

Abstract
Effective intracellular delivery is a significant impediment to research and therapeutic applications at all processing scales. Physical delivery methods have long demonstrated the ability to deliver cargo molecules directly to the cytoplasm or nucleus, and the mechanisms underlying the most common approaches (microinjection, electroporation, and sonoporation) have been extensively investigated. In this review, we discuss established approaches, as well as emerging techniques (magnetofection, optoinjection, and combined modalities). In addition to operating principles and implementation strategies, we address applicability and limitations of various in vitro, ex vivo, and in vivo platforms. Importantly, we perform critical assessments regarding (1) treatment efficacy with diverse cell types and delivered cargo molecules, (2) suitability to different processing scales (from single cell to large populations), (3) suitability for automation/integration with existing workflows, and (4) multiplexing potential and flexibility/adaptability to enable rapid changeover between treatments of varied cell types. Existing techniques typically fall short in one or more of these criteria; however, introduction of micro-/nanotechnology concepts, as well as synergistic coupling of complementary method(s), can improve performance and applicability of a particular approach, overcoming barriers to practical implementation. For this reason, we emphasize these strategies in examining recent advances in development of delivery systems.
Keywords: intracellular delivery, transfection, lab-on-a-chip, microfluidics, micro-/nanotechnology
Introduction
Delivery of small and macromolecules—including DNA, drug molecules, imaging agents, peptides, antibodies, and enzymes—into cells is a major hurdle to realizing their full potential in a range of research and therapeutic applications, from experimental, bench-scale scientific discovery to clinical use and industrial bioproduction. Uniform treatment of a cell population (or tissue) on an individual cell basis is necessary to achieve effective delivery; however, scalability and exquisite control of treatment parameters (in time and space) are conflicting requirements. This is especially true for biological systems with significant inherent heterogeneity. Approaches to intracellular delivery are generally separated into two categories: (1) those that use biological (viral) vectors and (2) those that rely on nonviral chemical vectors or physical techniques to access the cell interior or specific organelles (including the nucleus).
To achieve a desired outcome, a vector must mediate delivery of cargo molecules to a specific intracellular target (e.g., the nuclear or mitochondrial genomes for gene therapy). Figure 1 illustrates the primary barriers to intracellular delivery and insertion of genes into the nucleus, as well as various viral and nonviral approaches that are used to overcome these barriers. In this case, an effective vector must (1) protect plasmid DNA from degradation in the extracellular matrix (if present), (2) provide a means of bypassing or disrupting the cell membrane, (3) transit the DNA through the cytoplasm to the nucleus while limiting degradation by intracellular nucleases, and (4) insert the DNA into the nucleus, across or through the nuclear envelope.1–5 Lechardeur et al.6,7 and Belting et al.8 provide detailed descriptions of the specific challenges associated with each process along the nuclear delivery pathway (including mechanisms exploited by biological vectors in nature).
Figure 1.

Barriers to intracellular delivery. (a) Various viral proteins enable viral vectors to exploit endocytic and other natural transport processes within the host cell to gain access to the interior of the nucleus. (b) Chemical-mediated delivery uses transport processes that are similar to viral gene delivery without inherent evolutionary advantages. Efficient delivery is impeded by endocytic recycling or maturation into lysosomes, degradation after DNA release, or nuclear exclusion. (c, d) Physical methods bypass multiple barriers to delivery (the focus of this review).
With regard to in vivo gene transfer, viral vectors are the favored delivery method, having been used in the vast majority of gene therapy preclinical and clinical trials.9 Their exceptional gene delivery efficiency (and further, the ability to induce permanent transduction of target cells and tissues) reflects a natural evolutionary development for that specific purpose. A number of in-depth reviews covering engineering of viral vectors and their application to gene therapy are found in the literature.9–12 Although viral delivery has proven very effective, the same characteristics that mediate infection of target cells for insertion of therapeutic genes may have adverse consequences. Adenoviral vectors have induced inflammatory immune responses and toxicity leading to patient death.13 Retroviral vectors carry the risks of transmission of replication-competent virus14 and, more importantly, of insertional mutagenesis leading to malignant transformation (e.g., patients developing cancer following treatment for genetic disease).15–17 Aside from biosafety concerns, viral vectors also possess a limited packaging capacity for incorporation of exogenous DNA.11
Chemical vectors provide some of the benefits of viral vectors without provoking a similar immune response. Unfortunately, efficiency and targeting can be inadequate, particularly in so-called difficult-to-transfect primary, progenitor, and stem cells. Unlike viral vectors, which are inherently able to achieve nuclear import, chemical vectors must overcome all biological barriers to delivery of cargo into the nucleus or other organelles (see Fig. 1). Regardless of these shortcomings, the use of cationic lipids and cationic polymers as nonviral vectors for gene transfer is well established, with numerous reviews discussing the many aspects of this approach.18–20 Strategies to improve chemical delivery focus on endosomal escape (to avoid degradation of cargo in lysosomes) or modification by cell-penetrating peptides or proteins (CPP) to deliver cargo molecules directly into the cytoplasm.20 Significant challenges remain and must be addressed for chemical vectors to gain acceptance as a viable in vitro and in vivo intracellular delivery technique for difficult-to-transfect cells and tissues.
Although viral and nonviral chemical methods maintain dominant positions in clinical and laboratory research applications, respectively, almost all physical approaches to intracellular delivery have seen significant advancement during the past decade, especially in applications involving nongene cargo molecules.21,22 An improved ability to predict and consistently realize desired treatment outcomes is attributed to a more thorough understanding of the phenomena underlying each method, as well as introduction of innovative implementation strategies. For example, detailed investigations into the relationships between treatment parameters and induced bioeffects have led to increased sonoporation efficacy without a corresponding increase in posttreatment mortality.23–25 Further, physical methods have shown more promise in treatment of difficult-to-transfect cells (primary, progenitor, and stem cells). The introduction of microfabrication and microfluidics concepts may enable use of long-established techniques such as microinjection and electroporation in emerging high-throughput applications.26–28 As performance concerns and practical deficiencies are addressed, physical delivery methods will experience increased acceptance and adoption in life sciences and biomedical research at all scales.
Physical methods are able to address the many challenges that are fundamental to delivery by chemical methods (see Fig. 1) while also minimizing or completely avoiding the side effects associated with viral vectors. Although these attributes contribute to the attractiveness of any physical approach to intracellular delivery, not all physical methods are equally suited to specific delivery applications. Indeed, direct comparison in terms of performance (e.g., transfection efficiency and cell viability) is difficult because specific techniques are often selected based on tissue or cell type, the nature of the cargo molecule to be delivered, desired treatment outcome, and even the skill of the researcher performing the experiment.29 In this review, we subdivide physical delivery methods into two categories: (1) methods that insert cargo molecules directly into the cytoplasm or nucleus and (2) methods that use a physical field to disrupt the cell membrane (electrical, mechanical, or thermal). Because magnetofection uses a magnetic field to drive carrier particles to cells (and not to achieve membrane permeabilization), this method is categorized as direct insertion. Operating principles and implementation strategies in various in vitro, ex vivo, and in vivo platforms are included for each method, as well as recent advances motivated by the introduction of micro-/nanotechnology approaches. Finally, we perform a critical assessment of each delivery method with regard to (1) treatment efficacy with diverse cell types and delivered cargo molecules, (2) suitability to different processing scales (from single cell to large populations), (3) suitability for automation/integration with existing workflows, and (4) multiplexing potential and flexibility/adaptability to enable rapid changeover between treatment of varied cell types, sizes, and morphologies.
Direct Insertion
Direct insertion methods bypass almost all biological barriers to delivery through an ability to control localization of delivered cargo within the cell (and nucleus). Unlike in field-induced membrane poration methods, the plasma membrane is not targeted for disruption in a systematic way (e.g., by forming pores prior to delivery); instead, each unit of cargo is transported across the cell membrane by a carrier liquid (e.g., microinjection) or particle (e.g., particle bombardment) as an integral part of the delivery process. Purely in vivo insertion techniques (hydrodynamic injection, conventional jet injection, and methods based on arrays of functionalized microneedles) are not discussed in this review, as these methods are tissue or organism based and do not achieve localization at the single-cell level. Reviews of these methods as applied to gene transfer may be found elsewhere.21,29,30
Microneedle and Jet Injection
Direct injection into the cytoplasm or nucleus is conceptually the most straightforward physical delivery method. In conventional microinjection, a solution of macromolecules is forced under pressure through a glass micropipette to a precise location within a single immobilized cell.31–33 An operator uses micromanipulators to control the movement and position of the small-diameter (0.1 to 5 μm) micropipette tip while observing the process under a microscope. Capecchi31 was first to demonstrate DNA microinjection into both the cytoplasm and nucleus of cultured mammalian cells. Intranuclear microinjection resulted in substantial gene expression; however, injection into the cytoplasm yielded no detectable activity, suggesting that plasmid DNA was either rapidly degraded or unable to gain access to the nucleus.
Although microinjection possesses the unique capability to directly access the nucleus (and other subcellular structures) on a consistent basis, its practical application is laborious and costly. In particular, the slow rate of delivery (injection of one cell at a time) has limited the adoption of microinjection techniques to low-throughput niche processes such as in vitro fertilization and production of transgenic animals.34 An experienced operator can inject up to 100 cells per hour. Efforts to improve the rate of delivery have focused on the development of expensive and sophisticated semi- or fully-automated systems incorporating either manual marking and automatic injection or full computer control of cell sorting, positioning/immobilization, injection, and collection.35,36 The use of automated systems has increased injection throughput significantly (up to 1500 cells per hour)35; however, throughput still lags behind that of alternative physical techniques.
Recent microfluidic approaches to microinjection promise to expand the range of applications for which this technique is viable. Both Adamo and Jensen37 and Zhang et al.26 reversed the typical microinjection strategy by moving cells onto stationary microneedles instead of positioning needles into immobilized cells. Adamo and Jensen37 used a microchannel to align and transport cells toward a single glass microcapillary loaded with a fluorescent marker compound (see Fig. 2a). Two valves controlled cell capture and release of cells to a collection reservoir. The authors estimate that a single-needle system could inject up to 3600 cells per hour. The possibility to control cell motion and/or microneedle actuation in individual treatment elements within an array suggests a route to multiplexing, although scale up to many thousands of samples may be difficult. Further, a simple design and fabrication using inexpensive and disposable parts are attractive features, resulting in potentially low cost implementation. Zhang et al.26 proposed an ultra-high-throughput microinjection platform comprising an array of capture sites with silicon penetrators (hollow or solid) for insertion of macromolecules into cells (see Fig. 2b–c).
Figure 2.

