementary Fig. 3). Confocal microscopy results suggest that overexpression of PRAS40 also attenuated reductions in MAP-2 protein levels (data not shown).
Gene transfer of PRAS40 promoted protein phosphorylation in the Akt and mTOR pathways
To identify proteins relevant to protective effects, we analyzed and compared gene transfer of PRAS40 on both phosphorylated and total protein levels of PTEN, Akt, GSK3β, PRAS40 and mTOR (Fig. 3). All protein bands were cut from the same gels (Supplementary Fig. 4). In the control group treated with lentiviral GFP vector, protein levels of p-PTEN, p-FKHR and p-GSK3β were decreased at both 5 and 24 h after stroke, and p-Akt and p-mTOR were also decreased at 24 h. Gene transfer of PRAS40 promoted p-Akt, p-FKHR, and p-mTOR protein levels as measured at 24 h, but did not alter p-PTEN or pGSK3β protein levels (Fig. 3).
Fig. 3.

The effects of gene transfer of PRAS40 on protein expression in the Akt/mTOR pathways 5 h and 24 h after stroke. A. Representative protein bands of critical molecules in the Akt/mTOR pathways, including phosphorylated and total protein expression, as measured by western blot. All protein bands presented are derived from the same gel, but were cut and re-arranged for convenient comparison (see Supplementary Fig. 5). B, C, D, E, F and G show the quantified protein levels of p-PTEN, p-Akt, p-FKHR, p-GSK3β, p-PRAS40 and p-mTOR, respectively. *, **P < 0.05, and 0.01, respectively, vs corresponding normal or ischemic brain transfected with control GFP vectors. Φ, ΦΦ show significance difference between the two indicated groups, P < 0.05, and 0.01, respectively.
PRAS40 KO resulted in larger infarction in two stroke models in mice
After demonstrating that PRAS40 overexpression reduced ischemic brain injury in rats and promoted protein phosphorylation in the Akt and mTOR pathways, we studied the effects of PRAS40 KO on brain injury after stroke in mice. All subjects were genotyped by PCR and confirmed before surgery (Fig. 4A). Western blot data suggest that PRAS40 was not expressed in the tissues of PRAS40 KO mice (Supplementary Fig. 4).
Fig. 4.
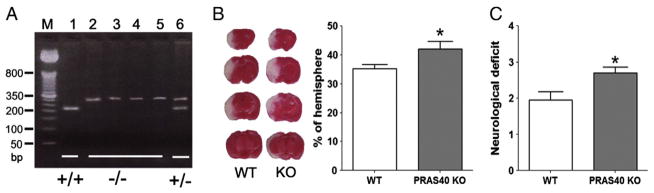
Confirmation of PRAS40 gene KO in mice and increased ischemic infarction and neurological score in PRAS40 KO mice compared to WT mice. A. PCR for genotyping PRAS40 KO mice. PCR product analysis on 4% agarose gel. Lane M: DNA size marker; Lane 1: C57BL/6 homozygote (+/+); Lanes 2, 3, 4, and 5: PRAS40 homozygote (−/−); Lane 6: PRAS40 heterozygote (+/−). B. Left: Representative infarction of coronal sections stained by TTC in WT and KO mice. Right: Bar graphs show average infarct sizes measured at 48 h post-stroke. C. Bar graphs show neurological deficit assessed 48 h after MCAO. n=9–10/group. *P < 0.05 vs WT.
We used 2 stroke models to evaluate the effects of PRAS40 KO on infarction. In one model, focal ischemia was induced by MCA suture occlusion for 60 min, which induces infarction in both the cortex and striatum. We confirmed that stroke resulted in similar changes in CBF during stroke and after reperfusion (Supplementary Fig. 5). Both infarct sizes and neurological scores were measured 2 days after stroke, and infarction was identified by TTC staining. Infarct sizes were significantly increased in PRAS40 KO mice compared to WT mice (Fig. 4B). PRAS40 KO also resulted in worsened neurological scores (Fig. 4C). In the other model, stroke was induced by permanent occlusion of both the distal MCA and ipsilateral CCA in order to generate infarction in the cortex only. Consistent with the MCA suture occlusion model, PRAS40 KO significantly enlarged infarct sizes in the cortex compared to WT mice (Supplementary Fig. 6). Note that the distal MCA occlusion model in both rats (Fig. 2) and mice (Supplementary Fig. 6) generated infarction in the cortex only, and the infarction sizes were expressed as percentages of the contralateral cortex.
