

Abstract Abstract
Intracellular pH (pHi) homeostasis is key to the functioning of vascular smooth muscle cells, including pulmonary artery smooth muscle cells (PASMCs). Sodium-hydrogen exchange (NHE) is an important contributor to pHi control in PASMCs. In this review, we examine the role of NHE in PASMC function, in both physiologic and pathologic conditions. In particular, we focus on the contribution of NHE to the PASMC response to hypoxia, considering both acute hypoxic pulmonary vasoconstriction and the development of pulmonary vascular remodeling and pulmonary hypertension in response to chronic hypoxia. Hypoxic pulmonary hypertension remains a disease with limited therapeutic options. Thus, this review explores past efforts at disrupting NHE signaling and discusses the therapeutic potential that such efforts may have in the field of pulmonary hypertension.
Keywords: hypoxia, pulmonary vascular smooth muscle, intracellular pH
Intracellular pH (pHi) modulation is vital to a variety of cellular functions, including migration, proliferation, vesicle trafficking, protein secretion, and volume regulation. In vascular smooth muscle, pHi also influences cellular contraction. Four transport mechanisms are known to be responsible for cellular pHi regulation: Na+-HCO3− cotransport, Na+-dependent Cl−/HCO3− exchange, Na+-independent Cl−/HCO3− exchange, and Na+/H+ exchange (NHE). While functional evidence clearly shows that NHE, Na+-dependent Cl−/HCO3− exchange, and Na+-HCO3− cotransport serve to alkalinize cells and that Na+-independent Cl−/HCO3− exchange effects acidification (Fig. 1), the relative significance of these various transport mechanisms to control of pHi under basal conditions appears to vary by experimental organism, tissue, and cell type. All four of these ion transporters have been described in various types of vascular smooth muscle1-4 and, with the exception of Na+-HCO3− cotransport, modulate pHi in pulmonary arterial smooth muscle cells (PASMCs).5-7 Of these, NHE plays a significant role in regulating PASMC pHi, under both resting and stimulated conditions. This review focuses on the ability of NHE to modulate PASMC function, describes the role of NHE in lung physiology and pathology, and points to potential areas of future research.
Figure 1.
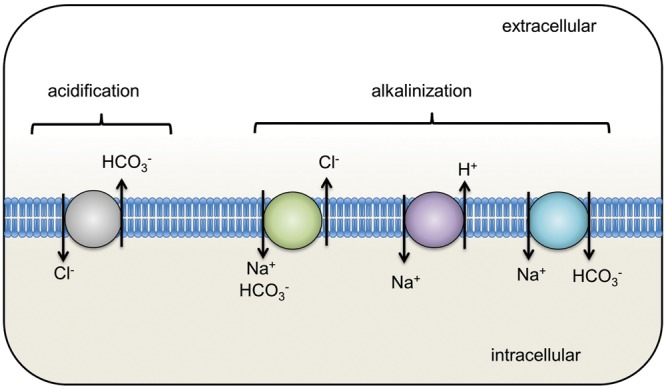
Ion transport mechanisms responsible for intracellular pH homeostasis. The Cl−/HCO3− exchanger is responsible for intracellular acidification, while the activities of all other exchangers/transporters cause intracellular alkalization.
The NHE isoforms
The NHE family of transmembrane proteins consists of at least 10 known isoforms. While the NHE isoforms are broadly grouped into 3 subtypes based on evolutionary homology, all NHEs share a similar structure, in which the membrane-spanning N-terminus effects import of an extracellular Na+ ion in exchange for export of an intracellular proton, while the cytosolic C-terminal domain contains various binding and phosphorylation sites that can modulate exchanger activity.8
Each NHE isoform is encoded by a distinct gene in the solute carrier 9 (SLC9) family, which is further divided into 3 subgroups. The SLC9A subgroup consists of NHEs 1–9, encoded by the genes Slc9a1–Slc9a9. NHE1, the first family member cloned in 1989,9 is ubiquitously expressed in all cell types and is thus considered a “housekeeping” isoform. Other NHEs show a more limited range of expression in human tissues and have yet to be studied as extensively as NHE1. NHE2 is expressed in the gastrointestinal tract and in kidney and endothelial cells and is thought to be involved in repair of damaged gastric epithelium.10 NHE3 is expressed in multiple organs and plays an important role in intestinal and renal sodium absorption.11 There is also evidence that NHE3 may regulate the respiratory drive.12 There is abundant expression of NHE4 in gastric parietal cells, where it is involved in regulating gastric acid secretion.13 NHE5 is expressed in brain and sperm and cycles between the plasma membrane and intracellular endosomes; it regulates dendritic spine growth.14 NHEs 1–5 exhibit plasma membrane localization, whereas NHEs 6, 7, and 9 are localized to intracellular organelles and are involved in regulating organellar pH.15 NHE8 is ubiquitously expressed in humans, and its localization to the plasma membrane or intracellular organelles varies developmentally by organ; its physiologic roles are still being defined.8
The SLC9B subgroup includes NHA1 (Na/H antiporter) and NHA2 (also known as NHE10), which are encoded by Slc9b1 and Scl9b2, respectively. These recently identified NHEs exhibit greater homology to prokaryotic NHEs, and their physiologic roles remain poorly characterized,8 with NHA1 apparently restricted to testis16 and NHA2 identified in bone17 and postulated to serve as a potential Na+/Li+ countertransporter.18 Finally, the SLC9C family contains proteins encoded by Slc9c1, which is believed to be sperm specific and to play an important role in sperm motility,19 and Slc9c2, about which little is known.
NHE1 and NHE2 were found to be expressed in whole-lung tissue preparations,20,21 whereas NHEs 3–5 were not.20,22,23 In contrast to the relatively wide distribution of expression seen with other NHE isoforms, NHEs 6–10 have significantly more restricted tissue and/or intracellular localization.15,24-26 Examination of the best-characterized NHEs revealed that NHE1, but not NHE2 or NHE3, was present in mouse27 and rat28 PASMCs, indicating that NHE1 is the primary isoform responsible for regulating cytosolic pH in this cell type.
The structure of NHE1
While the complete crystal structures of all NHE isoforms remain to be solved, modeling suggests a high degree of structural homology among the NHEs. Several topographical models were initially proposed, indicating that the NHE1 protein contained multiple transmembrane-spanning helices, although these models varied slightly as to the number and which parts of the protein composed these domains.9,20,29 More recently, a model structure of NHE1, which is highly conserved across species, has been constructed on the basis of the solved structure of the Escherichia coli NhaA Na+/H+ antiporter (Fig. 2A).30,31 Structural modeling of the human NHE1 predicts a 500–amino acid N-terminal domain consisting of 12 transmembrane segments (TMs) that contain the region responsible for ion transport and a short N-terminal cytoplasmic tail (Fig. 2B). The remaining 315 amino acids form a long C-terminal tail, located in the cytoplasm, that acts as a regulatory domain.32 While a portion of the protein required for transport of cations across the membrane has been localized to a handful of amino acid residues within the membrane domain,34 the exact mechanism by which cation transport occurs is still under investigation. In one model, TMs 4 and 11 form a central core, while TM2 creates two funnels that shape the path for transmembrane cation transport and residues in TM5 serve as the cation-binding site.30 This model suggests a transport mechanism whereby intracellular acidification induces conformational changes that transform NHE1 from an inactive to an active state, while further conformational changes within the active state allow for cation transport in a concentration gradient–dependent manner. In another model, TM domains 4 and 11 form a central core.33 Binding of H+ ions at TM9 causes a conformational change and direct contact between TM4 and TM9. As a result, TM4 and TM11 are reoriented, exposing a Na+-binding site to the extracellular space. Binding of Na+ triggers a crossover conformational change of TM4 and TM11, with the Na+ ion now exposed to the cytoplasm. Release of the Na+ ion allows protonation of the Na+-binding site and rearrangement back to the original conformation.33
Figure 2.
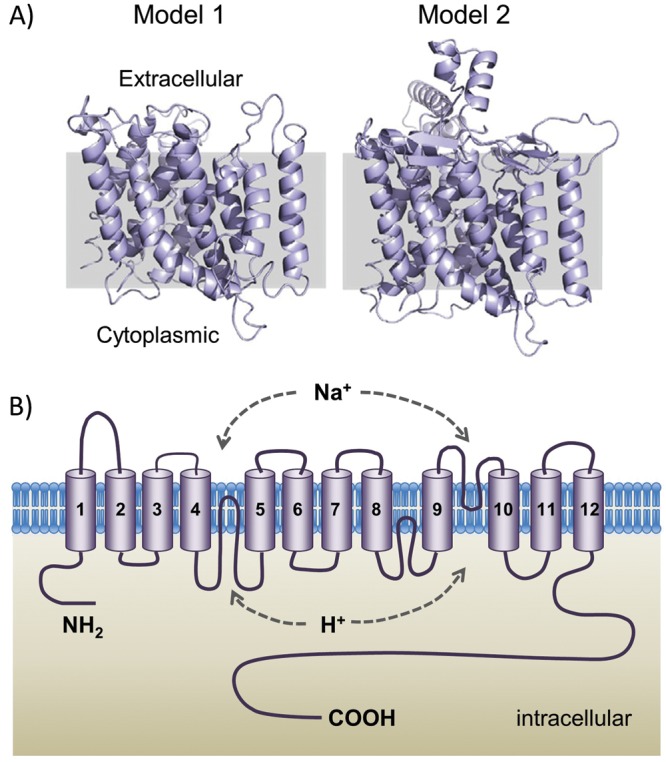
A, Three-dimensional structural models of the NHE1 transmembrane domains (reproduced with permission from Hendus-Altenburger et al.32). Model 1 represents the structure predicted by Landau et al.30 and is based on the crystal structure of Escherichia coli NHaA. Model 2, predicted by Nygaard et al.,33 is based on E. coli NhaA, experimental models of NHE1, and electron paramagnetic resonance spectroscopy. B, Predicted topology of NHE1. Ion translocation is believed to involve transmembrane domains 4 and 9 as well as 11.
The cytosolic C-terminal domain of NHE1 contains a number of sites important for phosphorylation and interaction with regulatory molecules. For example, calmodulin, a protein whose activation is dependent on intracellular Ca2+ levels, binds at amino acids 636–656.35 Other proteins that interact with the NHE1 C-terminus in a regulatory manner include the calcineurin homologous proteins (CHPs) 1–3, which bind at amino acids 515–530,36,37 and plasmalemmal phosphatidylinositol 4,5-biphosphate (PIP2), which is believed to bind at putative PIP2-binding domains.38
NHE1 also has multiple identified phosphorylation sites localized to serines and threonines in the C-terminal tail.39 Several protein kinases have been shown to be involved in the phosphorylation of NHE1, including protein kinase C (PKC),40,41 Rho kinase (ROCK),42,43 extracellular-signal-related kinases (ERKS),44 mitogen-activated-protein kinase (MAPK)–dependent pathways,45 p38 MAPK,46 and p90 ribosomal S6 kinase (p90rsk).45,47
Physiological functions of NHE1
Since NHE1 is found in almost all mammalian cells, its ubiquitous expression raises the possibility that it could be important in the function of multiple organ systems as well as in the pathophysiology of varied disease processes. The first substantial insight into the role of NHE1 in vivo was provided by the finding that mice with slow-wave epilepsy had a spontaneous genetic mutation in Slc9a1, resulting in a null allele.48 Subsequent targeted disruption of NHE1 in mice yielded animals that exhibited high mortality during weaning, growth retardation, ataxia, and seizures, with a life span of only a few weeks.49
The role of NHE1 in myocardial injury has been extensively studied. Ischemia causes intracellular acidification of cardiac myocytes, with reperfusion resulting in restoration of physiologic extracellular pH and creating a H+ gradient prompting efflux of H+, with concomitant Na+ influx through NHE1.50 The resultant rise in intracellular Na+ then prompts an increase in intracellular Ca2+ through the Na+/Ca2+ exchange system. Finally, elevated intracellular Ca2+ triggers deleterious downstream effects, including initiation of apoptotic pathways. NHE1 also plays a critical role in cardiac hypertrophy and remodeling after injury; indeed, cardiac-specific overexpression of NHE1 is sufficient to induce cardiac hypertrophy and heart failure in mice.51
There has also been extensive exploration of the role of NHE1 in cancer. NHE1-dependent cellular alkalinization has been shown to be necessary for oncogenic transformation.52 Beyond playing a role in malignant transformation, NHE1 activation promotes increased motility and invasion of breast cancer cells.53 Clearly, NHE1 expression/activity is critical for numerous physiological and pathologic processes. In the following sections, we discuss the role of NHE1 in PASMCs.
pHi homeostasis and NHE in PASMCs
With respect to maintenance of basal pHi, functional data in cat,6 guinea pig,7 and murine27 PASMCs demonstrated that removal of bicarbonate (HCO3−) from the extracellular solution resulted in a reduction in pHi, indicating that either Cl−/HCO3− exchange or Na+-HCO3− cotransport plays a role in alkalinizing PASMCs under basal conditions. Similarly, in ferret PASMCs, pHi in cells perfused with HCO3−-free buffer was substantially lower than what would typically be considered “normal.”5 These results would suggest that HCO3− transport also contributes to basal PASMC pHi in the ferret, although since no measurements were made in HCO3−-containing solution, the exact extent of that contribution is unknown. On the other hand, when ferret PASMCs were switched to a HCO3−- and Na+-free solution, pHi decreased even further, suggesting a potential role for NHE in maintenance of basal pHi.5 More-direct evidence for a contribution from NHE to PASMC pHi homeostasis under basal conditions comes from the use of NHE inhibitors. In physiologic solution containing extracellular HCO3− (and thus allowing for ion exchange through other pH-regulating channels), treating mouse27 or cultured guinea pig7 PASMCs with the NHE inhibitors ethylisopropyl amiloride (EIPA) or dimethyl amiloride (DMA), respectively, significantly decreased pHi, indicating that the role that NHE plays in PASMC pHi homeostasis is a significant one. Interestingly, application of DMA to cat PASMCs had little effect on basal pHi, which was substantially more acidic than that observed in other species, perhaps reflecting low baseline activity of NHE in this animal.6 These results indicate that while NHE can participate in regulation of resting pHi in PASMCs, the exact magnitude of the contribution may be species specific.
