

Summary
The Vibrio cholerae MARTXVc toxin delivers three effector domains to eukaryotic cells. To study toxin delivery and function of individual domains, the rtxA gene was modified to encode toxin with an in-frame beta-lactamase (Bla) fusion. The hybrid RtxA::Bla toxin was Type I secreted from bacteria; and then Bla was translocated into eukaryotic cells and delivered by autoprocessing, demonstrating the MARTXVc toxin is capable of heterologous protein transfer. Strains that produce hybrid RtxA::Bla toxins that carry one effector domain in addition to Bla were found to more efficiently translocate Bla. In cell biological assays, the actin cross-linking domain (ACD) and Rho-inactivation domain (RID) are found to crosslink actin and inactivate RhoA, respectively, when other effector domains are absent, with toxin autoprocessing required for high efficiency. The previously unstudied alpha-beta hydrolase domain (ABH) is shown here to activate CDC42, although the effect is ameliorated when RID is also present. Despite all effector domains acting on cytoskeleton assembly, the ACD was sufficient to rapidly inhibit macrophage phagocytosis. Both the ACD and RID independently disrupted polarized epithelial tight junction integrity. The sufficiency of ACD but strong selection for retention of RID and ABH suggest these two domains may primarily function by modulating cell signaling.
Introduction
Multifunctional-Autoprocessing Repeats-in-Toxin (MARTX) toxins are large bacterial proteins secreted from bacteria that function as a delivery platform for cytopathic and cytotoxic effector domains (Satchell, 2011). The MARTXVc toxin produced by the human pathogenic El Tor O1 strains of Vibrio cholerae is 4545 aa and is secreted from the bacterium by Type I secretion (Lin et al., 1999, Boardman & Satchell, 2004, Dolores & Satchell, 2013). The amino- and carboxyl-termini are comprised of glycine rich repeat regions that are proposed to form a pore for translocation of the central portion of the toxin across the eukaryotic cell plasma membrane to access the cytosol (Sheahan et al., 2004, Satchell, 2007). Within the cytosol, a translocated cysteine protease domain (CPD) is activated by binding inositol hexakisphosphate, a small molecule present at high concentrations in the eukaryotic cytosol, but absent from bacteria (Prochazkova & Satchell, 2008, Egerer & Satchell, 2010). The activated CPD then autoprocesses the MARTXVc holotoxin at four sites found in unstructured regions that flank three “effector domains”, releasing these domains to the cell cytosol (Prochazkova et al., 2009, Shen et al., 2009, Egerer & Satchell, 2010). Upon release, the effector domains are no longer associated with the holotoxin and are therefore bona fide toxin “effectors”. The first effector domain is the actin cross-linking domain (ACD) that introduces an isopeptide bond between actin protomers resulting in actin multimers that are not functional for actin assembly (Sheahan et al., 2004, Cordero et al., 2006, Kudryashov et al., 2008, Satchell, 2009). The second effector domain is a Rho-inactivation domain (RID) that is targeted to the plasma membrane where it modifies the Rho activation pathway resulting in loss of GTP-bound Rho from the cell and downstream cytoskeletal disassembly (Sheahan & Satchell, 2007, Geissler et al., 2010, Ahrens et al., 2013). The third effector domain is a member of the alpha/beta hydrolase (ABH) family of esterases/lipases/proteases (Satchell, 2011), but the function of this domain has not as of yet been determined.
The function of the V. cholerae MARTXVc toxin during infection of the small intestine is to promote colonization by evading the bacterial innate immune response (Olivier et al., 2007, Olivier et al., 2009). The toxin works in concert with the pore-forming toxin hemolysin (HlyA) and cholera toxin (CT) to avoid neutrophil clearance of bacteria from the small intestine (Queen & Satchell, 2012, Queen & Satchell, 2013). The toxin has also been shown in vitro to inhibit macrophage phagocytosis (Ma et al., 2009) and to affect the integrity of the tight junctions of polarized cellular monolayers (Fullner et al., 2001).
A major question regarding MARTXVc and MARTX toxins of other species is why they are multifunctional and carry so many diverse effector domains. One plausible hypothesis is that the released effectors function synergistically and benefit from delivery in equal molar concentrations. Alternatively, the effectors could function independently in distinct cell biological pathways; or, they could function in similar pathways, but in different target cells.
Addressing the contribution of a single effector domain to a cellular phenotype is complicated as the effectors originate as domains of a large holotoxin and the function of the released effector must be dissociated from contributions of other effector domains, as well as the contribution of the putative pore and autoprotease. This problem has been addressed in the past by introducing effector domains to cells by alternate delivery mechanisms, such as fusion to amino-terminus of anthrax toxin lethal factor and delivery to cells using anthrax toxin protective antigen or by transient-transfection (Sheahan et al., 2004, Cordero et al., 2006, Sheahan & Satchell, 2007, Ahrens et al., 2013, Antic et al., 2014, Ziolo et al., 2014). Alternatively, deletions and point mutations have been generated that alter just one or two effector domains leaving the remainder intact (Sheahan et al., 2004, Ahrens et al., 2013,). An issue with these approaches that routinely arises is whether a change of delivery strategy alters the localization of the toxin and thereby its function, or if genetic manipulation of the toxin gene ultimately affected toxin production, secretion, and/or translocation.
In this study, we developed a system to modify the MARTXVc toxin gene rtxA on the chromosome of V. cholerae to express fully functional MARTXVc toxins able to be secreted from bacteria and translocated to cells, but that carry either no effector domains or just a single effector domain. This provides a means to identify the contribution of a single effector to cell biological processes, independent of the other effector domains. Using this system, we demonstrate that the conserved repeat regions and CPD alone are sufficient for effector domain translocation by demonstrating that the MARTXVc toxin can deliver the heterologous protein beta-lactamase (Bla). Next, it is shown that each effector domain functions independently in cytoskeleton disassembly, but that RID and ABH have conflicting contributions to the activation state of the small GTPase CDC42. The optimal function of each effector domain depends on an active CPD, providing evidence that autoprocessing to release effectors from the holotoxin is essential for MARTXVc intoxication during natural delivery. The ability of V. cholerae MARTXVc to affect the integrity of the junctions in polarized intestinal cells is then found to be due independently to ACD and RID, whereas the ability to paralyze phagocytosis is linked only to cross-linking of actin by the ACD. These data reveal that MARTX toxin effector domains have differing contributions to relevant cell biological activities depending upon the cell type and reveal that the activity of one effector domain can be influenced by another in some cases, although they can also function completely independent of each other.
Results
V. cholerae ampicillin resistance due to secretion of a MARTXVc toxin converted to carry Bla
In this study, we sought to generate modified V. cholerae strains that either produce a MARTXVc toxin with no active effector domains or that deliver only a single effector. To accomplish this, a plasmid was constructed that has fused portions of the rtxA gene encompassing the region upstream of the acd and the region corresponding to the cpd, but absent the intervening effector domain gene sequences. The codons for the natural autoprocessing sites in front of ACD and CPD were retained in the cloned fragments to preserve the natural cleavage process. Between these two fragments was cloned a promoterless bla sequence. When the plasmid was exchanged into V. cholerae strain KFV119 (N16961ΔhapAΔhlyA) by double homologous recombination, the rtxA gene produces a toxin with an in-frame fusion to Bla (RtxA::Bla) replacing the ACD, RID, and ABH in the MARTXVc toxin (Fig. 1, Table 1). The resulting strain JD1 was resistant to the beta-lactam antibiotic ampicillin (Fig. 2), indicating the gain of the beta-lactam antibiotic cleavage activity of Bla. In comparison, a similar exchange of the rtxA::bla plasmid into a mutant with an insertion in the Type I secretion gene rtxB generated strain JD4, generated a strain that was now ampicillin sensitive. Thus, the gain of ampicillin resistance in the wild-type strain carrying rtxA::bla is not just an assay for toxin production, but also demonstrates the ability of the toxin to bypass the periplasm and to be Type I secreted into the medium, where it inactivates the bacteriostatic antibiotic. RtxA::Bla was also secreted resulting in ampicillin resistance from a strain JD5, which is isogenic with JD1 except that it has a C3568A point mutation in the catalytic site of the CPD (Sheahan et al., 2007), demonstrating that growth on ampicillin does not require autoprocessing of the toxin in the agar media.
Fig. 1. Schematic representation of MARTX toxins expressed by V. cholerae strains generated for this study.
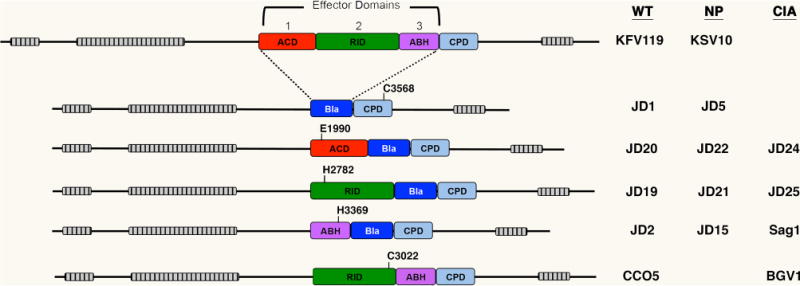
Domain organization of each toxin is shown to scale with grey-hatched bars representing the locations of the MARTX repeat regions. Effector domains are: the actin cross-linking domain (ACD, red), the Rho-inactivation domain (RID, green), and the alpha-beta hydrolase domain (ABH, purple). The location of surrogate effector domains beta-lactamase (Bla without secretion signal) is shown in blue. The cysteine protease domain (CPD) required for autoprocessing to release effector domains and/or Bla to eukaryotic cells after translocation is depicted in light blue. The relative position of catalytic residues for each effector domain and the CPD are shown numbered according to the full-length 4545 aa RtxA protein as annotated by Lin et al. (1999) and verified by Dolores & Satchell (2013). Key at right indicates V. cholerae strain designations for rtxA arrangements in isogenic ΔhapAΔhlyA background that is defined as wild type (WT) in this study. Those that carry the C3568A mutation in CPD for non-processing (NP) or catalytically inactivating (CIA) mutations as indicated in the diagram. Detailed genetics of strains are listed in Table 1.
