

A Cre/LoxP-based luciferase reporter system and biophotonic imaging was used to detect fusion of transplanted human pluripotent stem cell-derived mesenchymal stem cells (MSCs) to cells of living mice. Human MSCs, with transient expression of a viral fusogen, were delivered to murine hearts via a collagen patch. Previously unknown migratory patterns of delivered human MSCs and associated fusion products in the healthy murine heart were found.
Keywords: Cell fusion, Mesenchymal stem cells, Cell transplantation, Cardiac patch, Cre/LoxP
Abstract
Evidence suggests that transplanted mesenchymal stem cells (MSCs) can aid recovery of damaged myocardium caused by myocardial infarction. One possible mechanism for MSC-mediated recovery is reprogramming after cell fusion between transplanted MSCs and recipient cardiac cells. We used a Cre/LoxP-based luciferase reporter system coupled to biophotonic imaging to detect fusion of transplanted human pluripotent stem cell-derived MSCs to cells of organs of living mice. Human MSCs, with transient expression of a viral fusogen, were delivered to the murine heart via a collagen patch. At 2 days and 1 week later, living mice were probed for bioluminescence indicative of cell fusion. Cell fusion was detected at the site of delivery (heart) and in distal tissues (i.e., stomach, small intestine, liver). Fusion was confirmed at the cellular scale via fluorescence in situ hybridization for human-specific and mouse-specific centromeres. Human cells in organs distal to the heart were typically located near the vasculature, suggesting MSCs and perhaps MSC fusion products have the ability to migrate via the circulatory system to distal organs and engraft with local cells. The present study reveals previously unknown migratory patterns of delivered human MSCs and associated fusion products in the healthy murine heart. The study also sets the stage for follow-on studies to determine the functional effects of cell fusion in a model of myocardial damage or disease.
Significance
Mesenchymal stem cells (MSCs) are transplanted to the heart, cartilage, and other tissues to recover lost function or at least limit overactive immune responses. Analysis of tissues after MSC transplantation shows evidence of fusion between MSCs and the cells of the recipient. To date, the biologic implications of cell fusion remain unclear. A newly developed in vivo tracking system was used to identify MSC fusion products in living mice. The migratory patterns of fusion products were determined both in the target organ (i.e., the heart) and in distal organs. This study shows, for the first time, evidence of fusion products at sites distal from the target organ and data to suggest that migration occurs via the vasculature. These results will inform and improve future, MSC-based therapeutics.
Introduction
More than 715,000 Americans experience a new or recurrent myocardial infarction annually, leading to cell death in cardiac tissue distal to the lesion and a progressive loss of cardiac function [1]. One promising therapy to recover or at least sustain cardiac function is the delivery of mesenchymal stem cells (MSCs) to the damaged myocardium [2–8]. MSCs home to injured tissues [9, 10], and several mechanisms for MSC-based recovery have been proposed. First, MSCs can differentiate into cardiac cells to replace the dead and damaged cells [11–14]. Alternatively, the transplanted MSCs could act through secretion of paracrine factors that limit immune responses [15] and/or increase angiogenesis [10, 16–22]. A third possible mechanism is nuclear reprogramming via fusion between the MSCs and cells of the pericardium, epicardium, or myocardium [23]. Recent studies have uncovered evidence of cell fusion between stem cells and cardiac cells [24–29]. However, the effect of cell fusion and subsequent reprogramming on cardiac function at the cellular and tissue scale is not well understood.
The biologic outcome of the fusion of stem cells at the tissue and organ level has been even more challenging to discern than at the cellular scale owing to the difficulty in detecting and tracking fusion products in living organisms. To date, all methods to assess fusion in vivo have relied on tissue procurement and histologic analysis. For example, to study the contribution of fusion of bone marrow (BM)-derived cells to liver regeneration, female mice expressing green fluorescent protein (GFP) in all hematopoietic-derived cells were used as bone marrow donors (GFP+ cells) for male mice with liver injury. Fusion in this case was defined as binucleated, GFP+ cells with one Y chromosome observed in the histologic tissue sections. The investigators showed that bone marrow-derived hepatocytes had a unique gene profile relative to both hematopoietic cells and hepatocytes of the host, suggesting reprogramming after cell fusion [30, 31]. Cell fusion of bone marrow-derived hematopoietic cells and cardiomyocytes has also been reported in vivo, albeit at a low frequency [23, 29, 32]. However, just as with the liver, these studies relied on tissue analysis after sacrificing the host animal (thus limiting studies of the kinetics and outcomes of fusion). Also, analysis has typically been performed on the target organs and not the entire animal (thus limiting knowledge of the potential breadth of cell fusion).
Lacking is a method to accurately detect fusion in vivo in the entire organism over time. To address this gap, we previously developed a molecular approach in which bioluminescence is induced on fusion [28]. We use a construct encoding the firefly luciferase (Photinus pyralis) gene adjacent to a floxed stop codon. When cells expressing this gene fuse with cells expressing the Cre recombinase protein, the LoxP sites are cleaved, excising the stop signal and thereby allowing transcription of luciferase. To increase the frequency of fusion, we simultaneously transfected the cells with a viral fusogen, vesicular stomatitis virus (the glycoprotein, VSV-G), along with the LoxP-luciferase reporter plasmid [33, 34]. Because bioluminescence can be reliably detected in living organisms, an inducible “living” detection signal is produced that can be tracked in real time.
In the present study, we used this Cre/LoxP-based molecular approach to detect fusion of transplanted cells to cells of organs of living mice. Using this approach, we found that human mesenchymal stem cells delivered to the murine heart via a collagen-based patch can fuse after delivery and that hybrids formed in this way can be detected in vivo in the target organ and in surrounding organ systems. We confirmed the inducible fusion detection system with fluorescence in situ hybridization (FISH) for human and mouse centromeres and immunohistochemistry for human leukocyte antigen (HLA-A,B,C). We report the potential of this molecular method applied to the study of fusion in living organisms, especially with cellular transplantation. In addition, we show for the first time the substantial dissemination of human mesenchymal stem cells and human-mouse hybrid cells to the stomach and small intestine after transplantation to a healthy murine heart. Follow-on studies will investigate the functional effects of cell fusion in a small animal model of myocardial damage or disease.
Materials and Methods
Transgenic Mice
We used C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, http://www.jax.org) (5 mice, Fig. 1) or transgenic mice that constitutively express Cre recombinase (B6.C-Tg[CMV-cre]1Cgn/J; Jackson Laboratory), such that deletion of LoxP-flanked genes occurs in all tissues, including germ cells. The Cre gene is under transcriptional control of the cytomegalovirus (CMV) minimal promoter and is X-linked. The Cre sequence was introduced to BALB/cJ derived BALB/c-I embryonic stem cells (ESCs). The resulting mice were backcrossed to the BALB/c background for 8 generations and then backcrossed to the C57BL/6J background for 10 generations [35]. Only male Cre mice (4 mice, 2 months old) were used for Figures 2–6 in the study owing to a false-positive signal detected when imaging the female transgenic mice (data not shown).