NHE has been shown to play an even more significant role in regulating pHi homeostasis in response to an acid load. The “ammonium-pulse” technique is commonly utilized to measure NHE activity (Fig. 3). Briefly exposing cells to ammonium chloride (NH4Cl) allows modulation of pHi while maintaining extracellular pH.54 Baseline pHi is measured before the ammonium pulse, which then induces cytoplasmic alkalinization due to the influx of NH3 while conserving intracellular H+ concentration. Washout of NH4Cl with a Na+- and NH4+-free solution leads to profound intracellular acidification due to removal of NH3 and retention of intracellular H+. Subsequent restoration of extracellular Na+ allows resumption of NHE activity, recovery from the acid load, and return to normal basal pHi. Thus, NHE activity can be quantified as the rate of Na+-dependent recovery from intracellular acidification. With this method, NHE activity was clearly measured in PASMCs from cat,6 cow,55 guinea pig,7 mouse,27 and rat.28
Figure 3.
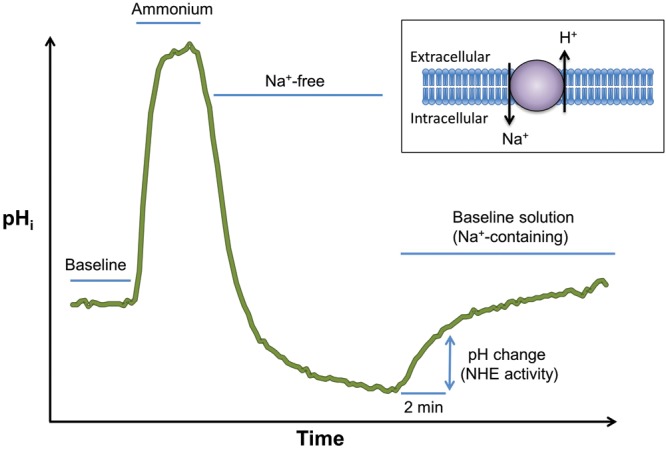
Ammonium pulse technique used to measure Na+/H+ exchange (NHE) activity. The pH is recorded in baseline solution. The cell is alkalinized with NH4Cl and then acidified with a solution that lacks both ammonium and sodium. Finally, the cell is placed back in the baseline solution, which contains sodium, allowing intracellular acid to be pumped out in exchange for sodium entry. The pH rise is thus a measure of NHE activity.
As with the studies examining the role of NHE in basal pHi regulation, the role of NHE in stimulated PASMC recovery from an intracellular acid load appears to vary among species. In ferret PASMCs, recovery from acid load was dependent on extracellular Na+ concentration and occurred even in the absence of extracellular HCO3−, a clear indication that NHE contributes to pHi control.5 Similarly, in rat and mouse PASMCs, recovery from acidification was largely prevented in the presence of NHE inhibitors.27,28 In guinea pig PASMCs, recovery from acid load was significantly slowed in the presence of DMA; however, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), an inhibitor of HCO3−/Cl− exchange, also significantly slowed the response, suggesting that NHE is not the only ion exchanger important to PASMC pHi homeostasis in this species.7 In contrast, in feline PASMCs, DMA slowed recovery from acid loading in HCO3−-free solutions but not in HCO3−-containing solutions, indicating that NHE is present but is likely not an important contributor to pHi homeostasis under physiologic conditions in the cat.6
Taken together, these studies indicate that, in PASMCs, resting pHi and recovery of pHi from acid loading are regulated by a mixture of transporters, with NHE playing a significant role in a majority of species, with the exception of the cat. While it appears clear that NHE exists and is active in PASMCs, the exact extent to which pHi homeostasis in these cells is dependent on NHE remains uncertain, given the varied results in different model organisms with different experimental techniques. Moreover, evaluation of the effect of NHE inhibitors on pHi in human PASMCs has yet to be performed and would lend further insight into the clinical relevance of NHE inhibition.
NHE1 and pulmonary vasculature function
Role of NHE1 in physiologic conditions
Plasma membrane NHEs play a critical role in regulating several cellular processes, including cell volume56 and signal transduction pathways involved in mediator release.57-59 However, the most extensively described function of NHEs in PASMCs is in regulation of pHi. As mentioned above, NHE1 is the only plasma membrane–localized NHE isoform known to be expressed in mouse and rat PASMCs,27,28 and while expression of the various NHE isoforms in PASMCs remains to be defined in humans, it is likely that the ubiquitous NHE1 is a major contributor to control of PASMC pHi across species.
Homeostasis of pHi regulates PASMC function in several ways, including control of vasomotor tone and cell proliferation and migration. With respect to the former, in isolated, perfused ferret lungs under normoxic conditions, acute intracellular alkalinization resulted in a rise in pulmonary arterial (PA) pressure, whereas acute acidification resulted in an even more pronounced elevation in PA pressure.5 Both acute alkalinization and acidification were also associated with a rise in intracellular Ca2+, suggesting the possibility that the acute change in vasomotor tone was mediated by Ca2+. In vascular smooth muscle cells, multiple pathways, including voltage-gated and store-operated Ca2+ channels, have been implicated in alkalosis-induced elevation of intracellular Ca2+.60 Interestingly, after the initial increase in PA pressure induced by acute intracellular alkalinization of ferret PASMCs, more prolonged alkalinization resulted in a further rise in PA pressure that was not associated with a rise in intracellular Ca2+.5 There is evidence that pHi exerts a direct (Ca2+-independent) effect on the contractile apparatus, through changes in calmodulin-dependent enzyme activity and ordering of the myofilament lattice.60 Intracellular alkalinization also resulted in canine PA contraction during isometric-tension studies and resulted in increased PA pressures in isolated perfused rat lungs under both normoxic and hypoxic conditions.54,61 Thus, data from multiple organisms indicate that a rise in pHi induces PASMC contraction, likely through both Ca2+-dependent and Ca2+-independent mechanisms.
It is important to note that the response of smooth muscle to an acute acid or base load may differ from the response to sustained acidification or alkalinization. In the absence of any data on how the pulmonary vasculature responds to sustained intracellular alkalinization, findings in the systemic vasculature may offer useful insight. Knockout of either the Na+/HCO3− cotransporter NBCn1, which alkalinizes cells by importing HCO3−, or NHE1 resulted in intracellular acidification in mouse vascular smooth muscle cells.62,63 In both of these knockout animals, the smooth muscle in the resistance vessels of the systemic vasculature was marked by reduced norepinephrine-stimulated, ROCK-dependent Ca2+ sensitivity. This suggests that ROCK activity may mediate changes in Ca2+ sensitivity, and thereby vascular tone, in response to prolonged changes in pHi, but the extent to which the same mechanisms apply in the pulmonary vasculature remains unclear.
There is also a link between pHi and cell proliferation. The correlation between alkalinization and cell proliferation was initially observed in fibroblasts64 and was later confirmed in bovine PASMCs, where challenge with either platelet-derived growth factor (PDGF) or epidermal growth factor induced alkalinization and proliferation.55 Moreover, inhibiting NHE with DMA attenuated both the alkalinization and the proliferation induced by these growth factors,55 indicating that NHE-mediated changes in pHi are important in controlling PASMC proliferation. Later studies using NHE1-deficient Chinese hamster ovary cells demonstrated that loss of NHE1 decreased the rate of proliferation and that proliferation could be restored with reintroduction of NHE1.65 Consistent with these findings, the NHE1-specific inhibitor sabiporide66 or knockdown of NHE1 with silencing RNA67 reduced proliferation rates in cultured human PASMCs, indicating a primary role for NHE1 in PASMC growth, although a direct link between intracellular alkalosis and PASMC proliferation remains to be proven, as NHE-mediated effects on cell volume or cytoskeletal rearrangement could also be contributing. Nonetheless, increased NHE activity, coinciding with increased pHi, is now considered to be an early event in cell proliferation.
Furthermore, NHE1 has been shown to play a role in PASMC migration. Initial work in neutrophils showed that pharmacologic activation of NHE induced neutrophil alkalinization and chemotaxis and that addition of an NHE inhibitor prevented these effects.68 Varying neutrophil pHi by altering Na+ and H+ gradients demonstrated that the chemotactic response correlated with the rise in pHi, suggesting that the migratory response was pH dependent. In addition, fibroblast migration was impaired in the setting of mutations to either the NHE1 ion translocation domain or the C-terminal cytoskeletal anchoring domain.69 Subsequent work in cultured human PASMCs revealed that inhibition of NHE1 by silencing RNA67 or sabiporide66 decreased cell migration. As with proliferation, these data indicate that NHE1 regulates PASMC migration, but the extent to which it does so through pHi modulation versus cytoskeletal rearrangement remains unclear.
Role of NHE1 in acute hypoxic pulmonary vasoconstriction (HPV)
While the preceding sections describe the role of NHE in modulating PASMC function under basal conditions, NHE1 also contributes to the pulmonary vascular response to pathologic stimuli, including hypoxia. The vasoconstrictor response of the pulmonary vasculature to hypoxia was first described in detail in the cat70 and was noted soon thereafter in humans.71 Since these early reports, HPV has been observed in all vertebrates, and an extensive literature on HPV has developed.72,73 HPV arises acutely upon exposure to hypoxia, with a significant increase in pulmonary vascular resistance (PVR) within 30 minutes and, if the hypoxic exposure remains brief (minutes to a few hours), largely reverses rapidly with return to normoxia.74
The onset of HPV occurs in phases, with an initial phase occurring within 5 minutes that reflects Ca2+-dependent smooth muscle contraction and a later phase of increasing PA pressure that appears to be Ca2+ independent.73 The initial hypoxic increase in intracellular Ca2+ in PASMCs is mediated by Ca2+ release from the sarcoplasmic reticulum and Ca2+ entry through voltage-gated L-type Ca2+ channels, Ca2+ permeable nonselective cation channels, and receptor-operated Ca2+ channels; in addition, hypoxia increases the sensitivity of the myofilament contractile apparatus to Ca2+.75 Factors involved in the later-phase Ca2+-independent rise in PVR include endothelial release of factors such as nitric oxide, prostacyclin, and endothelin-1 (ET-1);76 modulation of nitric oxide (NO) and reactive oxygen species within the pulmonary vasculature by red blood cells;77-80 and a potential role of neural modulation.81-83 The contribution of pHi, and of NHE in particular, to HPV remains a field of active inquiry.
