

Abstract
The first commercially approved human gene therapy in the Western world is Glybera (alipogene tiparvovec), which is an adenoassociated viral vector encoding the lipoprotein lipase gene. Glybera was recommended for marketing authorization by the European Medicines Agency in 2012. The European Medicines Agency had only ever reviewed three marketing authorization applications for gene therapy medicinal products. Unlike in the case of Glybera, the applications of the first two products, Cerepro and Contusugene Ladenovec Gendux/Advexin, both of which were for cancer diseases, were withdrawn. In this report, we studied the European public assessment reports of the three gene therapy products. During the assessment process, Glybera was re-examined and reviewed for a fourth time. We therefore researched the re-examination procedure of the European Union regulatory process. Approximately 25% of the new medicinal products initially given negative opinions from the Committee for Medicinal Products for Human Use were ultimately approved after re-examination from 2009 to 2013. The indications of most medicines were changed during the re-examination procedure, and the products were later approved with a mode of approval. These results suggested that the re-examination system in the European Union contributed to the approval of both several new drugs and the first gene therapy product.
Introduction
The first commercially approved gene therapy product in the Western world was Glybera (alipogene tiparvovec), which is based on an adenoassociated viral (AAV) vector for the treatment of the inherited metabolic disorder lipoprotein lipase (LPL) deficiency (LPLD).1,2 The marketing authorization for Glybera, which was valid throughout the European Union (EU), was issued on 25 October 2012. This approval made a great impact on the world, and it opened the door for a gene therapy market in the near future.
In the EU, advanced-therapy medicinal products (ATMPs) are medicines for human use that are based on gene therapy, somatic-cell therapy or tissue engineering. The regulatory framework for ATMPs was established in 2007,3 and the Committee for Advanced Therapies (CAT) was in place in the European Medicines Agency (EMA) in 2009. The CAT is a multidisciplinary committee of the best experts in Europe tasked to assess the quality, safety, and efficacy of ATMPs and to prepare a draft opinion on each ATMP application before the Committee for Medicinal Products for Human Use (CHMP) adopts their opinion. Several researchers have noted that this EU regulatory framework for advanced therapies contributed to their approval of the first gene therapy product.4 In addition, some groups have shown that many factors such as animal models and other regulatory aspects also encouraged the marketing approval of Glybera.5–10 From the viewpoint of the recapitulation of the features of human LPLD by animal models, the spontaneously generated LPL catalytically inactive mutant cats was critical for the approval of Glybera,5,7,10 though only 27 human patients were involved in the clinical trials.6,8 Glybera was designated as an orphan medicinal product in the EU.11 In their review article, Melchiorri et al. stated that all of the orphan drugs for ultra-rare diseases, which they defined as a prevalence of less than 1 in 100,000, ever assessed by CHMP were for disease conditions that follow either a continuously progressive course of deterioration or a very active disease course if left untreated.9 In contrast, LPLD results in pancreatitis, which exhibits a much more unpredictable and fluctuating disease course. Therefore, Melchiorri et al. explained that either a large clinical study or a long follow-up would be required as the clinical trial for Glybera. Because there were limitations incurred by its ultra-rare designation, Glybera was approved under exceptional circumstances after a re-examination of the CHMP’s opinion.5,8,9 “Under exceptional circumstances”12 and “conditional marketing authorization”13 are types of approval of a drug for early market access in the EU.14
Before Glybera, the marketing authorization applications (MAAs) for other gene therapies, Cerepro15,16 and Contusugene Ladenovec Gendux (CLG)17 (formerly named Advexin),18 were submitted to the EMA but were withdrawn at the end of the process. Cerepro was intended for the treatment of high-grade glioma. CLG and Advexin were the same products but had different indications. CLG was for recurrent or refractory squamous cell carcinoma of the head and neck (SCCHN), and Advexin was for Li-Fraumeni cancer.
