

Abstract Abstract
Increased peripheral resistance of small distal pulmonary arteries is a hallmark signature of pulmonary hypertension (PH) and is believed to be the consequence of enhanced vasoconstriction to agonists, thickening of the arterial wall due to remodeling, and increased thrombosis. The elevation in arterial tone in PH is attributable, at least in part, to smooth muscle cells of PH patients being more depolarized and displaying higher intracellular Ca2+ levels than cells from normal subjects. It is now clear that downregulation of voltage-dependent K+ channels (e.g., Kv1.5) and increased expression and activity of voltage-dependent (Cav1.2) and voltage-independent (e.g., canonical and vanilloid transient receptor potential [TRPC and TRPV]) Ca2+ channels play an important role in the functional remodeling of pulmonary arteries in PH. This review focuses on an anion-permeable channel that is now considered a novel excitatory mechanism in the systemic and pulmonary circulations. It is permeable to Cl− and is activated by a rise in intracellular Ca2+ concentration (Ca2+-activated Cl− channel, or CaCC). The first section outlines the biophysical and pharmacological properties of the channel and ends with a description of the molecular candidate genes postulated to encode for CaCCs, with particular emphasis on the bestrophin and the newly discovered TMEM16 and anoctamin families of genes. The second section provides a review of the various sources of Ca2+ activating CaCCs, which include stimulation by mobilization from intracellular Ca2+ stores and Ca2+ entry through voltage-dependent and voltage-independent Ca2+ channels. The third and final section summarizes recent findings that suggest a potentially important role for CaCCs and the gene TMEM16A in PH.
Keywords: calcium-activated chloride channels, pulmonary hypertension, TMEM16A, anoctamin, bestrophin
Ca2+-activated Cl− channels (ClCa or CaCCs) are Cl−-permeable channels activated by a rise in intracellular Ca2+. They are widely distributed small-conductance channels located in the plasma membrane of many cell types and are hypothesized to serve important functions in sperm maturation, fluid transport across epithelial tissues, cardiac repolarization, olfactory transduction, photoreceptor stimulation, neuronal and skeletal muscle excitation, cardiac repolarization, and vascular smooth muscle excitability and contraction. For more information on the general properties and role of CaCCs in various cell types and tissues, the reader is invited to consult excellent reviews on this topic.1-11
Byrne and Large12 were the first to report the existence of a Ca2+-activated Cl− conductance in smooth muscle by describing its general properties in rat anococcygeus smooth muscle. Since this initial discovery, that group and others have described this conductance in a variety of smooth muscle cells, including vascular smooth muscle cells (VSMCs).1,4,7 The identification in 2008 by three independent groups of the TMEM16, or anoctamin, family of genes as novel candidates for CaCCs13-15 sparked a renewed interest in refining our understanding about their function in various tissues and whether they could serve as novel therapeutic targets. VSMCs, and pulmonary arterial smooth muscle cells (PASMCs) in particular, exhibit a significant “classical” CaCC conductance that now appears to be encoded by TMEM16A (anoctamin-1, or ANO1) and has been suggested to play a major role in vascular tone. After a description of the biophysical, regulation, and molecular properties of CaCCs in PASMCs and other vascular myocytes, this review focuses on the various sources of Ca2+ triggering CaCCs and how they might affect the control of membrane potential and pulmonary arterial tone under physiological and pathophysiological conditions.
Biophysical properties of CaCCs in PASMCs
General macroscopic properties of Ca2+-activated Cl− currents
CaCCs exhibit complex biophysical properties dictated by the interplay of Ca2+-, voltage-, and time-dependent gating mechanisms. Adding to this complexity are recent findings demonstrating that the channels in VSMCs are also regulated by phosphorylation involving both Ca2+-dependent and Ca2+-independent kinases and phosphatases. Initial studies describing the properties of CaCCs in smooth muscle and other cell types investigated with the whole-cell recording variant of the patch-clamp technique used transient sources of Ca2+ to activate the channels (e.g., Ca2+ entry via Ca2+ channels or Ca2+ release from internal stores). Although such methods suggested that Ca2+ plays an obligatory role in activating CaCCs, they provided little information on the biophysical properties of CaCCs, because only one of the three main variables controlling CaCC gating, voltage, was clamped, while internal Ca2+ levels and time-dependent kinetics were continuously changing. Setting intracellular [Ca2+] ([Ca2+]i) to a known fixed value by using a Ca2+ chelating agent such as EGTA or BAPTA in the pipette solution allowed for studying Ca2+-activated Cl− currents (ICl(Ca)) under well-controlled conditions, such that time-dependent gating mechanisms could be investigated in detail under voltage-clamp conditions.7,16-18
Figure 1A shows a typical family of ICl(Ca) recorded in a rabbit PASMC dialyzed with a Cs+-containing pipette solution consisting of 10 mM BAPTA and 7.3 mM CaCl2 to clamp [Ca2+]i to 500 nM free Ca2+, with free [Mg2+] set to 1 mM. The elicited current reversed near the expected equilibrium potential for Cl− of ∼0 mV (Fig. 1B) under the conditions of this experiment and displayed pronounced outward rectification (Fig. 1B), meaning that the underlying open channels carried more current in the outward direction (>0 mV; Cl− influx) than in the inward direction (<0 mV; Cl− efflux). In addition, the current exhibited typical time-dependent activation kinetics at positive membrane potentials and slow deactivation upon returning to the negative holding potential (Fig. 1A). Figures 1C and 1D illustrate another example, showing that the outward rectification is not a property of the pore but is rather dictated by voltage-dependent gating that results in channel closure at negative potentials. In this experiment, the rabbit PASMC was also dialyzed with a pipette solution containing 500 nM [Ca2+]i. The protocol necessitated activating ICl(Ca) with an initial constant step to +100 mV from a holding potential of −50 mV, followed by a second step to a variable voltage to determine the magnitude and polarity of the ICl(Ca) tail current (Fig. 1C). The tail current measured immediately after repolarization reflects the behavior of the channel opened to a constant and stable open probability and thus that of the open pore. Plotting the magnitude of the tail current as a function of the voltage during the second step revealed a linear voltage relationship (Fig. 1D), as opposed to the strong outward rectification seen under near-steady-state conditions (Fig. 1B). This and many similar experiments by our group and other investigators confirmed that the open pore of CaCC in vascular myocytes conducts inward and outward currents equally.