Figure 1.

Frequency of fusion after delivery of MSCs to infarcted murine heart via TM collagen-based patch. (A): Representative optical section (multiphoton microscopy) of human MSCs after 2 days with the TM patch and stained for MSC marker CD105 (green) and DAPI (blue). More than 95% of cells imaged were CD105 positive. Regions with high intensity (CD105) indicate cells aligned with planes other than the focal plane, perhaps indicative of cells migrating into the patch. Scale bars = 50 µm. (B): MSCs and vMSCs fused with murine cells in an infarcted heart after delivery via TM patch. A significant increase was seen in the percentage of fusion products present in the TM for untransfected MSCs (TM + MSCs), 22% ± 17% (10 images/area), compared with the TM only control with no cells, 2% ± 2% (10 images/area). ∗, p < .05. The percentage of fusion products present in the BorderZone also increased for untransfected MSCs (14% ± 9%, 10 images/area), but it was not significantly different than that for the TM only control (0.2% ± 0.5%, 10 images/area). The percentage of fusion products in the TM and BorderZone increased (albeit not significantly) to 24% ± 16% and 23% ± 15% when the MSCs were transfected with VSV-G (TM + vMSC) before delivery (10 images/area). ∗∗, p < .01 compared with the TM only control. No significant difference was found between the TM control and both MSC and vMSCs in the unhealthy heart and healthy heart regions. All percentages represent 10 randomly selected images for each region in the tissue sections. (C): Representative images of murine heart after transplantation stained for human (red) and mouse (green) centromeres using fluorescence in situ hybridization. Top row: A field of view from the TM patch. Bottom row: A field of view from the BorderZone between the TM and infarcted myocardium. Scale bars = 50 µm. Abbreviations: BorderZone, area between patch and myocardium; DAPI, 4′,6-diamidino-2-phenylindole; MSCs, mesenchymal stem cells; TM, TissueMend; vMSCs, VSV-G-transfected MSCs.
Figure 2.

Detection of cell fusion in living mice. (A): Average bioluminescent radiance (p s−1 cm−2 sr−1) of the chest and abdomen of mice receiving MSCs 2 and 8 days after transplantation to the heart. (B): Representative IVIS imaging of one control and two treated mice (Mouse 1, Mouse 3). (C): Average bioluminescent radiance (p s−1 cm−2 sr−1) of heart, stomach, small intestine, liver and kidney (n = 4 mice). Signal from heart, stomach, and small intestine was significantly higher than that of corresponding control organs and kidney tissue of treated mice (∗∗, p < .01, ∗, p < .05). (D): Representative images for each organ. From top to bottom: photograph, bioluminescence emission, overlay. Scale bar = 10 mm. Abbreviation: p s−1 cm−2 sr−1, photons per second per cm2 per steradian.
Figure 6.

Detection of fusion products near the vasculature in the murine stomach. Fusion products (white arrowhead) were detected adjacent to the vasculature in the murine stomach using a human centromere probe (red) and a murine centromere probe (green) and an antibody for vWF, a marker for endothelial cells. (A): Bright field cross section of the stomach. (B): Immunohistochemistry for vWF (red) and nuclei (blue). Scale bar = 100 µm. (C): Fluorescence in situ hybridization staining for mouse centromeres (green), human centromeres (red), and nuclei (blue). Insets: Magnified views of a representative fusion product near a blood vessel (dashed line). Scale bar = 50 µm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; vWF, von Willebrand factor.
Cell Culture
Human MSCs derived from human embryonic stem cells (hMSCs from WA-01 or WA-09 [Fig. 1], a gift from Dr. Peiman Hematti, University of Wisconsin-Madison, Madison, WI) were expanded and cultured, as previously described [36]. In brief, hMSCs were cultured on a 0.1% gelatin (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com) pretreated flask containing α-minimum essential medium (MEM)-complete. α-MEM-complete consisted of α-MEM (Invitrogen, Carlsbad, CA, http://www.invitrogen.com), 10% fetal bovine serum (HyClone Laboratories, Logan, UT, http://www.hyclone.com), 0.1 mM nonessential amino acids (Invitrogen), and 2 mM l-glutamine (Invitrogen). hMSC cultures were allowed to grow to 60%–70% confluence and were replated at a concentration of 1,500 cells per cm2. These human ESC-derived MSCs have cell surface markers, differentiation potential, and immunologic properties in vitro that are similar to those of adult BM-derived MSCs [36].
Gene Transfer
hMSCs were transiently transfected with viral fusogen VSV-G [24, 37] to promote cell-cell fusion. In addition, they were simultaneously transfected with the luciferase gene adjacent to a floxed stop codon (p231 pCMVe-betaAc-STOP-luc; Addgene, Cambridge, MA, https://www.addgene.org) [28]. Transfection was accomplished using the Neon Transfection System (Invitrogen), as previously described [38]. All recombinant DNA research was conducted according to NIH guidelines and in accordance with the University of Wisconsin-Madison and University of Minnesota-Twin Cities institutional biosafety committees.
Myocardial Infarction and Cell Delivery
Mice underwent an infarction procedure by left coronary artery ligation (Fig. 1), such as is routinely performed at the University of Wisconsin Cardiovascular Physiology Core Facility [28, 39, 40]. All animal procedures were performed in accordance with the guidelines of the American Association for Laboratory Animal Science and the University of Wisconsin-Madison Animal Care and Use Committee.
Delivery of Transfected hMSCs via the TissueMend Matrix to the Murine Myocardium
Cotransfected hMSCs were delivered to the myocardium of mice 2 days after infarction (Fig. 1) or to a healthy heart (Figs. 2–6) via a collagen patch (TissueMend; TEI Biosciences, Boston, MA, http://www.teibio.com), as previously described [24, 39]. TissueMend matrices (2 × 2 × 0.8 mm) were placed in a 24-well plate (Falcon; Thermo Fisher Scientific, Pittsburgh, PA, http://www.thermofisher.com) and hydrated with α-MEM-complete culture medium. After electroporation, hMSCs were seeded on the TissueMend sections at a concentration of 1 × 106 cells per milliliter (Fig. 1A). The medium was changed at 24 and 48 hours, at which point the TissueMend matrix, containing ∼1 × 105 transfected hMSCs, was attached to the myocardium with a single suture (7-0 Prolene; Ethicon, Johnson & Johnson, New Brunswick, NJ, http://www.ethicon.com) at each corner of the matrix. A matrix was placed such that it was in contact with both the infarct and the peri-infarct regions of the myocardium (Fig. 1) [24, 39]. In the follow-up study, two matrices were placed on a healthy, noninfarcted mouse heart such that they were in contact with the myocardium (Figs. 2–6).