(a) Microfluidics-based single-cell microinjection system. 1: Cell is moved toward fixed microneedle. 2: Cell impinges on the needle. 3: Cell is lifted off of the needle and carried away.37 (b) Ultra-high-throughput (UHT) mechanoporation concept. 1: Cells are captured on a microneedle array. 2: Cargo is inserted into cells. 3: Cells are released. (c) SEM of 100 × 100 UHT capture site array with ~200 nm tip diameter solid penetrators.26 (d) Miniaturized jet injector concept.38
Similar to needle injection, jet injection employs a carrier fluid. High-velocity, ultrafine streams of a macromolecule solution are used to penetrate cells or target tissue. Unlike microinjection, which is a purely in vitro technique, jet injection is typically performed ex vivo or in vivo and has been most successful in transfection of muscle, skin, and tumors.39–41 Introduction of microfluidics, which demonstrate single-cell scale control without sacrificing throughput, may allow jet injection to become commonplace in vitro. Adamo et al.38 presented a prototype microfluidics-based jet injection system with the potential to treat 500 to 1000 cells per minute continuously (see Fig. 2d). Although these recent advances are substantial, there seems to be a fundamental limit to the number of samples that can be simultaneously microinjected in parallel due to external fluidic infrastructure constraints on sample delivery to multiple treatment channels and the rate at which jets containing cargo molecules can be actuated.
Particle-Mediated Delivery
In contrast with needle and jet injections, which use a carrier fluid, particle-mediated delivery transports macromolecules into target cells on an accelerated particle carrier. Particle bombardment (the biolistic method or gene gun) was originally used to deliver nucleic acids into intact plant cells.42 Having achieved effective insertion of DNA directly into inherently difficult-to-transform plant cells and tissues, the method was refined for use with smaller targets including mammalian cells.43 In brief, (1) heavy metal particles (typically gold or tungsten, diameter 1–1.5 μm) are coated with the macromolecule of interest and placed in solution on the face of a projectile, (2) the projectile is rapidly accelerated by a gas shock (e.g., from a chemical explosion, high-voltage [HV] electric spark, or helium discharge), and (3) the projectile is halted suddenly (e.g., by a mesh) releasing the microparticles at high velocity toward the target cells or tissue. Sanford et al.44 provided detailed strategies for optimization of the biolistic method. Commercial handheld and bench-top systems are available (Helios gene gun and PDS-1000/He; Bio-Rad Laboratories, Hercules, CA).
Although particle bombardment continues to be used predominantly for plant transformation, it has gained acceptance as an effective method of in vitro and in vivo nucleic acid delivery into mammalian cells and tissues, particularly for application to gene therapy and genetic immunization, where the target tissue is the skin.45 The shallow depth of penetration renders particle bombardment most effective for single-cell layers in vitro or superficial tissues (e.g., skin and mucosa) in vivo45,46; however, in vivo gene transfer into muscle and liver tissue has also been demonstrated.47,48 Like microinjection, the biolistic method can deliver DNA directly to the nucleus; for example, Shestopalov et al49 found that each green fluorescent protein (GFP)–expressing fiber cell of an intact lens contained a gold particle in its nucleus. Unfortunately, the range of outcomes within a field of treatment is unpredictable because of limited control over the distribution and penetration velocity of the microparticles. Cells containing no microparticles will exhibit no effect, whereas cells that are penetrated by a large number of particles are significantly damaged. The resultant correspondence between gene delivery and toxicity is not unique to particle bombardment among physical methods (see the Field-Induced Membrane Poration section), but an inability to consistently predict treatment outcomes based on the large number of experimental parameters is a disadvantage.44 Modifications to the conventional gene gun design have been proposed to enhance accuracy and gain access to deeper tissues. Specifically, O’Brien et al.50 redesigned the accelerator channel to generate a more tightly focused shot of gold particles, even at reduced gas pressure. Dileo et al.51 presented an entirely new gene gun using DNA-coated gold beads suspended in a liquid as the carrier. Although interesting, these improvements represent small evolutionary changes in operation and applicability and have had little influence on wider adoption.
In the strictest sense, magnetofection is not a direct insertion method as defined previously but a technique that enhances introduction of viral and nonviral gene vectors into cells.52,53 Particles are prepared by associating conventional gene vectors with magnetic nanoparticles (typically iron oxide coated with a polyelectrolyte). An external magnetic gradient field then pulls the magnetic particle-vector complexes toward the cells to be transfected (see Fig. 3a). It is possible that the magnetic sedimentation effect pulls the particles through the cell membrane52; however, it is more likely that in delivering the gene vectors directly to the cell surface, the opportunity for delivery via the endo-/lysosomal pathway is greatly increased.52–54
Figure 3.
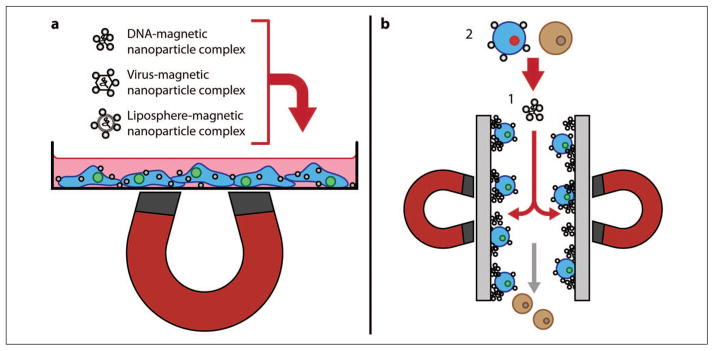
(a) Schematic overview of the magnetofection process. (b) Magselectofection. 1: A magnetic-activated cell sorting column is loaded with magnetic vectors. 2: Magnetically-labeled cells are associated with magnetic vectors using a high-gradient magnetic field.55
Regardless of the specific delivery mechanism, magnetofection has demonstrated improved gene transfer kinetics even at low dosage and the ability to localize delivery to a specific area under the influence of a magnetic field. These attributes have led to rapid growth in the number of studies involving magnetic field–mediated gene transfer both in vitro and in vivo.55 Scherer et al.53 report high-efficiency nonviral gene transfer and adenoviral magnetofection in vitro in cell lines (mouse fibroblast NIH3T3 and human erythroleukemia K562) and primary human peripheral blood lymphocytes that produce little or no Coxsackie and adenovirus receptor (i.e., cells that typically resist transduction by adenovirus). This result indicates that magnetic nanoparticles can mediate binding and internalization in cells that are otherwise resistant to a specific gene vector. High transduction efficiency was also observed in vivo with local transfection in the gastrointestinal tract and in blood vessels.53 Del Pino et al.56 used magnetic acoustically active lipospheres (MAALs) tagged with small interfering RNA (siRNA) to demonstrate the targeting abilities of magnetofection in fluidic conditions mimicking the bloodstream. Although application of ultrasound did not result in significant improvement in efficiency, further investigation is warranted as MAALs offer the ability to magnetically target specific tissues for sonoporation.55,56 Sanchez-Antequera et al.57 proposed a technique termed magselectofection that may enable a larger-scale implementation of the magnetofection process in an automated cell separation device (see Fig. 3b). In brief, (1) a commercially available magnetic-activated cell sorting column was first loaded with magnetic transfection/transduction complexes and (2) magnetically-labeled cells (T lymphocyte Jurkat, K562, and hematopoietic stem cells) were then associated with these immobilized magnetic vectors under a high-gradient magnetic field. Enhanced contact and internalization of the magnetic vectors in the cells resulted in high transfection/transduction efficiency.55,57 This technique is particularly appealing as it allows for cost-effective separation and genetic modification of cells in a single system with a minimum number of handling steps and low vector consumption. More generally, a primary advantage of magnetofection is its amenability to integration with other methods (whether as a complementary technique for enhancement of transfection outcomes or for simplification of an existing multistep process, e.g., magselectofection). In most cases, limitations on multiplexing, throughput, and changeover rate would be associated with the companion method. Plank et al.55 provided an extensive summary of magnetofection progress during its initial decade of existence, as well as prospects for adoption to various application areas.
Field-Induced Membrane Poration
While direct insertion methods seek to deliver exogenous molecules directly into the cell cytoplasm or nucleus, field-induced membrane poration techniques act to transiently disrupt the plasma membrane by creating pores through which molecules can enter cells (either actively or passively). Electroporation and sonoporation represent the most widely used nonviral physical transfection methods for effective treatment of mammalian cells both in vitro and in vivo; however, as with each of the other methods discussed in this review, there remain important obstacles to their adoption as viable replacements for chemical-mediated and viral methods. Here, advantages and limitations of each of these methods are presented, as well as strategies to address current deficiencies. Optical injection (or optical transfection if used for delivery of nucleic acids), which uses thermal or laser-induced cavitation-mediated mechanical fields to disrupt the cell membrane, is also briefly discussed.
Electroporation
Electric field–mediated permeabilization (electropermeabilization, electroporation, or electrotransfer) exposes cells to short HV pulses to achieve transient and reversible destabilization of the cell membrane. While in the permeabilized state, a variety of different molecules are able to enter the cells, either by diffusion (small molecules) or through an electrophoretically driven process (macromolecules including DNA). Although the technique was introduced by Neumann et al.58 as a method to transfect murine (mouse) lyoma cells, it proved early on to be better suited for DNA transfer to bacteria, an application that places less importance on viability after electroporation. As additional studies on the use of electroporation in mammalian cells were completed, protocol optimization led to improved treatment outcomes in vitro.59 In vivo gene transfer has been ongoing since the early 1990s, with demonstrated success in skin, skeletal muscle, liver, and solid tumor tissues.21,34,60 Wide adoption as a laboratory research tool has been motivated by refinement and to some extent standardization of commercially available electroporation systems (Bio-Rad Gene Pulser, Lonza Nucleofector, Invitrogen Neon, and BTX-Harvard Apparatus ECM systems, among others).
Although electropermeabilization is well established as an effective and broadly applicable gene transfer method, the mechanisms underlying pore formation and molecular delivery have been the subject of much debate. Detailed theoretical treatments are discussed in a number of reviews, including those by Gehl,59 Golzio et al.,61 and Escoffre et al.,62 and there is now a high degree of consensus regarding the first stage of electrotransfer, permeabilization of the plasma membrane by application of an electric field. Electroporation is achieved when the transmembrane potential induced in a cell by an external field exceeds a threshold value. Although the threshold for destabilization of the cell membrane is similar for various cell types, it generally increases with decreasing cell radius. Thus, electroporation of bacterial cells requires greater electric field strength than that required to permeabilize mammalian cells. Because of the negative resting potential of the cell, initial pore development occurs on the side of the cell facing the positive electrode (anode), followed by permeabilization of the side facing the negative electrode (cathode).63 Investigating electric field–mediated uptake of two fluorescent dyes, Gabriel and Teissie63 found that field strength dictated the area over which permeabilization was observed and that the degree of permeabilization was controlled by the duration and number of applied electric pulses. Importantly, both experimental observation and mathematical models suggest that the cell pole facing the anode exhibits a greater extent of permeabilization (i.e., poration occurs over a larger area), whereas the cell pole facing the cathode is porated to a larger degree (i.e., more pores per unit area).59,64 Membrane poration takes just a few microseconds; however, recovery and resealing occur on the order of minutes. Persistent destabilization cannot be exploited for large-molecule uptake, as Krassowska and Filev64 report that only small (<1 nm diameter) pores remain open beyond the application of the electric field.