As noted above, induction of intracellular alkalosis in PASMCs increased intracellular Ca2+ levels,5 contracted isolated pulmonary arteries,54 and increased PA pressure in isolated lungs,5,54 findings consistent with the possibility that alterations in pHi could contribute to HPV. Supporting this hypothesis, manipulating pHi by inducing intracellular acidification inhibited,61 while intracellular alkalinization enhanced,61,84 HPV. However, these reports stand in contrast to other studies, in which HPV was attenuated by hypocapnic alkalosis85-89 and enhanced by hypercapnic acidosis.88-90 It should be noted, however, that the magnitude of the change in pHi under these conditions was not measured and could vary considerably from that in experiments where pHi was manipulated directly. Nonetheless, these studies provide substantial evidence that changes in pHi during hypoxia could modulate HPV.
Unfortunately, only a few investigators have measured the effect of acute hypoxia on PASMC pHi, with conflicting results. In large-diameter porcine pulmonary arteries mounted for isometric-tension recording, hypoxia induced an initial, transient contraction without changes in pHi.91 The initial contraction was followed by dilation and decreased pHi and a later, sustained hypoxic contraction that was associated with pHi increasing back to normal levels. In isolated PASMCs where pHi was directly measured in real time with fluorescent indicators, exposure to acute hypoxia increased pHi in PASMCs derived from small-diameter cat pulmonary arteries, although hypoxia induced acidification in PASMCs from large-diameter cat pulmonary arteries.6 Adding to the confusion, acute hypoxia had no effect on pHi in PASMCs from distal rat intrapulmonary arteries.92 To be certain, variations in the severity of hypoxia, location of the vessels within the vascular tree, or species could account for the differences in findings. Interestingly, the alkalinization of small-diameter feline PASMCs was attributed to changes in Cl−/HCO3− exchange, not to NHE. Thus, whether acute hypoxia alters the activity of NHE1 in PASMCs from other species, whether this depends on the section of the vascular tree studied, and whether alterations in NHE1 activity exert effects on pHi during hypoxia remain to be fully resolved.
Role of NHE1 in hypoxia-induced pulmonary hypertension
Hypoxic pulmonary hypertension
While acute exposure to hypoxia results in a rapid, sustained contraction of the pulmonary vasculature that is immediately reversible with return to oxygenated conditions, prolonged hypoxia, as can occur with residence at high altitude or numerous chronic lung diseases, results in the development of pulmonary hypertension (PH). Ultimately, development of PH leads to enlargement of the right heart and, in some cases, eventual right heart failure. In humans, PH is defined by a mean resting PA pressure of ≥25 mmHg. This elevation in PA pressure can arise through multiple mechanisms, which has led to the disease being divided into 5 etiologic categories: (1) pulmonary arterial hypertension (PAH), (2) PH due to left heart disease, (3) PH due to hypoxemia, (4) chronic thromboembolic PH, and (5) miscellaneous.93
In all cases, a combination of increased pulmonary vascular tone and structural remodeling contributes to the development of PH. The concept that pulmonary vascular remodeling is an important contributor to hypoxia-induced PH stems from autopsy series that analyzed the pulmonary arteries of human subjects living in high-altitude areas of the Peruvian Andes, compared to controls living at sea level.94 High-altitude subjects showed greater muscularization of their distal PA trees, combined with increased thickening of the media and adventitia in the proximal pulmonary arteries. Furthermore, when normal humans were subjected to progressive hypobaric hypoxic over the course of 40 days during Operation Everest II, they developed an increase in PVR that did not respond to acute treatment with 100% oxygen, suggesting that the development of pulmonary vascular remodeling occurs over a relatively short time interval of hypoxic exposure and differs from the vasoconstriction observed with acute hypoxic challenge, which is rapidly reversible with reoxygenation.95 Subsequent to the initial human studies, detailed histologic examination of the pulmonary circulation from rats exposed to chronic hypoxia revealed the temporal progression of remodeling,96,97 the characteristics of which appeared to mimic the remodeling observed in humans with hypoxia-induced PH.
Understanding the cellular and molecular mechanisms underlying both the sustained contraction and the remodeling of the pulmonary vasculature in response to hypoxia has been the subject of intense efforts.98 Given that extension of smooth muscle into the distal pulmonary vasculature is the hallmark of hypoxia-induced remodeling, substantial focus has been placed on the growth and survival responses of PASMCs to hypoxia. Although the proliferative response is still not completely understood, it is clearly complex, involving a multiplicity of pathways. While it is beyond the scope of this review to fully delve into the profusion of pathways that have been implicated in the development of hypoxic PH, several factors that are associated with NHE1 appear to be involved in the pathogenesis of this disease, as is discussed in detail below.
The role of NHE1 in hypoxic PH
The finding that NHE was required for PASMC alkalinization and proliferation in response to physiologic stimuli, such as growth factors, raised the question of whether this exchanger was also involved in the PASMC growth responses related to pathologic stimuli, such as hypoxia. Initial studies demonstrated that PASMCs isolated from chronically hypoxic mice, a widely used model of hypoxic PH, exhibited an alkaline shift in resting pHi that continued to be evident days after the cells had been removed from the animals and the hypoxic stimulus.27 That the elevation in basal pHi was still observed when the cells were perfused with HCO3−-free buffer and could be significantly reduced by addition of an NHE inhibitor was consistent with NHE mediating the hypoxia-induced alkaline shift in pHi. Moreover, the elevation of pHi in PASMCs isolated from chronically hypoxic mice was correlated with augmented NHE activity.27 Furthermore, NHE1 messenger RNA (mRNA) and protein levels were increased in PASMCs isolated from chronically hypoxic mice, indicating that the PASMC response to prolonged hypoxia includes upregulation of NHE1 expression, leading to intracellular alkalinization. In these studies, the change in basal pHi induced by chronic hypoxia was substantially larger than that observed with acute hypoxia in cat PASMCs, where the alkalinization of PASMCs was found to be due to altered activity of Cl−/HCO3− exchange,6 suggesting that different mechanisms may be responsible for changes in basal pHi during acute and chronic hypoxic conditions.
A question that arises is whether the hypoxia-induced upregulation of NHE1 expression and activity is a consequence of a direct effect of hypoxia on PASMCs or is secondary to other hypoxia-induced changes, such as elevated pulmonary vascular pressure or circulating factors that are induced by hypoxia. The answer can be found in data derived from PASMCs isolated from normoxic animals that were subjected to prolonged hypoxic exposure ex vivo.28 Exposure to 4% O2 for 60 hours resulted in upregulation of NHE1 mRNA and protein in rat PASMCs that was accompanied by an alkaline shift in pHi and an increase in NHE activity. Qualitatively comparable changes in NHE1 expression and activity with in vivo and ex vivo hypoxic exposures indicate that the mechanism by which hypoxia induced these effects was intrinsic to the PASMCs.
PASMC alkalinization in response to prolonged hypoxia raised the possibility that NHE1 could be at the crossroads of the pathway from chronic hypoxia to pathologic pulmonary vascular remodeling. Initial experiments to test this hypothesis used pharmacologic inhibitors of NHE. Rats treated with either DMA or EIPA during exposure to 14 days of 10% O2 experienced a significant reduction in pulmonary vascular remodeling, PVR, and PA pressure, when compared to untreated control rats.99 To specifically target the role of NHE1, investigators turned to NHE1-null (Nhe1−/−) mice.100 In mice deficient for NHE1, the hypoxia-induced rise in right ventricular systolic pressure (RVSP) and right ventricular hypertrophy (RVH) was nearly prevented. In addition, Nhe1−/− mice exhibited an absence of pulmonary vascular remodeling, with significantly attenuated hypoxia-induced increases in pulmonary arteriolar wall thickness, vascular muscularization, and cell proliferation in the medial wall of the vasculature.100 These data provided key evidence confirming that NHE1 was necessary for the development of hypoxia-induced pulmonary vascular remodeling and PH in mice. While the role of NHE1 in humans clearly cannot be assessed via knockout, in vitro silencing of NHE1 in human PASMCs with short, interfering RNA reduced hypoxia-induced hypertrophy, proliferation, and migration, suggesting that NHE1 is important in the induction of pathologic PASMC responses to hypoxia in humans as well.67
The role of NHE1 in the development of other forms of PH remains to be evaluated in detail. Preliminary work has been performed in a rat model in which SU5416, a vascular endothelial growth factor receptor inhibitor, is given to rats in combination with chronic hypoxia exposure (SuHx model). The SuHx model recapitulates the pathology of human PAH better than other models, in that it results in the development of severe PH characterized by the presence of vaso-occlusive lesions (the pathologic hallmark of human PAH, which other rodent models of PH do not produce) that do not resolve when the rats are returned to normoxia.101 NHE activity was increased in PASMCs isolated from SuHx rats, in comparison with controls.102 Consistent with the findings in SuHx rats, NHE activity was increased in cultured PASMCs derived from human subjects with idiopathic PAH, compared to that in controls.102 Thus, while there is some indication that NHE1 may be involved in severe PAH, additional investigation is required to clarify its role.
Mechanisms controlling NHE1 activity and expression in PASMCs
Acute regulation of NHE activity
Non-pH-dependent regulation of NHE1 expression and activity occurs, in large part, through modifications to its cytosolic C-terminal domain. NHE1 turnover at the plasma membrane is controlled by C-terminal ubiquitination, which targets the exchanger for endocytosis.103 Factors influencing activation of NHE1 include phosphorylation and interaction with regulatory molecules. For example, binding to calmodulin, a protein whose activation is dependent on intracellular Ca2+ levels, activates NHE1.35 In the setting of low Ca2+, the high-affinity calmodulin binding site in the NHE1 C-terminus interacts with a region of the transmembrane domain, inhibiting proton exchange; in high-Ca2+ conditions, calmodulin binding prevents this autoinhibitory interaction.104 In addition, both PIP2 and CHP1 are essential cofactors for proper NHE1 function, as inhibition of the NHE1-CHP1 interaction36,37 or mutating the putative PIP2-binding domains38 markedly decreases NHE1 ion exchange activity.
NHE1 is further regulated by phosphorylation, with all known phosphorylation targets localized to serines and threonines in the C-terminal tail.39 Several protein kinases have been shown to be involved in the phosphorylation of NHE1, including PKC,40,41 ROCK,42,43 MAPK-dependent pathways,44-46 and p90rsk.45,47 The impact of phosphorylation on activity varies, in some cases increasing exchange activity or shifting the pH-dependent activation of NHE1 to more alkaline pHi.105 In contrast, phosphorylation of the high-affinity calmodulin binding site at Ser-648 by protein kinase B/Akt prevents the NHE1-calmodulin interaction and thus inhibits NHE1 ion transport activity.106 In addition to direct effects on the NHE1 protein, phosphorylation of NHE1 binding partners can influence NHE1 activity. For example, phosphorylation of calmodulin by Janus kinase 2 increases NHE1-calmodulin binding and thus activates NHE1.107
Interestingly, as with calmodulin, many of the kinases and regulatory proteins known to alter NHE1 activity exhibit Ca2+ sensitivity (such as PKC, ROCK, and CHP), suggesting the possibility that changes in intracellular Ca2+ concentration could act to indirectly modulate NHE1 activity. Clearly, the mechanisms that regulate NHE activity are complex and have yet to be exhaustively delineated. Moreover, while the regulation of NHE1 by phosphorylation and regulatory proteins has been described in a variety of cell types, which of these mechanisms regulates NHE1 activity in PASMCs is currently still under investigation.
Several factors known to be involved in the pathogenesis of PH have been shown to influence NHE activity. For instance, PDGF, which is released by circulating platelets, is implicated in PH, in that increased levels of PDGF are present in the blood and lungs of patients with various forms of PH. PDGF has been shown to stimulate rat108 and bovine55 PASMC proliferation, as well as human PASMC proliferation and migration,109 through pathways involving upregulated capacitative Ca2+ entry108,110 and increased NHE activity.55
Similarly, the peptide ET-1, a potent endothelial-derived vasoconstricting agent released primarily by endothelial cells, is upregulated in multiple forms of PH, including hypoxia-induced PH, and ET-1 receptor inhibitors have been successful in preventing and partially reversing hypoxic PH in animal models.111-113 Acute incubation with ET-1 increased NHE activity in a dose-dependent fashion in normoxic rat PASMCs.114 Furthermore, in the same study, pretreatment of PASMCs with a ROCK inhibitor prevented ET-1-induced upregulation of NHE activity.114 ROCK is activated in multiple models of PH, perhaps secondarily to increased circulating factors such as ET-1, and is implicated in signaling leading to both vasoconstriction and vascular remodeling.115-118 ROCK inhibitors have been shown to decrease PA pressures and PVR both in experimental models of PH and in PAH patients.119-121 The best-characterized action of ROCK is the phosphorylation of the myosin-binding subunit of myosin light chain phosphatase (MLCP), thereby inhibiting MLCP and thus increasing phosphorylation of myosin light chains, which serves to augment contraction. However, since ROCK inhibition also prevents ET-1-induced activation of NHE in normoxic settings, it is likely that the ET-1/ROCK pathway contributes to remodeling as well.
