

Abstract
Bone biomineralization is a well-regulated protein-mediated process where hydroxylapatite (HAP) crystals are nucleated with preferred orientation within self-assembled protein matrix. Mimicking this process is a promising approach to the production of bone-like protein/mineral nanocomposites for bone repair and regeneration. Towards the goal of fabricating such nanocomposites from sericin, a protein spun by Bombyx mori (B.mori) silkworm, and bone mineral HAP, for the first time we investigated the chemical mechanism underpinning the synergistic processes of the conformational change/self-assembly of B.mori sericin (BS) as well as the nucleation of HAP on the resultant self-assembled BS matrix. We found that BS, rich in anionic amino acid residues, could bind Ca2+ ions from the HAP precursor solution through electrostatic attraction. The Ca2+binding drove the conformational change of BS from random coils into β-sheets and its concomitant self-assembly into interconnected nanofibrous network-like protein matrix, which initiated the nucleation and growth of HAP crystals. HAP crystals directed by the resultant self-assembled BS matrix grew preferentially along their crystallographic c-axis, leading to the formation of HAP nano-needles. The HAP nano-needles in the self-assembled BS matrix were subsequently aggregated into globules, probably driven by the hydrogen bonding between C=O groups of BS and O-H groups of HAP nano-needles. The present work sheds light on the chemical mechanisms of BS self-assembly and the controlled mineralization directed by the self-assembled matrix. We also found that the resultant nanocomposites could promote the osteogenic differentiation of human bone marrow-derived mesenchymal stem cells. Thus our work also generates a biomimetic approach to bone-like silk protein/mineral nanocomposite scaffolds that can find potential applications in bone repair and regeneration.
Introduction
In the recent decades, fabrication of novel biomaterials for bone regeneration by mimicking the composition of natural bones is a hot area in the bioengineering field. Natural bone is a hierarchically structured nanocomposite material assembled under organism's fine control. During the natural bone biomineralization, type I collagen protein self-assembles to form an organic matrix and the needle-like hydroxylapatite (HAP) nanocrystals are nucleated and oriented within the self-assembled protein matrix, generating an extracellular matrix (ECM) for supporting the growth and differentiation of bone cells.1,2 The basic organizational unit of the ECM is mineralized type I collagen fibrils formed as a result of the oriented nucleation and growth of HAP crystals on the collagen fibrils. Inspired from bone biomineralization, using self-assembled protein-based matrix to direct HAP formation is a promising approach to the synthesis of bone-like protein/mineral nanocomposite biomaterials for bone repair and regeneration. Therefore, choosing proteins that can not only self-assemble into a matrix but also regulate the nucleation of HAP crystals within the matrix is the key to the success of this biomimetic approach. Among all of the protein-based systems such as silks,3,4 phage,5-7 and collagen8,9 being studied, silk proteins are one of the most promising candidates because silk proteins have properties desired in bone repair such as excellent biocompatibility and mechanical properties. Among silk proteins, B. mori sericin (BS), which is synthesized in the middle silk gland of B. mori silkworm, exhibits properties that are sought in biomedical applications, such as resistance to oxidation and bacteria as well as increased hydrophilicity and biodegradation.10-12
BS in the form of preformed solid films or on the surface of raw silk fibers has been used as a template to grow HAP crystals.13-17 However, these studies ignored the synergistic processes of BS molecular self-assembly and protein-directed HAP nucleation in solution because they used pre-formed dry solid BS substrates. We hypothesize that BS will experience conformational change and self-assembly in the presence of HAP precursor ions, which will be simultaneously coupled with the HAP nucleation to form mineralized structures. To test this hypothesis and understand the chemical mechanisms governing the synergistic process of BS self-assembly and HAP nucleation, this study examined the conformational change and self-assembly of BS as well as the HAP crystallization process initiated by incubating BS in an HAP precursor solution, namely, a modified simulated body fluid (1.5 SBF).
