

Abstract
DNA vectors for human gene therapy have to meet the efficacy and safety requirements. Minicircles (MCs), a class of optimized DNA vectors free of plasmid backbone (PB) DNAs, have emerged as promising candidates because of their superior transgene expression profiles. However, the existence of impure DNAs, including the unrecombined MC producing plasmid (PP) and PB circle, in the MC products made using the current technologies exceed the safety limit. Here, we report the development of an enhanced triplex DNA (TriD) technology to eliminate almost all the impure DNAs from the MC products. To do this, a pair of optimized TriD forming sequences was placed to flank the kanamycin resistance gene in the PP. The MC products were incubated with a biotinylated TriD forming DNA oligonucleotide (olig), and the resulted TriDs were removed by binding to streptovidin-coated magnetic beads. Consequently, the residual impure DNAs were 0.03% or less in the final MC products. The reproducibility of this technique was confirmed with MCs of various transgene expression cassettes, sizes, and quantities, suggesting its great potential in making high quality MC for human gene therapy.
Introduction
DNA vectors for human gene therapy application have to meet the efficacy and safety requirements. Minicircle (MC) DNA vectors have emerged as a class of optimized non-viral DNA vectors capable of meeting these requirements. MCs are made by elimination of the plasmid backbone (PB) DNAs from MC producing plasmids (PP) through DNA recombination in bacteria. The PB DNAs, including the plasmid replication origin and the antibiotic resistance gene, are essential for DNA cloning and plasmid amplification in bacteria, but become useless in mammalian cells. Furthermore, PB DNAs can silence transgene expression, trigger inflammation reaction and have the potential to shield the bacteria in mammalian cells.1–5 Free of the detrimental PB DNAs, the MCs are capable of expressing 10- to 1,000-fold higher therapeutic transgene products in vitro6 and in vivo.7 Different DNA recombinases have been used to make the MCs, including the Cre derived from P1 bacteriophage,8 ΦC31 from streptomyce bacteriophage,7 and lambda derived from lambda phage.9 Technologies for isolating MC DNA from bacteria include triplex DNA (TriD) technology, cesium chloride gradient centrifugation,4 and affinity column purification.7 The problems of the existed technologies were the low yield of MCs, high costs, labor demanding, and involvement of toxic materials. Recently, we have partially solved these problems by developing a genetically engineered Escherichia coli MC production system, which allows simple, rapid, and inexpensive production of MC in large quantity with more than twofold increase in the yield and 10-fold reduction in impure DNAs.10 Nevertheless, there remain about 0.5–1.5% of PP and PB, which fluctuate to higher occasionally, in the MC product, that have to be further eliminated to meet the most restrictive regulatory specifications set by World Health Organization,11 US Food and Drug Administration,12 and European Medicines Agency.13 These contaminants exist in that the reactions mediated by either recombinase ΦC31, which mediates the recombination reaction to produce the MC, or the endonuclease I-SceI, which mediates the degradation of PB circle (Figure 1a), do not reach 100% efficiency even under optimal reaction conditions. Furthermore, the current technologies are unable to separate the MCs from PP and PB, because all of them are circular DNAs without significant difference in size. Although other technologies, such as protein-based direct DNA capturing from bacterial lysate, can be used to make high-quality MC, they are very costly and have the difficulty to scale-up the production.14,15 The TriD technology has also been used to prepare high-quality plasmid DNA previously.16–18 The TriD technology is based on the formation of a reversible triple-helical structure between a single-stranded poly-pyrimidine oligonucleotide (olig) and a corresponding double-stranded poly-purine sequence in the circular DNA target. The high specificity of the triple-helix structure is mediated by Hoogsteen hydrogen bonding between triplets of the T+-AT or C+-GC. After formation of TriD complex between the biotinylated olig and plasmid in the bacterial lysate, the TriD is captured by streptovidine-coated beads and isolated by magnet. After extensive washing, the plasmid is released from the TriD complex and the olig-streptovidine-coated bead is recyclable (Figure 1, 2). This DNA base technology is more convenient to work with. Therefore, we set off to modify this technology for making high-quality MC. We used the MC product prepared by affinity column as the starting material, and the residual PP and PB circle became the impure DNAs to be captured and eliminated. Importantly, we determined and integrated the factors that increase the TriD formation efficiency into our TriD system. Consequently, the resulted TriD-mediated MC purification system allows elimination of almost all PP and PB DNAs from MC products conveniently and cost-effectively with ease to scale-up MC production. In concert with our previous engineered bacterium base MC production system,10 the present work will further facilitate clinical application of the MC technology.