In addition, proliferation of cultured bovine PASMCs in response to hypoxic stimuli involves the activation of PKC.122 Activation of PKC increased NHE expression and ion-exchange activity in vascular smooth muscle cells,40,123,124 and, in nonvascular cells, PKC was a required intermediary in ET-1-induced upregulation of NHE activity.125-127 However, in normoxic rat PASMCs, pretreatment with PKC inhibitors did not prevent ET-1-mediated augmentation of NHE,114 suggesting that, while NHE induction by ET-1 involves PKC signaling in certain conditions, this does not appear to be a global phenomenon. Further experiments will be required to determine whether PKC plays any role in NHE activation in PASMCs during chronic hypoxia.
Regulation of NHE1 expression
Role of hypoxia-inducible factors
The finding that chronic hypoxia increased NHE1 expression naturally raised questions as to the mechanism underlying this upregulation. An early candidate that emerged as a potential regulator of NHE1 expression was hypoxia-inducible factor 1 (HIF-1), a transcription factor that regulates the expression of a multitude of genes in response to changes in oxygen concentration.128 HIF-1 is composed of 2 subunits, HIF-1α and HIF-1β. While HIF-1β is constitutively expressed, HIF-1α is ubiquitinated, leading to proteasomal degradation, under normoxic conditions. HIF-1α ubiquitination is dependent on proline hydroxylation of HIF-1α, which in the setting of normoxia is catalyzed by prolyl hydroxylase domain proteins using O2 as a substrate.129,130 Since hypoxic conditions result in stabilization of the HIF-1α subunit, the accumulation of HIF-1α protein controls the sensitivity and selectivity of induction of the HIF-1 transcriptional complex.
Studies aimed at determining the effect of loss of HIF-1 function were initially limited by the fact that mice with complete deficiency for HIF-1α (Hif1a−/−) died during gestation.131 However, mice heterozygous for the null allele (Hif1a+/−) exhibited attenuated increases in right ventricular pressure, RVH, and pulmonary vascular remodeling in response to chronic hypoxia, when compared to their wild-type littermates.132 Thus, absence of full HIF-1 expression impaired the development of hypoxic PH.
NHE1 has been shown to be one of the genes regulated by HIF-1. In vivo chronic hypoxia exposure (10% O2 for 3 weeks) in Hif1a+/− heterozygous mice resulted in severely blunted upregulation of NHE1 mRNA and protein expression in PASMCs, compared to those in wild types.28 As would be expected in the setting of impaired NHE1 expression, PASMCs isolated from Hif1a+/− heterozygous mice also showed less hypoxia-induced alkalinization. Cells derived from Hif1a+/− mice also exhibited blunted hypoxia-induced proliferation.133 Finally, in vitro forced expression of HIF-1α in PASMCs isolated from normoxic rats resulted in increased NHE1 mRNA and protein expression as well as increased NHE activity. These data suggest that upregulation of NHE1 expression is one of the mechanisms whereby HIF-1 generates pathologic pulmonary vascular remodeling and PH in response to hypoxia.
Interestingly, HIF-1 appears to exert independent effects on the pulmonary vasculature and the right ventricle. Homozygous conditional deletion of HIF-1α in smooth muscle during chronic hypoxia exposure attenuated the development of hypoxia-induced pulmonary vascular remodeling and PH in mice.134 However, even in the absence of elevated PA pressures, the degree of hypoxia-induced RVH was unchanged in these mice. In Hif1a+/− mice with systemic heterozygous HIF-1α deficiency (which therefore have decreased HIF-1α expression in right ventricular myocytes), chronic hypoxia resulted in an attenuated increase in both PH and RVH.132 Considered together, these results suggest that HIF-1 activation in right ventricular myocytes contributes to hypoxic right ventricular remodeling, independent of PA pressures. While NHE1 activation has been clearly established as a mechanism for left ventricular hypertrophy, the contribution of HIF-dependent NHE1 expression in cardiac myocytes to right ventricular remodeling in chronic hypoxia remains unresolved.
Role of ET-1
In addition to acutely regulating NHE activity in PASMCs, in vitro treatment of normoxic rat PASMCs with ET-1 for 48 hours increased NHE1 mRNA expression.135 Exogenous application of ET-1 also concomitantly increased HIF-1α protein expression and decreased expression of prolyl hydroxylase 2, the enzyme responsible for targeting HIF-1α for degradation via hydroxylation.135 While application of exogenous ET-1 under nonhypoxic conditions indicated that increased ET-1 was sufficient to induce NHE1 expression, further evidence supporting a role for ET-1 in modulating NHE1 expression in PASMCs during hypoxia was provided by experiments in which rat PASMCs were pretreated with BQ-123, an antagonist of ET-1 receptor subtype A (ETA) during hypoxia.135 BQ-123 abrogated the upregulation of both HIF-1α protein and NHE1 mRNA that is normally induced by incubation in 4% O2. Given that ET-1 levels are known to be upregulated in the setting of chronic hypoxia, these data indicate that HIF-1α expression, which is already directly induced by low oxygen tension, and consequent NHE1 expression are further amplified by ET-1 during hypoxia.
Mechanisms of NHE1 signaling
Ca2+ signaling
In many cell types, changes in pHi and intracellular Ca2+ are tightly coupled.60 Elevated intracellular Ca2+ levels are implicated not only in pulmonary vasoconstriction but also in vascular remodeling.136 Thus, it is worthwhile to examine in further depth the effects of alkalinization on intracellular Ca2+.
In porcine coronary arteries and rat aortas, intracellular alkalinization induced a rise in intracellular Ca2+ that was attenuated by nifedipine, an inhibitor of L-type voltage-gated Ca2+ channels.137,138 Alkalinization also increased intracellular Ca2+ levels in ferret PASMCs and potentiated contraction in response to potassium chloride,5 indicating that L-type voltage-gated Ca2+ channels contributed to pH-mediated Ca2+ entry. Furthermore, intracellular alkalinization inhibited voltage-gated K+ currents, resulting in depolarization of canine PASMCs,139 which may serve as one of the mechanisms by which an alkaline shift in pH activates L-type Ca2+ channels. It is likely that other Ca2+ entry pathways can also be modulated by pHi. For example, in rat aortic smooth muscle cells, alkalinization potentiated Ca2+ entry induced by thapsigargin (which depletes stores of Ca2+ in the sarcoplasmic/endoplasmic reticula and thereby induces plasmalemmal Ca2+ entry through store-operated Ca2+ channels), suggesting that store-operated Ca2+ channels can also contribute to pH-mediated Ca2+ entry.140 There is also evidence of increased release of Ca2+ from intracellular stores in the setting of alkalinization. In smooth muscle cells from guinea pig portal veins, elevated pH was associated with increased inositol triphosphate–induced Ca2+ release from the endoplasmic reticulum.141 Thus, it is possible that increased NHE1 activity and alkalinization during chronic hypoxia could enhance Ca2+ entry into PASMCs.
ROCK, p27, and cyclin D1 signaling
Experiments utilizing Nhe1−/− mice and in vitro NHE1 silencing have yielded further insights into downstream effectors of NHE1 signaling. In Nhe1−/− mice, there was a decrease in ROCK expression and activity, an increase in the expression of p27 (a cyclin-dependent kinase inhibitor and known downstream factor of ROCK), and a decrease in cyclin D1 expression.100 Moreover, expression of E2F1, a transcription factor important in cell cycle progression that is known to be downstream of p27, was upregulated by hypoxia in human PASMCs, and this upregulation was inhibited in the setting of in vitro NHE1 silencing.67 The role of E2F1 as a downstream effector of NHE1 was further confirmed by the fact that overexpression of E2F1 prevented the decrease in PASMC proliferation and migration observed when NHE1 was silenced.67 Thus, hypoxia-induced upregulation of NHE1 leads to downstream ROCK activation, followed by increased p27 and subsequent E2F1 expression, which in turn mediates cell cycle progression and increased PASMC proliferation.
While the studies in Nhe1−/− mice placed ROCK as a downstream effector of NHE1 signaling, the interaction between NHE1 and ROCK is clearly more complex. As noted above, ET-1 induced NHE activity acutely in normoxic PASMCs via a mechanism involving ROCK, thus placing ROCK upstream of NHE.114 Consistent with the results obtained with acute exposure to ET-1 in PASMCs, NHE has also been shown to be downstream of ROCK and RhoA (Ras homolog A, a GTPase [guanosine triphosphatase] that increases ROCK activity) in nonvascular cell types.42,43,142,143 Given the temporal differences in these studies, the most likely scenario is that ROCK activation acutely phosphorylates and enhances NHE1 activity, with prolonged NHE1 upregulation in turn increasing ROCK expression. However, the exact mechanism by which NHE1 increases ROCK expression remains to be determined.
NHE1 and cytoskeletal anchoring
It has been recognized for some time that the C-terminal tail of NHE1, independent of the ion-transport domain, is involved in regulation of cell shape and cell function. Fibroblasts that express an NHE1 variant with a mutation in the ezrin-binding motif have impaired organization of actin stress fibers and an irregular cell shape, while fibroblasts expressing an NHE1 mutant with disrupted ion translocation function have normal cytoskeletal organization and cell shape.33 Furthermore, fibroblasts containing NHE1 with mutations in the C-terminal ezrin-binding motif also show evidence of impaired cell migration.69
As is implied in the name, mutations in the C-terminal ezrin-binding motif impair the ability of NHE1 to bind with ezrin,33 a member of a family of proteins referred to as ERM (ezrin, radixin, moesin) proteins.144 ERM proteins are characterized by their shared N-terminal FERM (4.1, ezrin, radixin, moesin) domain, which binds to various membrane proteins, and a shared C-terminal filamentous-actin binding site. As their shared structure suggests, this family of proteins plays an important role in the cross-linking of membrane proteins to the actin cytoskeleton, thereby facilitating changes in cell shape, membrane protein localization, and signal transduction. The ERM family is also characterized by a shared C-ERMAD (ERM-associated domain), which is able to intramolecularly bind to the N-terminal FERM domain. As the C-ERMAD domain overlaps the C-terminal actin-binding site, this intramolecular association prevents association of ezrin with actin and thus renders ezrin functionally inactive. ROCK regulates the activity of ezrin by phosphorylating a C-terminal threonine (T567).145 T567 phosphorylation prevents ezrin intramolecular association and thus allows activated ezrin to serve as a membrane-actin cross-linker (Fig. 4). Thus, phosphorylated ezrin serves as a structural link between the C-terminal tail of transmembrane NHE1 and the actin cytoskeleton, and it is through this interaction with phosphorylated ezrin that NHE1, acting as a membrane anchor for actin filaments, is able to regulate PASMC cell shape and migration.
Figure 4.

A, Schematic illustrating proposed mechanism by which NHE1/ezrin interactions controls pulmonary arterial smooth muscle cell (PASMC) shape changes. Ezrin phosphorylation by Rho kinase (ROCK) allows dissolution of intramolecular association between the head (FERM; [4.1, ezrin, radixin, moesin]) and tail (C-ERMAD [ERM (ezrin-radixin-moesin)-associated domain]) of ezrin. Active (phosphorylated) ezrin (P) then binds to and forms a crosslink between NHE1 and actin filaments (F-actin). B, Schematic illustrating pathways whereby chronic hypoxia (CH) leads to pathologic PASMC behavior via increased NHE1-actin cross-linking. Upregulation of the transcription factor hypoxia-inducible factor 1 (HIF-1) results in increased NHE1 expression. Activation of ROCK by CH results in phosphorylation of ezrin (P-ezrin) and binding between NHE1 and F-actin, tethering actin filaments to the cell membrane and facilitating cell movement necessary for migration and proliferation.
Therapeutic options for inhibiting NHE
Currently, oxygen therapy remains the most widely used treatment for hypoxia-induced PH, although supplemental oxygen is not a cure and can only slow the progression of the disease. The few pharmacologic therapies that have been approved for treatment of PAH focus on the inhibition of pulmonary vascular vasoconstriction.146 Despite these current therapies, the morbidity and mortality from both PAH and PH associated with hypoxia remain high.147 Thus, there is a significant interest in the discovery of agents that interfere with the processes that contribute to pulmonary vascular remodeling and in the effects of those agents on the prevention and reversal of PH. In that light, inhibition of NHE1 is potentially of great clinical interest. Several classes of drugs that have effects on NHE have been evaluated to date, although their effect in PH has so far been largely limited to animal models and in vitro studies.