BS is a soluble protein synthesized in the silk gland of silkworm. It has abundant carboxyl groups of acidic amino acids such as Glu and Asp (Table 1). The carboxyl groups of acidic amino acids play an important role in the nucleation of HAP crystals.18-21 The isoelectric point (pI) of BS is determined to be 4.1 (Figure S1). Hence, when BS is exposed in the mineralization solution buffer containing the ions of Ca2+ and PO43- (Figure 1A), we expect that the negatively charged carboxyl groups of BS will attract the positively charged Ca2+ to form a complex through electrostatic interaction, and the binding of the Ca2+ by BS will consequently trigger the conformational change of BS from random coils to β-sheets through hydrogen bonding and the concomitant self-assembly into nanofibrous matrix (Figure 1B). Our assumption is on the basis of the fact that Ca2+ as well as other divalent ions such as Mg2+ and Cu2+ could induce the structural transition of another silk protein, fibroin, from random coil to β-sheet supports.22-24 In turn, the Ca2+ ions enriched on the surface of the β-sheet structures will further attract the PO43- from the solution buffer, initiating the preferential nucleation of the HAP crystals on the β-sheet structures and the consequent growth of HAP nuclei preferentially along their c-axis (i.e., [001] direction) to form nano-needles (Figure 1C). In addition, the C=O groups in the carboxyl groups in the β-sheet structures and O-H groups of HAP nuclei will interact by forming OH•••O hydrogen bond because the two lone pairs of electrons on the C=O groups can act as “proton receptor site” to attract the “proton donor site” of the O-H groups.25-26 Moreover, the direction of the linear O-H bond of HAP crystals will extend along the lone electron pair on the oxygen atom of the C=O groups in –COO ions to ensure that the OH•••O hydrogen bond is linear (Figure 1C and D). Since the O-H bond in HAP is parallel to the c-axis of the HAP crystals,25-27 the OH•••O hydrogen bonding will drive the HAP-BS complex to aggregate into globule structures (Figure 1D).
Table 1.
The molar percentage of amino acids in BS
| Amino acid | % | Amino acid | % |
|---|---|---|---|
| Ala | 4.05 | Val | 3.22 |
| Gly | 14.48 | Leu | 0.99 |
| Tyr | 3.39 | Ile | 0.76 |
| Ser | 35.63 | Phe | 0.56 |
| Asp | 15.65 | Pro | 0.57 |
| Arg | 3.10 | Thr | 8.14 |
| His | 1.55 | Met | 0.14 |
| Glu | 4.74 | Cys | 0.29 |
| Lys | 2.72 |
Figure 1.
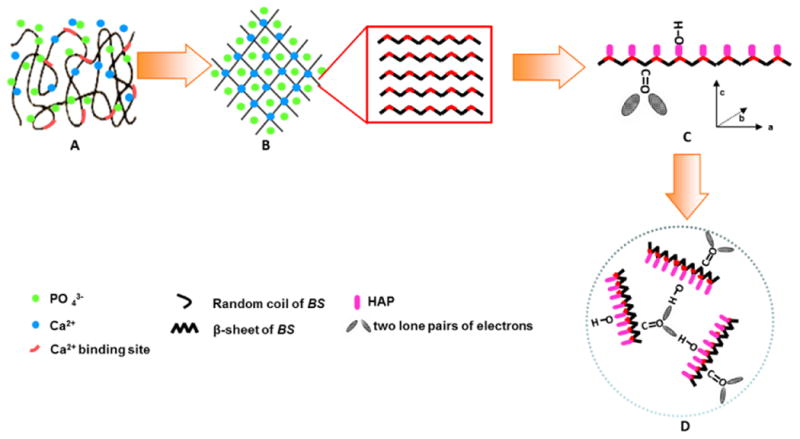
Schematic illustration of BS-mediated biomineralization when BS is incubated in 1.5 SBF. (A) BS with a random coil structure is exposed in the environment containing Ca2+ and PO43-, (B) Ca2+ binding by the anionic side-chains of BS drives the hydrogen bonding and triggers the conformational change of BS into β-sheet structure and the concomitant self-assembly into a nanofibrous network-like matrix; (C) The nanofibrous network-like matrix with β-sheet structure in turn initiates the nucleation of crystalline HAP nano-needles. The two lone pairs of electrons in the C=O groups of -COO- ion of BS acted as a “proton receptor site” to attract the “proton donor site” of O-H in HAP crystals, resulting in the formation of O— H•••O hydrogen bond. It should be noted that the O-H bond in HAP crystal structure is parallel to c-axis of HAP crystals.25-27 (D) The HAP nano-needles in the self-assembled BS matrix are further assembled into globule-like clusters due to the hydrogen bonding between C=O group in BS and O-H group in HAP crystals. BS triggers and mediates the oriented nucleation of HAP crystals during the whole process of the mineralization.