The accepted explanations of the pore formation and delivery processes, as well as experimental observations, suggest that small molecules are taken up by diffusion alone; however, the process by which DNA is delivered into the cell (and nucleus) is less well understood. An electrophoretic effect has long been implicated as the driver of the negatively charged DNA molecules across the permeabilized membrane and toward the cell interior.65,66 Further, experimental observations indicate that DNA enters only the more destabilized side of the cell facing the cathode.67 Thus, the surface area over which interactions between DNA and the plasma membrane occur can be increased by changing the polarity and orientation of the electric field, resulting in significant increases in gene expression.67 It is unclear whether another mechanism (DNA/membrane interaction, plasmid translocation, and finally diffusion in the cytosol) is necessary for insertion of DNA.62 The utility of electrophoresis for in vivo DNA transport into cells remains a point of contention.68–70
A thorough understanding of the electropermeabilization and molecular delivery processes is important to protocol development and optimization of treatment parameters for electroporation and electric field–mediated transfection in particular. The exponentially decaying pulses generated by early electroporators can be decomposed into a high-amplitude, short-duration (microsecond) pulse followed by a smaller-amplitude, long-duration (milliseconds) tail. This pulse shape is well suited to membrane poration followed by DNA electrophoresis to and into cells, which accounts for its continued use to this day. Recent introduction of square wave pulse generators provides additional flexibility in optimization of treatment outcomes. Various combinations of low-voltage (LV; ~50–100 V/cm) and HV (~0.7–1.5 kV/cm) pulses have been investigated in vitro and in vivo.70–72 Optimal transfection was observed for short 100 to 200 μs HV followed by longer (up to hundreds of milliseconds) LV pulses. Larger interelectrode distances and a greater need to avoid collateral damage are additional considerations for in vivo electrotransfer that limit field strengths to low hundreds of volts per centimeter.73 Further enhancement of electric field–mediated gene transfer has been achieved through exposure to additional electric fields at larger time intervals (up to 30 min). Ultrashort (nanosecond) pulsed electric fields of up to 300 kV/cm are found to interact with subcellular structures (including the nucleus) without affecting the plasma membrane.74 Schoenbach et al.74 observed GFP expression in essentially all cells (human promyelocytic leukemia HL-60) exposed to a conventional (long) electroporation pulse followed 30 min later by a short (nanosecond) pulse. GFP expression was seen in only a third of cells exposed to the conventional pulse alone. Although the technique is relatively new, this result suggests that nanosecond-pulsed electric fields enable the nuclear membrane barrier to be overcome.
Most commercial pulse generators are designed for use with cuvettes incorporating parallel plate electrodes separated by a fixed gap width (1, 2, or 4 mm) or multiwell plates with incorporated electrodes. Although some manufacturers limit protocols to those that they recommend for various cell types, most allow the user a level of direct control over signal parameters (e.g., exponential decay or square wave, time constant or pulse duration, pulse count, etc.). Manufacturers also offer a range of additional needle and plate electrode configurations for electroporation of tissues (in vitro or in vivo) and adherent cells in culture. In vitro transfection results are highly dependent on the electroporation buffer, with each manufacturer also supplying proprietary, typically low-conductivity buffer systems to allow for optimal field generation. Further, buffers may include additives (e.g., peptides or proteins bound to nuclear localizing sequences), which complex with plasmid DNA to facilitate intracellular transport to and into the nucleus after electropermeabilization and uptake into the cell interior.
Microfluidic electropermeabilization approaches provide two benefits over conventional cuvette-based and multiwell plate systems: (1) a drastically reduced interelectrode distance eliminating the need for HV sources to generate sufficient electric field strength and (2) a fairly broad range of throughput capability (from down to single cell up to tens of millions of cells per minute) with exquisite environmental control. Fundamentally, the upper limit on throughput is set by the minimum residence time required for electroporation to take place and the ability to pump fluid through microchannels at desired volumetric flow rates. At single-cell and small population levels, microfluidics-based electroporation can be used to study the fundamental mechanisms underlying various cellular processes (e.g., by monitoring electroporation dynamics and intracellular transport).75–80 Single cells are typically trapped during analysis, whereas small-volume samples can be manipulated (e.g., to induce cell lysis) under flow conditions so that intracellular contents are released for further analysis downstream.76,77,81 For example, Henslee et al.77 determined the minimum applied electric field required for permeabilization of cells (K562, NIH3T3, and mouse embryonic stem cells) in suspension by first trapping single cells using optical tweezers. Interestingly, the minimum field required to permeabilize cells of the same cell line was found not to depend on cell size, but instead, the permeabilization threshold was cell-line specific. These results suggest that further investigations of accepted mechanisms underlying cell electropermeabilization in the context of microfluidic single-cell analysis are warranted and may provide a path to higher efficiency and broader applicability of electroporation.
High-throughput microfluidic electrotransfer technologies take one of two forms: (1) parallel arrays of individual treatment elements or (2) continuous flow-through systems. Many single-cell trapping and analysis solutions have been expanded to include multiple trapping sites (see, for example, Ionescu-Zanetti et al.82). Arrays of electroporation elements have also been designed to interface with the 96-well plate format. Guignet and Meyer83 demonstrated parallel delivery of siRNA and cDNA into various difficult-to-transfect cells (e.g., primary neurons and differentiated neutrophils) using a device consisting of 96 suspended electrode pairs. In this approach, (1) electrode pairs were top loaded with small volumes of sample containing cells and macromolecules, (2) surface tension held the samples in place during electroporation, and (3) samples were displaced into a 96-well plate by addition of cell culture medium. Reported results (e.g., up to 45% eGFP cDNA transfection efficiency in differentiated HL-60 cells) are impressive considering the difficulty associated with transfecting these cells by nonviral conventional methods.
Microfluidic continuous flow electroporation systems have also been developed for high-throughput delivery and transfection. Wang and Lu28,84 exposed Chinese hamster ovary (CHO) cells to a high local field (~400 V/cm) by applying a constant direct current (DC) electric bias to a microfluidic channel with geometric variation. Electropermeabilization parameters were dictated by the sample flow rate and the length of a constriction in the channel, which defined the field exposure time as well as the constriction width and voltage amplitude, which defined the local field strength.84 The same concept was used to simulate application of single or multiple pulses by flowing cells through channels with several constrictions in series (see Fig. 4a). Devices with multiple constrictions (pulses) achieved better efficiency than those with a single narrow section after optimization of treatment parameters (residence time and field intensity in the constrictions).28 Zhan et al.85 extended this concept further by applying a low-frequency (10 Hz–10 kHz) alternating current (AC) signal to CHO cells in a flow-through device with four 35 μm wide, 150 μm long constrictions. The observed transfection efficiency (up to ~71%) was comparable to that of DC electroporation in similar devices. Adamo et al.86 used a comb-type electrode pattern and AC signal to achieve a similar effect (i.e., as a cell traveled along a microchannel, it experienced both a field variation because of the externally imposed AC voltage and due to the electrode spacing along the channel). Wang et al.87,88 incorporated a spiral electroporation section into a microfluidic device to establish a transverse vortex flow superimposed over the axially directed pressure-driven flow. Cells were subjected to the combined flow field during transit along the spiral section, experiencing changes in orientation with respect to the electric field, which caused a much larger fraction of the total cell surface to become permeabilized. Electroporation of CHO cells in a curved spiral-shaped geometry yielded a twofold increase in transfection efficiency compared with electroporation in a straight microchannel of identical length.
Figure 4.

(a) Continuous flow electroporation via geometric field amplification. 1: Single constriction. 2: Multiple “pulses” achieved by additional constrictions.28 (b) Electroporation using hydrodynamic focusing.89 (c) High-throughput continuous flow electroporation concept.27
Other approaches to microfluidic flow-through electroporation incorporate hydrodynamic focusing to isolate the electrodes from the cell solution (see Fig. 4b), avoiding many unfavorable effects associated with electrode reactions (e.g., water electrolysis leading to bubble formation, electric field distortion, and Joule heating).89,90 Wei et al.89 reported significantly improved transfection efficacy (both efficiency and viability) for laminar flow electroporation (with hydrodynamic focusing) versus conventional flow electroporation (sample directly exposed to electrodes) under similar flow electroporation conditions. Selmeczi et al.27 presented a high-throughput system for continuous electroporation of human dendritic cells in suspension. Figure 4c illustrates the device, which comprises two electrode meshes with microscopic holes (~70 μm hydraulic diameter) arranged perpendicular to the flow at a mesh-to-mesh distance of 400 μm. The mesh arrangement generates a homogeneous flow field for uniform and effective (comparable to cuvette-based systems) treatment of cells at a rate of 4 million cells/min. Further, treatment of hundreds (and potentially thousands) of millions of cells—a level of throughput that is extremely difficult to meet by other methods—is achievable.
Sono-/Mechanoporation
Ultrasound-mediated membrane poration (or sonoporation) has proven to be an effective method for molecular delivery and transfection. Whether due to the perception that sonoporation is less violent than electroporation or due to the level of comfort and familiarity with ultrasound use in clinical settings, sonoporation is the preferred physical delivery method for in vivo applications. A relatively young technology, in vitro gene transfer into mammalian cells by sonoporation was first reported in the mid-1990s.91 The addition of echocontrast microbubbles (e.g., Albumex) was found to greatly enhance transfection efficiency, suggesting that acoustic cavitation was the most likely mechanism underlying the sonoporation process.92,93 The relationships between various ultrasound signal parameters and treatment outcomes were also investigated at an early stage. Using ~1 MHz ultrasound without contrast agents, Tata et al.94 found that transfection efficiency was strongly dependent on ultrasound tone-burst repetition frequency between 10 Hz and 10 kHz. Regardless of the specific ultrasound conditions used, almost all early researchers observed a correlation between cell viability and expression levels after treatment (i.e., higher transfection efficiency was associated with increased levels of cell death).
Similar to electroporation, the exact mechanisms by which ultrasound mediates delivery and transfection are not fully understood. Experimental and theoretical analyses have implicated cavitation as the primary driver of membrane destabilization and permeabilization during sonoporation.95–97 Stable (periodic oscillation of gas bubbles) and transient (violent growth and collapse of gas bubbles) cavitation may induce reversible poration, but the rapid bubble expansion, collapse, and subsequent shock wave formation characteristic of transient cavitation are found to be more effective for delivery of exogenous molecules.97 A theoretical treatment of the role of transient cavitation in membrane permeabilization at low frequencies (20–100 kHz) was performed by Sundaram et al.97 Membrane disruption was attributed to both shock wave formation and shear stresses induced by oscillating bubble-induced fluid motion during transient cavitation. Schlicher et al.25 suggested that unlike electroporation, which generates a large number of subnanometer pores, sonoporation more closely resembles a wounding process. Evaluation of cavitation-induced morphologic changes using electron and confocal microscopy indicated outcomes ranging from formation of blebs to nonlytic necrotic death to nuclear ejection and instant cell lysis.25 A strong correlation between bubble destruction and transfection efficiency also exists for sonoporation at higher frequencies (~1–2.5 MHz) in the presence of echocontrast agents, although microscopic imaging suggests that damage regimes are different.23,98 Inclusion of contrast agents results in a significant increase in pitting of the cell membrane just after treatment, possibly explaining observed improvements in treatment efficacy (1.5 to 3-fold increase in expression).23 Investigation of ultrasound-mediated permeabilization in the presence of small fluorescent molecules has indicated that delivery is heterogeneous, inducing minimal, low, or high levels of uptake among cells in a treated population.99 The observed lack of uniformity, which is not seen following electroporation, is likely due to the temporal and spatial heterogeneity of the cavitation process itself, especially when performed in a bulk environment.