Early studies
Amiloride is a drug that was initially found to inhibit the sodium channel in urinary epithelia but has since been recognized to inhibit numerous ion channels. Thousands of amiloride analogs have been synthesized that inhibit Na+-conducting channels with varying specificity.148 DMA and EIPA are two amiloride analogs exhibiting relatively more specific inhibition of NHE ion transport. Initially, DMA was found to inhibit growth factor–induced PASMC proliferation in vitro.55 In vivo, it was subsequently determined that both DMA and EIPA, when given during a hypoxic exposure of 10% O2 for 14 days, inhibited the development of PH in rats.99 The attenuated development of PH with DMA and EIPA was achieved through decreased hypoxia-induced pulmonary vascular remodeling, as drug-treated animals showed decreased wall thickness of intra-acinous vessels, compared to untreated hypoxic controls.
NHE1-specific inhibitors
Inhibitors exhibiting high specificity for the NHE1 isoform have been developed and pursued primarily for their potential cardioprotective effects. In animal studies, these agents have consistently demonstrated cardiogenic benefits in the setting of myocardial ischemia reperfusion injury, especially when the agents are given before the ischemic insult.149 To date, 2 members of a family of specific NHE1 inhibitors, cariporide and eniporide, have advanced to human clinical trials for the treatment of ischemia reperfusion injury.150-153 Unfortunately, the results of these studies have been disappointing, with only the GUARDIAN study showing benefit of NHE1 inhibition in a single subgroup of patients, those who were reperfused via coronary artery bypass grafting (CABG).152 Furthermore, the subsequent EXPEDITION study, while showing decreased myocardial infarction in cariporide-treated patients undergoing high-risk CABG, also revealed an overall increase in mortality in the cariporide treatment group due to an increased rate of cerebrovascular events.153 Because of the ubiquitous nature of NHE1 and the concerns raised by these studies, clinical trials of NHE1 inhibitors have largely been discontinued.
Exploration of the potential benefits of NHE1-specific inhibitors in PH has been limited. Sabiporide, another member of the NHE1 inhibitor family, was shown to inhibit proliferation and migration of human PASMCs in vitro.66 In vivo, cariporide has been shown to attenuate the development of right heart failure in monocrotaline-treated rats.154 In that study, cariporide-treated rats experienced significant blunting of the monocrotaline-induced increase in RVSP and RVH as well as blunting of monocrotaline-induced necrosis, fibrosis, and mononuclear infiltration of right ventricular myocardial cells. The authors claimed that the beneficial effects of cariporide in this model of PH were mediated through direct protection of right ventricular myocardium, because they argued that cariporide had no effect on pulmonary vascular remodeling. However, their conclusion that cariporide did not inhibit pulmonary vascular remodeling is tempered by the small number of vessels in which pulmonary artery wall thickness was measured as well as by the absence of measurement of distal vascular neomuscularization via smooth muscle–specific actin staining. Thus, further efforts at characterizing the effect of NHE1 inhibitors on the pulmonary vasculature in vivo are needed, especially in the setting of chronic hypoxia.
It should be noted that the mechanism whereby NHE1-specific inhibitors act on the exchanger remains undefined. Cariporide was characterized as an inhibitor of NHE1 ion transport activity.155 However, the effect of known NHE inhibitors on the NHE-ezrin-actin interaction or on NHE-dependent cytoskeletal rearrangement remains unexamined. Given that the C-terminal tail of NHE is required for cytoskeletal interactions and that the NHE-ezrin interaction is important for regulating cell shape and migration, it would be of interest to assess the differential effects on the development of hypoxic PH of drugs targeted to inhibition of NHE ion exchange and drugs targeted to inhibition of NHE-mediated cytoskeletal rearrangement. Importantly, cariporide was found to inhibit monocrotaline-induced increase in NHE1 mRNA expression in right ventricular myocytes.154 Thus, NHE1-specific inhibitors could impair all mechanisms of NHE1 signaling merely by depleting NHE1 expression. The effect of NHE1 inhibitors on NHE1 mRNA or protein expression in PASMCs remains undetermined, and future work on the effect of NHE1 inhibitors on the pulmonary vasculature will have to interrogate their mechanism of action in greater detail.
HIF inhibitors
An additional mechanism of NHE inhibition is via upstream inhibition of HIF-1. As noted above, HIF-1 is a transcription factor that upregulates many genes, including NHE1, in response to hypoxia. Cardiac glycosides, including digoxin, have been found to decrease HIF-1α protein expression via inhibition of HIF-1α mRNA translation.156 Acriflavine, which historically was used as a topical antiseptic, also inhibited HIF-1 function, though via a different mechanism: it inhibits dimerization of HIF-1α with HIF-1β, thus preventing formation of a functional HIF-1 unit.157
Both medications have been found to have beneficial effects on PH in animal models. Digoxin, when given to mice during a 3-week exposure to hypoxia, successfully attenuated hypoxia-induced increases in RVSP, RVH, and pulmonary vascular remodeling.158 In the same study, when mice were allowed to develop hypoxia-induced PH over 3 weeks of hypoxic exposure and were then treated with digoxin during 2 weeks of further hypoxic exposure, digoxin-treated mice developed a lower RVSP than vehicle-treated mice, indicating that digoxin therapy can slow the progression of established hypoxic PH. In these mice, digoxin therapy prevented hypoxia-induced increase in NHE1 expression in PASMCs, suggesting that NHE inhibition may be one pathway through which digoxin protects against the development of PH. It is possible that inhibition of HIF-1 targets other than NHE1 may play a role in the prevention of PH or that digoxin may have HIF-1-independent effects that protect against PH; however, the fact that acriflavine, which inhibits HIF-1 via a different mechanism, also prevented the development of hypoxia-induced PH158 suggests that the protective effect of digoxin is likely through an HIF-dependent mechanism.
ET-1 receptor antagonists
Because ET-1 acutely increases NHE activity114 and is also implicated in upregulation of NHE1 expression in the setting of hypoxia,135 inhibition of ET-1 may serve as another upstream method of NHE inhibition. ET-1 signaling is effected through 2 G protein–coupled receptors, ETA and ETB. While ETA stimulation contributes to vasoconstriction, ETB receptor stimulation results in more ambiguous effects, because it prompts release of both vasodilatory NO and prostacyclin from endothelial cells as well as proconstrictory agents such as thromboxane A2.159 However, both receptors promote human PASMC proliferation and thus may contribute to vascular remodeling.160 Treatment of rats with BQ-123, a selective antagonist of the ETA receptor, during a 2-week exposure to 10% O2 prevented pulmonary vascular remodeling and PH113 and significantly reversed established hypoxic PH when started in the middle of a 4-week exposure to 10% O2.113 Nonselective inhibition of both ETA and ETB with bosentan has been shown to be similarly protective in the development of hypoxic PH.161 While evaluation of bosentan in patients with hypoxic PH has been limited but disappointing,162 the use of ET receptor antagonists in PAH has proven more promising.163-167 To date, no experiments have been performed to evaluate whether NHE expression and/or activity are altered in PASMCs from chronically hypoxic animals treated with ET receptor antagonists, and thus, as with digoxin, it remains unclear to what extent the effects of ET-1 receptor antagonists are mediated through NHE inhibition versus effects on other signaling pathways.
Future Directions
A fundamental question raised by the finding that NHE1 is necessary for the development of hypoxia-induced PH in animal models is whether NHE1 signaling plays a similarly important role in human disease. Evaluation of NHE1 expression and activity and NHE1-cytoskeletal interactions in sample tissue from human subjects with hypoxic PH, compared to those in control samples, would clarify the relevance of this line of work to humans.
While this review has focused primarily on hypoxia-induced PH, it is also conceivable that the NHE1 signaling pathway may have relevance to the development of other classes of PH. HIF-1α expression is increased in normoxic PASMCs isolated from patients with PAH, compared to that in controls,168 although the mechanism underlying this upregulation of HIF-1α in normoxic conditions remains incompletely understood. This raises the possibility that NHE1 expression could be increased in PAH and could contribute to cytoskeletal rearrangement and pathologic PASMC function. Exploration of these pathways in tissue from PAH patients, compared to tissue from control patients, would help to shed light on the role of NHE1 in this disease. Given the paucity of available PA tissue from PAH patients, animal models such as the SuHx rat model might also be used to provide insight into the role of NHE1 in the development of PAH.
Further exploration of inhibitors of NHE1-mediated cytoskeletal rearrangement would also prove valuable. Current NHE1 inhibitors are plagued by justifiable concern about neurologic side effects. Drugs that specifically disrupt the NHE1-ezrin interaction, if developed, could prove beneficial in hypoxia-induced PH, with the potential for a more benign side-effect profile. Alternatively, inhalational delivery of NHE1 inhibitors could prove effective, with a lower likelihood of systemic toxicity.
Finally, digoxin, which for years has been successfully used for the treatment of atrial fibrillation and left heart failure in humans, has been shown to be effective in both prevention and reversal of hypoxic PH in mice. The potential benefits of digoxin therapy in humans with hypoxic PH remain underexplored. A small case series of 5 patients with cor pulmonale (right heart failure due to underlying lung disease) treated with digoxin resulted in near normalization of PA pressures, accompanied by marked clinical improvement.169 Whether the benefits of digoxin are mediated through direct improvement of right ventricular contractility, decrease of PVR, or a combination of the two remains to be clarified. Further exploration of the potential benefits of digoxin in cor pulmonale has been limited because of concerns for the possibility of increased risk of digoxin toxicity in patients with lung disease.170 Initial retrospective cohort studies examining benefits and risks in patients with hypoxic PH who have received digoxin in the past would be useful and could provide the basis for future prospective randomized controlled trials.
Source of Support: This work was funded by National Institutes of Health grants T32 HL007534, R03 HL114902, and R01 HL073859.
Conflict of Interest: None declared.