Bioluminescence Imaging
Recipient mice constitutively expressed Cre recombinase; therefore, when transplanted human MSCs fused with cells of the recipient, the LoxP sites were cleaved, and the stop signal was excised, allowing expression of luciferase. Luciferase expression was detected 2 and 8 days after cell transplantation in living mice using an in vivo illuminessence system (IVIS) (IVIS Spectrum; Caliper Life Sciences, Hopkinton, MA, http://www.caliperis.com) imaging system, as previously described [28]. The average radiance was determined by measuring the emitted photons per second per cm2 per steradian of each organ using the Living Image In Vivo Imaging Software (PerkinElmer, Life and Analytical Sciences, Waltham, MA, http://www.perkinelmer.com).
Optical Analysis of Heart/Tissue Explants
Murine hearts, stomachs, small intestines, livers, and kidneys were harvested 8 days after matrix implantation to determine the incidence of fusion at the cellular level. After excision, the hearts were bisected longitudinally through the matrix, and the other organs were bisected longitudinally near the site of the bioluminescent signal. The tissues were immediately placed into 10% buffered formalin (pH 7.2; Thermo Fisher Scientific) for 24 hours, followed by 24 hours of fresh 10% buffered formalin, and a final 24-hour incubation in 70% ethanol. The samples were further processed for paraffin embedding and sectioning, as previously described [41]. A tissue digestion kit with an all human centromere probe (PlatinumBright550) and all mouse centromere probe (PlatinumBright495; Kreatech Diagnostics, Amsterdam, The Netherlands, http://www.kreatech.com) was used to perform FISH on the sections [24]. The samples were processed by the Cytogenetics Laboratory (WiCell Research Institute, Madison, WI, http://www.wicell.org), according to the manufacturer’s protocol. In brief, slides with paraffin-embedded sections were baked for 4 hours at 56°C. The specimens were incubated with pepsin (Thermo Fischer Scientific) for 70 minutes for tissue digestion before sequential hybridization of the human probe followed by the mouse probe. After hybridization, the slides were stained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Images were acquired with a 60× UPlanSApo (NA = 1.35 Oil), DAPI, green, and orange filters, on an Olympus BX41 upright fluorescence microscope (Olympus, Center Valley, PA, http://www.olympusamerica.com), and analyzed with FISHView, version 5.5, software (Applied Spectral Imaging, Vista, CA, http://www.spectral-imaging.com). In the studies with myocardial infarction (Fig. 1), 3 different hearts were stained with FISH, and 10 images per region were analyzed (∼40–150 cells per image, depending on the heart region). The control mice received either the TissueMend patch (without cells) or the TissueMend patch with untransfected MSCs after induction of infarction, as described in the preceding section. In the cases without myocardial infarction, 3 different hearts were stained with FISH, and 6 images per region were analyzed (∼20–100 cells per image, depending on the heart region). The small intestine and stomach from the mouse with the greatest abdominal bioluminescence emission were stained with FISH, and at least 8 images per organ were analyzed (∼100 cells per image). Fusion events were defined as nuclei with positive staining for both human centromeres (red) and mouse centromeres (green). The frequency of fusion was quantified for each image and defined as the number of fusion events divided by the total number of nuclei and reported as a percentage (% fusion products).
For immunohistochemistry (IHC) analysis, organ sections were deparaffinized by incubating at 60°C for 1 hour and then washed for 6 minutes in Xylene twice. The sections were rehydrated by dipping the sections 15 times each in 100% ethanol, 100% ethanol, 95% ethanol, and, finally, ultrapure water. Antigen retrieval was accomplished by incubating the sections for 20 minutes at 37°C in 0.5% pepsin (Thermo Fischer Scientific) in 5 mM HCl. The sections were removed and allowed to cool for 10 minutes at room temperature. The sections were rinsed in 1× phosphate-buffered saline (PBS) twice for 3 minutes. The sections were incubated with 1:10 dilution of unconjugated AffiniPure Fab fragment goat anti-mouse IgG (H+L) (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, http://www.jacksonimmuno.com) in PBS for 1 hour at room temperature. The sections were rinsed again in 1× PBS twice for 3 minutes. A 1:50 dilution of the anti-HLA-A,B,C (EMR8-5; MBL International Corp., Woburn, MA, http://www.mblintl.com) antibody or a 1:25 dilution of anti-von Willebrand factor (F8/86, Thermo Fisher Scientific, Minneapolis, MN, http://www.thermofisher.com) was made with dilution buffer containing 5% bovine serum albumin (HyClone), 2% goat serum (MP Biomedical, Solon, OH, http://www.mpbio.com), 1% glycine (Sigma-Aldrich), and 0.1% triton-X (MP Biomedical). Next, 40 µl of this antibody solution was placed on each tissue section overnight at 4°C. The sections were washed with 1× PBS and incubated for 45 minutes at 4°C with 40 µl of a 1:200 dilution of the secondary antibody (AF647 goat anti-mouse; Invitrogen) in dilution buffer. The sections were washed with 1× PBS and mounted using 1,4-diazabicyclo[2.2.2]octane (Dabco)/DAPI solution composed of 5% Dabco (Sigma-Aldrich) and 0.01% DAPI (Sigma-Aldrich) in a mixture of 50% glycerol (Thermo Fischer Scientific) and 50% 2× PBS on a microscope coverslip sealed with nail polish. Fluorescence emission was detected using an IX71 inverted deconvolution fluorescence microscope (Olympus). The images were acquired with a 20× UPlanFluor objective (NA = 0.5), using Slidebook software (Intelligent Imaging Innovations, Denver, CO, http://www.intelligent-imaging.com) and analyzed using ImageJ (Fiji; open source software, http://pacific.mpi-cbg.de/wiki/index.php/Fiji). Background fluorescence was determined using a secondary antibody-only control.