Table 1.
Strains and plasmids used in this study
| Designation | Relevant genotype | Reference |
|---|---|---|
| Vibrio cholerae | ||
| KFV119 | N16961 ΔhapA, ΔhlyA, SmR | (Sheahan et al., 2004) |
| JD23b | KFV80 [N16961 ΔhapA ΔrtxDBCA] ΔhlyA, SmR | (Boardman & Satchell, 2004), This study |
| CCO5 | KFV119 rtxAΔacd, SmR | (Sheahan et al., 2004) |
| RtxA::Bla strains | ||
| JD1 | KFV119 rtxA::bla, SmR | This study |
| JD20 | JD1 rtxA::acd-bla, SmR | This study |
| JD19 | JD1 rtxA::rid-bla, SmR | This study |
| JD2 | JD1 rtxA::abh-bla, SmR | This study |
| Catalytically-inactive strains | ||
| JD24 | JD20 rtxA::acd-bla E1990A, SmR | This study |
| JD25 | JD1 rtxA::rid-bla H2782A, SmR | This study |
| Sag1 | JD1 rtxA::abh-bla H3369A, SmR | This study |
| BGV1 | CCO5 rid-C3022A, SmR | (Ahrens et al., 2013) |
| CPD-autoprocessing defective strains | ||
| KSV10 | KFV119 rtxA-C3568A (non-processing mutant), SmR | (Sheahan et al., 2007) |
| JD5 | KSV10 rtxA::bla C3568A, SmR | This study |
| JD22 | JD5 rtxA::acd-bla C3568A, SmR | This study |
| JD21 | JD5 rtxA::rid-bla C3568A, SmR | This study |
| JD15 | JD5 rtxA::abh-bla C3568A, SmR | This study |
| Type I secretion defective mutants | ||
| JD3b | BBV16 [N16961ΔhapA, rtxB::nptII] ΔhlyA, KmR, SmR | (Boardman & Satchell, 2004), This study |
| JD4 | JD3 rtxA::bla, KmR, SmR | This study |
| Conjugation plasmids | ||
| pDS132 | oriR6K, sacB, mob, cat, CmR | (Philippe et al., 2004) |
| pJD1 | pDS132 with rtxA::bla recombination insert | This study |
| pJD34 | pDS132 with rtxA::acd-bla recombination insert | This study |
| pJD47 | pDS132 with rtxA::acd-E1990A-bla recombination insert | This study |
| pJD35 | pDS132 with rtxA::rid-bla recombination insert | This study |
| pJD49 | pDS132 with rtxA::rid-H2782A-bla recombination insert | This study |
| pJD4 | pDS132 with rtxA::abh-bla recombination insert | This study |
| pSAG1 | pDS132 with rtxA::abh-H3369A-bla recombination insert | This study |
Strains JD3 and JD23 were made isogenic with other strains in the study from referenced parent strain by deletion of the hlyA gene using plasmids and methods as previously described Fullner et al., (2002).
Fig. 2. RtxA::Bla strains are ampicillin resistant but do not induce HeLa cell lysis.
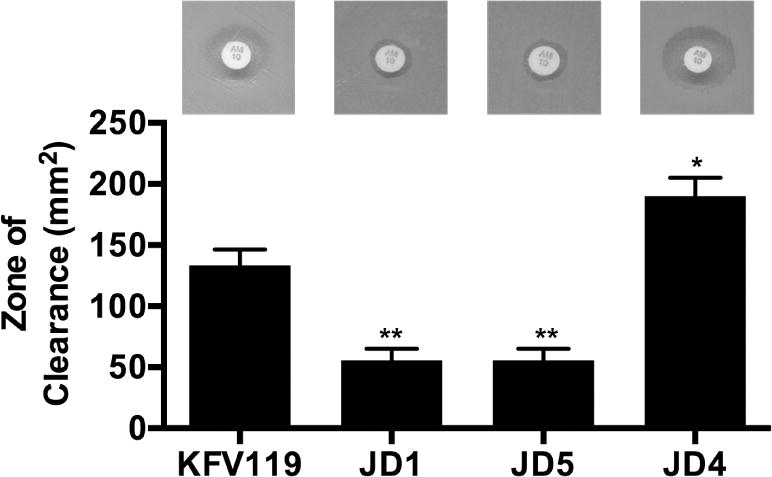
(A) A BBL Sensi-Disc (ampicillin, AM10) was placed in the center of an LB agar plate evenly spread with V. cholerae strain as indicated. Plates were photographed after overnight incubation at 37°C and the area of the zone of clearance was calculated according to A=πr2. V. cholerae strains used (in order shown) are KFV119 (wild-type), JD1 (rtxA::bla), JD5 (rtxA::bla C3658A), JD4 (rtxA::bla rtxB::km). Statistical significance compared to KFV119 is indicated as determined by multiple comparisons after one-way ANOVA (**p<0.005)
Heterologous translocation of Bla to HeLa epithelial cells
Previous studies have established that HeLa epithelial cells are susceptible to MARTXVc toxin dependent covalent cross-linking of actin (Geissler et al., 2009) and to inactivation of RhoA (Geissler et al., 2010, Ahrens et al., 2013), indicating HeLa cells are a useful cell line for studying MARTXVc delivery of effector domains. Delivery of Bla to the cytosol of HeLa cells was quantifed using CCF2-AM, a membrane-permeant fluorescent reagent based on the beta-lactam antibiotic cephalosporin. Within eukaryotic cells, endogenous esterases rapidly convert CCF2-AM into its negatively charged form CCF2 that can undergo fluorescence resonance energy transfer (FRET) to release green fluorescence under violet laser excitation (Zlokarnik et al., 1998). When CCF2 is cleaved by Bla within the host cytosol, FRET is disrupted and the reagent fluoresces blue. Since the de-esterification of CCF2-AM within the host cytosol is necessary to observe fluorescence, any extracellular CCF2-AM cleaved by the toxin outside of the host cells will not be detected.
For these assays, HeLa cells were incubated with various strains of V. cholerae for 60 min at an MOI of 100 and then treated with gentamicin to kill the bacteria and loaded with CCF2-AM. Stained uninfected cells were >98% green compared with 0% for unstained cells, indicating successful loading of CCF2 into HeLa cells (Fig. 3A). More than 98% of the cells co-incubated with rtxA::bla containing V. cholerae strains JD1, JD4, and JD5 were all successfully loaded with CCF2-AM and emitted green fluorescence indicating conversion to CCF2. Among these CCF2+ cells treated with JD1, 31% of the cells showed blue fluorescence indicating that these cells contained Bla that was delivered from the bacterium to the eukaryotic cell cytoplasm where CCF2 was cleaved (Fig. 3A). The presence of CCF2+Bla+ cells depended on the Type 1 secretion of the RtxA::Bla toxin from the bacterium as cells treated with the rtxA::bla rtxB::km strain JD4 emitted no blue fluorescence. Successful delivery of Bla also depended on a catalytically active CPD for autoprocessing release of Bla into the cytosol, since none of the cells treated with the non-processing strain JD5 emitted blue fluorescence (Fig. 3A). Over the course of the assay, none of the strains induced cell lysis as measured by release of lactate dehydrogenase (LDH) (data not shown), consistent with previous data that the MARTXVc toxin does not induce epithelial cell lysis (Fullner & Mekalanos, 2000).
Fig. 3. Translocation of Bla to HeLa cells is more efficient when an effector domain is present.
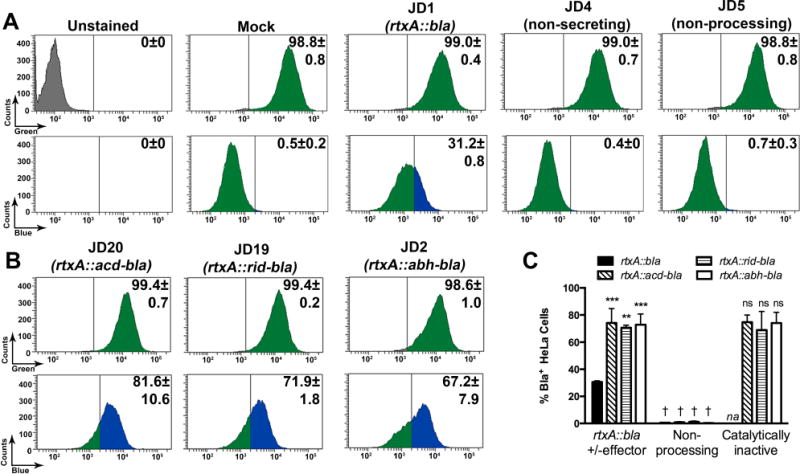
Translocation of Bla activity was determined using CCF2 fluorescence from V. cholerae treated cells quantified by flow cytometry. (A,B) Number of cells and percentage of cells from 10,000 events were determined using the AmCyan emission channel for CCF2+ cells (green peaks, upper panels) and then gated for those that also shifted to positive for emission in the Pacific Blue channel for Bla+ cells (blue peaks). The threshold for assignment as CCF2+ was determined by gating according to unstained, untreated cells (grey peaks) and for Bla+ cells by gates set according to mock (PBS) treated cells (second set of panels from right in row A. Strains used are as indicated in figure. (C) Percent Bla+ cells for additional samples quantified by the same method are shown as histograms alongside data from panels A and B. Additional strains for this panel are non-processing strains JD22, JD21, and JD15 and catalytically inactive JD24, JD25, and SAG1 that produce carry variant forms of the toxin gene as shown in the legend and described in Fig. 1 and Table 1. Data shown are the average and standard deviation of two biological replicates. Statistical significance in panel C was determined by multiple comparisons after one-way ANOVA (**p<0.005 and ***p<0.001 compared to unmodified rtxA::bla; †p<0.05 compared to paired sample without mutation; ns is not significant compared to paired sample without mutation). na indicates not applicable.