Figure 1.

Typical macroscopic Ca2+-activated Cl− currents (ICl(Ca)) recorded in a rabbit pulmonary artery smooth muscle cell with the whole-cell variant of the patch-clamp technique. The cell was dialyzed with a pipette solution containing CsCl and tetraethylammonium chloride with Ca2+ concentration set to 500 nM with the inclusion of the Ca2+ chelator BAPTA and an appropriate concentration of CaCl2.18 A, The top traces represent a representative family of ICl(Ca) elicited by the voltage-clamp protocol displayed below the traces. B, Current-voltage relationships for the currents in A measured immediately after the capacitative current surge (instantaneous ICl(Ca), labeled 1 in A; black squares) and at the end of 1-s pulses (late ICl(Ca), labeled 2 in A; red circles). The experiment shows the slight outward rectification of instantaneous ICl(Ca), while the quasi-steady-state ICl(Ca) displays pronounced outward rectification due to voltage-dependent gating. C, Double-pulse protocol examining the properties of the fully activated ICl(Ca). Currents (top traces) were evoked by the voltage-clamp protocol depicted below the traces. D, Amplitude of the tail current in C measured immediately after the capacitative currents during the second step (arrow in C) as a function of voltage, showing that the fully activated current is linear. This suggests that the open pore of the underlying Ca2+-activated Cl− channel carries inward and outward Cl− currents equally well.
Unitary currents through native CaCCs
Single-channel experiments in smooth muscle cells (SMCs) from human mesenteric artery,19 mouse, rat, and rabbit aorta,20-22 and rabbit pulmonary artery22-24 established that CaCCs are small-conductance anion channels with a single-channel conductance ranging from 1.2 to 3.5 pS. In agreement with studies describing macroscopic ICl(Ca), excised inside-out patch experiments in mouse aortic SMCs showed that single CaCCs are quickly activated by an elevation of Ca2+ on the cytoplasmic side of the membrane (Fig. 2A–2D). In addition, it was demonstrated in rabbit PASMCs that the unitary currents produced by these channels display ohmic behavior in the open state (Fig. 2E–2I), as well as voltage dependence (Fig. 2E, 2F, 2H). Another interesting property was the observation that single CaCCs display multiple conductance states, a behavior noted in both aortic20,21 and pulmonary artery23,24 SMCs (Fig. 2H–2J). The possible implication of this channel property for modes of activation by Ca2+ and voltage is discussed further below.
Figure 2.

Complex behavior of unitary currents through single Ca2+-activated Cl− channels (CaCCs) recorded in mouse aortic and rabbit pulmonary artery myocytes with the inside-out configuration of the patch-clamp technique. A–D, Inside-out patch recordings showing the Ca2+ dependence of single CaCCs recorded from mouse aortic smooth muscle cells (SMCs). The bar above each plot illustrates the change in [Ca2+] in the bath. Each plot reflects open probability (NPo) as a function of time after patch excision (indicated by a vertical bar labeled “Excise”). Below each NPo-versus-time plot are selected segments of original single-channel recording traces. E–J, Inside-out patch recordings from rabbit pulmonary artery SMCs (PASMCs), illustrating the Ca2+ and voltage dependence of single CaCCs as well as the existence of multiple subconductance states. E–H show sample recordings obtained at −100 mV (bottom) and +100 mV (top). E, F, and H were respectively obtained with 50 nM, 250 nM, and 1 μM Ca2+ in the bath. G and I show expanded traces from F and H, respectively, demarcated by horizontal bracketed bars. J, Mean current-voltage relationships of unitary CaCCs for the fully conducting and two subconductance states. A–D were reproduced from Hirakawa et al.21 with permission from the American Physiological Society. E–J were reproduced from Piper and Large23 with permission from the Physiological Society.
Selectivity of CaCCs
Replacement of Na+, the main cation in the extracellular medium, with less permeable cations such as N-methyl-d-glucamine had little effect on the magnitude and reversal potential of macroscopic or unitary ICl(Ca) in vascular myocytes,1,7,19-21 indicating that CaCCs are much more permeable to anions than to cations. Anion replacement experiments showed that ICl(Ca) in VSMCs exhibit an anion permeability sequence consistent with permeation driven primarily by the hydration energy of individual ions in a conduction pathway that exhibits a low electric field strength, with SCN− ≫ I≫ > NO3− > Br− > Cl− aspartate ≅ glutamate.1,4,7,18,23,25-27
Kinetic models predicting the Ca2+ and voltage dependence of native CaCCs
Quantitative analysis of the Ca2+ and voltage dependence of macroscopic and unitary ICl(Ca) in parotid acinar cells,16
Xenopus oocytes,17 and VSMCs23,28 showed that the affinity of the Ca2+ binding site located on the internal side of the membrane is voltage dependent, with membrane depolarization increasing the efficacy of Ca2+ in activating the channel. On the basis of a Hill coefficient consistently higher than 1, the above studies also proposed that a minimum of 2 Ca2+ are required to cooperatively open the channel. Arreola et al.16 were the first to propose a kinetic model describing the Ca2+ and voltage dependence of CaCCs recorded in parotid acinar cells: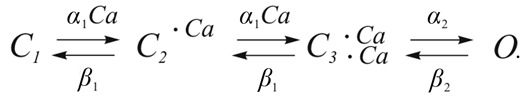 In this 4-state model, 2 calcium ions bind sequentially, which then leads to channel opening from a closed, fully bound state. The Ca2+-binding steps are defined by two reversible equilibrium reactions, each defined by an equilibrium constant K1 that is equal to β1/α1, where α1 is the first-order binding-rate constant and β1 is the unbinding-rate constant. The equilibrium transition from C3·2Ca to O was defined by the equilibrium constant K2 being equal to β2/α2, where α2 is the opening-rate constant and β2 is the closing-rate constant. In this scheme, both K1 and K2 are sensitive to voltage, each defined by a monotonic exponential function. The direct implication of this model is that Ca2+ binding is voltage dependent and therefore that the binding sites would lie within the transmembrane electric field (∼13% distance from the inside when the Woodhull model16 is applied). Although the kinetic model reasonably predicted the steady-state properties and kinetics of ICl(Ca) at low to intermediate [Ca2+]i (100–250 nM), the voltage dependence of the kinetics of the simulated currents significantly deviated from actual data at higher [Ca2+]i (1,000 nM).