To confirm that the MSCs retained their phenotype after seeding onto the TissueMend patch and before transplantation, the MSCs were seeded onto TissueMend patches at a concentration of 1 × 106 cells per milliliter and allowed to attach overnight. The medium was changed at 24 and 48 hours, at which point, the cells were fixed with 4% paraformaldehyde for 15 minutes and then washed twice with 1× PBS. A 1:25 dilution of goat anti-CD105 (GKY02; R&D Systems, Minneapolis, MN, http://www.rndsystems.com) was made with dilution buffer containing 5% bovine serum albumin (HyClone), 2% goat serum (MP Biomedical), 1% glycine (Sigma-Aldrich), and 0.1% triton-X (MP Biomedical). Next, 200 µl of this antibody solution was placed on the patches seeded with MSCs overnight at 4°C. The patches seeded with MSCs were washed with 1× PBS and incubated for 45 minutes at 4°C with 200 µl of a 1:200 dilution of the secondary antibody (AF488 donkey anti-goat; Invitrogen) in dilution buffer. The patches seeded with MSCs were washed with 1× PBS and stained with Dabco/DAPI solution. Fluorescence emission was detected using a multiphoton fluorescence microscope (Prairie Technologies, Madison, WI, http://www.prairie-technologies.com). Images were acquired with a 40× objective (NA = 0.80), using Prairie View, version 5.0, software and analyzed with ImageJ. Background fluorescence was determined using a secondary antibody-only control.
Statistical Analysis
Statistical analyses were performed using analysis of variance with Tukey’s honest significant difference post hoc test for multiple comparisons or Student’s t test for 2 independent samples; p < .05 was considered significant. Data were analyzed with SigmaPlot (Systat Software Inc, San Jose, CA, http://www.systat.com).
Results
Fusion of MSCs Occurs Both Spontaneously and With the Aid of Viral Fusogens After Transplantation to the Murine Heart
Previously, we showed that fusion of transplanted MSCs with recipient cells occurs spontaneously in the murine heart [24]. In the present study, a myocardial infarction was induced in C57BL/6 mice and then treated with either a TissueMend patch without cells (TM only) or a TissueMend patch with human MSCs (TM + MSCs). MSCs seeded on the patch and incubated for 2 days before transplantation maintained expression of CD105 (>95% of cells imaged; Fig. 1A). The mice were sacrificed after 3 weeks, and the heart of each was fixed and sectioned. The histologic sections were probed using FISH for human-specific and mouse-specific centromeres, and all nuclei containing both probes were considered fusion products. Fusion products were identified in the TissueMend patch, BorderZone (area between patch and myocardium), unhealthy heart (the infarct scar), and healthy heart (areas distant from the infarct). Recently, we have conducted a more comprehensive quantitative analysis of these tissues by systematically counting 10 images, selected randomly for each region. In each region, the percentage of fusion products was determined by dividing the number of fusion products by the total number of cells imaged. We detected substantial numbers of fusion products in the TissueMend patch group, (TM + MSCs, 22% ± 17%), relative to the TM only control (2% ± 2%, p < .05; Fig. 1B; supplemental online Table 1). A similar result was observed in the BorderZone group (TM + MSCs, 14% ± 9%; TM only, 0.2% ± 0.5%). Representative images of the TissueMend and BorderZone regions can be seen in Figure 1C (inset shows magnified view).
In the same study, we questioned whether transfection of MSCs with the fusogen of the vesicular stomatitis virus (VSV-G) before transplantation augments fusion. The percentage of fusion products in the TissueMend patch and BorderZone was higher (although not statistically significantly higher) with addition of the fusogen (24% ± 16% and 23% ± 15%; Fig. 1B; supplemental online Table 1). Because of this augmented analysis of tissues from previous studies and in an effort to realize as much cell fusion as possible in a healthy mouse model, we elected to implement our in vivo tracking method using MSCs transfected with the VSV-G viral fusogen. The present study could have been conducted without the fusogen (just as is the case for most preclinical and clinical MSC grafts).
Cell Fusion Can Be Detected at the Delivery Site and at Distal Organs in Living Mice
To test the hypothesis that cell fusion between hMSCs and cells of the heart occurs and can be detected in living animals, we transplanted hMSCs expressing viral fusogen VSV-G and floxed luciferase to the healthy murine myocardium. To determine whether transplanted cells could fuse with the cells of the ventricle and persist for at least 1 week, live animal imaging for bioluminescence (and therefore cell fusion) was conducted at 2 and 8 days after transplantation. Bioluminescence was detected both in the chest region and in the mid/lower abdomen (Fig. 2A, 2B), suggesting the transplanted cells could fuse with the recipient cells both in the heart (chest region) and in organs distal to the heart (abdominal region). The average radiance was unchanged or increasing from 2 to 8 days, suggesting either that fusion can occur over an extended period or that cells that fuse early after transplantation are capable of proliferation. A similar increase in average radiance owing to MSC proliferation in vivo has recently been shown to occur in mice who received MSCs injected subcutaneously [42].
At 8 days, the bioluminescence peaked before decreasing (supplemental online Fig. 1); therefore, the mice were euthanized, and the heart and organs in the vicinity of the bioluminescent signal were extracted and imaged independently. A distinct bioluminescent emission was detected in the excised heart of all (n = 4) mice receiving transplanted cells (2,090 ± 1,008 photons per second per cm2 per steradian [p s−1 cm−2 sr−1]; p < .05 compared with the average radiance in nonbioluminescent organs [e.g., kidney]; Fig. 2C, 2D). Bioluminescence was also detected in the stomach and small intestine of the transplanted mice, at levels similar to that observed in the heart. The stomach had the highest emission (3,673 ± 996.8 p s−1 cm−2 sr−1), and the small intestine emission was slightly lower (2,723 ± 590.6 p s−1 cm−2 sr−1; n = 4 mice; p < .01, compared with average radiance in nonbioluminescent organs [e.g., kidney]). Some of the mice receiving cell transplants also exhibited low levels of bioluminescence in the liver (1,149 ± 119.0 p s−1 cm−2 sr−1; n = 4; not significant compared with average radiance in nonbioluminescent organs [e.g., kidney]). Other distal organs exhibited no bioluminescence (e.g., kidneys, spleen, and lungs) and were not excised for quantification. However, the kidneys were used as a negative control organ for comparison (399.4 ± 102.1 p s−1 cm−2 sr−1). The detected luciferase activity is a reflection of the number of cells expressing luciferase and therefore a reflection of the relative number of cell fusion events.