These data show that a heterologous protein Bla can function as a surrogate effector domain of the MARTXVc toxin and can be provided to cells by the natural MARTXVc delivery route of (i) Type 1 secretion from the bacterium, (ii) translocation across the eukaryotic plasma membrane, and (iii) CPD autoprocessing to release effector domain into the cytosol.
MARTXVc toxins with a single effector domain have improved efficiency of translocation
Having demonstrated Bla translocation and delivery to the cytosol, we next determined if the RtxA::Bla toxin could be exploited to carry and deliver a single effector domain. The gene sequences corresponding to the rtxA acd, rid, and abh regions were separately amplified along with sequences for their natural processing sites and cloned into the rtxA::bla plasmid placing the sequence for the effector domains in front of bla (Fig. 1). Subsequently, the effector domain sequences were modified by site-directed mutagenesis to change an essential residue. Specifically, the codons for the ACD catalytic residue E1990 (Geissler et al., 2009, Kudryashova et al., 2012) and the RID catalytic residue H2782 (Ahrens et al., 2013) were changed to Ala. Although the function of ABH remains unknown, H3369 was changed to Ala based on a putative Ser-Asp-His catalytic triad defined by strong structural homology with other members of the alpha-beta hydrolase enzyme family. Both catalytically active and inactive effector domain sequences fused to bla were exchanged into the strain JD1, such that the resulting toxins would be a “gain of function” modification compared to the effector-less strain.
In the CCF2 effector delivery assay, the efficiency of transfer of Bla from rtxA::acd-bla strain JD20 was much higher than observed for effector-less JD1 with 82% of cells showing blue fluorescence and these differences were consistent across multiple experiments. Similarly, the presence of RID or ABH in the first position in strains JD19 and JD2, respectively, also increased the efficiency of Bla delivery into HeLa cells (Fig. 3B and 3C). There was a numerical trend for ACD > RID > ABH matching the sequence of the effector domains in the holotoxin, but these differences are not statistically significant. In all cases, positive detection of Bla in eukaryotic cells depended upon the presence of a catalytically active CPD for autoprocessing, but not on the catalytic activity of the effectors themselves (Fig. 3C).
These data show several new findings on effector translocation. First, the MARTX toxins can perform heterologous protein transfer into eukaryotic cells. Further, while the presence of an effector domain can increase delivery of Bla, the catalytic activity of any of the effector domains is not required. Finally, the release of Bla from the holotoxin to the cytoplasm due to CPD autoprocessing is essential for Bla to access the large cytosolic pool of CCF2 to generate a threshold of blue fluorescence sufficient for detection in flow cytometry. Thus, the CCF2 assay detects together translocation across the membrane and successful autoprocessing.
MARTXVc toxin that can deliver only the ACD can covalently crosslink actin
The effector domain ACD has been shown to introduce a covalent crosslink into actin resulting in “laddering” of actin when separated on SDS-PAGE and detected with anti-actin antibody (Fig. 4)(Sheahan et al., 2004). HeLa cells treated with JD1 that secretes the effector-less RtxA::Bla showed no actin laddering, whereas cells treated with JD20 that secretes RtxA::ACD-Bla showed laddering equivalent to the wild-type KFV119 control and the catalysis depended upon the E1990 catalytic residue. Cells treated with V. cholerae JD22 that cannot autoprocess to release the ACD, showed a dramatic decrease in the amount of crosslinked actin, with only dimer detected (Fig. 4). This result shows that while release of the effector domain from the holotoxin is not absolutely essential, potentially only actin close to the site of toxin uptake at the membrane is crosslinked, but the larger pool of cellular actin is never accessed.
Fig. 4. The ACD is sufficient for actin cross-linking and requires CPD autoprocessing.
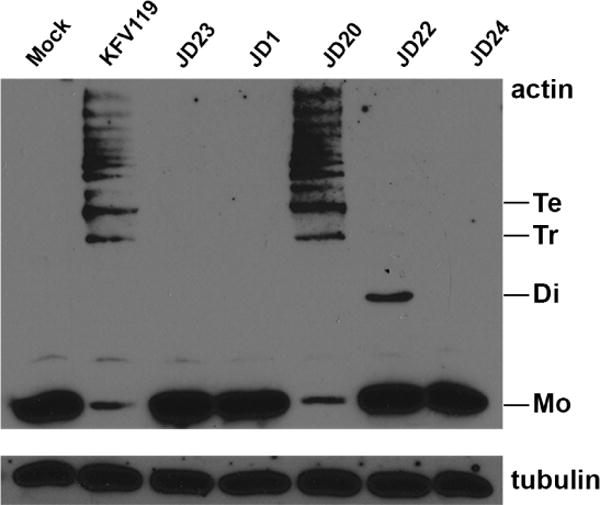
Proteins in cell lysates prepared from HeLa cells treated with V. cholerae or mock-treated with PBS for 90 min were separated by 8% SDS-PAGE and actin visualized by western blotting with anti-actin antibody. Detection of tubulin is shown as a loading control. Monomeric (Mo) and cross-linked dimers (Di), trimers (Tr), and tetramers (Te) are indicated. V. cholerae strains used (in order shown) are KFV119 (wild-type), JD23 (ΔrtxABCD), JD1 (rtxA::bla), JD20 (rtxA::acd-bla), JD22 (acd-bla C3568A), and JD24 (acd-bla E1990A). Experiment shown is representative of two independent experiments.
MARTXVc toxin that can deliver only the RID can inactive RhoA GTPase
The effector domain RID causes inactivation of RhoA, a small GTPase that can be detected by a G-LISA™ assay that detects only the active GTP bound conformation of RhoA in cell extracts (Sheahan & Satchell, 2007, Ahrens et al., 2013). The G-LISA™ assay was not technically feasible with the wild-type strain KFV119 harboring full-length toxins as the long co-incubation results in severe cell rounding due to ACD and hence poor recovery of the lysates. Thus, the isogenic Δacd mutant CCO5 was substituted. We confirmed previous data that CCO5 induces a significant loss in levels of active RhoA and this is due to RID as a mutation in the catalytic cysteine of RID significantly reduced this activity; although, as previously shown, BGV1 does retain some RhoA inactivation activity as it is believed this particular mutant in C3022 maintains the ability to bind and partially inhibit its intracellular target (Ahrens et al., 2013).
The amount of active RhoA in cells treated with strain JD1 that secretes effectorless RtxA::Bla was not significantly different from rtxA null strain JD23 (Fig. 5). There was, however, a >50% decrease in active RhoA levels in cells treated with RtxA::RID-Bla secreting strain JD19 and this decrease is comparable to that observed for CCO5, indicating that decreased Rho-GTP is due solely to RID without a contribution from ABH. This inactivation by RID was fully reversed by the H2782A catalytic site mutation in RID (Fig. 5), which unlike the C3022A mutation, is a complete inactivating mutation (Ahrens et al., 2013).
Fig. 5. Reduction of Rho-GTP by RID is only partially dependent on autoprocessing.
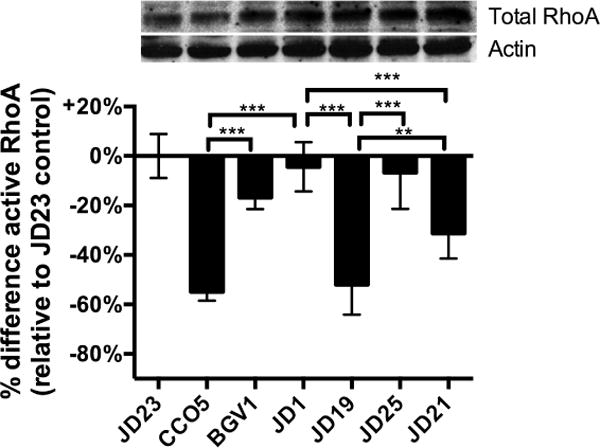
Active RhoA in cell lysates prepared from HeLa cells treated with V. cholerae or mock-treated with PBS for 4 h was measured by G-LISA. Absorbance data are normalized as percent difference compared to the average absorbance in each experiment for cells treated with rtxA null strain JD23 to limit data to effects due to the toxin and not due solely to exposure of cells to bacteria. Upper panel shows total RhoA and actin loading control in cell lysates from a representative experiment by western blotting. V. cholerae strains used (in order shown) are JD23 (ΔrtxABCD), CCO5 (rtxAΔacd), BGV1 (rtxAΔacd C3022A), JD1 (rtxA::bla), JD19 (rtxA::rid-bla), JD25 (rid-bla H2782A), and JD21 (rid-bla C3568A). Three to nine pooled data points are from three experiments performed with three biological replicates. Statistical significance between samples indicated as determined by multiple comparisons after one-way ANOVA (**p<0.005, ***P<0.001).