References
- 1.Aalkjær C, Cragoe EJ Jr. Intracellular pH regulation in resting and contracting segments of rat mesenteric resistance vessels. J Physiol 1988;402(1):391–410. [DOI] [PMC free article] [PubMed]
- 2.Aalkjær C, Peng HL. pH and smooth muscle. Acta Physiol Scand 1997;161(4):557–566. [DOI] [PubMed]
- 3.Lucchesi PA, Berk BC. Regulation of sodium-hydrogen exchange in vascular smooth muscle. Cardiovasc Res 1995;29(2):172–177. [PubMed]
- 4.Boedtkjer E, Aalkjaer C. Intracellular pH in the resistance vasculature: regulation and functional implications. J Vasc Res 2012;49(6):479–496. [DOI] [PubMed]
- 5.Farrukh IS, Hoidal JR, Barry WH. Effect of intracellular pH on ferret pulmonary arterial smooth muscle cell calcium homeostasis and pressure. J Appl Physiol 1996;80(2):496–505. [DOI] [PubMed]
- 6.Madden JA, Ray DE, Keller PA, Kleinman JG. Ion exchange activity in pulmonary artery smooth muscle cells: the response to hypoxia. Am J Physiol Lung Cell Mol Physiol 2001;280(2):L264–L271. [DOI] [PubMed]
- 7.Quinn DA, Honeyman TW, Joseph PM, Thompson BT, Hales CA, Scheid CR. Contribution of Na+/H+ exchange to pH regulation in pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 1991;5(6):586–591. [DOI] [PubMed]
- 8.Donowitz M, Tse CM, Fuster D. SLC9/NHE gene family, a plasma membrane and organellar family of Na+/H+ exchangers. Mol Asp Med 2013;34(2–3):236–251. [DOI] [PMC free article] [PubMed]
- 9.Sardet C, Franchi A, Pouysségur J. Molecular cloning, primary structure, and expression of the human growth factor–activatable Na+/H+ antiporter. Cell 1989;56(2):271–280. [DOI] [PubMed]
- 10.Xue L, Aihara E, Wang TC, Montrose MH. Trefoil factor 2 requires Na/H exchanger 2 activity to enhance mouse gastric epithelial repair. J Biol Chem 2011;286(44):38375–38382. [DOI] [PMC free article] [PubMed]
- 11.Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol 2005;67:411–443. [DOI] [PubMed]
- 12.Wiemann M, Schwark JR, Bonnet U, Jansen HW, Grinstein S, Baker RE, Lang HJ, Wirth K, Bingmann D. Selective inhibition of the Na+/H+ exchanger type 3 activates CO2/H+-sensitive medullary neurones. Pfluegers Arch 1999;438(3):255–262. [DOI] [PubMed]
- 13.Gawenis LR, Greeb JM, Prasad V, Grisham C, Sanford LP, Doetschman T, Andringa A, Miller ML, Shull GE. Impaired gastric acid secretion in mice with a targeted disruption of the NHE4 Na+/H+ exchanger. J Biol Chem 2005;280(13):12781–12789. [DOI] [PubMed]
- 14.Diering GH, Mills F, Bamji SX, Numata M. Regulation of dendritic spine growth through activity-dependent recruitment of the brain-enriched Na+/H+ exchanger NHE5. Mol Biol Cell 2011;22(13):2246–2257. [DOI] [PMC free article] [PubMed]
- 15.Nakamura N, Tanaka S, Teko Y, Mitsui K, Kanazawa H. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J Biol Chem 2005;280(2):1561–1572. [DOI] [PubMed]
- 16.Ye G, Chen C, Han D, Xiong X, Kong Y, Wan B, Yu L. Cloning of a novel human NHEDC1 (Na+/H+ exchanger like domain containing 1) gene expressed specifically in testis. Mol Biol Rep 2006;33(3):175–180. [DOI] [PubMed]
- 17.Lee SH, Kim T, Park ES, Yang S, Jeong D, Choi Y, Rho J. NHE10, an osteoclast-specific member of the Na+/H+ exchanger family, regulates osteoclast differentiation and survival [corrected]. Biochem Biophys Res Commun 2008;369(2):320–326. [DOI] [PubMed]
- 18.Xiang M, Feng M, Muend S, Rao R. A human Na+/H+ antiporter sharing evolutionary origins with bacterial NhaA may be a candidate gene for essential hypertension. Proc Natl Acad Sci USA 2007;104(47):18677–18681. [DOI] [PMC free article] [PubMed]
- 19.Wang D, King SM, Quill TA, Doolittle LK, Garbers DL. A new sperm-specific Na+/H+ exchanger required for sperm motility and fertility. Nat Cell Biol 2003;5(12):1117–1122. [DOI] [PubMed]
- 20.Orlowski J, Kandasamy RA, Shull GE. Molecular cloning of putative members of the Na/H exchanger gene family: cDNA cloning, deduced amino acid sequence, and mRNA tissue expression of the rat Na/H exchanger NHE-1 and two structurally related proteins. J Biol Chem 1992;267(13):9331–9339. [PubMed]
- 21.Wang Z, Orlowski J, Shull GE. Primary structure and functional expression of a novel gastrointestinal isoform of the rat Na/H exchanger. J Biol Chem 1993;268(16):11925–11928. [PubMed]
- 22.Brant SR, Yun CH, Donowitz M, Tse CM. Cloning, tissue distribution, and functional analysis of the human Na+/N+ exchanger isoform, NHE3. Am J Physiol Cell Physiol 1995;269(1):C198–C206. [DOI] [PubMed]
- 23.Attaphitaya S, Park K, Melvin JE. Molecular cloning and functional expression of a rat Na+/H+ exchanger (NHE5) highly expressed in brain. J Biol Chem 1999;274(7):4383–4388. [DOI] [PubMed]
- 24.Miyazaki E, Sakaguchi M, Wakabayashi S, Shigekawa M, Mihara K. NHE6 protein possesses a signal peptide destined for endoplasmic reticulum membrane and localizes in secretory organelles of the cell. J Biol Chem 2001;276(52):49221–49227. [DOI] [PubMed]
- 25.Numata M, Petrecca K, Lake N, Orlowski J. Identification of a mitochondrial Na+/H+ exchanger. J Biol Chem 1998;273(12):6951–6959. [DOI] [PubMed]
- 26.Numata M, Orlowski J. Molecular cloning and characterization of a novel (Na+,K+)/H+ exchanger localized to the trans-Golgi network. J Biol Chem 2001;276(20):17387–17394. [DOI] [PubMed]
- 27.Rios EJ, Fallon M, Wang J, Shimoda LA. Chronic hypoxia elevates intracellular pH and activates Na+/H+ exchange in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2005;289(5):L867–L874. [DOI] [PubMed]
- 28.Shimoda LA, Fallon M, Pisarcik S, Wang J, Semenza GL. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 2006;291(5):L941–L949. [DOI] [PubMed]
- 29.Wakabayashi S, Pang T, Su X, Shigekawa M. A novel topology model of the human Na+/H+ exchanger isoform 1. J Biol Chem 2000;275(11):7942–7949. [DOI] [PubMed]
- 30.Landau M, Herz K, Padan E, Ben-Tal N. Model structure of the Na+/H+ exchanger 1 (NHE1): functional and clinical implications. J Biol Chem 2007;282(52):37854–37863. [DOI] [PubMed]
- 31.Padan E, Kozachkov L, Herz K, Rimon A. NhaA crystal structure: functional-structural insights. J Exp Biol 2009;212(11):1593–1603. [DOI] [PubMed]
- 32.Hendus-Altenburger R, Kragelund BB, Pedersen SF. Structural dynamics and regulation of the mammalian SLC9A family of Na+/H+ exchangers. Curr Top Membr 2014;73:69–148. [DOI] [PubMed]
- 33.Nygaard EB, Lagerstedt JO, Bjerre G, Shi B, Budamagunta M, Poulsen KA, Meinild S, et al. Structural modeling and electron paramagnetic resonance spectroscopy of the human Na+/H+ exchanger isoform 1, NHE1. J Biol Chem 2011;286(1):634–648. [DOI] [PMC free article] [PubMed]
- 34.Denker SP, Huang DC, Orlowski J, Furthmayr H, Barber DL. Direct binding of the Na-H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H+ translocation. Mol Cell 2000;6(6):1425–1436. [DOI] [PubMed]
- 35.Köster S, Pavkov-Keller T, Kühlbrandt W, Yildiz Ö. Structure of human Na+/H+ exchanger NHE1 regulatory region in complex with calmodulin and Ca2+. J Biol Chem 2011;286(47):40954–40961. [DOI] [PMC free article] [PubMed]
- 36.Pang T, Su X, Wakabayashi S, Shigekawa M. Calcineurin homologous protein as an essential cofactor for Na+/H+ exchangers. J Biol Chem 2001;276(20):17367–17372. [DOI] [PubMed]
- 37.Mishima M, Wakabayashi S, Kojima C. Solution structure of the cytoplasmic region of Na+/H+ exchanger 1 complexed with essential cofactor calcineurin B homologous protein 1. J Biol Chem 2007;282(4):2741–2751. [DOI] [PubMed]
- 38.Aharonovitz O, Zaun HC, Balla T, York JD, Orlowski J, Grinstein S. Intracellular pH regulation by Na+/H+ exchange requires phosphatidylinositol 4,5-bisphosphate. J Cell Biol 2000;150(1):213–224. [DOI] [PMC free article] [PubMed]
- 39.Sardet C, Counillon L, Franchi A, Pouysségur J. Growth factors induce phosphorylation of the Na+/H+ antiporter, glycoprotein of 110 kD. Science 1990;247(4943):723–726. [DOI] [PubMed]
- 40.Touyz RM, Schiffrin EL. Growth factors mediate intracellular signaling in vascular smooth muscle cells through protein kinase C–linked pathways. Hypertension 1997;30(6):1440–1447. [DOI] [PubMed]
- 41.Ober SS, Pardee AB. Both protein kinase C and calcium mediate activation of the Na+/H+ antiporter in Chinese hamster embryo fibroblasts. J Cell Physiol 1987;132(2):311–317. [DOI] [PubMed]
- 42.Tominaga T, Barber DL. Na-H exchange acts downstream of RhoA to regulate integrin-induced cell adhesion and spreading. Mol Biol Cell 1998;9(8):2287–2303. [DOI] [PMC free article] [PubMed]
- 43.Tominaga T, Ishizaki T, Narumiya S, Barber DL. p160ROCK mediates RhoA activation of Na-H exchange. EMBO J 1998;17(16):4712–4722. [DOI] [PMC free article] [PubMed]
- 44.Wallert MA, Thronson HL, Korpi NL, Olmschenk SM, McCoy AC, Funfar MR, Provost JJ. Two G protein-coupled receptors activate Na+/H+ exchanger isoform 1 in Chinese hamster lung fibroblasts through an ERK-dependent pathway. Cell Signal 2005;17(2):231–242. [DOI] [PubMed]
- 45.Moor AN, Fliegel L. Protein kinase-mediated regulation of the Na+/H+ exchanger in the rat myocardium by mitogen-activated protein kinase-dependent pathways. J Biol Chem 1999;274(33):22985–22992. [DOI] [PubMed]
- 46.Khaled AR, Moor AN, Li A, Kim K, Ferris DK, Muegge K, Fisher RJ, Fliegel L, Durum SK. Trophic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which induces intracellular alkalinization. Mol Cell Biol 2001;21(22):7545–7557. [DOI] [PMC free article] [PubMed]
- 47.Maekawa N, Abe J, Shishido T, Itoh S, Ding B, Sharma VK, Sheu SS, Blaxall BC, Berk BC. Inhibiting p90 ribosomal S6 kinase prevents Na+/H+ exchanger–mediated cardiac ischemia-reperfusion injury. Circulation 2006;113(21):2516–2523. [DOI] [PubMed]
- 48.Cox GA, Lutz CM, Yang CL, Biemesderfer D, Bronson RT, Fu A, Aronson PS, Noebels JL, Frankel WN. Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice. Cell 1997;91(1):139–148. [DOI] [PubMed]
- 49.Bell SM, Schreiner CM, Schultheis PJ, Miller ML, Evans RL, Vorhees CV, Shull GE, Scott WJ. Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures. Am J Physiol Cell Physiol 1999;276(4):C788–C795. [DOI] [PubMed]
- 50.Lazdunski M, Frelin C, Vigne P. The sodium/hydrogen exchange system in cardiac cells: its biochemical and pharmacological properties and its role in regulating internal concentrations of sodium and internal pH. J Mol Cell Cardiol 1985;17(11):1029–1042. [DOI] [PubMed]
- 51.Nakamura TY, Iwata Y, Arai Y, Komamura K, Wakabayashi S. Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ Res 2008;103(8):891–899. [DOI] [PubMed]
- 52.Reshkin SJ, Bellizzi A, Caldeira S, Albarani V, Malanchi I, Poignee M, Alunni-Fabbroni M, Casavol V, Tommasino M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J 2000;14(14):2185–2197. [DOI] [PubMed]
- 53.Cardone RA, Bagorda A, Bellizzi A, Busco G, Guerra L, Paradiso A, Casavola V, Zaccolo M, Reshkin SJ. Protein kinase A gating of a pseudopodial-located RhoA/ROCK/p38/NHE1 signal module regulates invasion in breast cancer cell lines. Mol Biol Cell 2005;16(7):3117–3127. [DOI] [PMC free article] [PubMed]
- 54.Krampetz IK, Rhoades RA. Intracellular pH: effect on pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 1991;260(6):L516–L521. [DOI] [PubMed]
- 55.Quinn DA, Dahlberg CG, Bonventre JP, Scheid CR, Honeyman T, Joseph PM, Thompson BT, Hales CA. The role of Na+/H+ exchange and growth factors in pulmonary artery smooth muscle cell proliferation. Am J Respir Cell Mol Biol 1996;14(2):139–145. [DOI] [PubMed]
- 56.Putney LK, Denker SP, Barber DL. The changing face of the Na+/H+ exchanger, NHE1: structure, regulation, and cellular actions. Annu Rev Pharmacol Toxicol 2002;42:527–552. [DOI] [PubMed]
- 57.Ayajiki K, Kindermann M, Hecker M, Fleming I, Busse R. Intracellular pH and tyrosine phosphorylation but not calcium determine shear stress–induced nitric oxide production in native endothelial cells. Circ Res 1996;78(5):750–758. [DOI] [PubMed]
- 58.Gerritsen ME, Perry CA, Moatter T, Cragoe EJ Jr., Medow MS. Agonist-specific role for Na+/H+ antiport in prostaglandin release from microvessel endothelium. Am J Physiol Cell Physiol 1989;256(4):C831–C839. [DOI] [PubMed]
- 59.Ghigo D, Bussolino F, Garbarino G, Heller R, Turrini F, Pescarmona G, Cragoe EJ Jr., Pegoraro L, Bosia A. Role of Na+/H+ exchange in thrombin-induced platelet-activating factor production by human endothelial cells. J Biol Chem 1988;263(36):19437–19446. [PubMed]
- 60.Wakabayashi I, Poteser M, Groschner K. Intracellular pH as a determinant of vascular smooth muscle function. J Vasc Res 2006;43(3):238–250. [DOI] [PubMed]
- 61.Raffestin B, McMurtry IF. Effects of intracellular pH on hypoxic vasoconstriction in rat lungs. J Appl Physiol 1987;63(6):2524–2531. [DOI] [PubMed]
- 62.Boedtkjer E, Praetorius J, Matchkov VV, Stankevicius E, Mogensen S, Füchtbauer AC, Simonsen U, Füchtbauer EM, Aalkjaer C. Disruption of Na+,HCO3− cotransporter NBCn1 (slc4a7) inhibits NO-mediated vasorelaxation, smooth muscle Ca2+ sensitivity, and hypertension development in mice. Circulation 2011;124(17):1819–1829. [DOI] [PubMed]
- 63.Boedtkjer E, Damkier HH, Aalkjaer C. NHE1 knockout reduces blood pressure and arterial media/lumen ratio with no effect on resting pHi in the vascular wall. J Physiol 2012;590(8):1895–1906. [DOI] [PMC free article] [PubMed]
- 64.L’Allemain G, Paris S, Pouysségur J. Role of a Na+-dependent Cl−/HCO3− exchange in regulation of intracellular pH in fibroblasts. J Biol Chem 1985;260(8):4877–4883. [PubMed]
- 65.Kapus A, Grinstein S, Wasan S, Kandasamy R, Orlowski J. Functional characterization of three isoforms of the Na+/H+ exchanger stably expressed in Chinese hamster ovary cells: ATP dependence, osmotic sensitivity, and role in cell proliferation. J Biol Chem 1994;269(38):23544–23552. [PubMed]
- 66.Wu D, Doods H, Stassen JM. Inhibition of human pulmonary artery smooth muscle cell proliferation and migration by sabiporide, a new specific NHE-1 inhibitor. J Cardiovasc Pharmacol 2006;48(2):34–40. [DOI] [PubMed]
- 67.Yu L, Hales CA. Silencing of sodium-hydrogen exchanger 1 attenuates the proliferation, hypertrophy, and migration of pulmonary artery smooth muscle cells via E2F1. Am J Respir Cell Mol Biol 2011;45(5):923–930. [DOI] [PMC free article] [PubMed]
- 68.Simchowitz L, Cragoe EJ Jr. Regulation of human neutrophil chemotaxis by intracellular pH. J Biol Chem 1986;261(14):6492–6500. [PubMed]
- 69.Denker SP, Barber DL. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J Cell Biol 2002;159(6):1087–1096. [DOI] [PMC free article] [PubMed]
- 70.von Euler US, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand 1946;12(4):301–320.