The kinetics of pore formation and resealing have also been studied extensively. Although estimates of membrane recovery time generally range from a few seconds to minutes, Duvshani-Eshet et al.23 found that plasmid added to cells up to 5 h after ultrasound treatment was able to enter a small percentage of the cell population. Even so, plasmid uptake dropped significantly (from 63% to 13%) when added immediately after versus just prior to ultrasound application.23 Sonoporation tends to produce larger pores than electroporation, with size estimates of 20 to 500 nm based on the physical diameter of successfully delivered markers, as well as electron microscopy images of cells exposed to ultrasound.100 The degree of membrane permeabilization is most sensitive to pulse repetition frequency and ultrasound intensity.95 Whether ultrasound can play a role in delivery of DNA to and across the nuclear membrane is the subject of some debate. Zarnitsyn and Prausnitz101 observed DNA expression in less than one-fourth of cells with DNA plasmid uptake, attributing this outcome to poor intracellular trafficking; however, the kinetics of protein expression is significantly faster for ultrasound-mediated DNA delivery (~3–5 h) than for Lipofectamine-mediated delivery (~20 h), suggesting that sonoporation can bypass certain barriers associated with chemical methods (e.g., the endo-/lysosomal pathway).23
Most in vitro studies involving ultrasound-mediated membrane poration have been performed using custom experimental setups comprising commercial electronics, commercial or custom ultrasound transducers, custom sample chambers, and micropositioning systems.24,25,98–100,102 Other implementations have used commercial therapeutic ultrasound probes immersed directly into cells cultured in multiwell plates.23,97 Although treatment outcomes are dependent on a large number of parameters, optimization strategies have been developed. Zarnitsyn and Prausnitz101 performed optimization of DNA uptake and transfection in human prostate cancer cell line DU145 while varying the acoustic energy density (10–30 J/cm2), cell concentration (106–108 cells/mL), and temperature (21 to 37 °C), as well as with (at 500 kHz) and without (at 24 kHz) the contrast agent Optison. Maximum efficiency was found at a cell concentration of 107 cells/mL, a temperature of 37 °C, and an ultrasound frequency of 500 kHz. In addition, both uptake and mortality were correlated with increasing energy exposure. Karshafian et al.24 were able to achieve up to 32% uptake of fluorescent FITC-dextran with 96% viability in murine fibrosarcoma cell line KHT-C at 500 kHz in the presence of Definity microbubbles. Again, efficacy and mortality were found to increase with acoustic energy exposure (as calculated from peak negative pressure, pulse repetition frequency, pulse duration, and total treatment time). Newman and Bettinger95 provided a detailed summary of in vivo ultrasound-enhanced gene transfer in various tissues (skeletal muscle, cardiac muscle, and kidney) including ultrasound conditions and optimal treatment outcomes.
The wavelengths (~1–100 mm) associated with low-frequency (tens of kHz to 1 MHz) conventional ultrasound are incompatible with most microfluidic devices, which have characteristic length scales on the order of tens of micrometers. For this reason, microfluidic sonoporation devices typically exploit standing wave characteristics of acoustic fields with at least one physical dimension in the 0.3 to 1 mm range, which corresponds to resonances at high kilohertz to low megahertz frequencies. Under these conditions, biological cells focus at the nodal plane(s) of the acoustic pressure field due to acoustic radiation forces.103,104 Lee and Peng105 report improved transfection of K562 by cationic polyethyleneimine (PEI)/DNA complexes during exposure to a 1 MHz ultrasound standing wave field at conventional scales. Multiple focal planes separated by 750 μm were observed in the 13 mm diameter, 38 mm tall sample chamber. Because cells were focused at pressure nodes where the radiation force was a minimum, ultrasound-mediated membrane poration was dismissed (a transfection efficiency of only 5% was observed with naked DNA); instead, the increased probability of collision (due to higher local concentrations) between cells and nonviral vectors at the nodal planes was thought to enhance association of the cells and PEI/DNA complexes. This result is in conflict with observations of cell sonoporation by standing wave fields in two different microfluidic geometries.106,107 Rodamporn et al.107 used a fixed sonoporation chamber (980 kHz resonance) to treat 20 μL suspensions of HeLa cells and DNA plasmid. Performance was assessed at various pressure amplitudes and total treatment times achieving GFP expression in up to 69% of cells with 80% viability. Carugo et al.106 performed continuous-flow sonoporation of H9c2 cardiomyoblast cells using a standing pressure wave (2.27 MHz resonance) to focus cells along the centerline of a microfluidic channel (see Fig. 5a). The exact mechanism of membrane permeabilization was not identified; however, based on uptake studies with molecules of varying molecular weight, there appeared to be a complex relationship between efficiency, pore size, and applied field strength (amplitude of voltage driving a piezoelectric transducer). Regardless of the delivery mechanism, high-efficiency (up to 100%) ultrasound-mediated delivery of large macromolecules without the use of contrast agents was achieved.106
Figure 5.

(a) Acoustic focusing sonoporation concept.106 (b) Micromachined ultrasonic ejector array for mechanoporation of cells via ejection from cell-sized orifices.108 (c) Microfluidic device concept for shear-induced membrane permeabilization in a constriction.109,110
In the absence of ultrasound, microfluidic devices can induce membrane permeabilization through exposure to mechanical stress fields (mechanoporation) under confined-flow conditions. Hallow et al.111 subjected DU145 cells to variable fluid shear stress by flowing cell suspensions under pressure through cylindrical (50–800 μm diameter) and conical (300 μm inlet diameter and 50 μm outlet diameter) microchannels, which were laser cut through Mylar plastic sheets of varying thickness. Short-duration, high-shear-stress exposure at the tapered outlet of conical microchannels produced the best results (36% uptake and 80% viability). Zarnitsyn et al.108 used a micromachined ultrasonic atomizer (see Fig. 5b) to eject a suspension of human malignant glioma cells (LN443) and uptake molecules from an array of 400 cell-sized (36–50 μm hydraulic diameter) square orifices. Operating at a device resonance of ~1 MHz, a maximum uptake efficiency of 85% with 80% viability was observed for ejection from 45 μm orifices. Ejection from smaller orifices resulted in significant (95%) cell mortality. Using a similar device, we have achieved transfection efficiency of ~45% in human embryonic kidney (HEK293) cells with similar viability (unpublished results). Lee et al.109 and Sharei et al.110 mechanically deformed cells in suspension by forcing them through a parallel array of microchannels with constrictions 30% to 80% smaller than the cell diameter (see Fig. 5c). Mechanoporation and delivery of proteins, siRNA, and quantum dots into cells were enabled by controlled application of compression and shear forces as the cells passed through the constrictions. High levels of uptake (up to 60% depending on the cell type and macromolecule delivered) were observed in several difficult-to-transfect cells (primary fibroblasts and dendritic cells, as well as murine embryonic stem cells) without a significant loss of cell viability.110
Optoporation
Although laser-mediated membrane permeabilization (optoporation, optoinjection, optical transfection, or laserfection) can target specific cells (in a similar manner to micro-injection) to achieve high efficiency, delivery of cargo molecules is enabled through localized or systematic disruption of the cell membrane (for example, by a thermal or mechanical field) as with electroporation and sonoporation. Various laser-based membrane permeabilization mechanisms have been exploited.112,113 The earliest demonstrations of laser-mediated transfection used a focused nanosecond pulsed laser (neodymium-doped yttrium-aluminum garnet (Nd:YAG), third harmonic 355 nm wavelength) to puncture cells (via a heating effect) at a particular location.114–116 Kurata et al.114 found that transfection efficiency was significantly improved by focusing the laser slightly within the cell near the nucleus (as opposed to into the cytoplasm). These early experiments used single and multiple pulses with spot sizes ranging from 0.3 to 2 μm.114–116 Near-infrared (titanium:sapphire, 800 nm wavelength) femtosecond lasers have been used for in vivo117 and in vitro118 gene transfer with great success. Tirlapur and Konig118 reported 100% efficiency with zero mortality in transfection of CHO and rat-kangaroo kidney (PtK2) epithelial cells. A multiphoton reaction that generates a low-density free electron plasma cloud has been implicated in membrane permeabilization using femtosecond lasers.113
Concerns over adverse effects and the destructive power of ultraviolet laser radiation with respect to damaging untargeted cellular and subcellular components have led to use of absorbing dyes and particles, as well as indirect membrane disruption by laser-induced stress waves (LISW). Palumbo et al.119 used light-absorbing phenol-red dye to achieve laser-mediated (argon-ion, 488 nm wavelength) membrane poration and transfection of NIH3T3 cells by localized heating of the cells. Use of membrane stains that absorb in the infrared spectrum (e.g., 1064 nm wavelength) potentially eliminates the possibility of damaging other cellular components. A major challenge with methods that rely on direct laser manipulation of the cell membrane is the precise alignment and positioning of the beam. In addition to the relatively sophisticated and expensive equipment required to generate and align the laser, low throughput is a significant disadvantage of these methods.
As an alternative to direct irradiation of a single-cell membrane, a laser can be directed at a larger target that is in communication with the cells or tissue (e.g., the black rubber disk attached to the bottom of a cell culture dish used by Terakawa et al.120) to generate so-called laser-induced stress waves (LISWs). Although the specific membrane permeabilization mechanism is unknown, acceleration of exogenous molecules or photomechanical stress waves (possibly due to cavitation) may lead to poration of the cell membrane. LISW-based gene transfer enables simultaneous treatment of a population of cells; however, transfection efficiency is not comparable (<10% efficiency) to direct laser injection.
Enhanced membrane permeability has been achieved by using light-absorbing particles to locally damage (by heat, laser-induced cavitation, or particle bombardment) the cell membrane. Pitsillides et al.121 labeled cells with micro- and nanoparticle absorbers prior to exposure to a pulsed (20 ns) laser and found that particle size played an important role in observed bioeffects, with larger particles having a greater damage range (i.e., unbound nanoparticles could cause no damage, whereas microparticles caused significant nonspecific damage). Lapotko et al.122 found that nanoparticle cluster size (prescribed by the number of particles in a cluster) must exceed a threshold for generation of cavitation bubbles nucleated on particle clusters. Further, the required laser energy threshold for cell membrane poration decreased with increasing cluster size, suggesting that effective particle-mediated optoporation is achievable through flood exposure to a nonfocused laser field. With sufficient energy, a laser beam could be expanded to cover an entire cell population (over millimeter- to centimeter-length scales) enabling higher throughput delivery. Chakravarty et al.123 demonstrated delivery of small molecules, proteins, and DNA into two cell lines (DU145 and GS-9L gliosarcoma) by activating carbon black nanoparticles with femtosecond laser pulses. Calcein uptake was observed in up to 90% of cells with >90% viability.