PRAS40 KO resulted in less phosphorylation of proteins in the mTOR pathway but not in the Akt pathway
Levels of p-Akt and p-PTEN in the Akt pathway, and p-P70S6K and p-S6 in the mTOR pathway were measured by western blot in WT and PRAS40 KO mice. WT mice showed significant though transient increases after stroke in p-Akt at 5 h, in p-PTEN at 24 and 48 h, in p-P70S6K at 5 and 24 h, and in p-S6 at 5 h. PRAS40 KO did not significantly alter protein levels of p-Akt and p-PTEN compared to WT mice at any measured time points (Fig. 5). PRAS40 KO did significantly downregulate protein levels after stroke of p-P70S6K at 5 and 24 h, and inhibited p-S6 protein levels at 5, 24 and 48 h (Fig. 5A).
Fig. 5.

PRAS40 KO did not alter protein levels of p-Akt and p-PTEN, but inhibited protein levels of p-P70S6K and p-S6, and inhibited interactive effects between mTOR with Akt and GSK3β. A. The left shows western blot of representative protein bands for p-Akt, p-PTEN, p-P70S6K, and p-S6. β-actin was probed to show even loading of protein samples. Bar graphs on the right show average protein levels, which were normalized to the amount of sham animals and expressed as percentages. B. Left: Representative protein bands derived from co-immunoprecipitation analysis for interactive effects between mTOR with Akt or GSK3β. Proteins were co-immunoprecipitated using anti-mTOR antibodies, and extracted proteins were analyzed by western blot. Anti-Akt and anti-GSK3β antibodies were used to measure Akt and GSK3β protein levels that physically interact with mTOR. Both positive (whole-brain extract) and negative controls (prepared with protein A/G plus agarose without mTOR antibody) measuring Akt and GSK3β proteins were used. Right: Bar graphs of Akt and GSK3β protein levels. Con, contralateral side; Ips, ipsilateral ischemic side; n.c., negative control, p.c., positive control. *, **, ***P < 0.05, 0.01, and 0.001, respectively, vs sham; ϕ, ϕϕ, ϕϕϕ,P < 0.05, 0.01, and 0.001, respectively, between two indicated groups.
PRAS40 KO resulted in less interaction of mTOR with both Akt and GSK3β
To investigate protein-to-protein interactions of mTOR with Akt and GSK3β, the mTOR antibody was used to co-immunoprecipitate these proteins, assessed by western blot. Results suggest that stroke increased protein interactions of mTOR with both Akt and GSK3β, but were blocked by PRAS40 KO (Fig. 5B).
Re-introduction of the PRAS40 gene protected against stroke
If PRAS40 is crucial for neuroprotection against stroke, its reintroduction into a PRAS40 KO animal should help recover its protective effects. Our last experiment therefore studied the effect on infarct size of PRAS40 gene transfer into PRAS40 KO mice. Lentiviral vectors expressing PRAS40 were stereotaxically delivered into the striatum 5 days before stroke and infarct size was examined using cresyl violet staining. The data show that gene transfer reduced infarct size in PRAS40 KO mice to a level comparable to WT mice (Fig. 6).