Confirmation of hMSC-Mouse Cell Fusion at the Cellular Level
To assess fusion at the cellular level, FISH was used to distinguish the centromeres of the donor (human) from those of the recipient (mouse). Cells exhibiting fluorescence signals indicative of both human and mouse centromeres were considered fusion products. Using this criterion, the fusion products were detected and quantified in organs emitting a bioluminescent signal. In the heart, the fusion products were 52% ± 24% of the total cells imaged in the collagen patch (n = 17 fields of view containing fusion products of interest, 358 total cells) and 44% ± 27% of total cells imaged in scar-like tissue (BorderZone) between the patch and damaged myocardium (n = 17 fields of view containing fusion products of interest, 1,693 total cells; Fig. 3A; Table 1). A few fusion products were localized to the healthy heart (3% ± 5% of total cells imaged, n = 4 fields of view containing fusion products of interest, 553 total cells). The background fluorescence signal (green) in the healthy heart image resulted from autofluorescence of the cardiac sarcomeres (cytoplasmic), which did not interfere with the nuclear staining of the centromeres (Fig. 3C). The result was not a function of the TissueMend delivery vehicle, because similar results were obtained after a bolus injection of VSV-G-transfected MSCs directly to the myocardium (supplemental online Fig. 2).
Figure 3.

Frequency of fusion after delivery of vMSCs to healthy, noninfarcted, murine myocardium via TM collagen-based patch. vMSCs fused with recipient cells in the murine heart after delivery via TM patch. (A): Significantly more fusion products were detected in the TM patch and BorderZone than in the healthy heart distant from the patch (∗∗, p < .01). (B): Bright field cross section of the heart. (C): Fluorescence in situ hybridization images are stained for mouse centromeres (green), human centromeres (red), and nuclei (blue). Representative fusion products, defined as dual color fluorescence within individual nuclei, are designated with a white arrowhead. Insets: Magnified views of representative fusion products. Background signal in the healthy heart resulted from autofluorescence of cardiac sarcomeres; left in place intentionally to appreciate the position of the fusion products relative to the healthy myocardial tissue. Scale bar = 50 µm. (D): Immunohistochemistry for HLA-A,B,C (green) and nuclei (blue). Scale bar = 50 µm. Abbreviations: BorderZone, area between patch and myocardium; BZ, BorderZone; DAPI, 4′,6-diamidino-2-phenylindole; MSCs, mesenchymal stem cells; Myo, myocardium; TM, TissueMend; vMSCs, VSV-G-transfected MSCs.
Table 1.
Frequency of fusion in organs and tissues of mice after transplantation of mesenchymal stem cells expressing viral fusogens
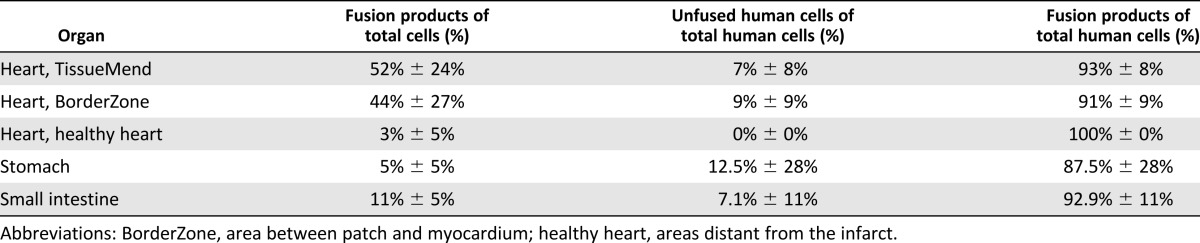
The small intestine exhibited extensive fusion (11% ± 5% of total cells imaged, n = 8 fields of view containing fusion products of interest, 880 total cells), with most of the fusion products localized to the lamina propria of the villi (34% ± 18% of the cells in the lamina propria, 222 total cells; Fig. 4; Table 1). No fusion products were detected in the epithelium of the small intestinal villi (n = 72 villi). Unfused human cells were seen in the small intestine (7.1% of human cells detected via FISH, n = 8 fields of view, 98 total human cells). In the stomach, the fusion products were 5% ± 5% of the total cells imaged (n = 14 fields of view containing fusion products of interest, 913 total cells), and the fusion products were primarily detected in regions close to the vasculature (Figs. 5, 6; Table 1). Unfused human cells were seen in the stomach (12.5% of human cells detected via FISH, n = 14 fields of view, 32 total human cells). These results confirmed the bioluminescent emission detected in the live animals in a variety of organs.
Figure 4.

Detection of fusion products in the murine small intestine using fluorescence in situ hybridization (FISH). Fusion products (white arrowhead) and unfused human cells (white arrow) were detected in the murine small intestine using a human centromere probe (red) and a murine centromere probe (green). (A): Bright field cross section of the small intestine. (B): FISH images are stained for mouse centromeres (green), human centromeres (red), and nuclei (blue). Insets: Magnified views of representative fusion products. Scale bar = 50 µm. (C): Immunohistochemistry for HLA-A,B,C (green) and nuclei (blue). Scale bar = 50 µm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; HLA, human leukocyte antigen.
Figure 5.

Detection of fusion hybrids in the murine stomach using fluorescence in situ hybridization (FISH). Fusion products (white arrowhead) and unfused human cells (white arrow) were detected in the murine stomach using a human centromere probe (red) and a murine centromere probe (green). (A): Bright field cross section of the stomach. (B): FISH images are stained for mouse centromeres (green), human centromeres (red), and nuclei (blue). Insets: Magnified views of representative fusion products. Scale bar = 50 µm. (C): Immunohistochemistry for HLA-A,B,C (green) and nuclei (blue). Scale bar = 50 µm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; HLA, human leukocyte antigen.
HLA-A,B,C Expressing Cells Are Detected at the Delivery Site and Distal Organs
To further confirm the translocation of human MSCs from the site of delivery (the heart) to the small intestine and stomach, IHC was conducted with an anti-HLA-A,B,C antibody. Using this method, human cells were found in tissues harboring a bioluminescent signal (Figs. 3D, 4C, 5C). Just as with the FISH analysis in the heart, the human cells were concentrated in the collagen patch and in the scar-like tissue between the patch and damaged myocardium. The small intestine proved difficult to probe owing to the high levels of autofluorescence. However, a few human cells expressing high levels of HLA-A,B,C (at an intensity level greater than the background) were detected in the lamina propria. In the stomach, the human cells were observed in regions near blood vessel-like structures, similar to what was seen in the FISH analysis. These results further confirmed the movement of human MSCs from the mouse heart to the small intestine and stomach.
Detection of Fusion Products Near the Vasculature in the Murine Stomach
To test whether the observed fusion products were located near blood vessels in a distal organ, the stomach was stained with an anti-von Willebrand factor (vWF) antibody, a marker for endothelial cells. By staining a stomach section adjacent to the section used for FISH analysis, the location of the fusion products relative to the blood vessels could be ascertained. Fusion products were usually found near the vWF-positive cells (Fig. 6). One fusion product (Fig. 6, inset) was located directly next to a blood vessels (Fig. 6, dashed line). This result supports the possibility that human cells or human cell hybrids transit via the vasculature to distal organs.