Combined Methods
Methods that synergistically combine mechanical (both direct insertion and field-based sonoporation) and electro/magnetic techniques show great promise for improved delivery performance (from an efficiency/viability standpoint and with regard to practical treatment aspects such as dynamic range of operation and cost). Intracellular delivery methods that exploit activated particles in suspension (e.g., microbubbles for use in sonoporation and magnetic micro-/nanoparticles for magnetofection) are well suited to integration in combined-mode systems. In fact, magnetofection as described above represents a combined method in which magnetic particles are associated with viral or nonviral chemical vectors for delivery and transduction/transfection. Creation of MAALs has been proposed to achieve targeted ultrasound (e.g., to guide contrast agent–like particles under the action of a magnetic field to a target tissue in vivo or to magnetically-labeled target cells in vitro).55,56,124 Interestingly, ultrasound has also been used in vitro to enhance retrovirus-mediated gene transfer without the use of contrast agents, although the mechanism of action was not identified.125
Electroporation has exhibited versatility as a complementary gene transfer method and as a primary method of membrane permeabilization and molecular insertion after targeting/localization is achieved by other techniques. Electrically active polymethylmethacrylate microneedles were proposed by Choi et al.126 as a method of intradermal delivery and electroporation-mediated transfection. The ability to penetrate human skin in vivo and electroporation of cells (red blood cells and DU145) in vitro demonstrated the potential for synergistic administration and transfer of DNA locally to the skin.126 Combinations of electroporation and sonoporation have also been found to significantly enhance transfection performance (over individual treatment modalities). Different physical mechanisms underlying each of these methods (e.g., DNA migration to and into cells under the action of an electric field and the possibility for cell wounding by sonoporation leading to larger and more long-lived pores) imply that there may be potential benefits to either treatment order (i.e., electrosonoporation or sonoelectroporation). Escoffre et al.127 demonstrated a sixfold increase in transfection efficiency for in vitro combined electrosonoporation of CHO cells versus exposure to an electric field alone. Exposure to the electric field was found to disrupt the plasma membrane and to induce electrophoretic migration of plasmid DNA toward the permeabilized membrane. Sonoporation in the presence of gas microbubbles then enabled the DNA associated with the membrane to enter the cytoplasm rapidly. Escoffre et al.127 suggested that increased performance was motivated by greater accessibility of DNA to the cytoplasm. Electrosonoporation has also been used to improve gene transfer outcomes in vivo by applying ultrasound and an electric field concurrently in murine muscle tissue.128
Because of the inherent difficulty associated with transfection, much of this review focuses on the applicability of various intracellular delivery methods to gene transfer. Unfortunately, superior transfection performance does not necessarily translate to efficient delivery of all cargo types whether due to the size of the cargo molecule or its sensitivity to treatment parameters. The largest pores generated by electropermeabilization limit the diameter of delivered cargo molecules to tens of nanometers. Further, electric fields can cause degradation and aggregation of proteins and quantum dots. Mechano- and optoporation are able to generate larger pores (hundreds of nanometers to more than 1 μm); however, insertion of large cargo molecules is ineffective because of the slow speed of cargo diffusion and decreased viability associated with increased cell wounding. Wu et al.129 introduced a combined optoporation/microinjection (via fluid carrier) method to overcome these issues. A titanium-coated microcapillary pipette (termed photothermal nanoblade) was heated rapidly with a short laser pulse to generate a cavitation bubble, which locally punctured the cell membrane without disturbing the rest of the structure. High-efficiency delivery of various cargos (from DNA and RNA to 200 nm polystyrene beads to 2 μm bacteria) under pressure was demonstrated without advancing the micropipette into the cell.129 Thus, large pores (for delivery of large cargo) were created using only gentle contact with the cell. Similarly, we have substantially enhanced the performance of the mechano-/sonoporation system illustrated in Figure 5b by including subsequent exposure of a sample that underwent mechanoporation to an electric field. When a low-strength electric field (~50 V/cm) was applied to mechanically treated cells within tens of seconds, significant improvement to transfection efficiency (e.g., up to 90% transfection in HEK293 cells) was observed with no corresponding change in viability (unpublished results). Enhancement was also obtained for other cell types through this staged combination of tuned (with respect to time and amplitude) acoustic/mechanical and electric fields. We believe that this combined method is no more violent than sonoporation or electroporation alone because DNA migration did not require exposure to an electric field of sufficient strength to permeabilize the cell membrane.
Discussion
Scalability and control of treatment on a single-cell basis (resulting in improved efficacy) are competing requirements, which are difficult to reconcile. As illustrated by various mechano-/sono- and electroporation systems described in this review, micro-/nanotechnologies allow for confinement of biophysical action and therefore improved control of treatment down to the scale of an individual cell. Unfortunately, this exquisite control of treatment parameters (in time and space) places a burden on scalability (defined as the number of cells processed per unit time), as it can be achieved only through parallelization (arraying of treatment elements) or increased throughput (i.e., the speed of treatment) of a given treatment channel. In these aspects, there is a substantial difference between methods that are continuous flow based and those that use a sequence of discrete steps. For example, electro- and sonoporation in microchannels belong to the first group and potentially could achieve much greater processing speed through design innovations in process organization, whereas direct insertion using microneedle or jet injection and particle-mediated delivery are executed through a sequence of steps with significant time overhead associated with sample handling and preparation between steps. These arguments notwithstanding, there are substantial balance-of-plant limitations to parallelization for continuous flow–based systems. Practical implementation requires that one develop and support a cumbersome infrastructure for controlled sample delivery at identical flow rate and composition (both in terms of cell density and drug/gene concentration) to each treatment channel of the array. Further, the system must handle a large overhead associated with management of pressure drop that scales proportional to the treated sample volumetric flow rate. The latter fundamental limitation is a key concern for all nonbatch (flow-based) physical methods of delivery within micro-/nano-geometries, thus limiting their applications to the laboratory scale (~10–100 millions of cells per minute) applications. In some regards, automation and robotic handling have the greatest potential to affect throughput of the discrete-step (batch) techniques, but this will necessarily come with increased complexity and therefore greater cost. Even with automation, the applicability of batch physical delivery techniques will likely be limited to smaller-scale applications, with delivery rates approaching ~1 to 10 thousands of cells per minute.
With respect to suitability of integration with existing workflows, flow-based methods are likely to require additional sample preparation steps before treatment, including cell separation based on size (if a treatment method efficacy depends on the cell size), suspension and buffer change (perhaps multiple times), dilution or preconcentration, and mixing with reagents/delivery molecules, among others. Performing these functions in a microfluidic format will help achieve greater flexibility and speed, as many of these steps are diffusion controlled and therefore enhanced dramatically with a scale down of the device characteristic feature size. Yet the need for these additional sample preparation steps in a typical workflow will bring about an increase in cost, which could be positively affected by taking advantage of laboratory automation techniques. The discrete-step (batch) methods typically have greater flexibility in handling heterogeneous cell populations and are less demanding of the specific environment. These characteristics afford them an advantage over flow-based methods with regard to simple and therefore less expensive sample preparation steps. On the other hand, if sample preparation and posttreatment monitoring (providing the potential for feedback) and analysis steps are required for assessment of treatment efficacy, a continuous flow–based method may be more suitable for incorporation with existing workflows. Process sequences that consist of both continuous-flow and discrete-step (batch) techniques are fundamentally more difficult to streamline and parallelize, suggesting that each step and transition be analyzed carefully in design of an optimal workflow concerning scalability and cost.
Flexibility/adaptability to rapid changeover between different cell types and suitability for multiplexing are complementary requirements, which are most relevant (and desirable) to the analytical workflows found in research and development (i.e., those that deal with small-size samples and heterogeneous cell populations) as opposed to the routine industrial/biomanufacturing environment. In this case, procedure complexity and cost per treatment are less of an issue, as significant value can be derived in being able to use the same method with uniformly high efficacy to treat a multitude of samples and perform “library-scale” analyses, which are critical to high value-added applications such as drug discovery. Techniques using an electrical or magnetic field to facilitate cell treatment are most readily amenable to individual control of each treatment channel and thus can be massively multiplexed when used in combination with microarray devices, whether continuous flow or batch based. On the other hand, techniques that use mechanical fields (e.g., sonoporation) are more difficult to multiplex on a large scale in a microarray format, owing to technological difficulties in controlled actuation and unfavorable scale down of mechanical transducers (especially those with moving parts) regarding the forces/displacements that they can generate. These methods (as well as needle insertion and jet/particle injection) are, however, more flexible and forgiving with respect to the environment in which the cells must reside during the treatment (e.g., without the need for a custom buffer). This is in contrast to electric and magnetic field–based methods, the efficacy of which is much more sensitive to the buffer physical properties. Therefore, techniques relying on mechanical action are better suited for rapid changeover between different cell types/media environments.
Interestingly, the above discussion of various competing requirements (e.g., scalability vs. efficacy/control or flexibility vs. multiplexing) and tradeoffs in the advantages and deficiencies of different treatment methods suggests that hybrid approaches, which synergistically combine advantages of the mechanical (both indirect field based and direct penetration) and electro/magnetic techniques, hold the greatest promise for high-efficacy treatment and practical implementation. Combined mode treatment offers the opportunity for effective multiplexing for treating multiple samples and suitability for treatment of a wide range of cell types/sizes with few modifications. With respect to a dynamic range of operation (scale up and down), flow-based micro-/nanofluidic methods may have an advantage over standard batch-based techniques, as with suitable automation they could be run with a variable duty cycle to effectively cover the full spectrum of operating modalities from near batch (at a vanishingly small duty cycle) to highest throughput (at 100% duty cycle). Lastly, the minimum sample size that can be processed by any flow-based treatment method is defined by the dead volume existing in the system/device components, which provides an additional motivation to pursue miniaturization and integration strategies enabled by use of emerging micro-/nano-manufacturing techniques.
Acknowledgments
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences (NIH-NIGMS; grant 8R44GM103448-03).
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J. M. Meacham, K. Durvasula, F. L. Degertekin, and A. G. Fedorov have financial interests in OpenCell Technologies, Inc.