By contrast to the ACD where loss of CPD dramatically decreased its actin crosslinking activity, loss of CPD autoprocessing produced only a modest effect on RhoA inactivation by RID and the levels of active RhoA was significantly different compared to cells treated with JD21 or JD19 (Fig. 5). This intermediate effect could indicate that autoprocessing is not absolutely required for RID activity as its target RhoA is located located at the membrane. However, the process likely occurs less efficiently perhaps due to the limited ability of the RID to reach RhoA molecules distant from the MARTXvc toxin translocation sites.
ABH domain activates CDC42 when other effector domains are absent
The third effector domain of the MARTXVc toxin is the ABH domain that is a member of the large alpha-beta hydrolase family (Satchell, 2011). The function of this domain has not been previously investigated. When HeLa cells treated with JD2 that secretes a toxin with only the ABH domain were observed by phase microscopy, no overt cellular phenotype was observed within 4 h (data not shown). As an alternative screen for effects on cytoskeleton dynamics, we assayed HeLa cells treated with JD2 for changes in the activation state of small GTPases. No effect on RhoA or Rac GTPases was observed (data not shown). However, in this initial screen, an effect on CDC42 was noted. When quantified, treatment of cells with JD2 for 4 h resulted in 50–70% increase in the levels of activated CDC42 compared to cells treated with V. cholerae that does not produce the MARTX toxin (Fig. 6). The ability of ABH to stimulate CDC42 was dependent upon the putative catalytic residue H3369 and on an active CPD (Fig. 6). This increase in active CDC42 was surprising as CCO5, which delivers RID and active ABH, showed a modest decrease in active CDC42 (Fig. 6), consistent with previous observations (Sheahan & Satchell, 2007). Given the strength of the CDC42 activation in the presence of ABH, we considered if this activity was masked in CCO5 by simultaneous delivery of the RID. In support of this possibility, CCO5 with an inactivating point mutation in the RID domain showed increased activation of CDC42, while delivery of RID alone from strain JD19 was found to induce a dramatic decrease in levels of active CDC42 that exceeded the decrease stimulated by CCO5 (Fig. 6).
Fig. 6. The ABH domain activates CDC42 dependent upon residues C3568 and H3369.
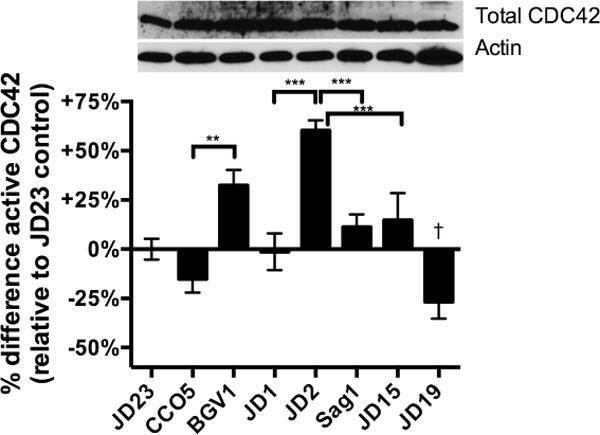
Active CDC42 in cell lysates prepared from HeLa cells treated with V. cholerae or mock-treated with PBS for 4 h was measured by G-LISA™. Absorbance data are normalized as percent difference compared to the average absorbance in each experiment for cells treated with rtxA null strain JD23 to limit data to effects due to the toxin and not due solely to exposure of cells to bacteria. Upper panel shows total CDC42 in the same cell lysates by western blotting. V. cholerae strains used (in order shown) are JD23 (ΔrtxABCD), CCO5 (rtxAΔacd), BGV1 (rtxAΔacd C3022A), JD1 (rtxA::bla), JD2 (rtxA::abh-bla), Sag1 (abh-bla H3369A) JD15 (abh-bla C3568A), and JD19 (rtxA::rid-bla). Three to nine pooled data points are from three experiments performed with three biological replicates. Statistical significance between samples indicated as determined by multiple comparisons after one-way ANOVA (**p<0.005, ***P<0.001; †p<0.001 compared to all other samples except CCO5)
Thus, these data on ABH mark the first evidence that ABH is functional in cell biological signaling resulting in activation of CDC42. This and potentially other activities associated with ABH have likely been previously masked in the context of the holotoxin as co-delivery of RID reverses the activation state of CDC42. This then further opens up new avenues to study the putative esterase activity of this domain that might influence cell signaling pathways that control CDC42.
Inhibition of J774 phagocytosis by MARTXVc toxin is primarily due to ACD
The MARTXVc toxin has been shown to inhibit phagocytosis by J774 mouse macrophages (Ma et al., 2009). To test if the inhibition of phagocytosis is a multifunctional phenotype or if one domain alone is sufficient to inhibit phagocytosis, J774 macrophages were incubated with V. cholerae for 45 min prior to killing of bacteria with gentamicin and loading with CCF2-AM. Under this experimental condition, all strains showed measureable LDH release (20–30%). However, this lysis is not due to the activity of the MARTXVc toxin as the rtxA null mutant also showed similar level of cell lysis (Fig. 7A). Under the same experimental condition, cells treated with effector-less RtxA::Bla secreting strain JD1 or Bla strains modified to deliver a single effector domain emitted blue fluorescence indicating that Bla can be successfully translocated into J774 cells. Similar to the HeLa cells, the efficiency of Bla translocation was improved when a natural effector domain was present in the MARTXVc toxin compared to the effector-less strain and was dependent upon the type I secretion of the toxin from the bacterium and its autoprocessing by the CPD (Fig. 7B).
Fig. 7. ACD is sufficient to inhibit phagocytosis by J774 macrophages.
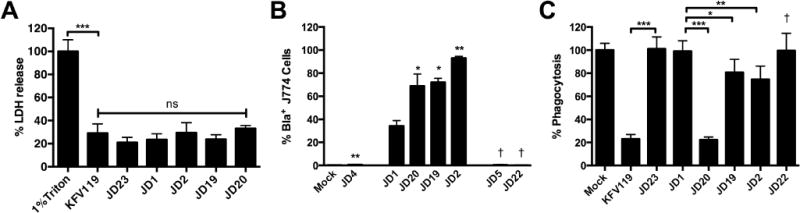
Cultured J774 macrophage cells were exposed to V. cholerae strain for 45 min and then assayed. (A) Percent cell lysis was determined as described in experimental procedures. (B) Translocation of Bla was quantified as shown in Fig. 3 with percent CCF2+/Bla+ cells from 10,000 cells shown as a histogram. (C) Percent phagocytosis was determined by uptake of fluorescent pHrodo Green E. coli Bioparticles. V. cholerae strains used in panel A (in order shown) are KFV119 (wild-type), JD23 (ΔrtxABCD), JD1 (rtxA::bla), JD20 (rtxA::acd-bla), JD19 (rtxA::rid-bla), and JD2 (rtxA::abh-bla). Additional strains in panels B and C are JD4 (rtxA::bla rtxB::km), JD5 (rtxA::bla C3568A), and JD22 (rtxA::acd-bla C3568A). Data shown in panels A and C are the average and standard deviation of three biological replicates while panel B represents biological duplicates. Statistical significance between samples indicated as determined by multiple comparisons after one-way ANOVA (**p<0.05 and ***P<0.005 are compared to rtxA::bla. †p<0.05 compared to paired sample without mutation)
In a separate experiment, J774 cells similarly treated with bacteria were treated with gentamicin to kill bacteria and simultaneously incubated with pHrodo beads coated with E. coli lysate. These beads fluoresce red only after phagocytosis and acidification of the phagosome. In this assay, wild type KFV119 inhibited phagocytosis by 80%, while the strain producing an effector-less RtxA::Bla toxin did not inhibit phagocytosis, indicating that effector domains are essential for the inhibition of phagocytosis (Fig. 7C). Strain JD20, which delivers RtxA::ACD-Bla, also inhibited phagocytosis by ~80% indicating that essentially all of the inhibition of phagocytosis is due to the ACD. RtxA::ACD-Bla with a mutation in the CPD did not show a similar inhibition demonstrating that delivery of the toxin to the cytosol is essential for inhibition of actin cross-linking (Fig. 4) as well as inhibition of phagocytosis (Fig. 7). In contrast to the overwhelming effect of ACD, RID and ABH each showed a modest albeit significant inhibition of phagocytosis. Thus, in the context of the holotoxin, inhibition of phagocytosis is likely due solely to the ACD.
ACD and RID, but not ABH, affect the integrity of polarized intestinal cells
The MARTXVc toxin has also been shown to affect polarized colonic epithelial cells resulting in the loss of trans-epithelial resistance (TEER) (Fullner et al., 2001), but the effector domains responsible for this effect have not been previously investigated. V. cholerae were added to the apical side of polarized T84 cell monolayer and TEER was measured. As shown previously, the resistance across the monolayer dropped over a 4 h period dependent upon the production of the MARTXVc toxin (Fig. 8A). This drop in TEER has previously been shown to be due to the loss of the integrity of the cellular tight junctions and not due to cell lysis or opening of ion channels (Fullner et al., 2001). V. cholerae expressing the RtxA::Bla toxin with no effector domains either with or without active CPD did not affect the integrity of the monolayers distinct from the rtxA null strain JD23, indicating that the loss of tight junction integrity requires effector domains (Fig. 8B) and is not due to ion flux from the putative apical pore formed by the repeat regions of the MARTXVc toxin.
Fig. 8. Both ACD and RID independently disrupt tight junctions in the polarized T84 monolayers.