In this 4-state model, 2 calcium ions bind sequentially, which then leads to channel opening from a closed, fully bound state. The Ca2+-binding steps are defined by two reversible equilibrium reactions, each defined by an equilibrium constant K1 that is equal to β1/α1, where α1 is the first-order binding-rate constant and β1 is the unbinding-rate constant. The equilibrium transition from C3·2Ca to O was defined by the equilibrium constant K2 being equal to β2/α2, where α2 is the opening-rate constant and β2 is the closing-rate constant. In this scheme, both K1 and K2 are sensitive to voltage, each defined by a monotonic exponential function. The direct implication of this model is that Ca2+ binding is voltage dependent and therefore that the binding sites would lie within the transmembrane electric field (∼13% distance from the inside when the Woodhull model16 is applied). Although the kinetic model reasonably predicted the steady-state properties and kinetics of ICl(Ca) at low to intermediate [Ca2+]i (100–250 nM), the voltage dependence of the kinetics of the simulated currents significantly deviated from actual data at higher [Ca2+]i (1,000 nM).
Kuruma and Hartzell17 performed a similar biophysical analysis of ICl(Ca) in Xenopus oocytes and proposed a radically different gating scheme based on an important observation also described by our group for ICl(Ca) in PASMCs.28
Figure 3 illustrates this observation, using the same data from the typical experiments shown in Figure 1. The two families of traces from Figure 1A and 1C were reproduced in Figure 3, showing only areas of interest. Time-dependent ICl(Ca) elicited by depolarizing steps more positive than +30 mV (right-hand set of traces) and deactivating ICl(Ca) tail currents (left-hand set of traces) were fitted with a single exponential function overlaid on the traces as a red or blue solid line. The estimated time constants of activation (τon; blue) and deactivation (τoff; red) were plotted as functions of voltage in the plot below the traces. The plot clearly shows that τon is not particularly sensitive to voltage, whereas τoff declines exponentially with membrane hyperpolarization. On the basis of very similar data and in contrast to the model proposed by Arreola et al.,16 Kuruma and Hartzell17 reasoned that voltage-dependent gating of CaCCs must involve a mechanism whereby channel closure, rather than channel opening, confers voltage sensitivity, and they proposed the following kinetic model: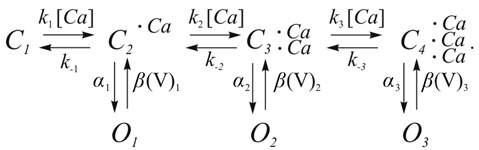 This 7-state model comprises 3 Ca2+ binding steps, each of which can lead to independent channel opening. While the binding-rate constants k1, k2, and k3 and the unbinding-rate constants k−1, k−2, and k−3 were set to identical values, the opening-rate constants α1, α2, and α3 were given progressively larger values. In this model, voltage sensitivity is governed by the closure-rate constants β(V)1, β(V)2, and β(V)3, each defined by a specific exponential function related to voltage. This model predicted well the Ca2+ and voltage dependence and accounted for most kinetic aspects of ICl(Ca) in Xenopus oocytes.17 Moreover, a separate analysis in rabbit pulmonary artery (PA) myocytes conformed to this model.28 In their detailed analysis of the gating properties of single CaCCs recorded from rabbit PASMCs, Piper and Large23 identified at least 3 subconductance state levels, ranging from ∼1.2 to 3.5 pS. Interestingly, the probability of detecting the lower subconductance states of 1.2 and 1.8 pS was high when the internal face of the membrane was exposed to 1 μM Ca2+, with no evidence of the fully conducting state at 3.5 pS. In contrast, only the fully conducting state of ∼3.5 pS and a subconductance level of ∼1.8 pS were apparent with 50 nM Ca2+ on the cytoplasmic side. Analysis of single-channel behavior with 250 or 500 nM Ca2+ revealed intermediate behaviors. Piper and Large23 speculated that the multiple subconductance states might correspond to the 3 open states in the kinetic model proposed by Kuruma and Hartzell.17 Sequential binding of multiple calcium ions would not only enhance the open probability of the channel but would also partially obstruct the permeation pathway leading to subconductance state levels.