References
- 1.Bureau MF, Naimi S, Ibad RT, et al. Intramuscular Plasmid DNA Electrotransfer Biodistribution and Degradation. Biochim Biophys Acta Gene Struct Expression. 2004;1676:138–148. doi: 10.1016/j.bbaexp.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Miller AM, Dean DA. Tissue-Specific and Transcription Factor-Mediated Nuclear Entry of DNA. Adv Drug Deliv Rev. 2009;61:603–613. doi: 10.1016/j.addr.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Lukacs GL, Haggie P, Seksek O, et al. Size-Dependent DNA Mobility in Cytoplasm and Nucleus. J Biol Chem. 2000;275:1625–1629. doi: 10.1074/jbc.275.3.1625. [DOI] [PubMed] [Google Scholar]
- 4.Stephens DJ, Pepperkok R. The Many Ways to Cross the Plasma Membrane. Proc Natl Acad Sci U S A. 2001;98:4295–4298. doi: 10.1073/pnas.081065198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaughan EE, DeGiulio JV, Dean DA. Intracellular Trafficking of Plasmids for Gene Therapy: Mechanisms of Cytoplasmic Movement and Nuclear Import. Curr Gene Ther. 2006;6:671–681. doi: 10.2174/156652306779010688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lechardeur D, Lukacs GL. Intracellular Barriers to Non-viral Gene Transfer. Curr Gene Ther. 2002;2:183–194. doi: 10.2174/1566523024605609. [DOI] [PubMed] [Google Scholar]
- 7.Lechardeur D, Verkman AS, Lukacs GL. Intracellular Routing of Plasmid DNA during Non-viral Gene Transfer. Adv Drug Deliv Rev. 2005;57:55–767. doi: 10.1016/j.addr.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 8.Belting M, Sandgren S, Wittrup A. Nuclear Delivery of Macromolecules: Barriers and Carriers. Adv Drug Deliv Rev. 2005;57:505–527. doi: 10.1016/j.addr.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Waehler R, Russell SJ, Curiel DT. Engineering Targeted Viral Vectors for Gene Therapy. Nat Rev Genet. 2007;8:573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kay MA, Glorioso JC, Naldini L. Viral Vectors for Gene Therapy: The Art of Turning Infectious Agents into Vehicles of Therapeutics. Nat Med. 2001;7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 11.Thomas CE, Ehrhardt A, Kay MA. Progress and Problems with the Use of Viral Vectors for Gene Therapy. Nat Rev Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 12.Walther W, Stein U. Viral Vectors for Gene Transfer: A Review of Their Use in the Treatment of Human Diseases. Drugs. 2000;60:249–271. doi: 10.2165/00003495-200060020-00002. [DOI] [PubMed] [Google Scholar]
- 13.Chuah MKL, Collen D, VandenDriessche T. Biosafety of Adenoviral Vectors. Curr Gene Ther. 2003;3:527–543. doi: 10.2174/1566523034578140. [DOI] [PubMed] [Google Scholar]
- 14.Van den Broeke A, Burny A. Retroviral Vector Biosafety: Lessons from Sheep. J Biomed Biotechnol. 2003;1:9–12. doi: 10.1155/S1110724303209128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hacein-Bey-Abina S, von Kalle C, Schmidt M, et al. A Serious Adverse Event after Successful Gene Therapy for x-Linked Severe Combined Immunodeficiency. N Engl J Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 16.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. Lmo2-Associated Clonal t Cell Proliferation in Two Patients after Gene Therapy for scid-x1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 17.VandenDriessehe T, Collen D, Chuah MKL. Biosafety of Onco-Retroviral Vectors. Curr Gene Ther. 2003;3:501–515. doi: 10.2174/1566523034578113. [DOI] [PubMed] [Google Scholar]
- 18.Elouahabi A, Ruysschaert JM. Formation and Intracellular Trafficking of Lipoplexes and Polyplexes. Mol Ther. 2005;11:336–347. doi: 10.1016/j.ymthe.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Morille M, Passirani C, Vonarbourg A, et al. Progress in Developing Cationic Vectors for Non-viral Systemic Gene Therapy against Cancer. Biomaterials. 2008;29:3477–3496. doi: 10.1016/j.biomaterials.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 20.Torchilin VP. Recent Approaches to Intracellular Delivery of Drugs and DNA and Organelle Targeting. Annu Rev of Biomed Eng. 2006;8:343–375. doi: 10.1146/annurev.bioeng.8.061505.095735. [DOI] [PubMed] [Google Scholar]
- 21.Mellott AJ, Forrest ML, Detamore MS. Physical Non-viral Gene Delivery Methods for Tissue Engineering. Ann Biomed Eng. 2013;41:446–468. doi: 10.1007/s10439-012-0678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Villemejane J, Mir LM. Physical Methods of Nucleic Acid Transfer: General Concepts and Applications. Br J Pharmacol. 2009;157:207–219. doi: 10.1111/j.1476-5381.2009.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duvshani-Eshet M, Baruch L, Kesselman E, et al. Therapeutic Ultrasound-Mediated DNA to Cell and Nucleus: Bioeffects Revealed by Confocal and Atomic Force Microscopy. Gene Ther. 2006;13:163–172. doi: 10.1038/sj.gt.3302642. [DOI] [PubMed] [Google Scholar]
- 24.Karshafian R, Bevan PD, Williams R, et al. Sonoporation by Ultrasound-Activated Microbubble Contrast Agents: Effect of Acoustic Exposure Parameters on Cell Membrane Permeability and Cell Viability. Ultrasound Med Biol. 2009;35:847–860. doi: 10.1016/j.ultrasmedbio.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Schlicher RK, Hutcheson JD, Radhakrishna H, et al. Changes in Cell Morphology due to Plasma Membrane Wounding by Acoustic Cavitation. Ultrasound Med Biol. 2010;36:677–692. doi: 10.1016/j.ultrasmedbio.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Ballas CB, Rao MP. Towards Ultrahigh Throughput Microinjection: Mems-Based Massively-Parallelized Mechanoporation. Conf Proc IEEE Eng Med Biol Soc. 2012;2012:594–597. doi: 10.1109/EMBC.2012.6346001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selmeczi D, Hansen TS, Met O, et al. Efficient Large Volume Electroporation of Dendritic Cells through Micrometer Scale Manipulation of Flow in a Disposable Polymer Chip. Biomed Microdevices. 2011;13:383–392. doi: 10.1007/s10544-010-9507-1. [DOI] [PubMed] [Google Scholar]
- 28.Wang HY, Lu C. Microfluidic Electroporation for Delivery of Small Molecules and Genes into Cells Using a Common DC Power Supply. Biotechnol Bioeng. 2008;100:579–586. doi: 10.1002/bit.21784. [DOI] [PubMed] [Google Scholar]
- 29.Escoffre JM, Teissie J, Rols MP. Gene Transfer: How Can the Biological Barriers Be Overcome? J Membr Biol. 2010;236:61–74. doi: 10.1007/s00232-010-9275-0. [DOI] [PubMed] [Google Scholar]
- 30.Al-Dosari MS, Gao X. Nonviral Gene Delivery: Principle, Limitations, and Recent Progress. AAPS J. 2009;11:671–681. doi: 10.1208/s12248-009-9143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Capecchi MR. High-efficiency transformation by direct micro-injection of DNA into cultured mammalian-cells. Cell. 1980;22(2):479–488. doi: 10.1016/0092-8674(80)90358-x. [DOI] [PubMed] [Google Scholar]
- 32.Gordon JW, Ruddle FH. Gene-Transfer into Mouse Embryos: Production of Transgenic Mice by Pronuclear Injection. Method Enzymol. 1983;101:411–433. doi: 10.1016/0076-6879(83)01031-9. [DOI] [PubMed] [Google Scholar]
- 33.Gordon JW, Scangos GA, Plotkin DJ, et al. Genetic-Transformation of Mouse Embryos by Micro-Injection of Purified DNA. Proc Natl Acad Sci U S A Biol Sci. 1980;77:7380–7384. doi: 10.1073/pnas.77.12.7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehier-Humbert S, Guy RH. Physical Methods for Gene Transfer: Improving the Kinetics of Gene Delivery into Cells. Adv Drug Deliv Rev. 2005;57:733–753. doi: 10.1016/j.addr.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 35.Ansorge W, Pepperkok R. Performance of an Automated-System for Capillary Microinjection into Living Cells. J Biochem Biophys Methods. 1988;16:283–292. doi: 10.1016/0165-022x(88)90062-0. [DOI] [PubMed] [Google Scholar]
- 36.Graf SF, Madigou T, Li RY, et al. Fully Automated Microinjection System for Xenopus Laevis Oocytes with Integrated Sorting and Collection. J Lab Autom. 2011;16:186–196. doi: 10.1016/j.jala.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Adamo A, Jensen KF. Microfluidic Based Single Cell Microinjection. Lab Chip. 2008;8:1258–1261. doi: 10.1039/b803212b. [DOI] [PubMed] [Google Scholar]
- 38.Adamo A, Roushdy O, Dokov R, et al. Microfluidic Jet Injection for Delivering Macromolecules into Cells. J Micromech Microeng. 2013;23:035026. doi: 10.1088/0960-1317/23/3/035026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furth PA, Shamay A, Hennighausen L. Gene-Transfer into Mammalian-Cells by Jet Injection. Hybridoma. 1995;14:149–152. doi: 10.1089/hyb.1995.14.149. [DOI] [PubMed] [Google Scholar]
- 40.Furth PA, Shamay A, Wall RJ, et al. Gene-Transfer into Somatic Tissues by Jet Injection. Anal Biochem. 1992;205:365–368. doi: 10.1016/0003-2697(92)90449-h. [DOI] [PubMed] [Google Scholar]
- 41.Walther W, Minow T, Martin R, et al. Uptake, Biodistribution, and Time Course of Naked Plasmid DNA Trafficking after Intratumoral In Vivo Jet Injection. Hum Gene Ther. 2006;17:611–624. doi: 10.1089/hum.2006.17.611. [DOI] [PubMed] [Google Scholar]
- 42.Klein TM, Wolf ED, Wu R, et al. High-Velocity Microprojectiles for Delivering Nucleic-Acids into Living Cells. Nature. 1987;327:70–73. [PubMed] [Google Scholar]
- 43.Yang NS, Burkholder J, Roberts B, et al. Invivo and Invitro Gene-Transfer to Mammalian Somatic-Cells by Particle Bombardment. Proc Natl Acad Sci U S A. 1990;87:9568–9572. doi: 10.1073/pnas.87.24.9568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanford JC, Smith FD, Russell JA. Optimizing the Biolistic Process for Different Biological Applications. Method Enzymol. 1993;217:483–509. doi: 10.1016/0076-6879(93)17086-k. [DOI] [PubMed] [Google Scholar]