Figure 1.
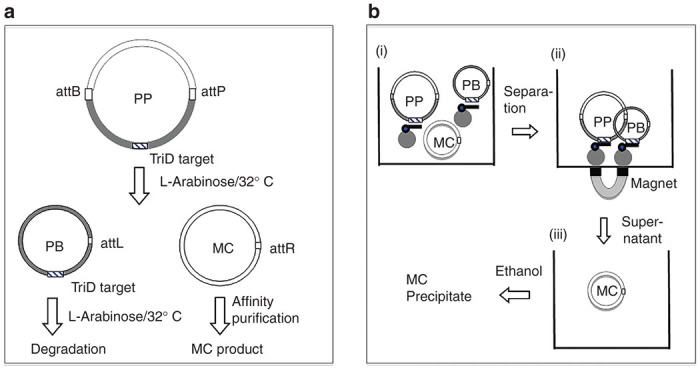
Schemes illustrating minicircle (MC) generation and triplex DNA (TriD)-mediated purification. (a) MC generation was conducted according to established protocol.10 Briefly, in the overnight culture of Escherichia coli strain ZYCY10P3S2T transfected with MC producing plasmid (PP), MC generation is induced by addition of L-arabinose which induces expression of both ΦC31 recombinase, which mediates DNA recombination between attB and attP, and endonuclease I-SceI, which mediates destruction of plasmid backbone circle (PB); consequently, MC becomes the only episomal circular DNA to be isolated using affinity column. (b) TriD-mediated MC Purification. (i) MC product is incubated with biotinylated DNA olgionucleotide (Olig), which forms triplex DNA complex with corresponding TriD forming sequences in the PP and PB. (ii) Subsequently, the TriD complex is removed by streptovidin-coated magnetic beads. (iii) The MC solution is collected and MC is precipitated with ethanol and reconstituted in TE. attB & attP, bacterial and bacteriophage attachment sites, respectively; attL & attR, the hybrid sequences; the tobacco pipe-like shape, biotinylated Olig; the solid circle, streptovidin bead; the diamond, TriD forming sequence.
Figure 2.

Construction of producing plasmids (PPs) encoding different transgenes and triplex DNA (TriD) target sequences. (a) Schemes illustrating the structure of PPs and the expression cassette; (b) the resulted PP5, 6, and 7, the promoters, transgen and poly A, and the PP size; (c) the TriD forming sequences in the resulted PP. KanR, kananmycin resistance gene; ISce1sx32, 32 ISce1 restriction sites; TriD FS, TriD forming sequence; LDLr, low density lipoprotein receptor gene or its promoter; DsRed, red fluorescent gene; Luc, luciferase gene; poly A, polyadenylational signal; bpA, bovine polyadenylational signal
Results
Enhancement of purification efficiency by increasing the length of TriD forming sequences
We determined the length of TriD forming sequences on the MC purification efficiency based on the well-characterized discrete Bio15 (ref. 17) and consecutive Bio21 (refs. 16,19) by others. We demonstrated that the TriD treatment was able to bring down the impure PPs in the MC product to far below the levels less than 1%, the limit as set by Food and Drug Administration12 in all reactions. Specifically, in the discrete Bio15 group, the residual impure DNAs was brought down to 0.63% from about 2.5% for the PP1 encoding an 15-bp TriD forming sequence, but was 0.16%, almost fourfold less, for the PP2 encoding two 45-bp TriD forming sequences (Figure 3b, PP1 versus PP3, P < 0.01). A similar trend was demonstrated in the consecutive Bio21 group, where the impure DNAs were brought down to 0.41-, 0.26-, and 0.10% with the PPs encoding 21-, 42- and 84-bp TriD forming sequences, respectively (Figure 3b, PP3 versus PP4, P < 0.01; PP3 versus PP5, P < 0.001; PP4 versus PP5, P < 0.001). These results were confirmed by gel electrophoresis, where the impure DNAs were obvious before TriD treatment but became undetectable in all the TriD-treated samples (Figure 3a).