In this article, we studied the assessment processes of Glybera, Cerepro, and CLG/Advexin to discuss issues for gene therapy products as these three are the only gene therapy products to be evaluated by the EMA so far. Furthermore, we summarized the recent results of the re-examination of the new medicinal products and ATMPs in the EU because, in our opinion, we thought that the re-examination procedure was one of the key events in the approval of Glybera; there is no such re-examination system in the United States or Japan. In addition, we investigated the modes of approval for the re-examined new medicinal products.
Results
Overview of gene therapy products in the EU
The information for Glybera, Cerepro, and CLG/Advexin is summarized in Table 1. The regulatory events for the products are illustrated in the timeline in Figure 1. Glybera, Cerepro, and Advexin were granted orphan designations in 2004, 2002, and 2006, respectively. CLG is the same product as Advexin but is not an orphan drug because its indication SCCHN is not a rare disease in the EU. The MAA of Advexin was submitted before the framework for ATMPs had been established. CLG is classified as an ATMP. The second application of Cerepro was assessed by the CAT as an ATMP and was withdrawn after an adoption of a negative opinion but before the re-examination was finished.
Table 1. Approved or withdrawn gene therapy product applications for marketing in the EU.
| Trade name (INN) | Applicant | Vector | Transgene | Approval/withdrawal date | Indication |
|---|---|---|---|---|---|
| Glybera (alipogene tiparvovec) | Amsterdam Molecular Therapeutics B.V./uniQure biopharma B.V.a | Adenoassociated virus | Human lipoprotein lipase (LPL) gene variant LPLS447X | Approved, 25 October 2012 | For adult patients diagnosed with familial LPL deficiency and suffering from severe or multiple pancreatitis attacks despite dietary fat restrictions |
| Cerepro (sitimagene ceradenovec) | Ark Therapeutics Ltd.b | Adenovirus | Herpes simplex virus-thymidine kinase gene | Withdrawn, 8 March 2010 | In combination with ganciclovir sodium for the treatment of operable high grade glioma |
| (Adenovirus-mediated herpes simplex virus-thymidine kinase gene) | Withdrawn, 13 July 2007 | ||||
| Contusugene Ladenovec Gendux (contusugene ladenovec) | Gendux Molecular Ltd. (Introgen Therapeutics Inc.)c | Adenovirus | Human p53 tumor suppressor gene | Withdrawn, 12 June 2009 | In adults for the treatment of patients with recurrent or refractory squamous cell carcinoma of the head and neck as monotherapy |
| Advexin (contusugene ladenovec) | Withdrawn, 17 December 2008 | For the treatment of Li-Fraumeni cancer patients |
EU, European Union; INN, International nonproprietary name.
Applicant for the marketing authorization for Glybera was changed from Amsterdam Molecular Therapeutics to uniQure biopharma B.V. on 12 July 2012.
Ark Therapeutics Group plc sold its operating subsidiaries, including Ark Therapeutics Ltd., to WKD Holding Oy on 15 March 2013.
Introgen Therapeutics, Inc. was a parent company of Gendux Molecular Ltd.
Figure 1.

Regulatory timeline of gene therapy products; Glybera, Cerepro, and Contusugene Ladenovec Gendux (CLG)/Advexin. Glybera was approved after the third negative opinion. Each marketing authorization application of Cerepro or CLG/Advexin was submitted twice and withdrawn. The adenovirus-mediated herpes simplex virus-thymidine kinase (Cerepro, first time) was submitted before the advanced-therapy medicinal products (ATMPs) framework started. CLG was not designated as an orphan medicine, whereas Advexin (the same product as CLG) was granted an orphan designation. AMT, Amsterdam Molecular Therapeutics Holding N.V.; CHMP, Committee for Medicinal Products for Human Use; CLG, Contusugene Ladenovec Gendux; EC, European Commission; HSV-tk, herpes simplex virus-thymidine kinase; MAA, Marketing Authorization Application; uniQure, uniQure N.V.; WKD, WKD Holding Oy.