To verify this hypothesis, we firstly investigated whether BS could preferentially bind to the Ca2+ ions due to the anionic status of the BS and whether such binding triggered its self-assembly into the β-sheet structures by means of circular dichroism spectroscopy (CD) and atomic force microscopy (AFM). 1.5 SBF was used as a mineralization buffer solution for BS-mediated HAP nucleation. BS was incubated in 1.5 SBF at 37.2°C for 5 days to initiate the nucleation and growth of HAP crystals. The mineralized structures were characterized by transmission electron microscopy (TEM), X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FT-IR). Moreover, the proliferation and osteogenic differentiation of human bone marrow-derived mesenchymal stem cells (BMSCs) on both non-mineralized and mineralized BS were investigated.
Experimental
Preparation of aqueous BS solution
The cocoons we purchased were spun from the five-instar B.mori silkworms. They were dried by treating them in hot air to kill pupas inside and to remove moisture for storage. The resultant cocoons contained BS that are soluble in boiling water, allowing us to extract aqueous BS solution from cocoons at higher temperature according to the reported procedure.28 Briefly, B. mori cocoons were cut into small pieces and boiled in the deionized water for 10 min. This process allowed the BS to be dissolved in the boiling deionized water. After cooling, an aqueous BS solution was obtained by collecting the resultant solution followed by centrifugation. The amino acid analysis of BS was performed using Hitachi L8900 Amino Acid Analyzer (Table 1). In addition, the pI of extracted BS solution was calculated by measuring its zeta potential at various pH values ranging from 2.5 to 7.5.
Self-assembly of BS in the CaCl2 solution
The Ca2+ binding ability of the aqueous BS solution was examined according to a previously reported method.29 The formation rate of turbid calcium carbonate precipitates was monitored by the absorption at 570 nm of a solution prepared by adding 1.5 mL of 100 mM CaCl2 solution to a mixture of 1.5 mL of 100 mM NaHCO3 (pH 8.7) and 300 μL of aqueous BS solution with concentrations of .2.5, 5.0, 7.5 and 10.0 mg/ml. Ultraviolet spectrophotometer (SHIMADZU 2450) was used to test the absorbance at 570 nm of the resultant mixture. The Tris-HCl solution (with NaHCO3 added but in the absence of BS) was used as a control.
To investigate the structure of BS in the aqueous BS solution, 5 ml of the aqueous BS solution with a concentration of 2 mg/L was incubated in 5 ml of 10 mM CaCl2 overnight and then the structures formed were characterized using CD and AFM. The CD measurement was performed between 190 and 250 nm with a MOS-450 Spectrometer (Biologic, France) by diluting the concentration of BS to be 0.1 mg/ml in the 5 mM CaCl2 solution in a 1 mm path-length cell at room temperature. Data were collected at the rate of 0.5 nm/s. Three consecutive scans were averaged and smoothed using the accompanying software program. For AFM, the concentration of the above solution was diluted to be 1×10-2 mg/ml by deionized water. 4 μL of this diluted solution was deposited on freshly cleaved mica and air-dried, followed by washing with 20 μL deionized water. The images were taken and processed by software (NanoScope Image). The aqueous BS solution was used as a control. In addition, to further investigate the effect of the concentration of CaCl2 on the structural transition of BS, the concentration of CaCl2 was varied from 1.25 to 5 mM.
Biomineralization of BS
Mineralization was performed by using the reported protocol.2 Briefly, 1.5 SBF was first prepared by dissolving CaCl2, NaHCO3, KCl, K2HO4•3H2O, MgCl2, CaCl2, and Na2SO4 in deionized water according to the previously reported protocol.30 The solution was then buffered at pH=7.25 by using tris-hydroxymethyl-aminomethane or hydrochloric acid. The mineralization of BS was initiated by placing 10 ml of the aqueous BS solution into a dialysis bag and dialyzing against 1.5 SBF for up to 5 days at 37.2°C. The resultant mineralization solution was termed BS/1.5SBF. The 1.5 SBF was refreshed every 12 hours.
TEM, XRD and FT-IR characterization
After mineralization for 3 and 5 days, respectively, BS/1.5SBF was sampled for TEM observation. To prepare samples for observation by TEM (FEI Tecnai G2F30), 10 μL of BS/1.5SBF was put onto a copper grid and allowed to dry. Then the sample was washed by deionized water twice to remove any soluble salts. Selected area electron diffraction (ED) was also performed. For comparison, TEM observation of non-mineralized aqueous BS solution was performed according to the previously reported.31 In brief, 10 μL of BS solution was dried on the copper grid and washed by 10 μL ddH2O. The sample was then negatively stained by 1% uranyl acetate (UA).