Figure 3.
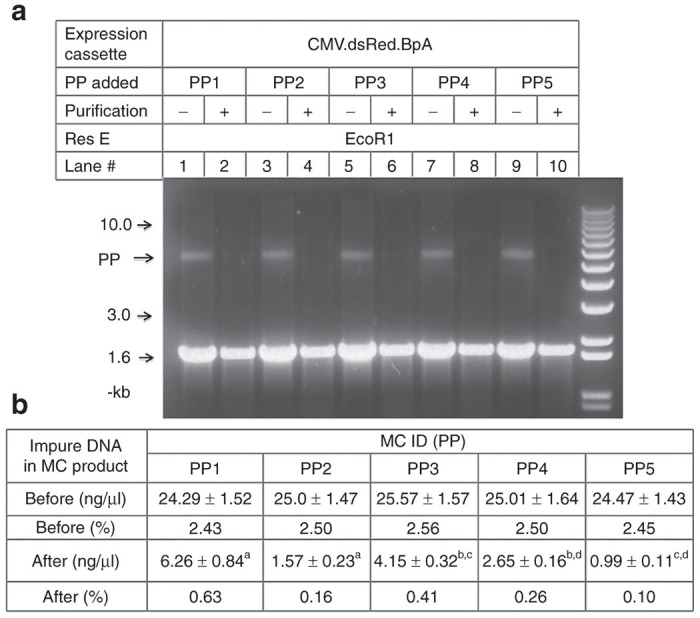
Influence of the composition of triplex DNA (TriD) forming sequence on the purification efficiency. About 2.5% of the PP1-5 (Figure 2c) encoding different TriD forming sequences were added to the minicircle (MC) products prepared with PP5 before TriD-mediated purification. (a) Gel electrophoresis photo illustrating the MC products before and after TriD-mediated purification; all lanes loaded with 1 µg of EcoR1-restricted MC samples; (b) Quantity of impure DNAs in the MC products before and after TriD-mediated purification as determined by qPCR. bP < 0.01; a,c,dP < 0.001. Res E, restriction enzyme. PP, producing plasmids.
Elimination efficiency in MC products reconstituted with different proportion of impure PPs
Because the proportion of the impure DNAs in the MC products varies greatly with different transgenes or MC production technologies, we conducted experiments to determine the purification efficiency of MC products reconstituted with variable amounts of PPs as impure DNAs. About 1-, 2-, 5- and 10% of the PP2 carrying either the discrete 90-bp target sequence or the PP5 encoding the consecutive 84-bp sequence, respectively, were added into MC products made using the corresponding PPs. We demonstrated that in the Bio21 group the TriD technology was able to eliminate 92- to 97% of the impure PPs and bring them down to 0.03- to 0.8% in the final MC products. The efficiency was a little lower in the Bio15 group, with the elimination efficiency ranging from 88- to 95% and the residual PPs from 0.05- to 1.2% (Figure 4a,b). In both groups, the residual PPs in the purified MC products were increased slightly with the increased percent of the PPs in the starting MCs, but all of them were far below the 1% limit set by Food and Drug Administration.12 It is obvious that the present TriD technology is so efficient that the impure PPs in the MC products can be eliminated to a levels lower than the most restrict requirements, even when the impure PPs levels is as high as 10% in the starting MC products. Furthermore, these results confirmed that the consecutive Bio21 was more efficient than the discrete Bio15, with statistically significant difference at all levels of PP contamination (Figure 4c).
Figure 4.