Glybera is indicated for LPLD. Cerepro and CLG/Advexin were indicated for cancer. The clinical trials of Glybera, Cerepro, and CLG/Advexin are summarized in Table 2. The clinical trials for Glybera used only historical controls. The clinical trials for Cerepro or CLG/Advexin were controlled to compare the efficacy with the standard therapy. There were no treatment approaches for LPLD, which is Glybera’s target, available for comparison, except for dietary restrictions that are difficult to maintain and do not always have a sufficient effect.11 The most severe complication associated with LPLD is pancreatitis, and the frequency of the attacks of pancreatitis varied across the patients. The frequency of pancreatitis is a patient-oriented outcome, i.e., its clinical relevance is high. This value is the possibly best measure of efficacy and can be used with a historical control to compare before and after administration of Glybera per each patient. Gene therapies must be established in regard to their efficacy, as for all other medicines. Glybera was shown to reduce the frequency of pancreatitis and was ultimately approved.
Table 2. Main clinical trials of approved or withdrawn gene therapy products.
| Trade name | Clinical trial phase | Patients number |
Investigational medicine |
Types of study | |
|---|---|---|---|---|---|
| Subjected | Control | ||||
| Glybera | 1/2 | 8 | 8 | 0 | Single-center (the Netherlands), two doses, open-label |
| 2/3 | 14 | 14 | 0 | Single-center (Canada), dose-escalation, open-label | |
| 2/3 | 5 | 5 | 0 | Dual-center (Canada), single dose, open-label | |
| (total 27) | |||||
| Cerepro | 1 | 12 | 0 | 12* | Single-center (Finland), dose defining |
| 1 | 14 | 7 | 7a, 7b | Single-center (Finland), single dose, nonrandomized, open-label | |
| 2 | 36 | 17 | 19 | Single-center (Finland), single dose, randomized, open-label | |
| 3c | 251d | 124 | 126 | Multicenter (Europe and Israel), single dose, randomized, open-label | |
| (total 313) | |||||
| CLG/Advexin | 1 | 34 | 34 | 0 | Single-center (USA), nonrandomized, open-label |
| 2 | 61 | 61 | 0 | Multicenter (USA), open-label | |
| 2 | 112 | 107 | 5 | Multicenter (USA), dose controlled (two doses), randomized, open-label | |
| 3 | 123 | 63 | 60 | Multicenter (USA), dosing study, randomized, open-label | |
| (total 330) | |||||
CLG, Contusugene Ladenovec Gendux.
Retroviral herpes simplex virus-thymidine kinase gene treated.
Historical control from the asterisked(*) clinical trial.
The data from this phase III clinical trial was used only for the second marketing authorization application submission.
One patient withdrew consent.
Re-examination procedure of Glybera
Glybera encountered negative opinions from the CHMP three times during its assessment process (Figure 1). The CAT and the CHMP noted these concerns in their negative opinions in June 2011: insufficient evidence of a persistent effect in lowering blood fats, insufficient evidence at this stage of a reduction in the pancreatitis rate, concern over the risk associated with the administration of Glybera into patients’ muscles, and the use of immunosuppressive medicines.11 During its first re-examination in October 2011, the CAT thought that the efficacy and safety of Glybera was sufficiently demonstrated in selected patients with LPLD suffering from at least one pancreatitis episode and concluded that the concerns could be addressed with additional postmarketing studies. However, the CHMP maintained their negative opinion, and the European Commission (EC) requested a re-evaluation of Glybera in a restricted indication of patients with severe or multiple pancreatitis attacks. Though the CHMP maintained a negative opinion in April 2012, Glybera was evaluated again for a formal opinion of the CAT and was finally recommended for approval in July 2012. This sequence of events suggests that the re-examination procedure gave the agency and the applicant an opportunity to find a subgroup in which the benefits outweigh its risks, which contributed to the drug’s approval.
Re-examination of new medicinal products
The legal basis for re-examination is under Regulation (EC) No 726/2004.19 The regulation emphasizes an enlargement of the scientific committees, scientific advice and evaluation by scientific experts, as well as re-examination procedures for a better assurance of applicants’ rights.
We counted the number of re-examined new medicinal products, including the ATMPs from 2009 to 2013 (refs. 20–22) (Figure 2), as the “Procedural advice on the re-examination of CHMP opinions” was last updated in February 2009. The CHMP adopted 204 positive first opinions23 and 28 negative first opinions for new medicines. Among these 28 negative first opinions for new medicines, 18 products were re-examined. As a result, the CHMP changed their opinion on 7 products from negative to positive, which is 25% (7 of 28) of all of their negative opinions.