Discussion
In the present study, we took advantage of a Cre/LoxP-based molecular approach to detect fusion of hMSCs after transplantation to the heart. Thus, we were able to determine the presence of fusion in the heart and in other tissues and organ systems of the mouse. We found (a) fusion of hMSCs with cells of the mouse can occur at the delivery site and at distal organs; (b) fusion is not a prerequisite nor does it appear to hinder movement of hMSCs away from the delivery site, because unfused human cells were found in the stomach and small intestine; and (c) fusion products in the distal organs were localized around the vasculature. This is the first study to specifically track fusion of MSCs after transplantation and to do so at the whole organism scale.
Migration of MSCs has been indentified in studies conducted to monitor homing and engraftment of MSCs. MSC migration in disease models or injured tissue has been reported in tumors [43], arthritic joints [44], middle cerebral artery occlusion [45], and myocardial infarction [46, 47]. In a disease model for Chagas disease, which affects heart function, MSCs injected intravenously migrated to a limited degree to the heart and to a greater extent to the liver, lungs, and spleen [48]. Similarly, other studies have shown that most of MSCs injected to the venous system were found in the liver, lung, and spleen [47, 49]. We also observed hMSC fusion products in the liver, although most hMSC fusion products were detected in the heart, stomach, and intestine. This difference from previous studies could reflect the delivery method or that transplanted cells expressed a viral fusogen. In our study, hMSCs were delivered on a myocardial patch rather than through the intravenous injection used in other studies. hMSC fusion products in the heart were found primarily in the collagen patch and in scar-like tissue between the patch and damaged myocardium. Homing of hMSCs or hMSC fusion products of our study could also have been affected by the expression of fusogen, VSV-G. The fusogen might enable fusion of hMSCs with circulating cells, thereby altering their migratory properties and subsequent tissue targets. Also, our analysis of tissue sections followed the detection of fusion at the organ level. Thus, it is possible we missed organs that contained only human cells or that contained fused human cells at levels too low to detect using our approach.
Interestingly, the hMSC fusion products in organs distal to the heart (i.e., stomach and small intestine) were found primarily near the vasculature, suggesting that blood vessels were the likely path for mobility. Another possibility is that hMSCs remained in the tissues in which the fusogen was most likely to be active (pH ∼5.5) and therefore most likely to fuse with the recipient cells. Similarly, the proximity of cells of the stomach to acidic digestion conditions could also have activated the fusogen, stimulating fusion. Yet unclear is whether hMSCs fuse with cells of the murine heart and then migrate or migrate first to then fuse with cells of the distal tissues. Unfused hMSCs were detected in the stomach and small intestine, suggesting that hMSCs can migrate to and infiltrate the distal organs without fusion. Also unclear is what cell type or types of the recipient serve as fusion partners. Possible candidates include those previously reported to fuse with transplanted MSCs, including cardiomyocytes of the heart [23, 29, 32] and hepatocytes of the liver [30, 31]. In addition, macrophages might fuse with transplanted MSCs, because they have well-defined fusion machinery used to fuse with other macrophages for the formation of giant cells [50].
The detection of fusion products was made possible by combining an inducible, Cre/LoxP-based molecular strategy with whole animal imaging using the IVIS system. The Cre/LoxP system uses the enzyme Cre recombinase, which only cuts at LoxP sites, to excise the floxed stop codon from the LoxP-luciferase construct, thus expressing the reporter only when both the Cre recombinase enzyme (mouse cells) and LoxP-luciferase plasmid (hMSCs) are present in the same cell. This inducible method ensures detection of only true fusion products (and their progeny) both in vitro and in vivo. Also, the IVIS system allows imaging of bioluminescence intensity of the entire animal, not just the regions of interest. We used this macro scale imaging technology to detect fusion in locations that were not necessarily expected after delivery of our transfected cells. In addition, imaging was conducted while the animal was still alive, making imaging at successive time points possible. A more in-depth study of the kinetics of fusion in vivo will be an important next step in applying this approach. In the present study, the LoxP-luciferase plasmid was expressed transiently; thus, the signal was lost once the plasmid has degraded within the cell, which occurs ∼1 week after transfection. If the LoxP-luciferase construct was redesigned to be expressed for a longer period or if it were incorporated into the genome, the kinetics experiment could be performed to examine long-term survival (>1 week) and migration of the fusion products.
The Cre/LoxP-based approach and the IVIS system are limited. First, the inducible Cre/LoxP system detects only true fusion products; however, the generation of a signal requires time to remove the stop codon and then transcription and translation of the reporter gene. This temporal delay prevents detection of fusion immediately after the event. Thus, the actual frequency of cell fusion is likely to be underrepresented in our findings. Second, the detection limit of the IVIS system for internal organs, including the heart, is ∼10,000 cells [28]. The resolution limit makes finding very rare events nearly impossible. For example, hMSC fusion products might have been present in other distal organs in the mouse; however, if the threshold were not greater than the background level, fusion would be undetectable. Recent developments in the field of live animal imaging could help address this limitation. For example, a 100-fold stronger synthetic luciferase substrate has been engineered that increases the sensitivity of the system and theoretically decreases the detection threshold to less than 100 events [51].
Conclusion
The results of the present study establish the efficacy of using a bioluminescent Cre/LoxP system, in conjunction with whole animal imaging, to examine cell fusion after transplantation in live animals. Not only were we able to detect fusion events in live mice, our results suggest that hMSCs, and possibly hMSC fusion products, have the ability to migrate to distal organs and engraft with local cells. This knowledge is important for the clinical transplantation of hMSCs, because it appears hMSCs can home, reside, and possibly fuse in unanticipated organs of the recipient. This could cause potential complications in patients. Similarly, although cell fusion occurs at a lower level without a fusogen, hMSCs are able to spontaneously fuse with other cells [52, 53]; therefore, additional information about cell fusion and its role in regeneration is essential when assessing the clinical potential and possible side effects of cell transplantation. The work we have presented provides not only additional insights into MSC cell fusion and translocation but also establishes a technological platform for additional study of cell fusion in vivo with other types of cellular grafts. These results also provide a rationale for future studies to determine how hMSC fusion products affect the functional profile of a diseased murine heart and the mechanisms that govern the functional capacity of fusion products.