In addition, BS/1.5SBF incubated for 5 days was freeze-dried. The obtained powder was rinsed with deionized water and then air dried. The powder was monitored with XRD (X′ Pert PRO) and FT-IR (SHIMADZU 8400s). For XRD, the samples were scanned from 10° to 60° with a scanning speed of 6°/min in situ. For FT-IR, the samples were pressed into discs with KBr at a mass ratio of 1:20. Infrared spectra were taken using a Perkin-Elmer system 2000 FT-IR spectrometer. The deconvolution of amide I region was performed by Origin 8.5 software.
Viability and alkaline phosphatase (ALP) activity of BMSCs on BS/HAP nanocomposites
We used BMSCs as testing cells to test the proliferation and osteogenic differentiation of the cells on the BS/HAP nanocomposites. Cells were cultured on the samples including BS as well as BS mineralized in 1.5 SBF for 3 and 5 days (denoted as BS3 and BS5, respectively), which were coated on the culture plates, respectively. After the cells were cultured for 1, 3 and 5 days, the cell morphology was observed according to the previously reported protocol.32 Cell Titer 96 Aqueous One Solution cell proliferation (MTS) assay was conducted to verify cell viability according to the manufacturer's protocol (Promega). In addition, we followed our published protocols33-35 to evaluate the osteogenic differentiation of BMSCs on BS, BS3 and BS5 by detecting the activity of ALP, which represents early stage marker of the osteogenic differentiation of BMSCs.
Results and discussion
Ca2+ binding ability, conformational change and self-assembly of BS
In this work, we tried to obtain the evidence on Ca2+ binding ability of BS by testing its capability in inhibiting the spontaneous precipitation of supersaturated CaCO3.29 Such inhibition indicated that BS could bind Ca2+ to reduce the concentration of free Ca2+ ions so that less CaCO3 tended to precipitate from the supersaturate solution. As seen in Figure 2A, CaCO3 was prone to precipitate upon the addition of Tris-HCl solution into the supersaturated solution containing NaHCO3 and CaCl2 whereas the presence of BS in the same solution displayed a dose-dependent inhibition of CaCO3 precipitation over a BS concentration range of 25, 5.0, 7.5 to 10.0 mg/mL, indicating the Ca2+ binding ability of BS. This proves our assumption that negatively charged carboxyl groups rich in BS (Table 1) can electrostatically attract the positively charged Ca2+. Then the structure of Ca2+-bound BS was investigated by CD measurement. The aqueous BS solution adopted a random coil conformation in the absence of Ca2+. However, addition of CaCl2 solution induced the conformational change of BS into β-sheet overnight (Figure 2B). We also found that the conformational change from random coil into β-sheet induced by Ca2+ was dependent on the concentration of CaCl2 (Figure S2); a higher concentration is needed to achieve obvious conformational change. It was reported that cations could induce the conformational change of another silk protein, fibroin, from random coil to β-sheet,22-24 further proving that electrostatic attraction between negatively charged carboxyl groups rich in BS and positively charged Ca2+ can result in the conformational change of β-sheet of BS. AFM images further showed that the morphology of BS was changed from separated well-defined nano-fibrils (Figure 2C) to interconnected nanofibrous network-like matrix (Figure 2D) when the secondary structure of BS was changed from random coils to β-sheets. This morphological change was probably because hydrogen bonding interaction between BS nanofibers also made them interconnected and aggregated to form networks. In addition, aggregation of nanofibers tended to increase in the diameter of the nanofibers in the network-like matrix (Figure 2D). These results confirmed that Ca2+ binding by the acidic amino acids rich in the BS triggered the conformational change of BS from random coils to β-sheets and the concomitant self-assembly of BS into a nanofibrous network-like matrix that could initiate the nucleation, growth and assembly of HAP crystals.
Figure 2.

Conformational change and self-assembly of BS in CaCl2 solution. (A) The Ca2+ binding ability of BS demonstrated by the decreasing absorbance at 570 nm of the mixture (made of 1.5 mL of 100 mM NaHCO3 (pH 8.7) and 300 μL of 20 mM Tris-HCl solution) in the presence of added BS with different concentration (0, 2.5, 5.0, 7.5, 10.0 mg/mL) after the addition of 1.5 mL of 100 mM CaCl2 (pH 8.7). The Tris-HCl solution (with NaHCO3 added but in the absence of BS) was used as a control. The stronger inhibition of precipitation of calcium carbonate, reflected by the reduced absorbance at 570 nm due to the inhibited calcium carbonate precipitation, indicated the higher Ca2+ binding activity. (B) CD spectra of aqueous BS solution before and after the addition of 5 mM CaCl2, respectively. The positive absorbance at 195 nm and negative absorbance at 217 nm indicated the appearance of β-sheet structure. (C-D) AFM image of the protein-based structures formed in aqueous BS solution before (C) and (D) after pre-incubation with 10 mM CaCl2 for 5 days.