- 45.Lin MTS, Pulkkinen L, Uitto J, et al. The Gene Gun: Current Applications in Cutaneous Gene Therapy. Int J Dermatol. 2000;39:161–170. doi: 10.1046/j.1365-4362.2000.00925.x. [DOI] [PubMed] [Google Scholar]
- 46.Wang J, Murakami T, Yoshida S, et al. Predominant Cell-Mediated Immunity in the Oral Mucosa: Gene Gun-Based Vaccination against Infectious Diseases. J Dermatol Sci. 2003;31:203–210. doi: 10.1016/s0923-1811(03)00027-6. [DOI] [PubMed] [Google Scholar]
- 47.Kuriyama S, Mitoro A, Tsujinoue H, et al. Particle-Mediated Gene Transfer into Murine Livers Using a Newly Developed Gene Gun. Gene Ther. 2000;7:1132–1136. doi: 10.1038/sj.gt.3301192. [DOI] [PubMed] [Google Scholar]
- 48.Zelenin AV, Kolesnikov VA, Tarasenko OA, et al. Human Dystrophin Genes Are Expressed in Mouse Skeletal Muscle Fibers after Ballistic Transfection. FEBS Lett. 1997;414:319–322. doi: 10.1016/s0014-5793(97)01019-3. [DOI] [PubMed] [Google Scholar]
- 49.Shestopalov VI, Missey H, Bassnett S. Delivery of Genes and Fluorescent Dyes into Cells of the Intact Lens by Particle Bombardment. Exp Eye Res. 2002;74:639–649. doi: 10.1006/exer.2002.1191. [DOI] [PubMed] [Google Scholar]
- 50.O’Brien JA, Holt M, Whiteside G, et al. Modifications to the Hand-Held Gene Gun: Improvements for In Vitro Biolistic Transfection of Organotypic Neuronal Tissue. J Neurosci Methods. 2001;112:57–64. doi: 10.1016/s0165-0270(01)00457-5. [DOI] [PubMed] [Google Scholar]
- 51.Dileo J, Miller TE, Chesnoy S, et al. Gene Transfer to Subdermal Tissues via a New Gene Gun Design. Hum Gene Ther. 2003;14:79–87. doi: 10.1089/10430340360464732. [DOI] [PubMed] [Google Scholar]
- 52.Plank C, Schillinger U, Scherer F, et al. The Magnetofection Method: Using Magnetic Force to Enhance Gene Delivery. Biol Chem. 2003;384:737–747. doi: 10.1515/BC.2003.082. [DOI] [PubMed] [Google Scholar]
- 53.Scherer F, Anton M, Schillinger U, et al. Magnetofection: Enhancing and Targeting Gene Delivery by Magnetic Force In Vitro and In Vivo. Gene Ther. 2002;9:102–109. doi: 10.1038/sj.gt.3301624. [DOI] [PubMed] [Google Scholar]
- 54.Huth S, Lausier J, Rudolph C, et al. Characterisation of the Mechanism of Magnetofection. Mol Ther. 2003;7:S372–S372. [Google Scholar]
- 55.Plank C, Zelphati O, Mykhaylyk O. Magnetically Enhanced Nucleic Acid Delivery: Ten Years of Magnetofection-Progress and Prospects. Adv Drug Deliv Rev. 2011;63:1300–1331. doi: 10.1016/j.addr.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.del Pino P, Munoz-Javier A, Vlaskou D, et al. Gene Silencing Mediated by Magnetic Lipospheres Tagged with Small Interfering RNA. Nano Lett. 2010;10:3914–3921. doi: 10.1021/nl102485v. [DOI] [PubMed] [Google Scholar]
- 57.Sanchez-Antequera Y, Mykhaylyk O, van Til NP, et al. Genetic Modification of Target Cells. Blood. 2011;117:E171–E181. doi: 10.1182/blood-2010-08-302646. [DOI] [PubMed] [Google Scholar]
- 58.Neumann E, Schaeferridder M, Wang Y, et al. Gene-Transfer into Mouse Lyoma Cells by Electroporation in High Electric-Fields. EMBO J. 1982;1:841–845. doi: 10.1002/j.1460-2075.1982.tb01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gehl J. Electroporation: Theory and Methods, Perspectives for Drug Delivery, Gene Therapy and Research. Acta Physiol Scand. 2003;177:437–447. doi: 10.1046/j.1365-201X.2003.01093.x. [DOI] [PubMed] [Google Scholar]
- 60.Wolff JA, Malone RW, Williams P, et al. Direct Gene-Transfer into Mouse Muscle Invivo. Science. 1990;247:1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 61.Golzio M, Rols MP, Teissie J. In Vitro and In Vivo Electric Field-Mediated Permeabilization, Gene Transfer, and Expression. Methods. 2004;33:126–135. doi: 10.1016/j.ymeth.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 62.Escoffre JM, Portet T, Wasungu L, et al. What Is (Still Not) Known of the Mechanism by Which Electroporation Mediates Gene Transfer and Expression in Cells and Tissues. Mol Biotechnol. 2009;41:286–295. doi: 10.1007/s12033-008-9121-0. [DOI] [PubMed] [Google Scholar]
- 63.Gabriel B, Teissie J. Direct Observation in the Millisecond Time Range of Fluorescent Molecule Asymmetrical Interaction with the Electropermeabilized Cell Membrane. Biophys J. 1997;73:2630–2637. doi: 10.1016/S0006-3495(97)78292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krassowska W, Filev PD. Modeling Electroporation in a Single Cell. Biophys J. 2007;92:404–417. doi: 10.1529/biophysj.106.094235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dimitrov DS, Sowers AE. Membrane Electroporation: Fast Molecular-Exchange by Electroosmosis. Biochim Biophys Acta. 1990;1022:381–392. doi: 10.1016/0005-2736(90)90289-z. [DOI] [PubMed] [Google Scholar]
- 66.Sukharev SI, Klenchin VA, Serov SM, et al. Electroporation and Electrophoretic DNA Transfer into Cells: The Effect of DNA Interaction with Electropores. Biophys J. 1992;63:1320–1327. doi: 10.1016/S0006-3495(92)81709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faurie C, Phez E, Golzio M, et al. Effect of Electric Field Vectoriality on Electrically Mediated Gene Delivery in Mammalian Cells. Biochim Biophys Acta Biomembr. 2004;1665:92–100. doi: 10.1016/j.bbamem.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 68.Liu F, Heston S, Shollenberger LM, et al. Mechanism of In Vivo DNA Transport into Cells by Etectroporation: Etectrophoresis across the Plasma Membrane May Not Be Involved. J Gene Med. 2006;8:353–361. doi: 10.1002/jgm.851. [DOI] [PubMed] [Google Scholar]
- 69.Andre FM, Gehl J, Sersa G, et al. Efficiency of High- and Low-Voltage Pulse Combinations for Gene Electrotransfer in Muscle, Liver, Tumor, and Skin. Hum Gene Ther. 2008;19:1261–1271. doi: 10.1089/hum.2008.060. [DOI] [PubMed] [Google Scholar]
- 70.Vandermeulen G, Staes E, Vanderhaeghen ML, et al. Optimisation of Intradennal DNA Electrotransfer for Immunisation. J Control Release. 2007;124:81–87. doi: 10.1016/j.jconrel.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 71.Kanduser M, Miklavcic D, Pavlin M. Mechanisms Involved in Gene Electrotransfer Using High- and Low-Voltage Pulses: An In Vitro Study. Bioelectrochemistry. 2009;74:265–271. doi: 10.1016/j.bioelechem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 72.Pavlin M, Flisar K, Kanduser M. The Role of Electrophoresis in Gene Electrotransfer. J Membr Biol. 2010;236:75–79. doi: 10.1007/s00232-010-9276-z. [DOI] [PubMed] [Google Scholar]
- 73.Molnar MJ, Gilbert R, Lu YF, et al. Longevity and Safety of Electroporation-Assisted Plasmid-Based Gene Transfer into Mouse Muscles. Mol Ther. 2004;10:447–455. doi: 10.1016/j.ymthe.2004.06.642. [DOI] [PubMed] [Google Scholar]
- 74.Schoenbach KH, Joshi RP, Kolb JF, et al. Ultrashort Electrical Pulses Open a New Gateway into Biological Cells. Proc IEEE. 2004;92:1122–1137. [Google Scholar]
- 75.He HQ, Chang DC, Lee YK. Nonlinear Current Response of Micro Electroporation and Resealing Dynamics for Human Cancer Cells. Bioelectrochemistry. 2008;72:161–168. doi: 10.1016/j.bioelechem.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 76.Khine M, Lau A, Ionescu-Zanetti C, et al. A Single Cell Electroporation Chip. Lab Chip. 2005;5:38–43. doi: 10.1039/b408352k. [DOI] [PubMed] [Google Scholar]
- 77.Henslee BE, Morss A, Hu X, et al. Electroporation Dependence on Cell Size: Optical Tweezers Study. Anal Chem. 2011;83:3998–4003. doi: 10.1021/ac1019649. [DOI] [PubMed] [Google Scholar]
- 78.Valero A, Merino F, Wolbers F, et al. Apoptotic Cell Death Dynamics of hl60 Cells Studied Using a Microfluidic Cell Trap Device. Lab Chip. 2005;5:49–55. doi: 10.1039/b415813j. [DOI] [PubMed] [Google Scholar]
- 79.Huang Y, Rubinsky B. Flow-Through Micro-Electroporation Chip for High Efficiency Single-Cell Genetic Manipulation. Sens Actuator A-Phys. 2003;104:205–212. [Google Scholar]
- 80.Huang Y, Sekhon NS, Borninski J, et al. Instantaneous, Quantitative Single-Cell Viability Assessment by Electrical Evaluation of Cell Membrane Integrity with Microfabricated Devices. Sens Actuat A-Phys. 2003;105:31–39. [Google Scholar]
- 81.Lu H, Schmidt MA, Jensen KF. A Microfluidic Electroporation Device for Cell Lysis. Lab Chip. 2005;5:23–29. doi: 10.1039/b406205a. [DOI] [PubMed] [Google Scholar]
- 82.Ionescu-Zanetti C, Blatz A, Khine M. Electrophoresis-Assisted Single-Cell Electroporation for Efficient Intracellular Delivery. Biomed Microdevices. 2008;10:113–116. doi: 10.1007/s10544-007-9115-x. [DOI] [PubMed] [Google Scholar]
- 83.Guignet EG, Meyer T. Suspended-Drop Electroporation for High-Throughput Delivery of Biomolecules into Cells. Nat Methods. 2008;5:393–395. doi: 10.1038/nmeth.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang HY, Lu C. Electroporation of Mammalian Cells in a Microfluidic Channel with Geometric Variation. Anal Chem. 2006;78:5158–5164. doi: 10.1021/ac060733n. [DOI] [PubMed] [Google Scholar]
- 85.Zhan YH, Cao ZN, Bao N, et al. Low-Frequency AC Electroporation Shows Strong Frequency Dependence and Yields Comparable Transfection Results to DC Electroporation. J Control Release. 2012;160:570–576. doi: 10.1016/j.jconrel.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 86.Adamo A, Arione A, Sharei A, et al. Flow-Through Comb Electroporation Device for Delivery of Macromolecules. Anal Chem. 2013;85:1637–1641. doi: 10.1021/ac302887a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang J, Zhan YH, Ugaz VM, et al. Vortex-Assisted DNA Delivery. Lab Chip. 2010;10:2057–2061. doi: 10.1039/c004472e. [DOI] [PubMed] [Google Scholar]