Discussion
We are the first to demonstrate that PRAS40 KO mice had larger infarctions than WT animals, and that re-introduction of PRAS40 lentiviral vectors into the ischemic brains of PRAS40 KO mice recovered the protective effects of PRAS40. We also performed PRAS40 gene transfer in rats to confirm that PRAS40 overexpression attenuates brain injury induced by stroke. In addition, we demonstrated that PRAS40 overexpression promoted phosphorylation of proteins in the Akt and mTOR pathways. It appears that the underlying pathological mechanisms of PRAS40 KO are related to the downregulated phosphorylation of proteins in the mTOR pathway but not the Akt pathway, and that PRAS40 KO inhibited protein-to-protein interactions between Akt and mTOR proteins.
Many studies, including our own, have demonstrated that the Akt pathway contributes to neuronal survival after stroke (Gao et al., 2008; Noshita et al., 2003; Ouyang et al., 1999; Zhao et al., 2005). We and others have previously shown that the level of Akt phosphorylation at Ser 473 (p-Akt) is transiently increased after cerebral ischemia (Friguls et al., 2001, 2002; Kamada et al., 2007; Noshita et al., 2001; Wick et al., 2002; Yano et al., 2001; Zhao et al., 2005, 2006) while the levels of other phosphorylated proteins in the Akt pathway are reduced, including p-PTEN, p-PDK1, p-FKHR and p-GSK3β (Zhao et al., 2005), along with p-PRAS40. Other studies have shown that brain injury is reduced through p-Akt protein promotion by neuroprotectants such as growth factors, estrogen, free radical scavengers, preconditioning, postconditioning and hypothermia (Gao et al., 2008, 2010; Zhao et al., 2005, 2006).
We recently demonstrated that the Akt isoform transfer protects against ischemic injury by increasing p-PRAS40 protein levels and that the mTOR inhibitor rapamycin blocked these protective effects (Xie et al., 2013a). In this study, we constructed lentiviral vectors expressing constitutively active Akt1 and Akt3, two major isoforms of Akt, which reduced brain infarction in rats (Xie et al., 2013a) We found that overexpression of Akt1 and Akt3 not only promoted protein phosphorylation in the Akt pathway, including p-Akt, p-PTEN, and p-FKHR, but also promoted mTOR phosphorylation in the mTOR pathway (Xie et al., 2013a). We also showed that p-PRAS40 protein levels were increased by Akt isoform gene transfer and that the mTOR inhibitor rapamycin blocked the protective effects of Akt isoform gene transfer (Xie et al., 2013a). We also reported that mTOR in nude rats is crucial for the long-term protective effects of ischemic postconditioning, that postconditioning attenuated brain injury measured at 1 month after stroke by promoting post-stroke p-mTOR expression, and that this protection was nullified by rapamycin (Xie et al., 2013b). All of these studies suggest that both Akt and mTOR pathways are important in protecting against brain injury, which motivated us to further clarify the role of PRAS40 in linking the Akt and mTOR pathways.
Indeed, our present study suggests that PRAS40 transfers neuroprotective cell signaling from Akt to its downstream proteins mTOR, P70S6K and S6. It is known that mTOR is present in two complexes (Guertin and Sabatini, 2005, 2007): mTORC1 and mTORC2. mTORC1 is composed of mTOR, raptor, mLST8 and PRAS40; mTORC2 also contains mTOR and mLST8, but it contains rictor and mSin1 instead of raptor and PRAS40. mTORC1 phosphorylates 4EBP1 and S6K1, thus modulating protein synthesis and cell growth. The function of mTORC2 is less understood. Recent studies suggest that some neuroprotectants inhibit brain injury by promoting mTOR activity (Koh, 2008; Koh et al., 2008; Wu et al., 2012). In our present study PRAS40 overexpression appeared to promote protein phosphorylation in both the Akt and mTOR pathways, although PRAS40 KO did not alter p-Akt levels but significantly inhibited protein levels of p-S6K and p-S6. We postulate that PRAS40 gene transfer spared ischemic tissues by non-specifically preserving p-Akt proteins, while the PRAS40 KO executed its effects on PRAS40’s downstream molecules in the mTOR pathway rather than its upstream molecules in the Akt pathway. Taken together, our results provide solid evidence that PRAS40 confers protection by promoting mTOR activity.