Biomineralization of self-assembled BS matrix
To test the formation and assembly of HAP crystals in the resultant self-assembled BS matrix, we incubated BS in 1.5 SBF at 37.2°C for up to 5 days. TEM observation confirmed the obvious growth of HAP crystals after mineralization for 3 and 5 days (Figure 3B-D). As a control, the structure formed in the aqueous BS solution (interacted with only Ca2+ ions but not with 1.5 SBF) was also observed with TEM (Figure 3A). TEM imaging showed that prior to mineralization, BS was indeed self-assembled into a nanofibrous network-like matrix with the constituent nanofibers measuring about 10 nm in diameter (Figure 3A). After mineralization for 3 days, nano-needles were found on the self-assembled network-like BS matrix (Figure 3B). The diffraction spots from the (002) and (212) planes of HAP nano-needles in the ED pattern proved the formation of polycrystalline HAP crystals. The two white arrows in Figure 3B highlight the fact that two populations of roughly orthogonal HAP crystals are present in the selected area. Each arrow represents the c-axis of one of the two HAP populations, confirming that BS mediates the nucleation of HAP crystals preferentially elongated along the c-axis. After mineralization for 5 days, HAP globules were observed (Figure 3C). Imaging one globule at a higher magnification showed that the globule was composed of HAP nano-needles (Figure 3D) as we expected in Figure 1D. The ED pattern from a small area of the globule verified that the nano-needles were crystalline HAP with their long-axis being the crystallographic c-axis (Figure 3D, inset).
Figure 3.
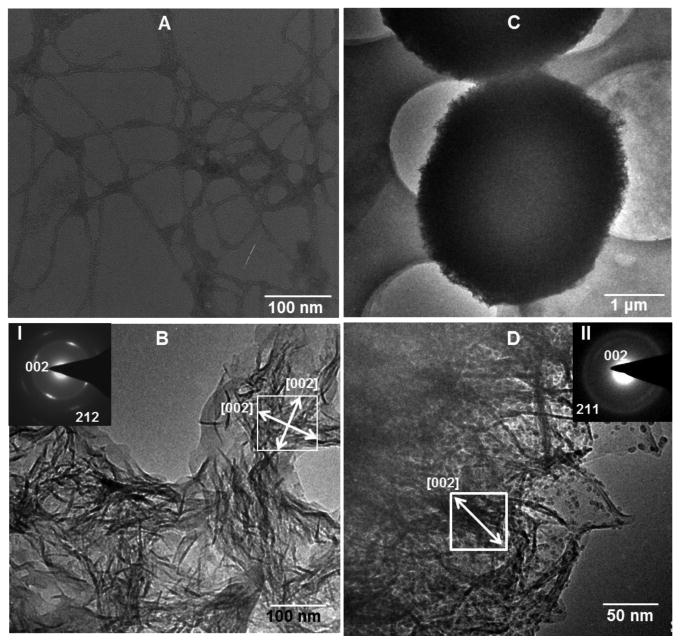
TEM images of BS before (A, only interacted with Ca2+) and after (B-D) mineralized in 1.5 SBF at 37.2°C for 3 days (B) and 5 days (C,D). D is the high magnification image corresponding to C. Insets in B and D (I and II) are electron diffraction patterns taken from the area highlighted by a white square in B and D, respectively. White arrows indicate the [002] direction (i.e., the c-axis direction) of the HAP crystals.
In addition, XRD and FT-IR spectra of the mineralized self-assembled BS matrix proved the nucleation of HAP crystals on the matrix (Figure 4). Two peaks at 25.8° and 32.8° in the XRD spectra (Figure 4A) could be assigned to (002) and (211) plane of HAP crystals, respectively, confirming that HAP crystals were nucleated on the self-assembled BS matrix after incubation in 1.5 SBF for 3 and 5 days. In the FT-IR spectra (Figure 4B), the prominent peaks at 600 and 1038 cm-1, which could be assigned to PO43- v4 bending and asymmetric PO43- v3 stretching, respectively, further confirmed the nucleation and growth of HAP crystals on the self-assembled BS matrix. In addition, we quantitatively determined the conformational change of BS by performing the deconvolution of the amide I band in the FT-IR spectra. The deconvolution results (Figure S3 and Table S1) indicated the gradual structural transition of BS from random coils and β-turns to β-sheets upon mineralization.