This 7-state model comprises 3 Ca2+ binding steps, each of which can lead to independent channel opening. While the binding-rate constants k1, k2, and k3 and the unbinding-rate constants k−1, k−2, and k−3 were set to identical values, the opening-rate constants α1, α2, and α3 were given progressively larger values. In this model, voltage sensitivity is governed by the closure-rate constants β(V)1, β(V)2, and β(V)3, each defined by a specific exponential function related to voltage. This model predicted well the Ca2+ and voltage dependence and accounted for most kinetic aspects of ICl(Ca) in Xenopus oocytes.17 Moreover, a separate analysis in rabbit pulmonary artery (PA) myocytes conformed to this model.28 In their detailed analysis of the gating properties of single CaCCs recorded from rabbit PASMCs, Piper and Large23 identified at least 3 subconductance state levels, ranging from ∼1.2 to 3.5 pS. Interestingly, the probability of detecting the lower subconductance states of 1.2 and 1.8 pS was high when the internal face of the membrane was exposed to 1 μM Ca2+, with no evidence of the fully conducting state at 3.5 pS. In contrast, only the fully conducting state of ∼3.5 pS and a subconductance level of ∼1.8 pS were apparent with 50 nM Ca2+ on the cytoplasmic side. Analysis of single-channel behavior with 250 or 500 nM Ca2+ revealed intermediate behaviors. Piper and Large23 speculated that the multiple subconductance states might correspond to the 3 open states in the kinetic model proposed by Kuruma and Hartzell.17 Sequential binding of multiple calcium ions would not only enhance the open probability of the channel but would also partially obstruct the permeation pathway leading to subconductance state levels.
Figure 3.

Voltage dependence of activation and deactivation kinetics of whole-cell Ca2+-activated Cl− currents, recorded from a rabbit pulmonary artery myocytes. The traces above the plot were reproduced from those shown in Figure 1. The corresponding voltage step protocols that elicited these currents are shown below the traces. Superimposed on the black original current traces are single exponential fits to the data delimited by the arrows. The plot below the traces shows the derived time constants of activation (τon; blue) and deactivation (τoff; red) plotted as functions of voltage. The line passing through the red data points represents a single exponential fit, whereas the line passing through the blue data points represents a linear fit whose slope is not significantly different from 0 (P > 0.05).
Pharmacology of CaCCs in vascular myocytes
The stilbene derivatives 4,4′-diisothiocyano-2,2′-stilbene (DIDS) and 4-acetamido-4′-isothiocyano-2,2′-stilbene (SITS), fenamates such as niflumic (NFA) and flufenamic acids, anthracene-9-carboxylic acid (A9C), diphenylamine-2-carboxylate, 5-Nitro-2-(3-phenylpropylamino)benzoic acid (NPPB), ethacrynic acid, frusemide, and indanyloxyacetic acid 94 (IAA-94) all inhibit CaCCs in VSMCs. However, the common denominator is that they inhibit the channels with low affinity (IC50 in the range of ∼10−6 to 10−3 M) and with relatively poor selectivity in discriminating the contribution of CaCCs from that of other types of Cl− channels (e.g., volume- or swelling-activated Cl− channels [ICl-Swell]), and many of these compounds also target cation channels. There are a number of excellent reviews and original articles that summarize the older literature on this topic.1,4,6,7,29-31
Of this “first generation” of CaCC inhibitors, NFA is the most potent blocker of these channels and the compound most frequently used to investigate the physiological role of CaCCs on vascular smooth muscle tone.1,4,7 In portal vein SMCs, NFA was reasonably more effective at blocking ICl(Ca) than ICl-Swell.32 The block produced by NFA is moderately voltage dependent, with membrane depolarization increasing its potency, possibly because of a preferential interaction of NFA with the open state. In several types of VSMCs, including PASMCs, NFA slows the deactivation of CaCCs at negative potentials,24,30,33-37 an observation in agreement with the idea that NFA preferentially binds the channel in the open state and remains bound to the channel for some time, delaying channel closure.1 Except for one report in rat aorta,38 NFA (≤100 μM) did not attenuate the dihydropyridine-sensitive contraction produced by KCl-induced depolarization,39-43 nor did it inhibit voltage-gated Ca2+ channels directly.34,41,44,45 However, NFA and other fenamates exert nonspecific effects that bear major implications for interpretation of physiological data, depending on the cell type in which they are tested. NFA is a nonsteroidal anti-inflammatory agent that could indirectly influence ion channels and vascular tone by altering the release of vasoactive by-products of arachidonic metabolism (e.g., prostaglandins) by the cyclo-oxygenase pathway. Fenamates were shown to stimulate the human slow delayed rectifier K+ channel (IsK) expressed in Xenopus oocytes,46 a delayed rectifier K+ current in canine jejunal SMCs,47 and large-conductance Ca2+-activated K+ channels (BKCa) in VSMCs.33,35,48 One study also reported the inhibition of a nonselective cation current in rat exocrine pancreatic cells.49 Cruickshank et al.50 demonstrated that NFA elicits Ca2+ release from internal stores in rat PASMCs, but this was not confirmed in two independent studies in the same preparation.41,43 Kato et al.51 described experiments showing that the relaxation by NFA or NPPB of rat pulmonary arteries precontracted with endothelin-1 (ET-1) was independent of their ability to inhibit CaCCs. Finally, two studies from our group reported that NFA exerts a paradoxical inhibition and stimulation of CaCCs in rabbit pulmonary37 and coronary30 artery myocytes. In these studies, the stimulation was more easily visualized after a rapid washout of the drug, although in PASMCs stimulation of ICl(Ca) was clearly evident at negative potentials in the presence of NFA. These experiments suggested the existence of multiple binding sites for NFA affecting both the pore and gating of the channel. A9C was also shown to produce peculiar bimodal effects on ICl(Ca) in rabbit PASMCs, inhibiting the steady-state current in a voltage-dependent manner while potently stimulating the inward tail current during repolarization.29 Interestingly, NFA and A9C produced dual effects on expressed TMEM16A CaCCs,52 the gene speculated to encode for CaCCs in VSMCs,53-55 very similar to those reported for ICl(Ca) in vascular myocytes. Taken together, this explains why investigators have to be extremely cautious in their interpretation of the effects of NFA and all other CaCC blockers when examining their effects on membrane potential, [Ca2+]i, and force in intact arteries.