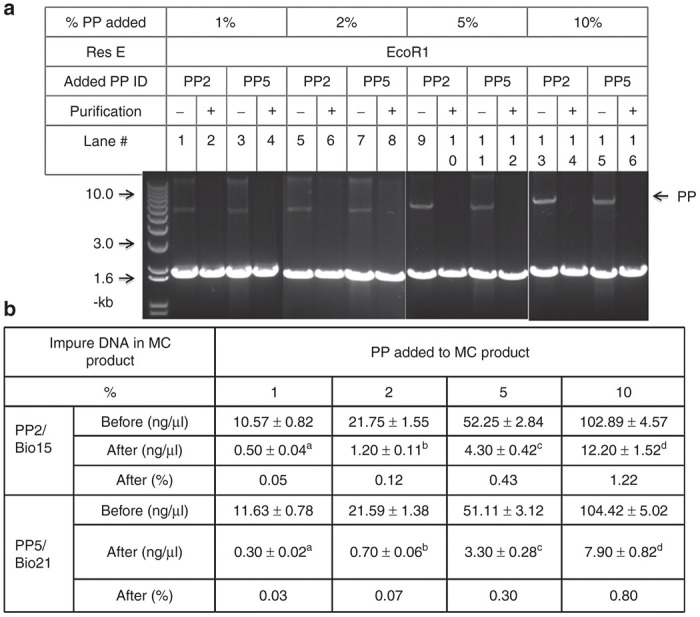
Efficiency of triplex DNA (TriD)-mediated purification with the minicircle (MC) products reconstituted with different proportion of impure producing plasmids (PPs). (a) Gel illustration of MC products before and after TriD-mediated purification. About 1-, 2-, 5-, and 10% PP5 were added into MCs made using PP2 or PP5 before purification. Each lane loaded with 1 μg of EcoR1-restricted MC product. (b) Amount of impure DNAs in the MC products as determined by qPCR before and after purification. c,dP < 0.05; a,bP < 0.01.
Purification efficiency with impure PPs of different size and in larger scale reactions
Because MCs may encode different expression cassettes so that the impure PPs may have different sizes, it is desirable to determine if the PP size influences the purification efficiency. Furthermore, in most preclinical studies, milligrams of MC are needed. Therefore, it is also interesting to determine the purification efficiency in larger scale reactions. To do these, PP5, PP6, and PP7, the size of which is 5.8-, 6.8-, and 9.0-kb (Figure 2a,b), respectively, were used to make MC in 400 ml overnight culture. All of these PPs encoded the same 82-bp consecutive TriD forming sequences so that the Bio21 was used in all the purification reactions. The size of the PPs covers most of the PPs used in the literatures. Consistent with previous study, the MC yields ranged from 4.0 to 5.5 mg per liter of overnight culture, which were 1.4–2.1-folds of that of their unrecombined PP counterparts in molar base.10 The increased molar amount of the MC was due to the continued amplification of the PPs during the additional 5 hours of incubation period. The MC products carried 1.50- to 1.95% of the impure PPs. The TriD reactions were expanded to 5 ml with 2 mg of MC products. As illustrated in Figure 5a,b, the residual PPs were brought down to about 0.03% in all the three final MC products independent of the PPs size. The MCs recovery rates ranged between 94- and 95%.
Figure 5.
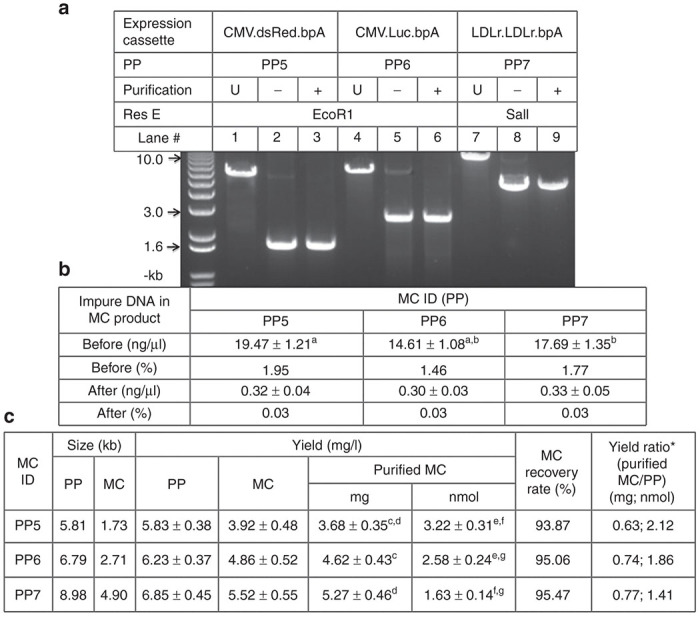
Effect of impure producing plasmid (PP) size on triplex DNA (TriD)-mediated purification efficiency. (a) Gel illustration of the purification efficiency. About 1.5–2.0% of the PP5, 6, and 7, the size of which is 5.8-, 6.8-, and 9.0-kb, were added to the corresponding minicircle (MC) products. Each lane was loaded with 1 μg of MC restricted with either EcoR1 or Sal1. (b) Impure PPs in the MC products as determined by qPCR. (c) The yield and recovery rate of the purified MCs. U, unrecombined PP. * The ratio of the yield based on weigh (mg) or mole (nmol). b,c,e P < 0.05; a,d,f,g P < 0.01.