- 71.Motley HL, Cournand A, Werko L, Himelstein A, Dresdale D. The influence of short periods of induced acute anoxia upon pulmonary artery pressures in man. Am J Physiol 1947;150(2):315–320. [DOI] [PubMed]
- 72.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 2012;92(1):367–520. [DOI] [PMC free article] [PubMed]
- 73.Swenson ER. Hypoxic pulmonary vasoconstriction. High Alt Med Biol 2013;14(2):101–110. [DOI] [PubMed]
- 74.Dorrington KL, Clar C, Young JD, Jonas M, Tansley JG, Robbins PA. Time course of the human pulmonary vascular response to 8 hours of isocapnic hypoxia. Am J Physiol Heart Circ Physiol 1997;273(3):H1126–H1134. [DOI] [PubMed]
- 75.Weigand L, Shimoda LA, Sylvester JT. Enhancement of myofilament calcium sensitivity by acute hypoxia in rat distal pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 2011;301(3):L380–L387. [DOI] [PMC free article] [PubMed]
- 76.Aaronson PI, Robertson TP, Ward JP. Endothelium-derived mediators and hypoxic pulmonary vasoconstriction. Respir Physiol Neurobiol 2002;132(1):107–120. [DOI] [PubMed]
- 77.Deem S, Swenson ER, Alberts MK, Hedges RG, Bishop MJ. Red-blood-cell augmentation of hypoxic pulmonary vasoconstriction: hematocrit dependence and the importance of nitric oxide. Am J Respir Crit Care Med 1998;157(4):1181–1186. [DOI] [PubMed]
- 78.Kiefmann R, Rifkind JM, Nagababu E, Bhattacharya J. Red blood cells induce hypoxic lung inflammation. Blood 2008;111(10):5205–5214. [DOI] [PMC free article] [PubMed]
- 79.Crawford JH, Isbell TS, Huang Z, Shiva S, Chacko BK, Schechter AN, Darley-Usmar VM, et al. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood 2006;107(2):566–574. [DOI] [PMC free article] [PubMed]
- 80.Sprague RS, Olearczyk JJ, Spence DM, Stephenson AH, Sprung RW, Lonigro AJ. Extracellular ATP signaling in the rabbit lung: erythrocytes as determinants of vascular resistance. Am J Physiol Heart Circ Physiol 2003;285(2):H693–H700. [DOI] [PubMed]
- 81.Lejeune P, Vachiéry JL, Leeman M, Brimioulle S, Hallemans R, Melot C, Naeije R. Absence of parasympathetic control of pulmonary vascular pressure-flow plots in hyperoxic and hypoxic dogs. Respir Physiol 1989;78(2):123–133. [DOI] [PubMed]
- 82.Lodato RF, Michael JR, Murray PA. Absence of neural modulation of hypoxic pulmonary vasoconstriction in conscious dogs. J Appl Physiol 1988;65(4):1481–1487. [DOI] [PubMed]
- 83.Marshall JM. Peripheral chemoreceptors and cardiovascular regulation. Physiol Rev 1994;74(3):543–594. [DOI] [PubMed]
- 84.Gordon JB, Martinez FR, Keller PA, Tod ML, Madden JA. Differing effects of acute and prolonged alkalosis on hypoxic pulmonary vasoconstriction. Am Rev Respir Dis 1993;148(6):1651–1656. [DOI] [PubMed]
- 85.Brimioulle S, Lejeune P, Vachiéry JL, Leeman M, Melot C, Naeije R. Effects of acidosis and alkalosis on hypoxic pulmonary vasoconstriction in dogs. Am J Physiol Heart Circ Physiol 1990;258(2):H347–H353. [DOI] [PubMed]
- 86.Fike CD, Hansen TN. The effect of alkalosis on hypoxia-induced pulmonary vasoconstriction in lungs of newborn rabbits. Pediatr Res 1989;25(4):383–388. [DOI] [PubMed]
- 87.Lloyd TC Jr. Influence of blood pH on hypoxic pulmonary vasoconstriction. J Appl Physiol 1966;21(2):358–364. [DOI] [PubMed]
- 88.Viles PH, Shepherd JT. Relationship between pH, Po2, and Pco2 on the pulmonary vascular bed of the cat. Am J Physiol 1968;215(5):1170–1176. [DOI] [PubMed]
- 89.Yamaguchi T, O’Brien RF, Hanson WL, Wagner WW Jr., McMurtry IF. Prostacyclin contributes to inhibition of hypoxic pulmonary vasoconstriction by alkalosis. Prostaglandins 1989;38(1):53–63. [DOI] [PubMed]
- 90.Silove ED, Inoue T, Grover RF. Comparison of hypoxia, pH, and sympathomimetic drugs on bovine pulmonary vasculature. J Appl Physiol 1968;24(3):355–365. [DOI] [PubMed]
- 91.Leach RM, Sheehan DW, Chacko VP, Sylvester JT. Energy state, pH, and vasomotor tone during hypoxia in precontracted pulmonary and femoral arteries. Am J Physiol Lung Cell Mol Physiol 2000;278(2):L294–L304. [DOI] [PubMed]
- 92.Shimoda LA, Luke T, Sylvester JT, Shih HW, Jain A, Swenson ER. Inhibition of hypoxia-induced calcium responses in pulmonary arterial smooth muscle by acetazolamide is independent of carbonic anhydrase inhibition. Am J Physiol Lung Cell Mol Physiol 2007;292(4):L1002–L1012. [DOI] [PubMed]
- 93.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009;54(1 suppl.):S43–S54. [DOI] [PubMed]
- 94.Arias-Stella J, Saldaña M. The terminal portion of the pulmonary arterial tree in people native to high altitudes. Circulation 1963;28(5):915–925. [DOI] [PubMed]
- 95.Groves BM, Reeves JT, Sutton JR, Wagner PD, Cymerman A, Malconian MK, Rock PB, Young PM, Houston CS. Operation Everest II: elevated high-altitude pulmonary resistance unresponsive to oxygen. J Appl Physiol 1987;63(2):521–530. [DOI] [PubMed]
- 96.Hislop A, Reid L. New findings in pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. Br J Exp Pathol 1976;57(5):542–554. [PMC free article] [PubMed]
- 97.Meyrick B, Reid L. The effect of continued hypoxia on rat pulmonary arterial circulation: an ultrastructural study. Lab Invest 1978;38(2):188–200. [PubMed]
- 98.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 2006;99(7):675–691. [DOI] [PubMed]
- 99.Quinn DA, Du HK, Thompson BT, Hales CA. Amiloride analogs inhibit chronic hypoxic pulmonary hypertension. Am J Respir Crit Care Med 1998;157(4):1263–1268. [DOI] [PubMed]
- 100.Yu L, Quinn DA, Garg HG, Hales CA. Deficiency of the NHE1 gene prevents hypoxia-induced pulmonary hypertension and vascular remodeling. Am J Respir Crit Care Med 2008;177(11):1276–1284. [DOI] [PMC free article] [PubMed]
- 101.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, et al. Hypoxia inducible-factor1α regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 2010;176(3):1130–1138. [DOI] [PMC free article] [PubMed]
- 102.Huetsch JC, Shimoda LA. The activity of the sodium-hydrogen exchanger in pulmonary arterial smooth muscle cells is increased in models of pulmonary arterial hypertension. Am J Respir Crit Care Med 2014;189(mtg. abstr.):A3342.