Figure 4.

(A) XRD patterns and (B) FT-IR spectra of BS before (a, only interacted with Ca2+) and after mineralized for different times (b, 3 days; c, 5 days). The appearance of the (203), (211) and (002) planes proved the nucleation of HAP crystals on the self-assembled BS matrix. The prominent peaks at 600 and 1038 cm-1 in B indicated the formation of HAP crystals on the BS.
Possible mechanism of HAP nucleation directed by the self-assembled BS matrix
BS can mediate the nucleation of HAP crystals preferentially with their long axis elongated along the c-axis (Figure 3B), consistent with our speculation in Figure 1C. After the formation of HAP nano-needles in the self-assembled BS matrix, BS and HAP crystals in the mineralized matrix can form hydrogen bonding by the interaction between the two lone pairs of electrons of C=O of BS and the OH of HAP nano-needles (Figure 1C),25-27 Such hydrogen bonding interaction is expected to force HAP nano-needles to be clustered into a globule-like structure (Figure 1D). This hypothesis was further confirmed by the fact that HAP globules were indeed observed after mineralization for 5 days (Figure 3C). Moreover, the broadening of O-H band as well as the shift of the O-H band to a lower wavenumber (Figure 5) is a direct evidence for the formation of hydrogen bonding (HO•••H) between OH in HAP and C=O group in BS with the β-structure as predicted in Figure 1. Collectively, the data from AFM and TEM images as well as XRD and FT-IR spectra all confirm our hypothesis on the BS-templated biomineralization proposed in Figure 1. Namely, Ca2+ ions in the HAP precursor solution (1.5 SBF) trigger the conformational change of BS from random coils to β-sheets and the concomitant self-assembly of BS into nanofibrous network-like matrix. Then the Ca2+-bearing self-assembled BS matrix electrostatically attracts PO43- to cause the super-saturation of HAP on the surface of the matrix and consequently initiates the nucleation of HAP nano-needles. Finally, the hydrogen bonding interaction between the C=O groups of BS and OH groups (parallel to c-axis of HAP crystals) of HAP nano-needles drives the assembly of HAP nano-needles into globules.
Figure 5.

FTIR spectra of O-H stretching vibrations of synthetic HAP crystals (A), BS (B), and BS/HAP (C). The O-H peak is shifted to a smaller wavenumber and becomes broader as a result of the hydrogen bonding between HAP and BS as predicted in Figure 1C.
Cell proliferation and differentiation on BS/HAP nanocomposites
To determine that HAP crystals can improve the proliferation and osteogenic differentiation of BMSCs on the BS-based materials, we seeded BMSCs on non-mineralized BS matrix as well as BS/HAP nanocomposites including BS3 and BS5, then examined the cell morphology by immunofluorescence (Figure 6A), cell proliferation by MTS assay (Figure 6B) and osteogenic differentiation by ALP activity assay (Figure 6C). The confocal micrographs indicated that cells spread widely on the BS3 and BS5 compared to those on the BS. This result illustrated that BS/HAP nanocomposites could support cell adhesion and spreading. After being cultured for 5 days, the cells exhibited a more stellate-patterned phenotype on BS3 and BS5, and more cells were observed on the BS5 than the BS. MTS assay further confirmed that the proliferation rate on BS3 and BS5 was higher than on BS, and BS5 shows higher proliferation rate than BS3 (Figure 6B). In addition, ALP assay (Figure 6C) showed that the ALP activity of BMSCs on both BS3 and BS5 was significantly higher than the BS (p<0.01), suggesting that BS/HAP nanocomposites promoted the early stage of osteogenic differentiation of BMSCs in comparison with BS without HAP crystals. Furthermore, it indicated that BS5 had a higher capacity in promoting the osteogenic differentiation than BS3. Therefore, the MTS assay and ALP assay results indicated that HAP crystals can promote the proliferation and osteogenic differentiation of BMSCs, and increasing mineralization time can also promote the cell proliferation and osteogenic differentiation. The promotion of the osteogenic capacity of biomolecular materials by HAP was in agreement with our recent findings.32,36 It suggested that BS/HAP nanocomposites can be used as a biocompatible and osteogenic biomaterial for bone regeneration and repair.
Figure 6.