The role of PRAS40 in the mTOR pathway has been a matter of debate (Wiza et al., 2012). Mainstream studies suggest that PRAS40 inhibits mTORC1, as mTOR activity in some cell lines is blocked by PRAS40 overexpression and promoted by PRAS40 knockdown (Basu et al., 2011; Rapley et al., 2011; Wang et al., 2007). Loss of PRAS40 also increased phosphorylation of mTOR, p70S6K, and 4EBP1 and prevented SH-SY5Y cell injury induced by OGD (Chong et al., 2012). In contrast, other studies have reported that PRAS40 is essential to induce mTOR activity (Wiza et al., 2012), and that silencing PRAS40 impairs the phosphorylation of 4E-BP1 and S6, two of mTOR’s downstream proteins (Fonseca et al., 2007). To our knowledge, the present study is the first to show in a mammalian animal model that PRAS40 KO downregulates mTOR activity in ischemic brains. Our results support the idea that PRAS40 is essential for mTOR activity at least in cerebral ischemia. However, we did not observe altered basal mTOR activity in PRAS40 KO animals without stroke. mTOR signaling in the brain is required for axon growth, dendritic arborization and synaptogenesis (Swiech et al., 2008) but is disturbed in neurodegenerative diseases, such as Parkinson’s (Malagelada et al., 2006; Pan et al., 2008), Alzheimer’s (Lafay-Chebassier et al., 2005; Li et al., 2005; Rosner et al., 2008) and Huntington’s diseases (Sarkar et al., 2007). We do not exclude the possibility that our findings are specific to cerebral ischemia and the tissues under study. How PRAS40 alters mTOR activity in other organs, tissues and pathological settings deserves more study.
There are some limitations in this current study. First, protein bands of some Western blots were cut and presented (Figs. 1 and 3), although all of them come from the same gels (Supplementary Figs. 3 and 4). Second, proteins in the Akt and mTOR pathways were not examined and compared in parallel in PRAS40 KO mice and in animals transfected with PRAS40 lentiviral vectors. For instance, protein levels of p-P70S6K and p-S6 in the mTOR pathway were shown to be decreased in PRAS40 KO mice (Fig. 5), but these two proteins were not examined in PRAS40 transfected brains, even though we have shown that PRAS40 vector transfection promoted mTOR activity (Fig. 3). Third, since PRAS40 gene transfer reaches limited brain tissue, measuring its protective effects through reduction of absolute infarct volumes is not practical. Instead, a relative percentage was calculated to show the protective effects of PRAS40 gene transfer at the level of the brain section with virus transfection.
Our findings provide insights for developing novel PRAS40-based treatments for stroke. As both Akt and mTOR are critical for a broad range of physiological activities and pathological mechanisms, and PRAS40 links the Akt and mTOR cell signaling pathways, it is plausible to consider the cell-signaling between Akt and mTOR via PRAS40 as an integrative target in the ischemic brain. Pharmacological, gene and protein therapies may be developed to increase levels and activities of these critical proteins, thus promoting neuronal survival and brain functional recovery after stroke. In conclusion, by using a PRAS40 KO model and lentiviral vectors of PRAS40, we provide strong evidence that PRAS40 expression plays a pivotal neuroprotective role through transfer of protective cell signaling from the Akt pathway to the mTOR pathway, and thus provide novel insights for the development of novel treatments for stroke.
Supplementary Material
Acknowledgments
The authors thank Dr. Richard Roth for providing the PRAS40 KO mice and other technical support, Elizabeth Hoyte for the figure preparation and Cindy H. Samos for the manuscript editing.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nbd.2014.02.006.
Footnotes
Available online on ScienceDirect (www.sciencedirect.com).
Conflict of interest
None.
Funding disclosure
This study was supported by the AHA grants in aid 10GRNT4200024 and 1R01NS064136-01 (HZ). RX was partly supported by the National Natural Science Foundation of China (Grant no. 81200890).