The recent discovery of the TMEM16, or anoctamin, family of CaCCs by three independent laboratories,13-15 combined with the development of high-throughput screening assays, allowed for the identification of novel CaCC modulators displaying greater potency and specificity. Screening of medium- to large-scale synthetic and natural compound libraries led to the discovery of small-molecule inhibitors56,57 and activators58 of CaCCs. Among the identified inhibitors, T16AInh-A01 and CACCInh-A01 were found to be the most potent and selective substances, with IC50 ≤ 10 μM52,56,57 and a block that was insensitive to membrane potential,52 a useful property for functional studies in cells and tissues. T16AInh-A01 relaxed precontracted arteries, in line with a role in vascular contraction, as outlined below. Of the activators discovered, Eact (activator) and Fact (potentiator) were shown to stimulate native and ANO1-induced CaCC activity (EC50 ≈ 3–6 μM) by respectively activating the channels in the absence of intracellular Ca2+ or enhancing their Ca2+ sensitivity.58 Tannins, which are particularly concentrated in green tea and red wine, were identified as good inhibitors of TMEM16A or ANO1,59 tannic acid being the most potent tannin, with an IC50 of ∼6 μM. Recently, these “second-generation” CaCC modulators were successfully used in physiological experiments examining the potential role of CaCCs on agonist-mediated tone, and they are discussed below.
Regulation of CaCCs in vascular myocytes
One important observation made at an early stage was that CaCCs in many, but not all, cell types display significant loss of activity or rundown after patch excision in the inside-out configuration or after achieving whole-cell recording mode. This led to the speculation that CaCCs may be subjected to tight regulation by various mechanisms, including phosphorylation. Wang and Kotlikoff60 were the first to demonstrate that CaCCs are inactivated by Ca2+/calmodulin-dependent protein kinase II (CaMKII)-mediated phosphorylation. These authors convincingly showed that in tracheal SMCs, caffeine-induced CaCC current declined more quickly than the underlying Ca2+ transient (measured with Fura-2/AM), an effect attributed to CaMKII-induced phosphorylation of CaCC or an associated regulatory subunit. A study from our group similarly indicated that CaCC activity is also downregulated by CaMKII-induced phosphorylation in rabbit PASMCs and coronary artery SMCs, through a process involving a decrease in voltage sensitivity.18 Subsequent reports showed that the effect of CaMKII on CaCCs is antagonized by the serine-threonine phosphatase calcineurin (CaN) in rabbit coronary61 and pulmonary27 artery myocytes. More recently, our group showed that in rabbit PASMCs, ICl(Ca) are also regulated by the Ca2+-independent protein phosphatases 1 (PP1) and 2A (PP2A).62 Ayon et al.62 proposed that CaN Aα and, most likely, PP1 act in a common upstream-downstream signaling pathway to regulate CaCCs. Clearly, there are many missing pieces to this puzzle. It is unknown whether CaMKII, CaN, and PP1/PP2A regulate phosphotransferase activity directly on the pore-forming subunit of the CaCC or on an accessory subunit. Indirect evidence suggests that CaMKII is likely not the only kinase involved. In rabbit PASMCs, ICl(Ca) were shown to run down ∼70%–80% within 5 minutes of cell dialysis with 3–5 mM adenosine triphosphate (ATP), and this rundown was obliterated by replacing internal ATP with the nonhydrolyzable ATP analog adenylyl imidodiphosphate (AMP-PNP), by omitting ATP28,62,63 or by dialyzing the cells with excess CaN Aα.27 In contrast, specific inhibition of CaMKII by KN-93 increased ICl(Ca) that had already run down to a steady-state level only by a factor of <2.18 Phosphorylation was suggested to inhibit ICl(Ca) by promoting state-dependent channel closure, by shifting the steady-state voltage dependence toward positive potentials.28 Although the physiological significance of the negative regulation by phosphorylation of CaCCs in PA myocytes remains unclear, we may speculate that because the anion channels offer a powerful depolarizing stimulus yielding sustained Ca2+ influx through L-type Ca2+ channels (CaL) and therefore reinforcement of CaCC activation, Ca2+-dependent downregulation by phosphorylation may provide a means to attenuate the impact of the positive-feedback loop between CaL and CaCCs. The phosphorylation-mediated CaCC inactivation was also shown to reduce the affinity of NFA to inhibit ICl(Ca) in PA myocytes, an observation consistent with the known open-state interaction of this compound with the channel,63 which may explain, at least in part, the discrepancy in IC50 values measured in different VSMCs by different groups.30
Very recently, our group reported that phosphatidylinositol 4,5-bisphosphate, or PIP2, inhibits native ICl(Ca) in rat PASMCs and TMEM16A-encoded ICl(Ca).64 Although the physiological relevance of this important observation remains to be determined, it highlights another potential mechanism of activation of the channel after stimulation of receptors coupled to phospholipase C (PLC)–mediated membrane phospholipid breakdown.
Molecular candidates proposed to encode for CaCCs in VSMCs
For several decades after the initial discovery of CACCs, research efforts to determine their physiological role in various tissues was hampered by the need to use pharmacological agents exhibiting poor potency and selectivity, by the existence of several types of CaCCs displaying distinct biophysical properties and modes of activation by intracellular Ca2+, and, more significantly, by the lack of a valid molecular candidate. Over the past 20 years, 5 structurally unrelated families of Cl− channel genes have been postulated to form the molecular basis of CaCCs: the CLCA family;65-69 the long human isoform variant of CLC-3, a member of the voltage-gated Cl− channel superfamily of Cl− genes that requires CaMKII for activation;6,70,71 the human gene related to the Drosophila flightless locus called tweety;72,73 the vitelliform macular dystrophy protein bestrophin;74,75 and the recently identified TMEM16 or anoctamin gene family.13-15,76 Among these genes, the last two families have recently emerged as better molecular candidates in many cell types.