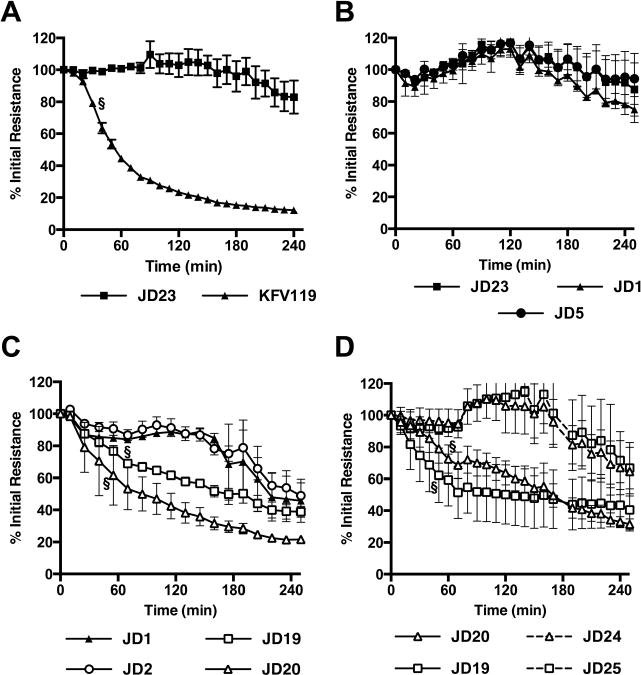
Polarized T84 colonic carcinoma cells were cultured to resistance ~1000 Ω cm2. V. cholerae was added to the apical side of the monolayer and resistance across the monolayer was measured every 10 min for a period of 250 min. Genotype of V. cholerae strains used are indicated in legends below panels and correspond to strains (in order A–D) KFV119 (wild type), JD23 (ΔrtxABCD), JD1 (rtxA::bla), JD5 (rtxA::bla C3568A), JD20 (rtxA::acd-bla), JD19 (rtxA::rid-bla), JD2 (rtxA::abh-bla), JD24 (rtxA::acd-bla E1990A), and JD21 (rtxA::rid-bla C3568A). Data shown are the average and standard deviation of three biological replicates and representative of at least two experiments for each of the strains. Symbol § designates the time point at which the percent sample initial resistance became statistically significant (p<0.05) from either the JD23 control (panels A and C) or the paired sample with a catalytically inactive mutation (panel D).
A screen of strains that translocate only one effector revealed that both ACD and RID disturb TEER, but ABH has no effect (Fig. 8C). Decreased activity of JD1 and JD2 was observed only after 180 min when it is common in all assays to observe loss of TEER due to degradation of the monolayer from bacterial exposure and repeated change in environment from frequent measurements. The relative effect of ACD compared to RID was variable by assay (compare Figs. 8C and 8D), although clearly both were able to independently contribute to the loss of TEER. In both cases, the loss depended upon a catalytically active domain (Fig. 8D) indicating that polarized cells are independently influenced by inactivation of small GTPases RhoA and CDC42 and by actin cross-linking. This is distinct from our results in the phagocytic cells where ACD had the predominating effect. Overall, this study reveals cell dependent changes in the role that different effectors may have in controlling cytoskeleton-dependent cell biological processes.
Discussion
MARTX toxins are large secreted bacterial toxins into which multiple effector domains are assembled in the core regions (Satchell, 2011). MARTX toxins in several bacterial species, most notably V. vulnificus, show extensive evidence of exchange of effector domains with other bacterial species resulting in distinct toxins in different strain isolates (Kwak et al., 2011, Roig et al., 2011, Satchell, 2011, Ziolo et al., 2014). By contrast, the MARTXVc toxin of V. cholerae is highly conserved showing little evidence of genetic exchange at the nucleotide level and essentially no change at the level of domain organization (Dolores & Satchell, 2013). Within the El Tor background responsible for the 7th cholera pandemic, the toxins are essentially 100% identical at the amino acid level. Even across a broad array of environmental isolates, the toxin gene sequences vary by less than 3% with sequence conservation particularly strong in the effector domain regions (Dolores & Satchell, 2013). This led us to consider if the MARTXVc toxin of V. cholerae was highly evolved by natural selection for the three effector domains to function together and/or in concert with the structural components of the toxin.
We started this project by considering if the novel MARTXVc toxin effector domain delivery system would allow the heterologous translocation of a protein other than its established effector domains and thus could provide a means to study translocation without disturbing the cytoskeleton. TEM-1 Bla was chosen as a surrogate effector domain as it is normally secreted by the Sec system to the bacterial periplasm (Minsky et al., 1986, Pradel et al., 2009) suggesting it could be (i) exported via a Type I system that secretes partially unfolded proteins (Koronakis et al., 2004) (ii) translocated via the putative MARTXVc pore that is also thought to translocate unfolded proteins (Kudryashova et al., 2014), and (iii) spontaneously refolded into an active enzyme in the eukaryotic cytosol where its activity could be detectable. TEM-1 Bla has also been successfully employed as a reporter for contact-dependent transfer of Type III effectors (Charpentier & Oswald, 2004, Marketon et al., 2005, Vossenkamper et al., 2010, Alam et al., 2011), Type VI secretion effectors (Ma et al., 2009, Broms et al., 2012), and Rhs proteins (Kung et al., 2012). Bla has also been used as a surrogate effector fused to the N-terminus of lethal factor to monitor effector delivery via protective antigen (Hu & Leppla, 2009). The easy unfolding of Bla for translocation was important also in studies of protective antigen as this protein is known not to translocate proteins that are resistant to unfolding, such a mCherry (Zornetta et al., 2010). A first observation of this study was that strains producing RtxA::Bla exhibited ampicillin resistance on agar plates. We were initially surprised to find that the RtxA::Bla-dependent inactivation of ampicillin required Type I secretion of the toxin. In retrospect, this finding should not have been unexpected. Beta-lactam antibiotics normally accumulate in the periplasm of Gram-negative bacteria after uptake through outer membrane porins and then are inactivated by TEM-1 Bla when it is secreted to the periplasmic space (Minsky et al., 1986). Unlike periplasmic TEM-1 Bla, the MARTX toxins like all other RTX toxins are produced in the cytosol and then secreted via Type I secretion to the medium without a periplasmic intermediate (Koronakis et al., 2004). The Type I secretion mutant used in this study was previously shown to accumulate toxin in the cytosol (Boardman & Satchell, 2004), and thus Bla from the intact RtxA::Bla toxin in the secretion mutant would not have access to periplasmic beta-lactam antibiotics and neither would RtxA::Bla toxin in a secretion competent bacteria as secretion bypasses the periplasm. Thus, the antibiotic disc assay can be exploited in the future as a means to confirm that modified toxins are successfully exported.
The next major finding was the successful detection of delivery of active Bla into HeLa and J774 cells. This shows for the first time that a MARTX toxin is capable of heterologous protein transfer and further that CPD autoprocessing is required for effector domain delivery. However, the efficiency of Bla delivery to eukaryotic cells (determined by the number of cells in which Bla cleaved CCF2 above the detection threshold), was significantly better when an effector domain was also present compared to the effectorless RtxA::Bla toxin. A simple explanation for this is that occupation of the space in front of Bla improves recognition of sites for processing and release by CPD increasing Bla detection. Thus inclusion of any protein content in this space could increase the efficiency of Bla delivery and therefore distance away from the repeat regions should be further investigated if the system is further developed for heterologous protein transfer. However, an intriguing alternative explanation is the MARTXVc holotoxin has undergone fine-tuning in its evolution for association of its effector domains with its translocation structure resulting either in more efficient translocation of effector domain across the plasma membrane or in improved association of the effector domains with the surface or active site of the CPD resulting in more efficient autoprocessing. It will be interesting to determine in the future if MARTX toxins from other bacterial species that are known to switch effector domains promiscuously by homologous recombination, such as V. vulnificus (Kwak et al., 2011, Roig et al., 2011), also show a similar trend or if they instead are evolved to produce a translocation structure that is less selective.
We next more closely examined cell biological activities for strains that would deliver just one effector domain. For all three MARTXVc effector domains, both the catalytically active and inactive effector domains were found to deliver Bla to cells with equivalent efficiency indicating that the translocation and autoprocessing do not require catalytically active effector domains. Using these toxin variants, we first show that actin cross-linking is a function solely of the ACD, which is not augmented or inhibited by other domains and is dependent on the critical residue E1990. This ACD activity is sufficient to inhibit phagocytosis in cells to the same level as macrophages treated with bacteria expressing the holotoxin. We also demonstrate for the first time that the ACD alone disturbs the integrity of the tight junctions in the polarized epithelial cells. Thus, the currently recognized physiological roles of the MARTXVc toxin can all be accounted for by the actin cross-linking ability of ACD.
This dominance of the ACD on cytoskeleton dynamics have prompted us to consider the need of having two other effector domains at all in the MARTXVc holotoxin. We surmise that while modification of cytoskeleton assembly is most easily studied in the laboratory due the dramatic effects on cell structure and shape, other cell biological processes might also be controlled by RID and ABH and these cell signaling events might be integral to the function of the holotoxin.
The RID is shown in this study to induce RhoA inactivation in the absence of other effector domains, just as previously shown in the context of the holotoxin and after delivery to cells by the anthrax toxin bioporter system (Sheahan & Satchell, 2007, Ahrens et al., 2013). The RID was intriguingly found to affect T84 polarized cells independent of the ACD with equivalent kinetics, suggesting that in this cell type the effector domains function either additively or redundantly to loosen the epithelial monolayer during V. cholerae infection, perhaps to enhance the release of nutrients in to the lumen.