Discussion
In this study, we report the development of an efficient, convenient and cost-effective TriD base technology capable of eliminating the impure DNAs, including the PP and the PB, from MC products almost completely. It was demonstrated repeatedly that this technology was able to eliminate more than 99% of the contaminated PPs and PBs from the MC products, and bring them down from as high as 10% to below 0.1% levels. This is far below the limits set by World Health Organization,11 Food and Drug Administration,12 and European Medicines Agency13 for clinical application of plasmid DNA in vaccination. Importantly, this enhanced TriD technology was very convenient because it needed only building in the TriD forming sequences in the PP using the standard DNA cloning technique, and the reactions from TriD formation through MC recovery could be finished in 4 hours. Furthermore the cost was very low because the major material biotinylated olig and other consumable materials were inexpensive. Although the cost of streptavidin bead was high, only a small amount was used, and furthermore, it was recyclable. Therefore, the TriD technology can be used to eliminate the major safety concern and bring MC technology one step closer to clinical application.
Others have used TriD technology to make high-quality plasmids by isolating them directly from bacterial lysate.16–18 Theoretically, high-quality MC can also be made by this technology. However, it will not be practicable for making clinical scale of MC due to the high cost. It has been found in several studies that the maximum magnetic bead capacity is 2–5 μg DNA per mg of bead for the target plasmids of around 5-kb. This is consistent with the specification of the manufacturer for the M280 streptovidin beads that 1 mg of the bead can bind 3–10 µg of plasmid of 2–4 kb in size. We found similar results in this study where 1 mg of streptavidin-coated bead was able to bind 3 μg of targeted plasmids of 5–8 kb. Therefore, 1 mg of MC produced by the direct isolating protocol would need 200–500 mg of the bead. In contrast, using the present TriD technology, it needs only about 5% of the beads to prepare 1 mg of highly pure MC from the MC product with up to 10% of impure PP and PB, and the bead amount can be cut down further for most MC preparations where less 2% impure PPs are found. The cost difference of the bead will become a significant factor when making MC in industrial scale.
We have conducted a series of experiments to optimize the TriD system using the 15- and 21-TriD sets, including the biotinylated oligs and their PP counterparts, which have been characterized previously.16,17,19 In the side-by-side experiments, we demonstrated that the 21-nt set worked significantly more efficient than the 15-nt one, and that the longer the TriD forming sequence in the PPs, the higher efficient to be eliminated. Up to fourfold increase in the purification efficiency has been achieved by simply increasing the length of the TriD forming sequence in the PP from 21- to 84-bp. Furthermore, we showed that the composition of the TriD formation sequence was an important factor. The TriD set comprised the consecutive sequence worked much more efficient than the one comprises the discrete sequence, even when the length of the TriD target sequences was about the same. This should be easy to understand because in the consecutive TriD set, the frequency of the TriD formation between the biotinylated olig and the PP is more than that in the discrete set. For example, there are eight potential TriD complexes generated by the Bio21 with each of the consecutive 42-bp corresponding PP target sequence, while only three in the Bio15 and the discrete 45-bp TriD target sequence. In summary, we have defined the factors that optimize the formation of the TriD complex between the oligs and the plasmids, and consequently have developed an enhanced TriD-mediated MC purification system that will facilitate clinical application of the MC technology.