Bestrophin was identified as the gene product of the vitelliform macular dystrophy type 2 gene (VMD2).75 Several mutations of this gene were associated with the vision disorder vitelliform macular dystrophy, or Best’s disease, an autosomal dominant abnormality characterized by an accumulation of lipofuscin-like material in the macular area of the eye. In 2002, Sun et al.74 were the first to report that the heterologous expression of the several highly conserved orthologs and paralogs of bestrophin genes in human (hBest1, hBest2), Caenorhabditis elegans (ceBest1), and Drosophila (dmBest1) produced ICl(Ca). Subsequent studies revealed the existence of 4 bestrophin genes in humans (hBest1–hBest4)77 and 3 in mouse (where Best4 is a pseudogene).78 Conserved orthologs have also been found in Xenopus.79 Bestrophins are ubiquitously expressed in excitable tissues and various epithelia.75 The membrane topology of bestrophins is currently unclear, as 474,80,81 or 679,82 putative hydrophobic transmembrane-spanning domains (TMDs) have been suggested on the basis of hydropathy plot analysis. Moreover, Tsunenari et al.83 performed a detailed structural analysis of hBest1 by site-mapping and cysteine-scanning experiments and alternatively proposed that the channel comprises 4 putative transmembrane domains (TMD1, TMD2, TMD4, and TMD6) and an additional hydrophobic loop (TMD5) partially reentering the membrane from the extracellular side. Finally, Milenkovic et al.81 suggested an alternative membrane topology for hBest1 whereby TMD1, TMD2, TMD5, and TMD6 would cross the membrane and TMD3 and TMD4 would be part of a long intracellular hydrophobic loop between TMD2 and TMD5. All proposed structural models predict that the N- and C-terminal ends lie in the cytoplasm.75
Although currents elicited by the expression of different bestrophin genes displayed variable rectification, with the exception of ceBest1 nearly all bestrophin products were shown to lack voltage- and time-dependent gating properties.74,75 Similar to native CaCCs in VSMCs, bestrophins are activated by physiological intracellular Ca2+ levels with an apparent dissociation constant Kd ≈ 200–400 nM, exhibit an anion permeability sequence of I− > Br− > Cl− > F−, and are inhibited by classical CaCC blockers such as DIDS, SITS, and NFA.74,75,79,84-86 A detailed mutational analysis of hBest1 identified a potential EF-hand Ca2+-sensing domain in the C-terminus of the protein that is analogous to those found on calmodulin (CaM).87 The single-channel conductance of endogenous and expressed bestrophins was reported to be similar to CaCCs in vascular myocytes19-21,23,24 laying in the range of 0.26 to ∼2 pS.88,89
In 2008, three groups of investigators independently showed that two members of the TMEM16 gene family, TMEM16A and TMEM16B, or anoctamin-1 and anoctamin-2 (ANO1, ANO2), encoded for CaCCs.8,13-15,76,90 ANO1 and ANO2 belong to a mammalian family of genes comprising 10 paralogs. Of these 10 members, only ANO1, ANO2, and ANO6 (TMEM16F) are Ca2+-activated ion channels. Although ANO1 and ANO2 are confirmed CaCCs, ANO6 was described as encoding for a CaCC91,92 or a nonselective Ca2+-activated cation channel permeable to Ca2+.93 ANO3, ANO4, ANO6, ANO7, and ANO10 have Ca2+-dependent phospholipid scramblase activity.94
Importantly, the biophysical properties and pharmacology of expressed ANO1 and ANO2 are very similar to those of CaCC currents recorded from native cells: (1) activation by physiological levels of intracellular Ca2+ (∼200–1,000 nM), (2) voltage and time dependence and outward rectification due to gating and an apparent Kd for Ca2+ that is voltage sensitive, (3) sensitivity to blocking by NFA, DIDS, and NPPB, (4) a relatively small single-channel conductance (8.3 pS, compared to ∼1–3 pS for native channels in VSMCs), and (5) an anion permeability sequence of SCN− > NO3− > I− > Br− > Cl− > F− > gluconate.13-15,53,54,59,95 ANO1 messenger RNA (mRNA) transcripts were shown to be ubiquitously expressed in cell types previously reported to exhibit CaCC activity, such as exocrine secretory epithelia (e.g., pancreatic acinar cells, salivary and parotid gland cells), several types of retinal cells, sensory neurons, liver, lung, placenta, trachea, vomeronasal organ, uterus, interstitial cells of Cajal in the gastrointestinal tract, sperm cells, heart, skeletal, and smooth muscles.13-15,76,90,96-102 ANO1 is robustly expressed at the mRNA and protein levels in VSMCs,53 and its knockdown by silencing RNA (siRNA) resulted in a reduced CaCC conductance in rat pulmonary54 and cerebral artery55 myocytes. Besides ANO1, high transcript levels were found for ANO6 and ANO10 in rat pulmonary arteries,54 but their role in this tissue remains undefined.
Anoctamins are predicted to exhibit 8 transmembrane domains with both N- and C-termini facing the cytoplasm (Fig. 4A).13,15,76,96 In this model, a portion of the large extracellular loop between TMD5 and TMD6 is proposed to reinsert in the membrane to form the pore, or P loop, of the channel. Mutations of several basic amino acid residues in this region altered the conductance and selectivity of the induced current15 (reviewed by Hartzell et al.76). However, this topology was recently challenged on the basis of site mapping by site-directed mutagenesis and accessibility of antibodies and sulfhydryl reagents on specific residues or domains of the protein.103 In this revised model (Fig. 4B), the stretch of residues thought to form an extracellular loop between TMD5 and TMD6 in the original model would instead be located in the cytoplasm, with the pore domain lying between TMD6 and TMD7. It also unveiled a new potential Ca2+ binding site, which is discussed further below. As for the quaternary structure of the protein, two recent studies provided convincing evidence for ANO1 forming a stable homodimer whose assembly is maintained by noncovalent interactions.108,109 Tien et al.110 confirmed this hypothesis and identified a stretch of 19 amino acid residues in the N-terminal domain located upstream of the first transmembrane domain that are critically involved in homodimerization of the protein and its functional expression. There is also evidence for the speculated pore loop region of ANO1 to play an important role in the trafficking of the channel to the plasma membrane and determining channel density.111
Figure 4.