(A) Morphology of human BMSCs on BS, BS3, and BS5 with actin stained in green and nuclei stained in blue by DAPI after being cultured for 1 day and 5 days. BMSCs were adherent more strongly and proliferated faster on BS5. (B) MTS assay of BMSCs proliferation on BS, BS3 and BS5 after being cultured for 1 day and 5 days. (C) Assay of the enzymatic activity of the early osteogenic differentiation marker, alkaline phosphatase (ALP), showing that BS3 and BS5 promoted the osteogenic differentiation of BMSCs in comparison to the BS. In addition, BS5 shows better capability in promoting the osteogenic differentiation of BMSCs than the BS3.* p< 0.05, ** p< 0.01. BS, BS3, and BS5 denote B. mori sericin, and B. mori sericin mineralized for 3 day and 5 days, respectively.
Conclusion
To gain an understanding on the synergetic process of BS self-assembly and HAP nucleation as well as the chemical mechanism of BS-templated biomineralization, we have studied how BS undergoes the conformational change and self-assembly process in the presence of HAP precursor ions, and then initiates the nucleation and assembly of HAP nano-needles. For the first time, we find that Ca2+ ions in the HAP precursor solution can trigger the conformational change of BS from random coils into β-sheets and the concomitant self-assembly from separated nanofibers into interconnected nanofibrous network-like matrix. The self-assembled BS matrix then induces the nucleation of HAP nano-needles with their long axis along the c-axis in the presence of PO43- ions. Due to the hydrogen bonding interaction between the C=O of BS and O-H groups of HAP nano-needles, the HAP nano-needles are forced to assemble into globules. These assembled BS/HAP globules significantly promote the proliferation and osteogenic differentiation of BMSCs when compared to the non-mineralized BS. Thus the mineralized self-assembled BS matrix can mimic the mineralized self-assembled collagen matrix in natural bone to some extent and thus potentially serve as building blocks for the development biocompatible protein/mineral scaffolds to support the growth and differentiation of stem cells for bone repair and regeneration.
Supplementary Material
Acknowledgments
M.Y. acknowledges the support of Zhejiang Provincial Natural Science Foundation of China (LZ12C17001) and Projects of Zhejiang Provincial Science and Technology Plans (2012C12910), National Natural Science Foundation of China (21172194), Silkworm Industry Science and Technology Innovation Team (2011R50028), National High Technology Research and Development Program 863 (2013AA102507). CBM would like to thank the financial support from National Institutes of Health (EB015190), National Science Foundation (DMR-0847758, CMMI-1234957 and CBET-0854414), Department of Defense Peer Reviewed Medical Research Program (W81XWH-12-1-0384), Oklahoma Center for Adult Stem Cell Research (434003) and Oklahoma Center for the Advancement of Science and Technology (HR14-160).
Contributor Information
Mingying Yang, Email: yangm@zju.edu.cn.
Chuanbin Mao, Email: cbmao@ou.edu.
Notes and references
- 1.Weiner S, Wagner HD. Ann Rev Mater Sci. 1998;28:271–298. [Google Scholar]
- 2.Nudelman F, Pieterse K, George A, Bomans PH, Friedrich H, Brylka LJ, Hilbers PA, de With G, Sommerdijk NA. Nature Mater. 2010;9:1004–1009. doi: 10.1038/nmat2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang MY, He W, Shuai YJ, Min SJ, Zhu LJ. J Polym Sci. 2013;51B:742–748. [Google Scholar]
- 4.Furuzono T, Taguchi T, Kishida A, Akashi M, Tamada Y. J Biomed Mater Res. 2000;50:344–352. doi: 10.1002/(sici)1097-4636(20000605)50:3<344::aid-jbm8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 5.Wang F, Cao B, Mao CB. Chem Mater. 2010;22:3630–3636. doi: 10.1021/cm902727s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He T, Abbineni G, Cao B, Mao CB. Small. 2010;6:2230–2235. doi: 10.1002/smll.201001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu H, Cao B, George A, Mao C. Biomacromolecules. 2011;12:2193–2199. doi: 10.1021/bm200274r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deshpande AS, Beniash E. Crystal Growth Des. 2008;8:3084–3090. doi: 10.1021/cg800252f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Yang C, Chen X, Zhao N. Adv Engi Mater. 2006;8:97–100. [Google Scholar]