Figure 2.
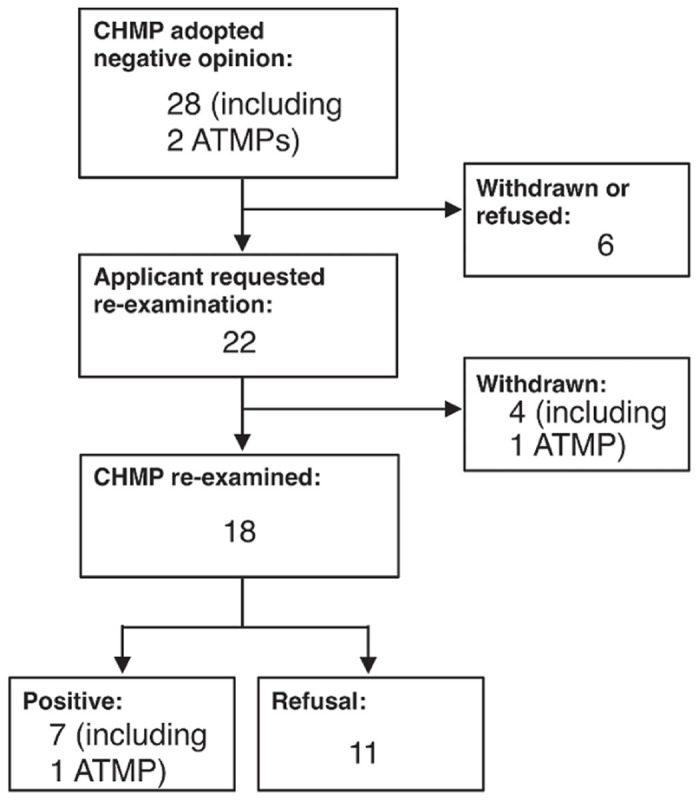
Summary of re-examination from 2009 to 2013. The numbers and results of the re-examination of the new medicinal products and ATMPs from January 2009 to December 2013 are shown as a flow chart. ATMP, advanced-therapy medicinal products; CHMP, Committee for Medicinal Products for Human Use.
Information about the seven re-examined and approved new medicinal products are shown in Table 3. It also shows that the indications were changed during the re-examination. Of the re-examined products, six of seven medicines (86%) were for rare diseases (Table 4). Of all of the new medicinal products assessed from 2009 to 2013, 32 of the 219 products (~15%) were orphan medicines.23 It was revealed that the ratio of orphan drugs in the re-examined and approved medicines is much higher than that of other medicines.
Table 3. Re-examined and approved new medicinal products in the EU from 2009 to 2013.
| Trade name (INN) | Therapeutic indication before re-examination | Approved indication | Orphan designation (prevalence in the EU) | Patient numbers in main clinical trials | Mode of approval |
|---|---|---|---|---|---|
| Deltyba (delamanid) | Multidrug-resistant tuberculosis | Use as part of an appropriate combination regimen for pulmonary multidrug resistant tuberculosis in adult patients when an effective treatment regimen cannot otherwise be composed for reasons of resistance or tolerability | Yes (2/10,000) | 481 | CMA |
| Defitelio (defibrotide) | Treatment and prevention of hepatic veno-occlusive disease in blood stem cell transplantation therapy | Severe hepatic veno-occlusive disease in hematopoietic stem-cell transplantation therapy | Yes (0.4/10,000) | 356 (prevention), 102 (treating) | UEC |
| Glybera (alipogene tiparvovec) | The long-term correction of lipoprotein lipase deficiency, to control or abolish symptoms and prevent complications in adult patients clinically diagnosed with lipoprotein lipase deficiency | Familial lipoprotein lipase deficiency and suffering from severe or multiple pancreatitis attacks despite dietary fat restrictions | Yes (0.02/10,000) | 27 | UEC |
| Bronchitol (mannitol) | Cystic fibrosis used either as add-on therapy to rhDNase (another treatment for cystic fibrosis) or on its own in patients who do not benefit from or cannot use rhDNase | Cystic fibrosis in adult patients as an add-on therapy to best standard of care | Yes (1.3/10,000) | 642 | N/A |
| Fampyra (fampridine) | Multiple sclerosis to improve their walking | To improve walking ability of adult patients suffering from multiple sclerosis with walking disability | No (8/10,000) | 540 | CMA |
| Cayston (aztreonam) | Treatment of chronic airway infections due to Pseudomonas aeruginosa in patients with cystic fibrosis aged 18 years and older to improve pulmonary function and respiratory symptoms | Suppressive therapy of chronic pulmonary infection due to Pseudomonas aeruginosa in patients with cystic fibrosis aged 18 years and oldera | Yes (1.3/10,000) | 375 | CMAb |
| Vedrop (tocofersolan) | Vitamin E deficiency due to digestive malabsorption in pediatric patients suffering from cystic fibrosis, congenital chronic cholestasis or hereditary chronic cholestasis | Vitamin E deficiency due to digestive malabsorption in pediatric patients suffering from congenital chronic cholestasis or hereditary chronic cholestasis, from birth (in term newborns) to 16 or 18 years of age, depending on the region | No (intended for rare diseases without orphan designation, prevalence is unknown) | 92 | UEC |