It was unexpected to discover that RID also has strong inhibitory effect on CDC42, as it was previously shown that the MARTXVc holotoxin has only a modest 10–20% inactivating effect on CDC42 (Sheahan & Satchell, 2007) and this modest effect was replicated in this study. However, recent studies have shown that when RID alone in fusion with LFN was delivered to the cells by the anthrax toxin bioporter system, it is also inactivated CDC42 by ~60% (Ahrens, 2014). At this point, we can argue that our previous inability to detect this dramatic effect on CDC42 inactivation was likely due to the presence of other effector domains within the holotoxin. This suggested that there must be an effector domain that tempers RID inhibition of CDC42.
Unique to this study, due to our ability to study the function of each of the effector domains independent of others, ABH was found to indeed have an activating effect on CDC42. This is the first ever association of ABH with a cell biological activity. Based on Fig. 5 and 6, the joint effect of RID and ABH as present in strain CCO5 results in a modest inactivation of CCO5. This suggests that the mechanism of CDC42 activation by ABH may occur upstream in the CDC42 regulatory pathway, while RID inactivates CDC42 at a downstream step, thereby exerting a net overall inhibitory effect when both domains are delivered simultaneously.
So why would ABH even be present in the holotoxin if its activity is nullified by another co-existing domain? This domain predicted to be an esterase does not yet have a known molecular function, but it is possible that its putative lipase, protease, or other activity is important in a key aspect of cell signaling that simply has a side effect of activating CDC42 due to interconnected pathways. The beneficial effect of the ABH must supercede its activation of CDC42 and this activity could be the selective pressure for retention of the ABH. Indeed, across all MARTX toxins, the ABH is the most frequently prevalent effector domain (Satchell, 2011) suggesting it is beneficial to a large number of bacterial species. Exploiting the system created here, we are now for the first time able to explore the mechanistic function of the ABH and it role in cell signaling, which was not previously possible due to lack of any measurable effects on the host cells.
It is interesting to note that while ACD manages to inhibit phagocytosis effectively, the co-presence of ABH and RID could simultaneously affect the innate response during V. cholerae infection. While CDC42 is noted most commonly for its role in activation of cytoskeleton assembly resulting in cell migration, it also plays a role in controlling MAP kinase signaling (Vojtek & Cooper, 1995). Thus, it is enticing to speculate that RID and ABH, while exerting control of the cytoskeleton via RhoA and CDC42, can also influence expression of cytokines (Sabio & Davis, 2014). Thereby, in phagocytic or epithelial cells incapacitated or damaged due to the ACD, the net neutral effect of RID+ABH on CDC42 and thus MAP kinase signaling would result in no additional recruitment of innate immune cells to replace cells disabled by ACD. It will be interesting to pursue the mechanism of ABH and RID in more detail now that there is an insight into a potential role of these effectors in cell signaling and the evidence of crosstalk of signaling pathways is apparent. Studies to reveal these mechanisms are on-going but this ability to generate MARTXVc toxins that can affect cells in the absence of other effector domains will certainly assist in revealing the molecular function of these domains in great depth. The role that each of these effectors play and how they complement or counteract each other when co-translocated can be an interesting area to investigate in the future.
Experimental procedures
Bacterial strains, reagents, and media
All bacterial strains used in this study are listed in Table 1. V. cholerae and E. coli strains were routinely grown in Difco Luria-Bertani (LB) broth with shaking at 30°C or 37°C, respectively, or on LB agar plates incubated at 37°C. Antibiotics were used at concentrations: streptomycin (Sm), 100 μg/ml; ampicillin (Ap), 100 μg/ml; and kanamycin (Km), 50 μg/ml. Chloramphenical (Cm) was used at 2 μg/ml for V. cholerae and 37.5 μg/ml for E. coli. All common reagents were obtained from Fisher Biosciences or Sigma-Aldrich. Enzymes for recombinant DNA were obtained from New England Biolabs. Dulbecco’s modified Eagle medium (DMEM), RPMI medium 1640 (RPMI), phosphate buffered saline (PBS), and Hank’s balanced salt solution (HBSS) were obtained from Life Technologies (Grand Island, NY). Complete DMEM or RPMI contained 10% fetal bovine serum and 1X Penicillin-Streptomycin solution (Life Technologies).
General cloning methods
Oligonucleotide primers (Suppl. Table 1) were obtained from Integrated DNA Technologies (Coralville, IA). All ligations using Life Technologies TOPO vectors were transformed into E. coli TOP10 cells provided with the TOPO cloning system and transformed E. coli bacteria were recovered as single colonies on LB+Km agar. All ligation of fragments into pDS132 were recovered in E. coli SM10λpir or DH5αλpir with transformed E. coli bacteria recovered as single colonies on LB+Cm agar.
Construction of pDS132 sacB-counterselectable plasmids
The rtxA gene corresponding to the region upstream of the acd through the codons for the natural ACD processing site was amplified from V. cholerae N16961 chromosomal DNA using primers ME12 and ME13. The rtxA gene region corresponding to the cpd and including codons for its natural processing site was amplified with primers ME16 and ME17. The promoterless gene for beta-lactamase (bla) was amplified from vector pAR1219 (Davanloo et al., 1984) with primers ME14 and ME15. All three PCR products were assembled into a single plasmid in multiple stages. The DNA sequence of the insert in the final vector pME31 is shown in Suppl. Fig. 1 with all the key characteristics annotated. The entire joined insert was released from the TOPO vector backbone using restriction enzymes XbaI and SacI and was cloned into the similarly digested vector pDS132 to generate the plasmid pJD1.
To generate acd-bla fusion plasmids, the region of rtxA from the SalI site upstream of the acd through the codons for the natural ACD downstream autoprocessing site was amplified using primers JD4 and JD5 and the 2.4 kb fragment was captured in TOPO vector to create pJD31. The fragment was excised with SalI and EcoRI and ligated into the similarly digested pME31 to create pJD32. The entire joined insert was released from the TOPO vector using restriction enzymes XbaI and SacI and was cloned into the similarly digested vector pDS132 to generate the plasmid pJD34. For the non-catalytic E1990A version, site-directed mutagenesis was conducted on plasmid pJD32 using primers JD36 and JD37. The insert was transferred to pDS132 as described above and the final plasmid was pJD47.
To generate rid-bla fusion plasmids, the region of rtxA corresponding to rid was amplified to include codons for both the upstream and downstream autoprocessing sites using primers JD6 and JD7 and the 2.1 kb fragment was captured in TOPO vector to create pJD23. The fragment was excised with MfeI and ligated into the compatible cohesive ends of EcoRI digested pME31 to create pJD24. The entire joined insert was released from the TOPO vector using restriction enzyme NruI and was cloned by blunt-end ligation into the Eco53kI site of pDS132 to generate the plasmid pJD35. For the non-catalytic H2782A version, site-directed mutagenesis was conducted on plasmid pJD24 using primers SA186 and SA187. The insert was transferred to pDS132 as described above and the final plasmid was pJD49.
To generate abh-bla fusion plasmids, the region of rtxA corresponding to ABH including both its upstream and downstream autoprocessing sites was amplified using primers JD2 and JD3 and the 1.0 kb fragment was captured in TOPO vector to create pJD2. The fragment was excised with EcoRI and ligated into EcoRI digested pME31 to create pJD3. The entire joined insert was released from the TOPO vector by first using restriction enzymes XbaI for 2 h followed by a partial digestion with diluted SacI incubated for only 4 min. The ligated 4 kB fragment was excised from a gel and was cloned into XbaI-SacI digested pDS132 to generate the plasmid pJD4. For the non-catalytic H3369A version, site-directed mutagenesis was conducted on plasmid pJD4 using primers SN1 and SN2 and the final plasmid was pSAG1.
Transfer of novel rtxA arrangements to V. cholerae chromosome
All pDS132-based plasmids were transferred to V. cholerae by conjugation from SM10λpir on LB agar plates. Single crossover recombination events resulting in plasmid integration into the rtxA gene were selected by streaking mating mixes to LB+Sm/Cm agar. Single colonies were purified by passage on LB+Sm/Cm and then passaged once on the LB+Sm(−Cm) to allow for secondary recombination to loop out the plasmid. Bacterial cells that excised the plasmid were selected on LB+Sm agar prepared without NaCl and supplemented with 5% sucrose and incubated at room temperature for 2 days. Single colonies were then screened for gain of the new rtxA arrangement and loss of the native arrangement by PCR.
Mating SM10λpir(pJD1) with KFV119, JD3, and KSV10 strains generated effectorless rtxA::bla strains JD1, JD4, and JD5, respectively. The plasmids introducing a single effector or catalytically inactive effectors were then transferred into JD1 or JD5 such that the single effector strains are “gain of function” arrangements compared to the effectorless strains as opposed to the “loss of function” had they been mated into strains with wild-type rtxA. All selected bacterial colonies were single colony purified, confirmed to have the proper construction by PCR, and when necessary sequenced across the junctions. All matings into JD5 were confirmed by restriction digestion of amplified DNA across the cpd region as previously described (Sheahan et al., 2007) to retain the cpd mutation from the parent strain rather than gain of the wild-type codon from the plasmid.
HeLa cell flow cytometry translocation assay
V. cholerae were inoculated from a single colony and grown overnight with shaking in LB+Sm media at 30°C. The culture was diluted 1:50 and then grown at 37°C with shaking until the culture reached an optical density of A600=~0.5. The bacteria were pelleted, washed 3 times in 1 ml PBS, and diluted to a concentration of 2×108 bacteria/ml.