- 103.Simonin A, Fuster D. Nedd4-1 and β-arrestin-1 are key regulators of Na+/H+ exchanger 1 ubiquitylation, endocytosis, and function. J Biol Chem 2010;285(49):38293–38303. [DOI] [PMC free article] [PubMed]
- 104.Bertrand B, Wakabayashi S, Ikeda T, Pouysségur J, Shigekawa M. The Na+/H+ exchanger isoform 1 (NHE1) is a novel member of the calmodulin-binding proteins: identification and characterization of calmodulin-binding sites. J Biol Chem 1994;269(18):13703–13709. [PubMed]
- 105.Sardet C, Fafournoux P, Pouysségur J. α-thrombin, epidermal growth factor, and okadaic acid activate the Na+/H+ exchanger, NHE-1, by phosphorylating a set of common sites. J Biol Chem 1991;266(29):19166–19171. [PubMed]
- 106.Snabaitis AK, Cuello F, Avkiran M. Protein kinase B/Akt phosphorylates and inhibits the cardiac Na+/H+ exchanger NHE1. Circ Res 2008;103(8):881–890. [DOI] [PubMed]
- 107.Mukhin YV, Vlasova T, Jaffa AA, Collinsworth G, Bell JL, Tholanikunnel BG, Pettus T, et al. Bradykinin B2 receptors activate Na+/H+ exchange in mIMCD-3 cells via Janus kinase 2 and Ca2+/calmodulin. J Biol Chem 2001;276(20):17339–17346. [DOI] [PubMed]
- 108.Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 2003;284(2):C316–C330. [DOI] [PubMed]
- 109.Perros F, Montani D, Dorfmüller P, Durand-Gasselin I, Tcherakian C, Le Pavec J, Mazmanian M. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2008;178(1):81–88. [DOI] [PubMed]
- 110.Ogawa A, Firth AL, Smith KA, Maliakal MV, Yuan JX. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 2012;302(2):C405–C411. [DOI] [PMC free article] [PubMed]
- 111.Rubens C, Ewert R, Halank M, Wensel R, Orzechowski HD, Schultheiss HP, Hoeffken G. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest 2001;120(5):1562–1569. [DOI] [PubMed]
- 112.Nakanishi K, Tajima F, Nakata Y, Osada H, Tachibana S, Kawai T, Torikata C, et al. Expression of endothelin-1 in rats developing hypobaric hypoxia-induced pulmonary hypertension. Lab Invest 1999;79(11):1347–1357. [PubMed]
- 113.DiCarlo VS, Chen SJ, Meng QC, Durand J, Yano M, Chen YF, Oparil S. ETA-receptor antagonist prevents and reverses chronic hypoxia-induced pulmonary hypertension in rat. Am J Physiol Lung Cell Mol Physiol 1995;269(5):L690–L697. [DOI] [PubMed]
- 114.Undem C, Rios EJ, Maylor J, Shimoda LA. Endothelin-1 augments Na+/H+ exchange activity in murine pulmonary arterial smooth muscle cells via Rho kinase. PLoS ONE 2012;7:e46303. doi:10.1371/journal.pone.0046303. [DOI] [PMC free article] [PubMed]
- 115.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 2007;100(6):923–929. [DOI] [PubMed]
- 116.Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol 2008;155(4):444–454. [DOI] [PMC free article] [PubMed]
- 117.Ward JP, McMurtry IF. Mechanisms of hypoxic pulmonary vasoconstriction and their roles in pulmonary hypertension: new findings for an old problem. Curr Opin Pharmacol 2009;9(3):287–296. [DOI] [PMC free article] [PubMed]
- 118.Firth AL, Choi IW, Park WS. Animal models of pulmonary hypertension: Rho kinase inhibition. Prog Biophys Mol Biol 2012;109(3):67–75. [DOI] [PubMed]
- 119.Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, Abe K, Takeshita A, Shimokawa H. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 2005;91(3):391–392. [DOI] [PMC free article] [PubMed]
- 120.Ishikura K, Yamada N, Ito M, Ota S, Nakamura M, Isaka N, Nakano T. Beneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circ J 2006;70(2):174–178. [DOI] [PubMed]
- 121.Fujita H, Fukumoto Y, Saji K, Sugimura K, Demachi J, Nawata J, Shimokawa H. Acute vasodilator effects of inhaled fasudil, a specific Rho-kinase inhibitor, in patients with pulmonary arterial hypertension. Heart Vessels 2010;25(2):144–149. [DOI] [PubMed]
- 122.Dempsey EC, McMurtry IF, O’Brien RF. Protein kinase C activation allows pulmonary artery smooth muscle cells to proliferate to hypoxia. Am J Physiol Lung Cell Mol Physiol 1991;260(2):L136–L145. [DOI] [PubMed]
- 123.Berk BC, Taubman MB, Cragoe EJ Jr., Fenton JW II, Griendling KK. Thrombin signal transduction mechanisms in rat vascular smooth muscle cells: calcium and protein kinase C-dependent and -independent pathways. J Biol Chem 1990;265(28):17334–17340. [PubMed]
- 124.Ebata S, Muto S, Okada K, Nemoto J, Amemiya M, Saito T, Asano Y. Aldosterone activates Na+/H+ exchange in vascular smooth muscle cells by nongenomic and genomic mechanisms. Kidney Int 1999;56(4):1400–1412. [DOI] [PubMed]
- 125.Koliakos G, Befani C, Paletas K, Kaloyianni M. Effect of endothelin on sodium/hydrogen exchanger activity of human monocytes and atherosclerosis-related functions. Ann NY Acad Sci 2007;1095:274–291. [DOI] [PubMed]
- 126.Guntupalli J, DuBose TD Jr. Effects of endothelin on rat renal proximal tubule Na+-Pi cotransport and Na+/H+ exchange. Am J Physiol Renal Physiol 1994;266(4):F658–F666. [DOI] [PubMed]
- 127.Walter R, Helmle-Kolb C, Forgo J, Binswanger U, Murer H. Stimulation of Na+/H+ exchange activity by endothelin in opossum kidney cells. Pfluegers Arch 1995;430(1):137–144. [DOI] [PubMed]
- 128.Shimoda LA, Semenza GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med 2011;183(2):152–156. [DOI] [PMC free article] [PubMed]
- 129.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001;292(5516):464–468. [DOI] [PubMed]
- 130.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001;292(5516):468–472. [DOI] [PubMed]
- 131.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev 1998;12(2):149–162. [DOI] [PMC free article] [PubMed]
- 132.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest 1999;103(5):691–696. [DOI] [PMC free article] [PubMed]
- 133.Shimoda LA. 55th Bowditch Lecture: effects of chronic hypoxia on the pulmonary circulation: role of HIF-1. J Appl Physiol 2012;113(9):1343–1352. [DOI] [PMC free article] [PubMed]
- 134.Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, Beussink L, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am J Respir Crit Care Med 2014;189(3):314–324. [DOI] [PMC free article] [PubMed]
- 135.Pisarcik S, Maylor J, Lu W, Yun X, Undem C, Sylvester JT, Semenza GL, Shimoda LA. Activation of hypoxia-inducible factor-1 in pulmonary arterial smooth muscle cells by endothelin-1. Am J Physiol Lung Cell Mol Physiol 2013;304(8):L549–L561. [DOI] [PMC free article] [PubMed]
- 136.Remillard CV, Yuan JX. High altitude pulmonary hypertension: role of K+ and Ca2+ channels. High Alt Med Biol 2005;6(2):133–146. [DOI] [PMC free article] [PubMed]
- 137.Wakabayashi I, Sakamoto K, Hatake K, Masui H, Yoshimoto S. Potentiating effect of NH4Cl on vasoconstriction in rat aorta. Biochem Biophys Res Commun 1991;178(3):808–814. [DOI] [PubMed]
- 138.Wakabayashi I, Kukovetz WR, Groschner K. NH4Cl-induced contraction of porcine coronary artery involves activation of dihydropyridine-sensitive Ca2+ entry. Eur J Pharmacol 1996;299(1–3):139–147. [DOI] [PubMed]
- 139.Ahn DS, Hume JR. pH regulation of voltage-dependent K+ channels in canine pulmonary arterial smooth muscle cells. Pfluegers Arch 1997;433(6):758–765. [DOI] [PubMed]
- 140.Wakabayashi I, Marumo M, Sotoda Y. Intracellular alkalinization augments capacitative Ca2+ entry in vascular smooth muscle cells. J Cardiovasc Pharmacol 2003;41(6):903–907. [DOI] [PubMed]
- 141.Tsukioka M, Iino M, Endo M. pH dependence of inositol 1,4,5-trisphosphate-induced Ca2+ release in permeabilized smooth muscle cells of the guinea-pig. J Physiol 1994;475(3):369–375. [DOI] [PMC free article] [PubMed]
- 142.Vexler ZS, Symons M, Barber DL. Activation of Na+/H+ exchange is necessary for RhoA-induced stress fiber formation. J Biol Chem 1996;271(37):22281–22284. [DOI] [PubMed]
- 143.Hooley R, Yu CY, Symons M, Barber DL. Gα13 stimulates Na+/H+ exchange through distinct Cdc42-dependent and RhoA-dependent pathways. J Biol Chem 1996;271(11):6152–6158. [DOI] [PubMed]
- 144.Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 2002;3(8):586–599. [DOI] [PubMed]
- 145.Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S, Tsukita S. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol 1998;140(3):647–657. [DOI] [PMC free article] [PubMed]
- 146.Barst RJ, Gibbs JS, Ghofrani HA, Hoeper MM, McLaughlin VV, Rubin LJ, Sitbon O, Tapson VF, Galiè N. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 2009;54(1 suppl.):S78–S84. [DOI] [PMC free article] [PubMed]
- 147.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, Howard LS, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012;186(8):790–796. [DOI] [PubMed]
- 148.Kleyman TR, Cragoe EJ Jr. Amiloride and its analogs as tools in the study of ion transport. J Membr Biol 1988;105(1):1–21. [DOI] [PubMed]
- 149.Karmazyn M. NHE-1: still a viable therapeutic target. J Mol Cell Cardiol 2013;61:77–82. [DOI] [PubMed]
- 150.Théroux P, Chaitman BR, Danchin N, Erhardt L, Meinertz T, Schroeder JS, Tognoni G, et al. Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations: main results of the GUARDIAN trial. Circulation 2000;102(25):3032–3038. [DOI] [PubMed]
- 151.Zeymer U, Suryapranata H, Monassier JP, Opolski G, Davies J, Rasmanis G, Linssen G, et al. The Na+/H+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction: results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J Am Coll Cardiol 2001;38(6):1644–1650. [DOI] [PubMed]
- 152.Boyce SW, Bartels C, Bolli R, Chaitman B, Chen JC, Chi E, Jessel A, et al. Impact of sodium-hydrogen exchange inhibition by cariporide on death or myocardial infarction in high-risk CABG surgery patients: results of the CABG surgery cohort of the GUARDIAN study. J Thorac Cardiovasc Surg 2003;126(2):420–427. [DOI] [PubMed]
- 153.Mentzer RM Jr., Bartels C, Bolli R, Boyce S, Buckberg GD, Chaitman B, Haverich A, et al. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann Thorac Surg 2008;85(4):1261–1270. [DOI] [PubMed]
- 154.Chen L, Gan XT, Haist JV, Feng Q, Lu X, Chakrabarti S, Karmazyn M. Attenuation of compensatory right ventricular hypertrophy and heart failure following monocrotaline-induced pulmonary vascular injury by the Na+/H+ exchange inhibitor cariporide. J Pharmacol Exp Ther 2001;298(2):469–476. [PubMed]
- 155.Scholz W, Albus U, Lang HJ, Linz W, Martorana PA, Englert HC, Schölkens BA. Hoe 694, a new Na+/H+ exchange inhibitor and its effects in cardiac ischaemia. Br J Pharmacol 1993;109(2):562–568. [DOI] [PMC free article] [PubMed]
- 156.Zhang H, Qian DZ, Tan YS, Lee KA, Gao P, Ren YR, Rey S, et al. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc Natl Acad Sci USA 2008;105(50):19579–19586. [DOI] [PMC free article] [PubMed]
- 157.Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA 2009;106(42):17910–17915. [DOI] [PMC free article] [PubMed] [Retracted]
- 158.Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, Sylvester JT, Semenza GL, Shimoda LA. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA 2012;109(4):1239–1244. [DOI] [PMC free article] [PubMed]
- 159.O’Callaghan DS, Savale L, Yaïci A, Natali D, Jaïs X, Parent F, Montani D, Humbert M, Simonneau G, Sitbon O. Endothelin receptor antagonists for the treatment of pulmonary arterial hypertension. Expert Opin Pharmacother 2011;12(10):1585–1596. [DOI] [PubMed]
- 160.Davie N, Haleen SJ, Upton PD, Polak JM, Yacoub MH, Morrell NW, Wharton J. ETA and ETB receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am J Respir Crit Care Med 2002;165(3):398–405. [DOI] [PubMed]
- 161.Eddahibi S, Raffestin B, Clozel M, Levame M, Adnot S. Protection from pulmonary hypertension with an orally active endothelin receptor antagonist in hypoxic rats. Am J Physiol Heart Circ Physiol 1995;268(2):H828–H835. [DOI] [PubMed]
- 162.Stolz D, Rasch H, Linka A, Di Valentino M, Meyer A, Brutsche M, Tamm M. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J 2008;32(3):619–628. [DOI] [PubMed]
- 163.Rubin LJ, Badesch DB, Barst RJ, Galiè N, Black CM, Keogh A, Pulido T, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346(12):896–903. [DOI] [PubMed]
- 164.Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, Badesch DB, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001;358(9288):1119–1123. [DOI] [PubMed]
- 165.Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, Badesch DB, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117(23):3010–3019. [DOI] [PubMed]
- 166.Barst RJ, Langleben D, Badesch D, Frost A, Lawrence EC, Shapiro S, Naeije R, Galiè N. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol 2006;47(10):2049–2056. [DOI] [PubMed]
- 167.Barst RJ, Langleben D, Frost A, Horn EM, Oudiz R, Shapiro S, McLaughlin V, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 2004;169(4):441–447. [DOI] [PubMed]
- 168.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang Y, Thenappan T, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 2012;110(11):1484–1497. [DOI] [PMC free article] [PubMed]
- 169.Ferrer MI, Harvey RM, Cathcart RT, Webster CA, Richards DW Jr., Cournand A. Some effects of digoxin upon the heart and circulation in man: digoxin in chronic cor pulmonale. Circulation 1950;1(2):161–186. [DOI] [PubMed]
- 170.Green LH, Smith TW. The use of digitalis in patients with pulmonary disease. Ann Intern Med 1977;87(4):459–465. [DOI] [PubMed]