Underlines in the “Therapeutic indication before re-examination” column highlight withdrawn part of the indication. Underlines in the “Approved indication” column highlight additional eligibility for the indication.
CMA, conditional marketing authorization; EU, European Union; INN, International Nonproprietary Name; N/A, not applicable; rhDNase, recombinant human deoxyribonuclease; UEC, under exceptional circumstances.
Extended of therapeutic indications to 6 years and older (June 2012).
Switched to full approval (September 2011).
Table 4. Ratio of orphan medicines on re-examination approval in the EU from 2009 to 2013.
| Total | Orphan medicines (per total) | |
|---|---|---|
| New medicines | 219 | 32 (15%) |
| No re-examined | 212 | 26 (12%) |
| Re-examined | 7 | 6 (86%) |
| Withdrawn/refused medicines | 21 | 7 (33%) |
EU, European Union.
Mode of approval of re-examined medicinal products
Marketing authorization “under exceptional circumstances” is granted to medicines for which the applicant is unable to provide comprehensive data on the efficacy and safety under normal states of use because the indication to be treated is rare or because the collection of comprehensive information is not possible or unethical.12 The medicine authorized under exceptional circumstances is reviewed annually to reassess the risk-benefit balance. Conditional marketing authorization is the approval of a medicine based on less complete clinical data than is normally required because it answers an unmet medical need.13 The conditional marketing authorization is valid for 1 year on a renewable basis. A “normal” marketing authorization can be attained when the applicant completes a full dossier.
Of the re-examined and approved medicines, three were approved under exceptional circumstances, and three were approved by conditional marketing authorization (Table 3). Specific mode of approval was applied according to the strength of evidence by the clinical trials. For example, Cayston, which is intended for a cystic fibrosis (CF) indication, was approved with a conditional marketing authorization because the long-term efficacy was not shown in the clinical trials; however, the drug addressed an unmet medical need for the CF indication.24 Bronchitol was approved without such mode of approval as an add-on therapy for the CF indication but not as a stand-alone therapy because the data only demonstrated efficacy as an add-on therapy.25 Thus, the strength of the evidence led to an approval of Bronchitol regardless of modes of approval for CF, though Cayston, also for CF, was approved by a conditional marketing authorization.
Overall, the re-examination system appears to facilitate the approvals of new medicinal products, including ATMPs, for rare diseases by opening the opportunity to determine the most appropriate indications and modes of approval in a transparent manner. Because the data from the clinical trials for a predetermined indication were often not sufficiently strong for that indication, the re-examination process allowed the regulatory boards to refine the studies to hone in on a specific indication. Orphan drugs are often developed for diseases without effective therapies, so appropriate indications may be difficult to determine before the clinical trial is complete. This may be why the re-examined and approved medicines were almost always orphan drugs that were approved with a mode of approval.