1×105 HeLa cells were seeded into a 24-well dish a day before the experiment. The media was changed to 1 ml incomplete DMEM (no supplements) and 50 μl PBS-washed bacteria was added to the media (MOI=100). Plates were spun at 500xg for 5 min and then incubated at 37°C/5%CO2 for 60 min. The media was then changed to 1 ml complete DMEM with 100 μg/ml gentamicin. Plates were incubated at 37°C/5%CO2 for 60 min then washed with 1 ml HBSS. Cells were covered with 500 μl HBSS and then 100 μl CCF2-AM (Life Technologies Molecular Probes) diluted according to manufacturer’s protocol was added, after which plates were kept at room temperature for 60 min. Cell were washed with 1 ml HBSS, collected in 500 μl HBSS with 1 mM EDTA. Fluorescence emission for 10,000 events per sample were collected using the Becton Dickson FACSCantoII cell analyzer with excitation at 405 nm (violet) and collection of data for green fluorescence using AmCyan 450/50 emission filter and for blue fluorescence using Pacific Blue 510/50 emission filter. All events were first gated to eliminate cell debris using forward (FSC) vs. side scatter (SSC) plots. Then Pacific Blue vs AmCyan plots for intact cells were plotted with AmCyan-positive counts indicating all cells that took up CCF2-AM and converted it to CCF2. Within the AmCyan-positive population, Pacific Blue-positive cells demonstrating successful translocation of Bla were gated within the same experiment based on plots of cells that were untreated with bacteria. All analysis was conducted with BD FACSDiva software.
J774 macrophage cell flow cytometry translocation assay
Assays were conducted as above except that incubation of bacteria was at MOI=10 and conducted with 105 J774 macrophage cells in incomplete RPMI medium followed by exchange into complete RPMI medium.
Cell lysis assays
105 HeLa or J774 cells/well were pre-seeded into a 12 well dish. Media was changed to 1 ml unsupplemented RPMI without phenol red (Life Technologies). Log-phase PBS washed V. cholerae cultures were prepared as indicated above and added to HeLa cells at an MOI=100 and to J774 cells at MOI=10 and plates were incubated at 37°C/5%CO2. HeLa cell culture media was sampled after 3 h and J774 culture media after 45 min. The amount of lactate dehydrogenase (LDH) released in to the medium by lysed cells was quantified using the Promega CytoTox96 non-radioactive cytotoxicity assay according to the manufacturer’s protocol. Percent lysis was calculated as A490(sample)/A490(100% lysis control)×100.
Actin cross-linking assay
105 HeLa cells were pre-seeded into a 12-well dish. Media was changed to incomplete DMEM for HeLa cells and to incomplete RPMI for J774 cells. Log-phase PBS washed V. cholerae cultures were prepared as described above and added to media over cells at an MOI=20 and incubated at 37°C/5%CO2 for 90 min for HeLa cells and 45 min for J774 cells. Cells were collected and boiled in 250 μl 2× SDS-PAGE buffer. Proteins were separated on an 8% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane (Sigma). Actin was visualized by Western blotting using Sigma monoclonal anti-actin antibody (Sigma) diluted 1:5000 and anti-mouse HRP secondary antibody (Sigma) diluted 1:5000 followed by detection of chemiluminescence (Thermo Scientific) based detection on x-ray film using reagents from Thermo Scientific. Samples were probed with anti-tubulin antibody (Sigma) as loading control.
G-LISA
For RhoA G-LISA, 1.5×106 HeLa cells were pre-seeded overnight into a 10 cm dish. For CDC42, 107 HeLa cells were pre-seeded into a 6 well dish. Media was changed to incomplete DMEM. Log-phase PBS washed V. cholerae cultures were prepared as described above and added to media over cells at an MOI=20 and incubated at 37°C/5%CO2. After incubation of bacteria with cells for 4 h, cells were collected, washed, and resuspended in 200 μl G-LISA lysis buffer with protease inhibitor added using reagents from Cytoskeleton, Inc. (Denver, CO). RhoA and CDC42 activation in lysates normalized for protein concentration were quantified using Cytoskeleton, Inc. G-LISA Activation assays according the manufacturer’s protocol and percent change in active RhoA or CDC42 was calculated as A490(sample)/A490(average mock control)×100.
GTPase western blots
Proteins in lysates prepared for G-LISA were boiled in SDS-PAGE sample buffer, separated on a 15% SDS-polyacrylamide gel, and transferred to nitrocellulose. RhoA or CDC42 were visualized by western blotting using Santa Cruz Biotechnology, Inc (Dallas, TX) monoclonal anti-RhoA antibody diluted 1:1000, polyclonal CDC42 antibody diluted to 1:2000 and anti-mouse horseradish peroxidase secondary antibody (Sigma) diluted 1:5000 followed by detection of chemiuminescence on x-ray film.
Phagocytosis assay
105 J774 cells were seeded into a 96-well dish in 90 μl incomplete RPMI without phenol red (Life Technologies). Log-phase PBS washed V. cholerae cultures were prepared as described above and 10 μl bacteria added to media over cells at an MOI=10. Plates were incubated at 37°C/5%CO2 for 45 min. The media was then changed to complete DMEM with 100 μg/ml gentamicin. Plates were incubated at 37°C/5%CO2 for 45 min and then 100 μl pHrodo Green E.coli Bioparticles (Life Technologies) were added and plates were incubated 37°C/5%CO2 for 1 hr. Plates were read in the SpectraMax M5 plate reader with excitation at 509 nm and emission at 533 nm. Percent phagocytosis was calculated as A533(sample)/A533(untreated control)×100.
T84 polarized epithelial cells
Polarized T84 colonic carcinoma cell monolayers were cultured on 0.33 cm2 trans-well filters until they reached ~1000 Ω cm2 at which point the media was exchange for media without fetal calf serum or antibiotics and V. cholerae bacteria were applied to the apical side of the monolayer as previously described (Fullner et al., 2001). Plates were kept at 37°C without CO2 except during resistance readings. The resistance across the monolayer was determined using World Precision Instruments Epithelial Volt-Ohm meter (EVOM). Experiments were terminated after 250 min since beginning often as early as 180 min, the monolayers deteriorate from lack of nutrition and mechanical disturbance from repeated measurements resulting in decreasing TEER of the buffer control treated samples.
Supplementary Material
Acknowledgments
We Kevin Ziolo for technical support and Gong Feng for assistance in establishing the T84 cell assays. FACS analysis was conducted using instrumentation and software provided by Northwestern Interdepartmental Immunobiology Center Flow Cytometry Facility. This work was supported by a fellowship from the Deutsche Forschungsgemeinshaft (to M.E.) and an Investigators in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund (awarded to K.J.F.S.) and by National Institutes of Health grants R01 AI051490, R01 AI092825, and R01 AI098369 (to K.J.F.S).