Supplementary Material
Acknowledgments
We thank Jayne Squirrel and Sean Palecek for critical comments on the paper; Erik M. McIntire, Seth M. Taapken, and Karen D. Montgomery of the WiCell Research Institute Cytogenetics Department; Timothy A. Hacker, Guoqing Song, and Jill Koch of the University of Wisconsin Cardiovascular Physiology Core Facility; Mohammed Farhoud for technical assistance with IVIS imaging; Philip Jung of the University of Minnesota for assistance with multiphoton imaging; and LeAnn Oseth for assistance with FISH imaging in the Cytogenomics Shared Resource at the University of Minnesota, with support from the comprehensive Masonic Cancer Center NIH Grant P30 CA077598. The present study was supported by the NIH (Award HL089679), National Science Foundation Graduate Research Fellowship Program, National Science Foundation (CAREER, Award 0844537).
Author Contributions
B.T.F.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; N.A.K.: conception and design, collection and/or assembly of data; B.M.O.: conception and design, financial support, administrative support, provision of study materials, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
References
- 1.Go AS, Mozaffarian D, Roger VL, et al. Executive summary: Heart disease and stroke statistics—2013 Update: A report from the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Latif A, Bolli R, Tleyjeh IM, et al. Adult bone marrow-derived cells for cardiac repair: A systematic review and meta-analysis. Arch Intern Med. 2007;167:989–997. doi: 10.1001/archinte.167.10.989. [DOI] [PubMed] [Google Scholar]
- 3.Amado LC, Saliaris AP, Schuleri KH, et al. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Natl Acad Sci USA. 2005;102:11474–11479. doi: 10.1073/pnas.0504388102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen S, Liu Z, Tian N, et al. Intracoronary transplantation of autologous bone marrow mesenchymal stem cells for ischemic cardiomyopathy due to isolated chronic occluded left anterior descending artery. J Invasive Cardiol. 2006;18:552–556. [PubMed] [Google Scholar]
- 5.Chen SL, Fang WW, Ye F, et al. Effect on left ventricular function of intracoronary transplantation of autologous bone marrow mesenchymal stem cell in patients with acute myocardial infarction. Am J Cardiol. 2004;94:92–95. doi: 10.1016/j.amjcard.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 6.Ma J, Ge J, Zhang S, et al. Time course of myocardial stromal cell-derived factor 1 expression and beneficial effects of intravenously administered bone marrow stem cells in rats with experimental myocardial infarction. Basic Res Cardiol. 2005;100:217–223. doi: 10.1007/s00395-005-0521-z. [DOI] [PubMed] [Google Scholar]
- 7.Shake JG, Gruber PJ, Baumgartner WA, et al. Mesenchymal stem cell implantation in a swine myocardial infarct model: Engraftment and functional effects. Ann Thorac Surg. 2002;73:1919–1926. doi: 10.1016/s0003-4975(02)03517-8. [DOI] [PubMed] [Google Scholar]
- 8.Tomita S, Li RK, Weisel RD, et al. Autologous transplantation of bone marrow cells improves damaged heart function. Circulation. 1999;100(suppl):II247–II256. doi: 10.1161/01.cir.100.suppl_2.ii-247. [DOI] [PubMed] [Google Scholar]
- 9.Mouiseddine M, François S, Semont A, et al. Human mesenchymal stem cells home specifically to radiation-injured tissues in a non-obese diabetes/severe combined immunodeficiency mouse model. Br J Radiol. 2007;80:S49–S55. doi: 10.1259/bjr/25927054. [DOI] [PubMed] [Google Scholar]
- 10.Nagaya N, Fujii T, Iwase T, et al. Intravenous administration of mesenchymal stem cells improves cardiac function in rats with acute myocardial infarction through angiogenesis and myogenesis. Am J Physiol Heart Circ Physiol. 2004;287:H2670–H2676. doi: 10.1152/ajpheart.01071.2003. [DOI] [PubMed] [Google Scholar]
- 11.Deb A, Wang S, Skelding KA, et al. Bone marrow-derived cardiomyocytes are present in adult human heart: A study of gender-mismatched bone marrow transplantation patients. Circulation. 2003;107:1247–1249. doi: 10.1161/01.cir.0000061910.39145.f0. [DOI] [PubMed] [Google Scholar]
- 12.Dimmeler S, Zeiher AM, Schneider MD. Unchain my heart: The scientific foundations of cardiac repair. J Clin Invest. 2005;115:572–583. doi: 10.1172/JCI24283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawada H, Fujita J, Kinjo K, et al. Nonhematopoietic mesenchymal stem cells can be mobilized and differentiate into cardiomyocytes after myocardial infarction. Blood. 2004;104:3581–3587. doi: 10.1182/blood-2004-04-1488. [DOI] [PubMed] [Google Scholar]
- 14.Yoon YS, Wecker A, Heyd L, et al. Clonally expanded novel multipotent stem cells from human bone marrow regenerate myocardium after myocardial infarction. J Clin Invest. 2005;115:326–338. doi: 10.1172/JCI22326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen G, Nayan M, Duong M, et al. Marrow stromal cells for cell-based therapy: The role of antiinflammatory cytokines in cellular cardiomyoplasty. Ann Thorac Surg. 2010;90:190–197. doi: 10.1016/j.athoracsur.2010.02.074. [DOI] [PubMed] [Google Scholar]
- 16.Caplan AI. Why are MSCs therapeutic? New data: New insight. J Pathol. 2009;217:318–324. doi: 10.1002/path.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006;98:1076–1084. doi: 10.1002/jcb.20886. [DOI] [PubMed] [Google Scholar]
- 18.Gnecchi M, He H, Liang OD, et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med. 2005;11:367–368. doi: 10.1038/nm0405-367. [DOI] [PubMed] [Google Scholar]
- 19.Gnecchi M, He H, Noiseux N, et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J. 2006;20:661–669. doi: 10.1096/fj.05-5211com. [DOI] [PubMed] [Google Scholar]
- 20.Kamihata H, Matsubara H, Nishiue T, et al. Implantation of bone marrow mononuclear cells into ischemic myocardium enhances collateral perfusion and regional function via side supply of angioblasts, angiogenic ligands, and cytokines. Circulation. 2001;104:1046–1052. doi: 10.1161/hc3501.093817. [DOI] [PubMed] [Google Scholar]
- 21.Kinnaird T, Stabile E, Burnett MS, et al. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ Res. 2004;94:678–685. doi: 10.1161/01.RES.0000118601.37875.AC. [DOI] [PubMed] [Google Scholar]
- 22.Linke A, Müller P, Nurzynska D, et al. Stem cells in the dog heart are self-renewing, clonogenic, and multipotent and regenerate infarcted myocardium, improving cardiac function. Proc Natl Acad Sci USA. 2005;102:8966–8971. doi: 10.1073/pnas.0502678102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nygren JM, Jovinge S, Breitbach M, et al. Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nat Med. 2004;10:494–501. doi: 10.1038/nm1040. [DOI] [PubMed] [Google Scholar]