Discussion
In this study, we reviewed the regulatory timeline of the first approved gene therapy products in the Western world, Glybera, and the two withdrawn gene therapy products, Cerepro and CLG/Advexin (Figure 1). Moreover, we focused on the re-examination procedure of the EMA because this system played an important role in the assessment of Glybera.
Glybera, Cerepro, and Advexin were designated as orphan medicines (Figure 1). The target diseases for gene therapy in this opening era are more likely to be rare diseases. As orphan diseases often have no approved therapies, researchers must try to cure the diseases with novel technologies such as gene therapy. It should be noted that ~80% of rare diseases are of genetic origin.26 However, these states have a small number of patients with severe symptoms, which might make clinical trials for gene therapy more difficult to conduct and the benefits of the medicines more difficult to show. Melchiorri et al. demonstrated that the Glybera’s case were unique as LPLD results in unpredictable and fluctuating pancreatitis,9 and the product was approved under exceptional circumstances with the implementation of a specific measures and conditions that would, after approval, allow for systematic enlargement of the database of safety and efficacy.
Regulation (EC) No 726/2004 outlined the maintenance of a centralized procedure for the authorization of medicines and is the legal basis for re-examination. This regulation emerged to consider the probable development of science and technology and the future enlargement of the EU. Thus, the EMA must retain a scientific assessment to ensure the consent of the members with different cultures. At the same time, this regulation guarantees the applicants’ rights to re-examination, as stated in the Whereas clause, the Recital 25 of the Regulation (EC) No 726/2004. The re-examination system was enacted to give opportunities for a new independent assessment system by different rapporteurs and to give the researchers an opportunity to change or refine the indication if the regulatory agency issues a negative opinion. Thus far, most of the products that were approved after re-examination were orphan medicinal products. For some orphan drugs like Glybera, to a priori build the application around a sound indication(s) with an appropriate dataset might have been difficult because of the extremely small number of patients, and the disease symptom. This might be the reason why the applicants started developing drugs for several or broader indications, and the appropriate indication(s) were determined during the re-examination procedure. As science and technology develop, medicines will be more personalized or for rare or severe diseases and will be difficult to evaluate initially. Both regulators and companies should engage to find an appropriate patient population where benefit/risk is positive already in the initial assessment procedure, thus avoiding a time- and resource consuming re-examination. The re-examination system and the modes of approval, however, contributed for the approvals of the medicinal products shown in this study at least as of the moment.
Materials and Methods
Information of approved/withdrawn gene therapy products
In this article, we refer to both gene therapy products and gene therapy product candidates (i.e., investigational medicines) as “gene therapy products.”
The information about the approved or withdrawn applications of gene therapy products was obtained from the appropriate website of the US Food and Drug Administration (FDA),27 the European Medicines Agency (EMA),28 the Pharmaceuticals and Medical Devices Agency (PMDA), Japan (in Japanese),29 and the National Institute of Health Sciences (NIHS), Japan (in Japanese).30 This study includes products comprising gene therapy medicinal products that were approved or withdrawn in the EU until December 2013, i.e., Glybera (international nonproprietary name (INN): alipogene tiparvovec),11 Cerepro (INN: adenovirus-mediated herpes simplex virus-thymidine kinase gene, sitimagene ceradenovec),15,16 Contusugene Ladenovec Gendux (CLG)/Advexin (INN: contusugene ladenovec).17,18 The information about the clinical trials was also found on the ClinicalTrials.gov database31 and in scientific papers.32–35 The year in which the clinical trial was completed was referred to by the European public assessment report or was substituted by the years of science reports for the clinical trial that was published.