Materials and Methods
Materials
Fifteen-nucleotide discrete biotinylated TriD forming olig 5′-TCTTCTTTCCTCTTT (Bio15), 21-nt consecutive TriD forming olig 5′-CTTCTTCTTCTTCTTCTTCTT (Bio21), and all other DNA oligs were synthesized by Invitrogen China (Shanghai, China); the streptavidin-coated magnetic beads (Dynabead M-280 Streptovidin) and magnet were purchased from Life Technologies China (Shanghai, China). Accoriding to the vendor, 1 mg of beads typically binds 700 pmoles of free biotin, 200 pmoles of biotinylated oligs, and 3–10 µg of a 2–4 kb double-stranded DNA.
Construction of PPs carrying TriD forming sequences
To determine the influence of the size and composition of the target DNA on the TriD forming efficiency, MC PP pMC.CMV.dsRed.bpA (Figure 2a; ref. 10) was used to make PPs encoding the TriD forming sequences flanking the kanamycin resistance genes. One group carries one 15- or two 45-bp of discrete homopyrimidine sequence(s) ((AGAAGAAAGGAGAAA)1 or 3) at the 5′ or both ends of the Kanamycin resistance gene, resulting in PP1 and PP2, respectively (Figure 2a). Both PPs were capable of forming TriD with Bio15. Another group carries one 21- or two 42-bp of the consecutive homopyrimidine ((GAA)7 or 14) at the 5′ or both ends of the kanamycin resistance gene, resulting in PP3, PP4, and PP5. All these PPs were capable of forming TriD with Bio21. All the polymerase chain reaction (PCR) primers generating these PPs were shown in Table 1. The kanamycin resistance gene of pMC.CMV.DsRed.bpA was used as the PCR template to generate kanamycin resistance gene with desired TriD forming sequences. The PCR products were cut with XhoI, and used to replace the Kanamycin resistance gene of the original plasmid, resulting in a series of PPs carrying different TriD forming sequences (Figure 2a,c).
Table 1. Oligonucleotides used for construction of TriD forming producing plasmids in this study.
|
DNA oligonucleotides | |
|---|---|
| Name | Sequence |
| DisO-15-F | CTGCTCGAG AGAAGAAAGGAGAAA TTAATAATTCAGAA GAACTCGTCAAGAA |
| DisO-90-F | CTGCTCGAG AGAAGAAAGGAGAAAAGAAGAAAGGAGAAAAGAAwGAAAGGAGAAATTAATAATTCAGAAGA ACTCGTCAAGAA |
| DisO-90-R | ACTGCTCGAGTTTCTCCTTTCTTCTTTTCTCCTTTCTTCTTTTCTCCTTTCTTCTCACGTAGAAAGCCAGTCCGCA |
| O-R | ACTGCTCGAGCACGTAGAAAGCCAGTCCGCA |
| ConO-21-F | CTGCTCGAG(GAA)7TTAATAATTCAGAAGAACTCGTCAAGAA |
| ConO-42-F | CTGCTCGAG(GAA)7TT(GAA)7TTAATAATTCAGAAGAACTCGTCAAG |
| ConO-84-F | CTGCTCGAG(GAA)14TTAATAATTCAGAAGAACTCGTCAAGAA |
| ConO-84-R | ACTGCTCGAG(TTC)14CACGTAGAAAGCCAGTCCGCA |
Triplex DNA (TriD) target sequences are in boldface and in italics.
Because the unrecombined PPs are one of the two impure DNAs in the MC products, it is desirable to determine if the size of the PPs could influence the TriD purification efficiency. To do this, two expression cassettes were used to replace that in PP5 (5.8-kb), resulted in additional two PPs, the PP6 (6.8-kb) and PP7 (9.0-kb), with different sizes (Figure 2b). All the three PPs encoding two (GAA)14 consecutive sequence and are able to form TriD with Bio21. The DNA oligs used to make these PPs were listed in Supplementary Data (Supplementary Table S1).