References
- Ahrens S. Micriobiology-Immunology. ProQuest, UMI Dissertations Publishing. Northwestern University; 2014. Identification of Essential Residues in the Rho-Inactivation Domain of the Multifunctional Autoprocessing RTX Toxin of Vibrio cholerae and its Interplay with Other Effectors; p. 143. [Google Scholar]
- Ahrens S, Geissler B, Satchell KJ. Identification of a His-Asp-Cys catalytic triad essential for function of the Rho inactivation domain (RID) of Vibrio cholerae MARTX toxin. J Biol Chem. 2013;288:1397–1408. doi: 10.1074/jbc.M112.396309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A, Miller KA, Chaand M, Butler JS, Dziejman M. Identification of V. cholerae Type Three Secretion System Effector Proteins. Infect Immun. 2011;79:1728–1740. doi: 10.1128/IAI.01194-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antic I, Biancucci M, Satchell KJ. Cytotoxicity of the Vibrio vulnificus MARTX toxin Effector DUF5 is linked to the C2A Subdomain. Proteins. 2014;82:2643–2656. doi: 10.1002/prot.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman BK, Satchell KJ. Vibrio cholerae strains with mutations in an atypical type I secretion system accumulate RTX toxin intracellularly. J Bacteriol. 2004;186:8137–8143. doi: 10.1128/JB.186.23.8137-8143.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broms JE, Meyer L, Sun K, Lavander M, Sjostedt A. Unique substrates secreted by the type VI secretion system of Francisella tularensis during intramacrophage infection. PLoS One. 2012;7:e50473. doi: 10.1371/journal.pone.0050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier X, Oswald E. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 beta-lactamase as a new fluorescence-based reporter. J Bacteriol. 2004;186:5486–5495. doi: 10.1128/JB.186.16.5486-5495.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero CL, Kudryashov DS, Reisler E, Satchell KJ. The actin cross-linking domain of the Vibrio cholerae RTX toxin directly catalyzes the covalent cross-linking of actin. J Biol Chem. 2006;281:32366–32374. doi: 10.1074/jbc.M605275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davanloo P, Rosenberg AH, Dunn JJ, Studier FW. Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1984;81:2035–2039. doi: 10.1073/pnas.81.7.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolores J, Satchell KJ. Analysis of Vibrio cholerae Genome Sequences Reveals Unique rtxA Variants in Environmental Strains and an rtxA-Null Mutation in Recent Altered El Tor Isolates. mBio. 2013;4:e00624–00612. doi: 10.1128/mBio.00624-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerer M, Satchell KJ. Inositol hexakisphosphate-induced autoprocessing of large bacterial protein toxins. PLoS Pathog. 2010;6:e1000942. doi: 10.1371/journal.ppat.1000942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullner KJ, Boucher JC, Hanes MA, Haines GK, 3rd, Meehan BM, Walchle C, Sansonetti PJ, Mekalanos JJ. The contribution of accessory toxins of Vibrio cholerae O1 El Tor to the proinflammatory response in a murine pulmonary cholera model. J Exp Med. 2002;195:1455–1462. doi: 10.1084/jem.20020318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullner KJ, Lencer WI, Mekalanos JJ. Vibrio cholerae-induced cellular responses of polarized T84 intestinal epithelial cells dependent of production of cholera toxin and the RTX toxin. Infect Immun. 2001;69:6310–6317. doi: 10.1128/IAI.69.10.6310-6317.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullner KJ, Mekalanos JJ. In vivo covalent crosslinking of actin by the RTX toxin of Vibrio cholerae. EMBO J. 2000;19:5315–5323. doi: 10.1093/emboj/19.20.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Bonebrake A, Sheahan KL, Walker ME, Satchell KJ. Genetic determination of essential residues of the Vibrio cholerae actin cross-linking domain reveals functional similarity with glutamine synthetases. Mol Microbiol. 2009;73:858–868. doi: 10.1111/j.1365-2958.2009.06810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Tungekar R, Satchell KJ. Identification of a conserved membrane localization domain within numerous large bacterial protein toxins. Proc Natl Acad Sci USA. 2010;107:5581–5586. doi: 10.1073/pnas.0908700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Leppla SH. Anthrax toxin uptake by primary immune cells as determined with a lethal factor-beta-lactamase fusion protein. PLoS One. 2009;4:e7946. doi: 10.1371/journal.pone.0007946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V, Eswaran J, Hughes C. Structure and function of TolC: the bacterial exit duct for proteins and drugs. Annu Rev Biochem. 2004;73:467–489. doi: 10.1146/annurev.biochem.73.011303.074104. [DOI] [PubMed] [Google Scholar]
- Kudryashov DS, Durer ZA, Ytterberg AJ, Sawaya MR, Pashkov I, Prochazkova K, Yeates TO, Loo RR, Loo JA, Satchell KJ, Reisler E. Connecting actin monomers by iso-peptide bond is a toxicity mechanism of the Vibrio cholerae MARTX toxin. Proc Natl Acad Sci USA. 2008;105:18537–18542. doi: 10.1073/pnas.0808082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryashova E, Heisler D, Zywiec A, Kudryashov DS. Thermodynamic properties of the effector domains of MARTX toxins suggest their unfolding for translocation across the host membrane. Mol Microbiol. 2014;92:1056–1071. doi: 10.1111/mmi.12615. [DOI] [PubMed] [Google Scholar]
- Kudryashova E, Kalda C, Kudryashov DS. Glutamyl phosphate is an activated intermediate in actin crosslinking by actin crosslinking domain (ACD) toxin. PloS one. 2012;7:e45721. doi: 10.1371/journal.pone.0045721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung VL, Khare S, Stehlik C, Bacon EM, Hughes AJ, Hauser AR. An rhs gene of Pseudomonas aeruginosa encodes a virulence protein that activates the inflammasome. Proc Natl Acad Sci USA. 2012;109:1275–1280. doi: 10.1073/pnas.1109285109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JS, Jeong HG, Satchell KJ. Vibrio vulnificus Proc Natl Acad Sci USA gene recombination generates toxin variants with altered potency during intestinal infection. Proc Natl Acad Sci USA. 2011;108:1645–1650. doi: 10.1073/pnas.1014339108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Fullner KJ, Clayton R, Sexton JA, Rogers MB, Calia KE, Calderwood SB, Fraser C, Mekalanos JJ. Identification of a Vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc Natl Acad Sci USA. 1999;96:1071–1076. doi: 10.1073/pnas.96.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma AT, McAuley S, Pukatzki S, Mekalanos JJ. Translocation of a Vibrio cholerae type VI secretion effector requires bacterial endocytosis by host cells. Cell Host Microbe. 2009;5:234–243. doi: 10.1016/j.chom.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O. Plague bacteria target immune cells during infection. Science. 2005;309:1739–1741. doi: 10.1126/science.1114580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minsky A, Summers RG, Knowles JR. Secretion of beta-lactamase into the periplasm of Escherichia coli: evidence for a distinct release step associated with a conformational change. Proc Natl Acad Sci USA. 1986;83:4180–4184. doi: 10.1073/pnas.83.12.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier V, Queen J, Satchell KJ. Successful small intestine colonization of adult mice by Vibrio cholerae requires ketamine anesthesia and accessory toxins. PloS One. 2009;4:e7352. doi: 10.1371/journal.pone.0007352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier V, Salzman NH, Satchell KJ. Prolonged colonization of mice by Vibrio cholerae El Tor O1 depends on accessory toxins. Infect Immun. 2007;75:5043–5051. doi: 10.1128/IAI.00508-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe N, Alcaraz JP, Coursange E, Geiselmann J, Schneider D. Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid. 2004;51:246–255. doi: 10.1016/j.plasmid.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Pradel N, Delmas J, Wu LF, Santini CL, Bonnet R. Sec- and Tat-dependent translocation of beta-lactamases across the Escherichia coli inner membrane. Antimicrob Agents Chemother. 2009;53:242–248. doi: 10.1128/AAC.00642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazkova K, Satchell KJ. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing of the Vibrio cholerae multifunctional autoprocessing RTX toxin. J Biol Chem. 2008;283:23656–23664. doi: 10.1074/jbc.M803334200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazkova K, Shuvalova LA, Minasov G, Voburka Z, Anderson WF, Satchell KJ. Structural and molecular mechanism for autoprocessing of MARTX Toxin of Vibrio cholerae at multiple sites. J Biol Chem. 2009;284:26557–26568. doi: 10.1074/jbc.M109.025510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queen J, Satchell KJ. Neutrophils Are essential for containment of Vibrio cholerae to the intestine during the proinflammatory phase of infection. Infect Immun. 2012;80:2905–2913. doi: 10.1128/IAI.00356-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queen J, Satchell KJ. Promotion of colonization and virulence by cholera toxin is dependent on neutrophils. Infect Immun. 2013;81:3338–3345. doi: 10.1128/IAI.00422-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig FJ, Gonzalez-Candelas F, Amaro C. Domain organization and evolution of multifunctional autoprocessing repeats-in-toxin (MARTX) toxin in Vibrio vulnificus. Appl Environ Microbiol. 2011;77:657–668. doi: 10.1128/AEM.01806-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Sem Immunol. 2014;26:237–245. doi: 10.1016/j.smim.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satchell KJ. MARTX: Multifunctional-Autoprocessing RTX Toxins. Infect Immun. 2007;75:5079–5084. doi: 10.1128/IAI.00525-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satchell KJ. Actin crosslinking toxins of Gram-negative bacteria. Toxins. 2009;1:123–133. doi: 10.3390/toxins1020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satchell KJ. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu Rev Microbiol. 2011;65:71–90. doi: 10.1146/annurev-micro-090110-102943. [DOI] [PubMed] [Google Scholar]
- Sheahan KL, Cordero CL, Satchell KJ. Identification of a domain within the multifunctional Vibrio cholerae RTX toxin that covalently cross-links actin. Proc Natl Acad Sci USA. 2004;101:9798–9803. doi: 10.1073/pnas.0401104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan KL, Cordero CL, Satchell KJ. Autoprocessing of the Vibrio cholerae RTX toxin by the cysteine protease domain. EMBO J. 2007;26:2552–2561. doi: 10.1038/sj.emboj.7601700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan KL, Satchell KJ. Inactivation of small Rho GTPases by the multifunctional RTX toxin from Vibrio cholerae. Cell Microbiol. 2007;9:1324–1335. doi: 10.1111/j.1462-5822.2006.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A, Lupardus PJ, Albrow VE, Guzzetta A, Powers JC, Garcia KC, Bogyo M. Mechanistic and structural insights into the proteolytic activation of Vibrio cholerae MARTX toxin. Nat Chem Biol. 2009;5:469–478. doi: 10.1038/nchembio.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojtek AB, Cooper JA. Rho family members: activators of MAP kinase cascades. Cell. 1995;82:527–529. doi: 10.1016/0092-8674(95)90023-3. [DOI] [PubMed] [Google Scholar]
- Vossenkamper A, Marches O, Fairclough PD, Warnes G, Stagg AJ, Lindsay JO, Evans PC, Luong le A, Croft NM, Naik S, Frankel G, MacDonald TT. Inhibition of NF-kappaB signaling in human dendritic cells by the enteropathogenic Escherichia coli effector protein NleE. J Immunol. 2010;185:4118–4127. doi: 10.4049/jimmunol.1000500. [DOI] [PubMed] [Google Scholar]
- Ziolo KJ, Jeong HG, Kwak JS, Yang S, Lavker RM, Satchell KJ. Vibrio vulnificus biotype 3 multifunctional autoprocessing RTX toxin is an adenylate cyclase toxin essential for virulence in mice. Infect Immun. 2014;82:2148–2157. doi: 10.1128/IAI.00017-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, Whitney M, Roemer K, Tsien RY. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science. 1998;279:84–88. doi: 10.1126/science.279.5347.84. [DOI] [PubMed] [Google Scholar]
- Zornetta I, Brandi L, Janowiak B, Dal Molin F, Tonello F, Collier RJ, Montecucco C. Imaging the cell entry of the anthrax oedema and lethal toxins with fluorescent protein chimeras. Cell Microbiol. 2010;12:1435–1445. doi: 10.1111/j.1462-5822.2010.01480.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.