Information of re-examined medicinal products
The information about the re-examined new medicinal products were obtained from the following documents: the CHMP monthly reports,20,21 the CAT monthly reports,36 and the European public assessment reports or the questions and answers for each product; Deltyba (INN: delamanid),37 Defitelio (INN: defibrotide),38 Bronchitol (INN: mannitol),25 Fampyra (INN: fampridine),39 Cayston (INN: aztreonam),24 and Vedrop (INN: tocofersolan).40
Acknowledgments
This work was supported by a grant from the Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems titled “Cell Sheet Tissue Engineering Center (CSTEC),” and the Global COE program, the Multidisciplinary Education and Research Center for Regenerative Medicine (MERCREM), from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. The authors are grateful to Mituo Umezu, Hiroshi Kasanuki, Yasuo Ikeda, Shinji Takeoka, Kiyotaka Iwasaki, Atsushi Aruga, Hiroshi Iseki, Toshiyuki Owaki, and Taisuke Ikawa for their helpful and critical discussions. This work was done in Tokyo, Japan.
K.Y. is an employee of Asahi Kasei Medical Co., Ltd. and Asahi Kasei Pharma Co., Ltd. and the holding company, Asahi Kasei Co., Ltd. (JAPAN). T.O. and M.Y. are shareholders of CellSeed Inc. (JAPAN). The other authors declare no conflict of interest.
References
- Wirth T, Parker N, Ylä-Herttuala S. History of gene therapy. Gene. 2013;525:162–169. doi: 10.1016/j.gene.2013.03.137. [DOI] [PubMed] [Google Scholar]
- Gaudet D, Méthot J, Kastelein J. Gene therapy for lipoprotein lipase deficiency. Curr Opin Lipidol. 2012;23:310–320. doi: 10.1097/MOL.0b013e3283555a7e. [DOI] [PubMed] [Google Scholar]
- Official Journal of the European Union Regulation (EC) No 1394/2007 of The European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004 . < http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:324:0121:0137:en:PDF > ( 2007
- Kassim SH, Somerville RP. From lipoproteins to chondrocytes: a brief summary of the European Medicines Agency’s regulatory guidelines for advanced therapy medicinal products. Hum Gene Ther. 2013;24:568–570. doi: 10.1089/hum.2013.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant LM, Christopher DM, Giles AR, Hinderer C, Rodriguez JL, Smith JB. Lessons learned from the clinical development and market authorization of Glybera. Hum Gene Ther Clin Dev. 2013;24:55–64. doi: 10.1089/humc.2013.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno MJ. Gene therapy coming of age – prevention of acute pancreatitis in lipoprotein lipase deficiency through alipogene tiparvovec. Eur Gastroenterol Hepatol Rev. 2010;6:48–53. [Google Scholar]
- Kastelein JJ, Ross CJ, Hayden MR. From mutation identification to therapy: discovery and origins of the first approved gene therapy in the Western world. Hum Gene Ther. 2013;24:472–478. doi: 10.1089/hum.2013.063. [DOI] [PubMed] [Google Scholar]
- Ylä-Herttuala S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol Ther. 2012;20:1831–1832. doi: 10.1038/mt.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchiorri D, Pani L, Gasparini P, Cossu G, Ancans J, Borg JJ. Regulatory evaluation of Glybera in Europe - two committees, one mission. Nat Rev Drug Discov. 2013;12:719. doi: 10.1038/nrd3835-c1. [DOI] [PubMed] [Google Scholar]
- Burnett JR, Hooper AJ. Alipogene tiparvovec, an adeno-associated virus encoding the Ser(447)X variant of the human lipoprotein lipase gene for the treatment of patients with lipoprotein lipase deficiency. Curr Opin Mol Ther. 2009;11:681–691. [PubMed] [Google Scholar]
- European Medicines Agency Assessment report for Glybera . < http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002145/WC500135476.pdf > ( 2012
- European Medicines Agency Guideline on procedures for the granting of a marketing authorisation under exceptional circumstances, pursuant to article 14 (8) of Regulation (EC) No 726/2004 . < http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004883.pdf > ( 2005
- European Medicines Agency Draft guideline on the scientific application and the practical arrangements necessary to implement commission regulation (EC) no 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of regulation (EC) no 726/2004 . < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500004908.pdf > ( 2006