Proposed secondary structures for anoctamin-1 (ANO1), or TMEM16A. A, Original model, based on predicted hydropathy profiles, describing the basic membrane topology of the protein, which comprises 8 transmembrane domains (TMDs), C- and N-terminal ends located in the cytoplasm, and a pore loop located between TMD5 and TMD6.13,15 B, Revised model proposing that the extracellular loop following TMD5 in the original model may instead face the cytoplasm.103 C, More detailed map of the revised membrane topology model of ANO1, revealing the locations and amino acid sequences of the 4 known spliced variants (a–d), the identification of 2 glutamate residues at positions 702 and 705 in mouse as potential Ca2+ binding sites,103 the location and hypothetical function of 3 classes of calmodulin (CaM)-binding domains (CaM-BD1 and CaM-BD2,104 CMB1 and CMB2,105 and regulatory CaM-binding motif [RCMB]106), and at least one N-glycosylation site (N-Glycos). The structure also exposes the location of 4 consecutive glutamate residues immediately proximal to spliced variant c, which were shown to be critical for Ca2+- and voltage-dependent gating of the channel.107
At least 4 exons of ANO1, but likely more,112 are alternatively spliced (identified as “a,” “b,” “c,” and “d”), producing different protein variants (Fig. 4C)13,76,98 that bear major significance for expression and channel function.13 Spliced variants b and d are alternatively spliced in pulmonary arterial smooth muscle, whereas spliced variant c is constitutively expressed.53,54,113 The a variant, which initiates the protein at the N-terminal end of the protein, is generally thought to be constitutively expressed; however, a minimal variant lacking this spliced exon was found in some tissues. It is also known that this particular exon is under the control of an alternative promoter. Ohshiro et al.114 recently confirmed that murine portal vein myocytes express heterodimers composed of distinct ANO1 spliced variants, further extending the level of functional complexity of this protein. A “minimal” isoform of ANO1 lacking all 4 spliced exons retained normal Ca2+ sensitivity but lacked voltage and time dependence.115 However, a recent study by the same group challenged that initial assertion by demonstrating that protein translation may be initiated at a noncanonical starting codon (non-ATG), which suggests that a significant fraction of the N-terminus is important for protein trafficking and insertion into the membrane as well as for channel function.116 A detailed analysis of spliced variants b (exon 6b in mouse; 22 amino acids) and c (exon 13 in mouse; 4 amino acids) suggested that the expression of the former reduces Ca2+ sensitivity by ∼4-fold at +80 mV, whereas omission of variant c (EAVK) attenuated the time dependence seen at positive potential, a process attributed to voltage-dependent gating.95 Another group interested in identifying the protein domains involved in the Ca2+ and voltage dependence of TMEM16A (see text below) revisited this question by analyzing the stretch of amino acids preceding and spanning variant c.107 While initial reports showed that spliced variant d (exon 15 in mouse; 26 amino acids) produced no detectable effect on the activity of expressed ANO1,13,95 a recent study showed that its inclusion slowed time-dependent activation and deactivation kinetics.117 Taken together, these observations highlight the profound influence of alternative splicing of ANO1 in different cell types and how the controlled expression of these variants may result in fine-tuning the activity of CaCCs and their role in cellular functions.
Heterologous expression studies of ANO1 have confirmed that channel gating is physiologically triggered by an elevation of Ca2+ on the cytoplasmic side, although the channel can also be opened by extreme positive potentials under conditions of strong Ca2+ buffering.106,107 Other divalent cations, such as Ba2+,107,118 Sr2+,118,119 and Ni2+,119 can also activate ANO1, albeit with a lower affinity (Ca2+ > Sr2+ ≈ Ni2+ > Ba2+); Mg2+, which competes with Ca2+, does not activate the channel,118 whereas Zn2+ blocks the channel.119 Whether ANO1 is directly gated by intracellular Ca2+ or has an obligatory requirement for an accessory Ca2+-binding protein, such as CaM or another unknown protein, is controversial. Neither a canonical EF-hand Ca2+-binding motif nor a CaM-binding domain (IQ) exists in ANO1. Analysis of the amino acid sequence revealed the existence of a stretch of 5 consecutive glutamate residues in the first intracellular loop between TMD1 and TMD2 (Fig. 4C).107 These residues were initially hypothesized to serve as a potential Ca2+-sensing domain, or “Ca2+ bowl,” by analogy to a series of aspartate residues identified in BKCa.120 The fifth glutamate residue is actually encoded by the first 3 base pairs of exon 13 in the mouse, a very short alternatively spliced exon (12 base pairs encoding for the c variant EAVK) that produces significant changes in the biophysical properties of the protein. Deletion of EAVK led to a profound decrease in the Ca2+ sensitivity of human ANO1, whereas mutating the 4 glutamate residues preceding EAVK to alanine residues produced anion currents lacking intrinsic voltage dependence. The same group also showed that voltage-dependent gating of ANO1 was allosterically facilitated by anions displaying a higher permeability than Cl− (SCN−, NO3−) or by high external Cl− concentration.107 Although this region is important in determining the Ca2+ and voltage dependence of ANO1, it is likely not the Ca2+ binding and trigger site for opening the channel. The most compelling evidence for the existence of a novel Ca2+-sensing domain came from a study by Yu et al.103 In addition to proposing a revised model of the secondary structure of ANO1, they showed that 2 glutamate residues at positions 702 and 705 of mouse ANO1 (Fig. 4C) were important for Ca2+ activation. Mutating both residues to glutamine led to a reduction in Ca2+ sensitivity of ∼2 orders of magnitude. In support of direct activation by Ca2+ is the recent demonstration that normal CaCC activity could be elicited by Ca2+ applied on the cytoplasmic side of purified ANO1 reconstituted in liposomes and that this process was lost by mutation of the same glutamate residues to glutamine, as discussed above,121 leading these investigators to suggest that ANO1 is necessary and sufficient to recapitulate the biophysical properties of CaCCs.