- 88.Geng T, Zhan YH, Wang J, et al. Transfection of Cells Using Flow-Through Electroporation Based on Constant Voltage. Nat Protoc. 2011;6:1192–1208. doi: 10.1038/nprot.2011.360. [DOI] [PubMed] [Google Scholar]
- 89.Wei ZW, Zhao DY, Li XM, et al. A Laminar Flow Electroporation System for Efficient DNA and siRNA Delivery. Anal Chem. 2011;83:5881–5887. doi: 10.1021/ac200625b. [DOI] [PubMed] [Google Scholar]
- 90.Zhu T, Luo CX, Huang JY, et al. Electroporation Based on Hydrodynamic Focusing of Microfluidics with Low DC Voltage. Biomed Microdevices. 2010;12:35–40. doi: 10.1007/s10544-009-9355-z. [DOI] [PubMed] [Google Scholar]
- 91.Kim HJ, Greenleaf JF, Kinnick RR, et al. Ultrasound-Mediated Transfection of Mammalian Cells. Hum Gene Ther. 1996;7:1339–1346. doi: 10.1089/hum.1996.7.11-1339. [DOI] [PubMed] [Google Scholar]
- 92.Bao SP, Thrall BD, Miller DL. Transfection of a Reporter Plasmid into Cultured Cells by Sonoporation In Vitro. Ultrasound Med Biol. 1997;23:953–959. doi: 10.1016/s0301-5629(97)00025-2. [DOI] [PubMed] [Google Scholar]
- 93.Greenleaf WJ, Bolander ME, Sarkar G, et al. Artificial Cavitation Nuclei Significantly Enhance Acoustically Induced Cell Transfection. Ultrasound Med Biol. 1998;24:587–595. doi: 10.1016/s0301-5629(98)00003-9. [DOI] [PubMed] [Google Scholar]
- 94.Tata DB, Dunn F, Tindall DJ. Selective Clinical Ultrasound Signals Mediate Differential Gene Transfer and Expression in Two Human Prostate Cancer Cell Lines: Lncap and pc-3. Biochem Biophys Res Commun. 1997;234:64–67. doi: 10.1006/bbrc.1997.6578. [DOI] [PubMed] [Google Scholar]
- 95.Newman CMH, Bettinger T. Gene Therapy Progress and Prospects: Ultrasound for Gene Transfer. Gene Ther. 2007;14:465–475. doi: 10.1038/sj.gt.3302925. [DOI] [PubMed] [Google Scholar]
- 96.Postema M, Gilja OH. Ultrasound-Directed Drug Delivery. Curr Pharm Biotechnol. 2007;8:355–361. doi: 10.2174/138920107783018453. [DOI] [PubMed] [Google Scholar]
- 97.Sundaram J, Mellein BR, Mitragotri S. An Experimental and Theoretical Analysis of Ultrasound-Induced Permeabilization of Cell Membranes. Biophys J. 2003;84:3087–3101. doi: 10.1016/S0006-3495(03)70034-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mehier-Humbert S, Yan F, Frinking P, et al. Ultrasound-Mediated Gene Delivery: Influence of Contrast Agent on Transfection. Bioconjugate Chem. 2007;18:652–662. doi: 10.1021/bc0602432. [DOI] [PubMed] [Google Scholar]
- 99.Guzman HR, Nguyen DX, Khan S, et al. Ultrasound-Mediated Disruption of Cell Membranes. II. Heterogeneous Effects on Cells. J Acoust Soc Am. 2001;110:597–606. doi: 10.1121/1.1376130. [DOI] [PubMed] [Google Scholar]
- 100.Karshafian R, Bevan PD, Burns PN, et al. Ultrasound-Induced Uptake of Different Size Markers in Mammalian Cells. 2005 Ieee Ultrasonics Symposium. 2005;1–4:13–16. [Google Scholar]
- 101.Zarnitsyn VG, Prausnitz MR. Physical Parameters Influencing Optimization of Ultrasound-Mediated DNA Transfection. Ultrasound Med Biol. 2004;30:527–538. doi: 10.1016/j.ultrasmedbio.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 102.Mehier-Humbert S, Bettinger T, Yan F, et al. Ultrasound-Mediated Gene Delivery: Kinetics of Plasmid Internalization and Gene Expression. J Control Release. 2005;104:203–211. doi: 10.1016/j.jconrel.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 103.Laurell T, Petersson F, Nilsson A. Chip Integrated Strategies for Acoustic Separation and Manipulation of Cells and Particles. Chem Soc Rev. 2007;36:492–506. doi: 10.1039/b601326k. [DOI] [PubMed] [Google Scholar]
- 104.Nilsson A, Petersson F, Jonsson H, et al. Acoustic Control of Suspended Particles in Micro Fluidic Chips. Lab Chip. 2004;4:131–135. doi: 10.1039/b313493h. [DOI] [PubMed] [Google Scholar]
- 105.Lee YH, Peng CA. Nonviral Transfection of Suspension Cells in Ultrasound Standing Wave Fields. Ultrasound Med Biol. 2007;33:734–742. doi: 10.1016/j.ultrasmedbio.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Carugo D, Ankrett DN, Glynne-Jones P, et al. Contrast Agent-Free Sonoporation: The Use of an Ultrasonic Standing Wave Microfluidic System for the Delivery of Pharmaceutical Agents. Biomicrofluidics. 2011;5(4):15. doi: 10.1063/1.3660352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rodamporn S, Harris NR, Beeby SP, et al. Hela Cell Transfection Using a Novel Sonoporation System. IEEE Trans Biomed Eng. 2011;58:927–934. doi: 10.1109/TBME.2010.2089521. [DOI] [PubMed] [Google Scholar]
- 108.Zarnitsyn VG, Meacham JM, Varady MJ, et al. Electrosonic Ejector Microarray for Drug and Gene Delivery. Biomed Microdevices. 2008;10:299–308. doi: 10.1007/s10544-007-9137-4. [DOI] [PubMed] [Google Scholar]
- 109.Lee J, Sharei A, Sim WY, et al. Nonendocytic Delivery of Functional Engineered Nanoparticles into the Cytoplasm of Live Cells Using a Novel, High-Throughput Microfluidic Device. Nano Lett. 2012;12:6322–6327. doi: 10.1021/nl303421h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sharei A, Zoldan J, Adamo A, et al. A Vector-Free Microfluidic Platform for Intracellular Delivery. Proc Natl Acad Sci U S A. 2013;110:2082–2087. doi: 10.1073/pnas.1218705110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hallow DM, Seeger RA, Kamaev PP, et al. Shear-Induced Intracellular Loading of Cells with Molecules by Controlled Microfluidics. Biotechnol Bioeng. 2008;99:846–854. doi: 10.1002/bit.21651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yao CP, Zhang ZX, Rahmanzadeh R, et al. Laser-Based Gene Transfection and Gene Therapy. IEEE Trans Nanobiosci. 2008;7:111–119. doi: 10.1109/TNB.2008.2000742. [DOI] [PubMed] [Google Scholar]
- 113.Stevenson DJ, Gunn-Moore FJ, Campbell P, et al. Single Cell Optical Transfection. J R Soc Interface. 2010;7:863–871. doi: 10.1098/rsif.2009.0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kurata S, Tsukakoshi M, Kasuya T, et al. The Laser Method for Efficient Introduction of Foreign DNA into Cultured-Cells. Exp Cell Res. 1986;162:372–378. doi: 10.1016/0014-4827(86)90342-3. [DOI] [PubMed] [Google Scholar]
- 115.Tao W, Wilkinson J, Stanbridge EJ, et al. Direct Gene-Transfer into Human Cultured-Cells Facilitated by Laser Micropuncture of the Cell-Membrane. Proc Natl Acad Sci U S A. 1987;84:4180–4184. doi: 10.1073/pnas.84.12.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tsukakoshi M, Kurata S, Nomiya Y, et al. A Novel Method of DNA Transfection by Laser Microbeam Cell Surgery. Appl Phys B-Photo. 1984;35:135–140. [Google Scholar]
- 117.Zeira E, Manevitch A, Khatchatouriants A, et al. Femtosecond Infrared Laser: An Efficient and Safe In Vivo Gene Delivery System for Prolonged Expression. Mol Ther. 2003;8:342–350. doi: 10.1016/s1525-0016(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 118.Tirlapur UK, Konig K. Cell Biology–Targeted Transfection by Femtosecond Laser. Nature. 2002;418:290–291. doi: 10.1038/418290a. [DOI] [PubMed] [Google Scholar]
- 119.Palumbo G, Caruso M, Crescenzi E, et al. Targeted Gene Transfer in Eucaryotic Cells by Dye-Assisted Laser Optoporation. J Photochem Photobiol B-Biol. 1996;36:41–46. doi: 10.1016/S1011-1344(96)07335-6. [DOI] [PubMed] [Google Scholar]
- 120.Terakawa M, Ogura M, Sato S, et al. Gene Transfer into Mammalian Cells by Use of a Nanosecond Pulsed Laser-Induced Stress Wave. Opt Lett. 2004;29:1227–1229. doi: 10.1364/ol.29.001227. [DOI] [PubMed] [Google Scholar]
- 121.Pitsillides CM, Joe EK, Wei XB, et al. Selective Cell Targeting with Light-Absorbing Microparticles and Nanoparticles. Biophys J. 2003;84:4023–4032. doi: 10.1016/S0006-3495(03)75128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lapotko DO, Lukianova E, Oraevsky AA. Selective Laser Nano-Thermolysis of Human Leukemia Cells with Microbubbles Generated around Clusters of Gold Nanoparticles. Laser Surg Med. 2006;38:631–642. doi: 10.1002/lsm.20359. [DOI] [PubMed] [Google Scholar]
- 123.Chakravarty P, Qian W, El-Sayed MA, et al. Delivery of Molecules into Cells Using Carbon Nanoparticles Activated by Femtosecond Laser Pulses. Nat Nanotechnol. 2010;5:607–611. doi: 10.1038/nnano.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vlaskou D, Mykhaylyk O, Krotz F, et al. Magnetic and Acoustically Active Lipospheres for Magnetically Targeted Nucleic Acid Delivery. Adv Funct Mater. 2010;20:3881–3894. [Google Scholar]
- 125.Naka T, Sakoda T, Doi T, et al. Ultrasound Enhances Retrovirus-Mediated Gene Transfer. Prep Biochem Biotechnol. 2007;37:87–99. doi: 10.1080/10826060701199007. [DOI] [PubMed] [Google Scholar]
- 126.Choi SO, Kim YC, Park JH, et al. An Electrically Active Microneedle Array for Electroporation. Biomed Microdevices. 2010;12:263–273. doi: 10.1007/s10544-009-9381-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Escoffre JM, Kaddur K, Rols MP, et al. In Vitro Gene Transfer by Electrosonoporation. Ultrasound Med Biol. 2010;36:1746–1755. doi: 10.1016/j.ultrasmedbio.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 128.Yamashita YI, Shimada M, Tachibana K, et al. In Vivo Gene Transfer into Muscle via Electro-Sonoporation. Hum Gene Ther. 2002;13:2079–2084. doi: 10.1089/10430340260395929. [DOI] [PubMed] [Google Scholar]
- 129.Wu TH, Teslaa T, Kalim S, et al. Photothermal Nanoblade for Large Cargo Delivery into Mammalian Cells. Anal Chem. 2011;83:1321–1327. doi: 10.1021/ac102532w. [DOI] [PMC free article] [PubMed] [Google Scholar]