- Boon WP, Moors EH, Meijer A, Schellekens H. Conditional approval and approval under exceptional circumstances as regulatory instruments for stimulating responsible drug innovation in Europe. Clin Pharmacol Ther. 2010;88:848–853. doi: 10.1038/clpt.2010.207. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency Withdrawal assessment report for Cerepro . < http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2010/01/WC500068119.pdf > ( 2009
- European Medicines Agency Withdrawal assessment report for Cerepro . < http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2011/02/WC500101545.pdf > ( 2011
- European Medicines Agency Withdrawal assessment report for Contusugene ladenovec gendux . < http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2010/01/WC500060811.pdf > ( 2009
- European Medicines Agency Withdrawal assessment report for Advexin . < http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2010/01/WC500063080.pdf > ( 2009
- The European Parliament and the Council of the European Union Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency . < http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:136:0001:0033:EN:PDF > ( 2004
- European Medicines Agency CHMP meeting highlights . < http://www.ema.europa.eu/ema/index.jsp?curl=pages/about_us/document_listing/document_listing_000378.jsp&mid=WC0b01ac0580028d2a > ( September 2011–December 2013
- European Medicines Agency CHMP monthly report . < http://www.ema.europa.eu/ema/index.jsp?curl=pages/about_us/document_listing/document_listing_000190.jsp&mid=WC0b01ac05802d6250 > ( 2001–July 2011
- European Medicines Agency Monthly statistics report . < http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/document_listing/document_listing_000256.jsp&mid=WC0b01ac0580099fbb > ( 2010–2013
- European Medicines Agency News: EMA recommends 81 medicines for marketing authorisation in 2013 . < http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2014/01/news_detail_002006.jsp&mid=WC0b01ac058004d5c1 > ( 2014
- European Medicines Agency Assessment report for Cayston . < http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000996/WC500019995.pdf > ( 2009
- European Medicines Agency Assessment report for Bronchitol . < http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001252/WC500130591.pdf > ( 2013
- EURORDIS About rare diseases . < http://www.eurordis.org/about-rare-diseases >.
- US Food and Drug Administration . < http://www.fda.gov >.
- European Medicines Agency . < http://www.ema.europa.eu >.
- Pharmaceuticals and Medical Devices Agency . < http://www.pmda.go.jp >.
- National Institute of Health Sciences (NIHS) List of clinical trials of gene therapy in Japan . < http://www.nihs.go.jp/cgtp/cgtp/sec1/gt_prtcl/prtcl-j3.html >.
- The U.S. National Institutes of Health ClinicalTrials.gov < http://clinicaltrials.gov/ >.
- Immonen A, Vapalahti M, Tyynelä K, Hurskainen H, Sandmair A, Vanninen R. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol Ther. 2004;10:967–972. doi: 10.1016/j.ymthe.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Sandmair AM, Loimas S, Puranen P, Immonen A, Kossila M, Puranen M. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Hum Gene Ther. 2000;11:2197–2205. doi: 10.1089/104303400750035726. [DOI] [PubMed] [Google Scholar]
- Puumalainen AM, Vapalahti M, Agrawal RS, Kossila M, Laukkanen J, Lehtolainen P. Beta-galactosidase gene transfer to human malignant glioma in vivo using replication-deficient retroviruses and adenoviruses. Hum Gene Ther. 1998;9:1769–1774. doi: 10.1089/hum.1998.9.12-1769. [DOI] [PubMed] [Google Scholar]
- Westphal M, Ylä-Herttuala S, Martin J, Warnke P, Menei P, Eckland D, ASPECT Study Group Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14:823–833. doi: 10.1016/S1470-2045(13)70274-2. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency CAT monthly report . < http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/document_listing/document_listing_000196.jsp&mid=WC0b01ac05800292a8 > ( 2009–2013
- European Medicines Agency Questions and answers - Positive opinion on the marketing authorisation for Deltyba . < http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/human/002552/WC500155462.pdf > ( 2013
- European Medicines Agency Assessment report for Defitelio . < http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002393/WC500153152.pdf > ( 2013
- European Medicines Agency Assessment report for Fampyra . < http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002097/WC500109957.pdf > ( 2011
- European Medicines Agency Assessment report for Vedrop . < http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000920/WC500047922.pdf > ( 2009