- 24.Kouris NA, Schaefer JA, Hatta M, et al. Directed fusion of mesenchymal stem cells with cardiomyocytes via VSV-G facilitates stem cell programming. Stem Cells Int. 2012;2012:414038. doi: 10.1155/2012/414038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin HP, Vincenz C, Eliceiri KW, et al. Bimolecular fluorescence complementation analysis of eukaryotic fusion products. Biol Cell. 2010;102:525–537. doi: 10.1042/BC20100033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuura K, Wada H, Nagai T, et al. Cardiomyocytes fuse with surrounding noncardiomyocytes and reenter the cell cycle. J Cell Biol. 2004;167:351–363. doi: 10.1083/jcb.200312111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh H, Bradfute SB, Gallardo TD, et al. Cardiac progenitor cells from adult myocardium: Homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sprangers AJ, Freeman BT, Kouris NA, et al. A Cre-Lox P recombination approach for the detection of cell fusion in vivo. J Vis Exp. 2012;51:e3581. doi: 10.3791/3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S, Wang D, Estrov Z, et al. Both cell fusion and transdifferentiation account for the transformation of human peripheral blood CD34-positive cells into cardiomyocytes in vivo. Circulation. 2004;110:3803–3807. doi: 10.1161/01.CIR.0000150796.18473.8E. [DOI] [PubMed] [Google Scholar]
- 30.Quintana-Bustamante O, Alvarez-Barrientos A, Kofman AV, et al. Hematopoietic mobilization in mice increases the presence of bone marrow-derived hepatocytes via in vivo cell fusion. Hepatology. 2006;43:108–116. doi: 10.1002/hep.21005. [DOI] [PubMed] [Google Scholar]
- 31.Quintana-Bustamante O, Grueso E, Garcia-Escudero R, et al. Cell fusion reprogramming leads to a specific hepatic expression pattern during mouse bone marrow derived hepatocyte formation in vivo. PLoS ONE. 2012;7:e33945. doi: 10.1371/journal.pone.0033945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, et al. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature. 2003;425:968–973. doi: 10.1038/nature02069. [DOI] [PubMed] [Google Scholar]
- 33.Jeetendra E, Robison CS, Albritton LM, et al. The membrane-proximal domain of vesicular stomatitis virus G protein functions as a membrane fusion potentiator and can induce hemifusion. J Virol. 2002;76:12300–12311. doi: 10.1128/JVI.76.23.12300-12311.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun X, Belouzard S, Whittaker GR. Molecular architecture of the bipartite fusion loops of vesicular stomatitis virus glycoprotein G, a class III viral fusion protein. J Biol Chem. 2008;283:6418–6427. doi: 10.1074/jbc.M708955200. [DOI] [PubMed] [Google Scholar]
- 35.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trivedi P, Hematti P. Derivation and immunological characterization of mesenchymal stromal cells from human embryonic stem cells. Exp Hematol. 2008;36:350–359. doi: 10.1016/j.exphem.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takada A, Robison C, Goto H, et al. A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci USA. 1997;94:14764–14769. doi: 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sprangers AJ, Freeman BT, Ogle BM. Electroporation can efficiently transfect hESC-derived mesenchymal stem cells without inducing differentiation. Open Stem Cell J. 2011;3:62–66. [Google Scholar]
- 39.Kouris NA, Squirrell JM, Jung JP, et al. A nondenatured, noncrosslinked collagen matrix to deliver stem cells to the heart. Regen Med. 2011;6:569–582. doi: 10.2217/rme.11.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michael LH, Entman ML, Hartley CJ, et al. Myocardial ischemia and reperfusion: A murine model. Am J Physiol. 1995;269:H2147–H2154. doi: 10.1152/ajpheart.1995.269.6.H2147. [DOI] [PubMed] [Google Scholar]
- 41.Ogle BM, Butters KA, Plummer TB, et al. Spontaneous fusion of cells between species yields transdifferentiation and retroviral transfer in vivo. FASEB J. 2004;18:548–550. doi: 10.1096/fj.03-0962fje. [DOI] [PubMed] [Google Scholar]
- 42.Preda MB, Rønningen T, Burlacu A, et al. Remote transplantation of mesenchymal stem cells protects the heart against ischemia-reperfusion injury. Stem Cells. 2014;32:2123–2134. doi: 10.1002/stem.1687. [DOI] [PubMed] [Google Scholar]
- 43.Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614–2623. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sutton EJ, Henning TD, Boddington S, et al. In vivo magnetic resonance imaging and optical imaging comparison of viable and nonviable mesenchymal stem cells with a bifunctional label. Mol Imaging. 2010;9:278–290. [PMC free article] [PubMed] [Google Scholar]
- 45.Jang KS, Lee KS, Yang SH, et al. In vivo tracking of transplanted bone marrow-derived mesenchymal stem cells in a murine model of stroke by bioluminescence imaging. J Korean Neurosurg Soc. 2010;48:391–398. doi: 10.3340/jkns.2010.48.5.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kraitchman DL, Heldman AW, Atalar E, et al. In vivo magnetic resonance imaging of mesenchymal stem cells in myocardial infarction. Circulation. 2003;107:2290–2293. doi: 10.1161/01.CIR.0000070931.62772.4E. [DOI] [PubMed] [Google Scholar]
- 47.Kraitchman DL, Tatsumi M, Gilson WD, et al. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 2005;112:1451–1461. doi: 10.1161/CIRCULATIONAHA.105.537480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jasmin, Jelicks LA, Koba W, et al. Mesenchymal bone marrow cell therapy in a mouse model of Chagas disease. Where do the cells go? PLoS Negl Trop Dis. 2012;6:e1971. doi: 10.1371/journal.pntd.0001971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gholamrezanezhad A, Mirpour S, Bagheri M, et al. In vivo tracking of 111In-oxine labeled mesenchymal stem cells following infusion in patients with advanced cirrhosis. Nucl Med Biol. 2011;38:961–967. doi: 10.1016/j.nucmedbio.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 50.Takeda Y, Tachibana I, Miyado K, et al. Tetraspanins CD9 and CD81 function to prevent the fusion of mononuclear phagocytes. J Cell Biol. 2003;161:945–956. doi: 10.1083/jcb.200212031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evans MS, Chaurette JP, Adams ST, Jr, et al. A synthetic luciferin improves bioluminescence imaging in live mice. Nat Methods. 2014;11:393–395. doi: 10.1038/nmeth.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spees JL, Olson SD, Ylostalo J, et al. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc Natl Acad Sci USA. 2003;100:2397–2402. doi: 10.1073/pnas.0437997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terada N, Hamazaki T, Oka M, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416:542–545. doi: 10.1038/nature730. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.