Production of MC DNA by two-stage fermentation
MCs were made using the previously established protocol10 with modification. Briefly, a seed culture was prepared using one transformed colony in 20 ml TB with kanamycin (50 μg/ml) in a 250 ml Conical flask and cultured at 37 °C with shaking at 250 rpm until reaching the logarithmic phase (OD A600 = 2.0). Subsequently, an expanded culture was carried out in a 500 ml flask containing 50 ml TB and kanamycin (50 μg/ml) with 2 ml of the seed culture growth. The culture was incubated at 37 °C with shaking at 250 rpm until reaching the stationary phage (OD A600 = 4.5) before combining with an equal volume of pre-made Induction Mix. The Induction Mix comprises 50 ml LB and 50 μl of 20% filtered L-arabinose. After mixing well, sodium hydroxide (1 mol/l) was added to adjust the Induction Mixture to pH 7.0. The culture continues at 30 °C and shaking at 250 rpm for additional 5 hours before the reaction is terminated. MC was isolated using Qiagen kit (Invitrogen, Catalogue Number 12183). Because the bacterial mass was greatly increased during the additional induction time, double volume of all buffers, including the suspension, lysate, and neutralization buffers, was used.10 In the scale-up experiment, the culture was carried out in a 2 l flask containing 400 ml growth medium TB and kanamycin (50 μg/ml) with 16 ml of the seed culture growth. The overnight culture was combined with the Induction Mix comprises 400 ml LB and 400 μl of 20% filtered L-arabinose before the incubation continued to induce MC production.
Purification of MC by TriD technology
The TriD-mediated purification was conducted using the MC products made using the Qiagen Kit as the starting material. As shown in Figure 1b, the MC product, biotinylated DNA olig, and streptavidin-coated magnetic beads were mixed in binding buffer comprising 0.2 mol/l acetate buffer (pH 4.5) containing either 2 mol/l NaCl or 0.1 mol/l MgCl2. The 2 ml reactions comprised 400 μg MC, 200 μMoles biotinylated olig, and 10 mg magnetic beads. The expanded 5 ml reactions comprised 2 mg MC, 1.0 mMoles biotinylated olig, and 50 mg magnetic beads. The mixure were incubated at room temperature (≈25 °C) for 2 hours with gentle stirring to promote TriD formation. At the end of the 2 hours incubation reaction, the TriD is removed by magnetic separation, and the purified MC was recovered from the supernatant by ethanol precipitation. The MC products were adjusted to 1 µg/µl before and after TriD-mediated purification. The magnetic bead-biotinylated olig was recycled after releasing the captured impure DNAs from the TriD complex. To do this, the bead-biotinylated olig-PP TriD was resuspended in cold borate dissociation buffer (pH 9.0) containing 0.5 mmol/l ethylenediaminetetraacetic acid and incubated for 0.5 hours. The resulted magnetic bead-biotinylated olig was recovered by magnet, washed with PBS buffer (pH 7.2) and stored at 4 °C until next use.
qPCR determination of purification efficiency of triplex affinity
The presence of impure DNAs (PBs and PPs) in the MC products was determined by absolute quantitative qPCR. The MC producer plasmid PP5 was used as a standard. The PCR primers kan-F (5′- GCCCAATAGCAGCCAGTCC) and kan-R (5′-TGATGCCGCCGTGTTCC) (Supplementary Table S1) were specific for kanamycin resistance gene shared by all PPs. qPCR was performed using the Qiagen QuantiFast SYBR Green PCR kit and the machine of Roche system 480 (Shanghai, China). The PCR program included a denaturing step of 94 °C for 5 minutes, followed by 40 cycles of 94 °C for 20 seconds, and 68 °C for 20 seconds each. Under these experimental conditions, the correlation equation was CT = −3.1226 lgX0 + 45.26 (R2 = 0.9988), where X0 is the copy number of DNA.
Statistics
Triplicate reactions were performed for each assay. Statistical analysis was performed by student’s t-test or analysis of variance test. Data are presented as means ± SD.
Acknowledgments
This work is funded by Shenzhen Government; the Grant No. are SFG 2012566 and SKC 2012237. Z-.Y.C. perceived the concept and worked with X.H.H. to design the experiments and write the manuscript. X.H.H., X.Y.G., Y.C., and C-.Y.H. conducted the experiments.
The authors declare no conflict of interest.