- 10.Nishida A, Naganuma T, Kanazawa T, Takashima Y, Yamada M, Okada H. Int J Pharm. 2011;407:44–52. doi: 10.1016/j.ijpharm.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y. Biotechnol Adv. 2002;20:91–100. doi: 10.1016/s0734-9750(02)00003-4. [DOI] [PubMed] [Google Scholar]
- 12.Nayak S, Talukdar S, Kundu S. Cell Tissue Res. 2012;347:783–794. doi: 10.1007/s00441-011-1269-4. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi A, Ohtsuki C, Miyazaki T, Kamitakahara M, Ogata S, Yamazaki M, Furutani Y, Kinoshita H, Tanihara M. J Royal Soc Interf. 2005;2:373–378. doi: 10.1098/rsif.2005.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeuchi A, Ohtsuki C, Kamitakahara M, Ogata S, Tanihara M, Miyazaki T, Yamazaki M, Furutani Y, Kinoshita H. Key Engi Mater. 2004;254:403–406. [Google Scholar]
- 15.Yang M, Mandal N, Shuai Y, Zhou G, Min S, Zhu L. Biomed Mater Engi. 2014;24:815–824. doi: 10.3233/BME-130873. [DOI] [PubMed] [Google Scholar]
- 16.Cai Y, Jin J, Mei D, Xia N, Yao J. J Mater Chem. 2009;19:5751–5758. [Google Scholar]
- 17.Cai Y, Mei D, Jiang T, Yao J. Mater Lett. 2010;64:2676–2678. [Google Scholar]
- 18.George A, Veis A. Chem Rev. 2008;108:4670–4693. doi: 10.1021/cr0782729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George A, Bannon L, Sabsay B, Dillon JW, Malone J, Veis A, Jenkins NA, Gilbert DJ, Copeland NG. J Biol Chem. 1996;271:32869–32873. doi: 10.1074/jbc.271.51.32869. [DOI] [PubMed] [Google Scholar]
- 20.He G, Dahl T, Veis A, George A. Nature Mater. 2003;2:552–558. doi: 10.1038/nmat945. [DOI] [PubMed] [Google Scholar]
- 21.He G, Dahl T, Veis A, George A. Tissue Res. 2003;44:240–245. [PubMed] [Google Scholar]
- 22.Ochi A, Hossain KS, Magoshi J, Nemoto N. Biomacromolecules. 2002;3:1187–1196. doi: 10.1021/bm020056g. [DOI] [PubMed] [Google Scholar]
- 23.Zhou L, Chen X, Shao Z, Zhou P, Knight DP, Vollrath F. FEBS Letters. 2003;554:337–341. doi: 10.1016/s0014-5793(03)01184-0. [DOI] [PubMed] [Google Scholar]
- 24.Zong XH, Zhou P, Shao ZZ, Chen SM, Chen X, Hu BW, Deng F, Yao WH. Biochemistry. 2004;43:11932–11941. doi: 10.1021/bi049455h. [DOI] [PubMed] [Google Scholar]
- 25.Mao CB, Li H, Cui F, Ma C, Feng Q. J Crystal Grow. 1999;206:308–321. [Google Scholar]
- 26.Mao CB, Li H, Cui F, Feng Q, Ma C. J Mater Chem. 1999;9:2573–2582. [Google Scholar]
- 27.Cao B, Mao CB. Langmuir. 2007;23:10701–10705. doi: 10.1021/la7014435. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Deng L, Yang M, Min S, Yang L, Zhu L. Intl J Mol Sci. 2011;12:3170–3181. doi: 10.3390/ijms12053170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang M, Muto T, Knight D, Collins AM, Asakura T. Biomacromolecules. 2008;9:416–420. doi: 10.1021/bm700665m. [DOI] [PubMed] [Google Scholar]
- 30.Takeuchi A, Ohtsuki C, Miyazaki T, Tanaka H, Yamazaki M, Tanihara M. J Biomed Mater Res. 2003;65A:283–289. doi: 10.1002/jbm.a.10456. [DOI] [PubMed] [Google Scholar]
- 31.Cao B, Xu H, Mao C. Microsc Res Tech. 2011;74:627–635. doi: 10.1002/jemt.21030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang M, Shuai Y, Zhang C, Chen Y, Zhu L, Mao C, OuYang H. Biomacromolecules. 2014;15:1185–1193. doi: 10.1021/bm401740x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Wang L, Li X, Mao C. Sci Rep. 2013;3:1242. doi: 10.1038/srep01242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu H, Cao B, Zhen Z, Laxmi AA, Li D, Liu S, Mao CB. Biomaterials. 2011;32:4744–4752. doi: 10.1016/j.biomaterials.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hao L, Yang H, Du C, Fu X, Zhao N, Xu S, Cui F, Mao CB. Y Wang J Mater Chem B. 2014;2:4794–4801. doi: 10.1039/C4TB00597J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang M, Zhou G, Castano H, Zhu Y. C B Mao J Biomed Nanotech. 2015;11:447–456. doi: 10.1166/jbn.2015.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.