References
- Apolonia L, et al. Stable gene transfer to muscle using non-integrating lentiviral vectors. Mol Ther. 2007;15:1947–1954. doi: 10.1038/sj.mt.6300281. [DOI] [PubMed] [Google Scholar]
- Basu A, et al. Calcineurin inhibitor-induced and Ras-mediated overexpression of VEGF in renal cancer cells involves mTOR through the regulation of PRAS40. PLoS One. 2011;6:e23919. doi: 10.1371/journal.pone.0023919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckpot K, et al. Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J Gene Med. 2003;5:654–667. doi: 10.1002/jgm.400. [DOI] [PubMed] [Google Scholar]
- Cheng MY, et al. Blocking glucocorticoid and enhancing estrogenic genomic signaling protects against cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:130–136. doi: 10.1038/jcbfm.2008.105. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, et al. PRAS40 is an integral regulatory component of erythropoietin mTOR signaling and cytoprotection. PLoS One. 2012;7:e45456. doi: 10.1371/journal.pone.0045456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan LL, et al. Glia modulate the response of murine cortical neurons to excitotoxicity: glia exacerbate AMPA neurotoxicity. J Neurosci. 1995;15:4545–4555. doi: 10.1523/JNEUROSCI.15-06-04545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca BD, et al. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem. 2007;282:24514–24524. doi: 10.1074/jbc.M704406200. [DOI] [PubMed] [Google Scholar]
- Friguls B, et al. Focal cerebral ischemia causes two temporal waves of Akt activation. Neuroreport. 2001;12:3381–3384. doi: 10.1097/00001756-200110290-00046. [DOI] [PubMed] [Google Scholar]
- Friguls B, et al. Activation of ERK and Akt signaling in focal cerebral ischemia: modulation by TGF-alpha and involvement of NMDA receptor. Neurobiol Dis. 2002;11:443–456. doi: 10.1006/nbdi.2002.0553. [DOI] [PubMed] [Google Scholar]
- Gao X, et al. The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J Neurochem. 2008;105:943–955. doi: 10.1111/j.1471-4159.2008.05218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, et al. The Akt pathway is involved in rapid ischemic tolerance in focal ischemia in Rats. Transl Stroke Res. 2010;1:202–209. doi: 10.1007/s12975-010-0017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, et al. Distinctive effects of T cell subsets in neuronal injury induced by cocultured splenocytes in vitro and by in vivo stroke in mice. Stroke. 2012;43:1941–1946. doi: 10.1161/STROKEAHA.112.656611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, et al. T cells contribute to stroke-induced lymphopenia in rats. PLoS One. 2013;8:e59602. doi: 10.1371/journal.pone.0059602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11:353–361. doi: 10.1016/j.molmed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hu Q, et al. Lentivirus-mediated transfer of MMP-9 shRNA provides neuroprotection following focal ischemic brain injury in rats. Brain Res. 2011;1367:347–359. doi: 10.1016/j.brainres.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Kamada H, et al. Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2007;27:521–533. doi: 10.1038/sj.jcbfm.9600367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh PO. Melatonin prevents ischemic brain injury through activation of the mTOR/p70S6 kinase signaling pathway. Neurosci Lett. 2008;444:74–78. doi: 10.1016/j.neulet.2008.08.024. [DOI] [PubMed] [Google Scholar]
- Koh PO, et al. Estradiol attenuates the focal cerebral ischemic injury through mTOR/p70S6 kinase signaling pathway. Neurosci Lett. 2008;436:62–66. doi: 10.1016/j.neulet.2008.02.061. [DOI] [PubMed] [Google Scholar]
- Kovacina KS, et al. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem. 2003;278:10189–10194. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- Lafay-Chebassier C, et al. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP–PS1 transgenic models and in patients with Alzheimer’s disease. J Neurochem. 2005;94:215–225. doi: 10.1111/j.1471-4159.2005.03187.x. [DOI] [PubMed] [Google Scholar]
- Li X, et al. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005;272:4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- Malagelada C, et al. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci. 2006;26:9996–10005. doi: 10.1523/JNEUROSCI.3292-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noshita N, et al. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:1442–1450. doi: 10.1097/00004647-200112000-00009. [DOI] [PubMed] [Google Scholar]