References
- Schleef M, Blaesen M, Schmeer M, Baier R, Marie C, Dickson G. Production of non viral DNA vectors. Curr Gene Ther. 2010;10:487–507. doi: 10.2174/156652310793797711. [DOI] [PubMed] [Google Scholar]
- Voss C. Production of plasmid DNA for pharmaceutical use. Biotechnol Annu Rev. 2007;13:201–222. doi: 10.1016/S1387-2656(07)13008-8. [DOI] [PubMed] [Google Scholar]
- Mayrhofer P, Schleef M, Jechlinger W. Use of minicircle plasmids for gene therapy. Methods Mol Biol. 2009;542:87–104. doi: 10.1007/978-1-59745-561-9_4. [DOI] [PubMed] [Google Scholar]
- Chen ZY, He CY, Meuse L, Kay MA. Silencing of episomal transgene expression by plasmid bacterial DNA elements in vivo. Gene Ther. 2004;11:856–864. doi: 10.1038/sj.gt.3302231. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Riu E, He CY, Xu H, Kay MA. Silencing of episomal transgene expression in liver by plasmid bacterial backbone DNA is independent of CpG methylation. Mol Ther. 2008;16:548–556. doi: 10.1038/sj.mt.6300399. [DOI] [PubMed] [Google Scholar]
- Jia F, Wilson KD, Sun N, Gupta DM, Huang M, Li Z. A nonviral minicircle vector for deriving human iPS cells. Nat Methods. 2010;7:197–199. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, He CY, Ehrhardt A, Kay MA. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther. 2003;8:495–500. doi: 10.1016/s1525-0016(03)00168-0. [DOI] [PubMed] [Google Scholar]
- Bigger BW, Tolmachov O, Collombet JM, Fragkos M, Palaszewski I, Coutelle C. An araC-controlled bacterial cre expression system to produce DNA minicircle vectors for nuclear and mitochondrial gene therapy. J Biol Chem. 2001;276:23018–23027. doi: 10.1074/jbc.M010873200. [DOI] [PubMed] [Google Scholar]
- Darquet AM, Cameron B, Wils P, Scherman D, Crouzet J. A new DNA vehicle for nonviral gene delivery: supercoiled minicircle. Gene Ther. 1997;4:1341–1349. doi: 10.1038/sj.gt.3300540. [DOI] [PubMed] [Google Scholar]
- Kay MA, He CY, Chen ZY. A robust system for production of minicircle DNA vectors. Nat Biotechnol. 2010;28:1287–1289. doi: 10.1038/nbt.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO 2007. The WHO Expert Committee on Biological Standardization 56th Report: Number 941. World Health Organization; Geneva. [PubMed] [Google Scholar]
- FDA 2007. Guidance for Industry: Considerations for Plasmid DNA Vaccines for Infectious Disease Indications.
- EMEA 2000. Note for Guidance on the Quality, Preclinical and Clinical Aspects of Gene Transfer Medicinal Products: CPMP/BWP/3088/3099.
- Jechlinger W, Azimpour Tabrizi C, Lubitz W, Mayrhofer P. Minicircle DNA immobilized in bacterial ghosts: in vivo production of safe non-viral DNA delivery vehicles. J Mol Microbiol Biotechnol. 2004;8:222–231. doi: 10.1159/000086703. [DOI] [PubMed] [Google Scholar]
- Mayrhofer P, Blaesen M, Schleef M, Jechlinger W. Minicircle-DNA production by site specific recombination and protein-DNA interaction chromatography. J Gene Med. 2008;10:1253–1269. doi: 10.1002/jgm.1243. [DOI] [PubMed] [Google Scholar]
- Costioli MD, Fisch I, Garret-Flaudy F, Hilbrig F, Freitag R. DNA purification by triple-helix affinity precipitation. Biotechnol Bioeng. 2003;81:535–545. doi: 10.1002/bit.10497. [DOI] [PubMed] [Google Scholar]
- Schluep T, Cooney CL. Purification of plasmids by triplex affinity interaction. Nucleic Acids Res. 1998;26:4524–4528. doi: 10.1093/nar/26.19.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Smith CL, Cantor CR. Sequence-specific DNA purification by triplex affinity capture. Proc Natl Acad Sci USA. 1992;89:495–498. doi: 10.1073/pnas.89.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils P, Escriou V, Warnery A, Lacroix F, Lagneaux D, Ollivier M. Efficient purification of plasmid DNA for gene transfer using triple-helix affinity chromatography. Gene Ther. 1997;4:323–330. doi: 10.1038/sj.gt.3300388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.