- Noshita N, et al. Copper–zinc superoxide dismutase affects Akt activation after transient focal cerebral ischemia in mice. Stroke. 2003;34:1513–1518. doi: 10.1161/01.STR.0000072986.46924.F4. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, et al. Survival- and death-promoting events after transient cerebral ischemia: phosphorylation of Akt, release of cytochrome C and activation of caspase-like proteases. J Cereb Blood Flow Metab. 1999;19:1126–1135. doi: 10.1097/00004647-199910000-00009. [DOI] [PubMed] [Google Scholar]
- Pan T, et al. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- Rapley J, et al. The mechanism of insulin-stimulated 4E-BP protein binding to mammalian target of rapamycin (mTOR) complex 1 and its contribution to mTOR complex 1 signaling. J Biol Chem. 2011;286:38043–38053. doi: 10.1074/jbc.M111.245449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner M, et al. The mTOR pathway and its role in human genetic diseases. Mutat Res. 2008;659:284–292. doi: 10.1016/j.mrrev.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Saito A, et al. Neuroprotective role of a proline-rich Akt substrate in apoptotic neuronal cell death after stroke: relationships with nerve growth factor. J Neurosci. 2004;24:1584–1593. doi: 10.1523/JNEUROSCI.5209-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, et al. Modulation of proline-rich akt substrate survival signaling pathways by oxidative stress in mouse brains after transient focal cerebral ischemia. Stroke. 2006;37:513–517. doi: 10.1161/01.STR.0000198826.56611.a2. [DOI] [PubMed] [Google Scholar]
- Sarkar S, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- Sun J, et al. Inhibition of mitochondrial permeability transition pore opening contributes to the neuroprotective effects of ischemic postconditioning in rats. Brain Res. 2012;1436:101–110. doi: 10.1016/j.brainres.2011.11.055. [DOI] [PubMed] [Google Scholar]
- Swiech L, et al. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784:116–132. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Vander Haar E, et al. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007;282:20036–20044. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J Biol Chem. 2008;283:15619–15627. doi: 10.1074/jbc.M800723200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick A, et al. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22:6401–6407. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiza C, et al. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am J Physiol Endocrinol Metab. 2012;302:E1453–E1460. doi: 10.1152/ajpendo.00660.2011. [DOI] [PubMed] [Google Scholar]
- Wu F, et al. Tissue-type plasminogen activator regulates the neuronal uptake of glucose in the ischemic brain. J Neurosci. 2012;32:9848–9858. doi: 10.1523/JNEUROSCI.1241-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R, et al. Akt isoforms differentially protect against stroke-induced neuronal injury by regulating mTOR activities. J Cereb Blood Flow Metab. 2013a;33:1875–1885. doi: 10.1038/jcbfm.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R, et al. Ischemic post-conditioning facilitates brain recovery after stroke by promoting Akt/mTOR activity in nude rats. J Neurochem. 2013b;127:723–732. doi: 10.1111/jnc.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, et al. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke. 2011;42:2026–2032. doi: 10.1161/STROKEAHA.110.593772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, et al. The protective effects of T cell deficiency against brain injury are ischemic model-dependent in rats. Neurochem Int. 2013;62:265–270. doi: 10.1016/j.neuint.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S, et al. Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab. 2001;21:351–360. doi: 10.1097/00004647-200104000-00004. [DOI] [PubMed] [Google Scholar]
- Zhao H, et al. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci. 2005;25:9794–9806. doi: 10.1523/JNEUROSCI.3163-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, et al. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol. 2006;34:249–270. doi: 10.1385/MN:34:3:249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.