Tian et al.104 proposed an alternative and indirect mechanism of activation of TMEM16A by internal Ca2+. In this model, which is analogous to small-conductance Ca2+-activated K+ channels,120 CaM physically interacts with ANO1, and this interaction is indispensable for channel activation in the presence of Ca2+. One problem with this finding is that the first of 2 putative CaM-binding domains (CaM-BD1) identified by bioinformatics in the N-terminus of ANO1 and shown to bind CaM overlaps spliced variant b (Fig. 4C). Yet robust Ca2+- and voltage-sensitive ICl(Ca) can be elicited by expression of TMEM16A lacking spliced variant b (Fig. 1).53,95,105-107,122 Moreover, inclusion of spliced variant b reduced the Ca2+ sensitivity of ANO1.95 In contrast, CaM-BD2 did not bind CaM and was found to play no role in channel activity.104 A potential role for activation of ANO1 by a phosphorylation step involving CaMKII or other kinases was also discarded,104,122 although one study showed that ATP promoted channel activation by an unknown mechanism.104
More recently, Vocke et al.106 identified a different regulatory CaM-binding motif (RCMB) in the N-terminus of ANO1 and ANO2 (Fig. 4C) that is distinct from CaM-BD1 and CaM-BD2. They suggested that RCMB is involved in activation of ANO1 and ANO2 by Ca2+ and in Ca2+-dependent inactivation of ANO2. Finally, a separate report published in 2013 suggested that CaM does not activate ANO1 but does regulate it by increasing its HCO3− permeability relative to Cl−,105 a mechanism suggested to be important in fluid secretion in submandibular acinar gland cells. In the latter study, specific peptides corresponding to 2 regions, one located immediately proximal to the first TMD on the N-terminal segment (CBM1) and the other within the intracellular loop between TMD6 and TMD7 (CBM2), were able to reduce the association of CaM to ANO1 and attenuate its effect on HCO3− permeability (Fig. 4C). However, this hypothesis will require further investigation, as a preliminary report from another group recently showed that CaM did not alter anion permeability under well-controlled voltage-clamp conditions.123 The potential role of CaM in activation of ANO1 was revisited recently by Yu et al.,122 who presented an elegant series of experiments arguing against a prerequisite role of CaM for activation of ANO1 by Ca2+. Although this group found that CaM could bind to ANO1, the interaction was very weak. Among several convincing arguments presented supporting direct activation of ANO1 by Ca2+ was the fact that Ba2+, which is unable to bind to CaM and trigger a conformational change,124 was able to activate ANO1, a result consistent with previous findings.107,118 Taken together, the above studies indicate that we still have a poor understanding of the molecular mechanisms involved in the activation of ANO1 by Ca2+ and voltage and of how phosphorylation regulates its activity. Future studies providing structural information will help clarify the mechanisms involved in gating.
Physiological role of bestrophin and anoctamin CaCCs in PASMCs
Very little is known about the functional role of bestrophins in VSMCs. Best1, Best2, and Best3 (Best4 is a pseudogene in mouse but not in human75) are expressed in the A7r5 SMC line; rat aorta, mesenteric arteries, and PASMCs;125 and the rabbit PA.7 The ICl(Ca) recorded from PASMCs dialyzed with a solution designed to clamp internal Ca2+ at a fixed elevated level (e.g., 500 nM) exhibit both instantaneous and time-dependent components following voltage-clamp steps to positive potentials (Fig. 1). It is generally interpreted that the instantaneous jump in membrane current reflects the activity of channels that are readily available at the holding potential before a depolarizing step eliciting the instantaneous current and that the channels responsible for such behavior correspond to a single population of channels.7,17,18,28,61 However, it is possible that a portion of the instantaneous component might be attributed, at least in part, to one or more bestrophin gene products, since the latter are voltage and time independent. Similar to bestrophin-induced currents, the current-voltage relationship of the instantaneous component of ICl(Ca) in PA myocytes is almost linear and is less sensitive to block by the putative CaCC inhibitor NFA.18
Two independent groups identified a Ca2+-activated Cl− conductance activated by the second messenger cyclic guanosine monophosphate and Ca2+ (IcGMP.Ca) in rat mesenteric arterial myocytes.126-128 Subsequent studies showed that Best3 encodes for this conductance and appears to play a major role in the rhythmic vasomotion but not in the tonic contraction of mesenteric arteries induced by vasoconstricting agonists.125,129 Interestingly, IcGMP.Ca is time and voltage independent and is relatively insensitive to NFA but can be blocked by Zn2+ (10 μM) applied externally.125-127 The potential role of Best3 in the pulmonary arterial circulation is unclear, as the expression of the protein is low.125
Because of similarities with ICl(Ca) in vascular myocytes and other cell types expressing CaCCs, anoctamins rapidly became the more promising candidate gene family suspected to encode for the still elusive native channel. Of the 10 anoctamin paralogs, ANO1 and ANO2 are confirmed CaCCs, while the evidence that the other family members display Ca2+-sensitive Cl− channel activity is unclear.99,130 ANO3–ANO7 appear to be distributed in the cytoplasm, most likely in the endoplasmic reticulum (ER).130
Anoctamins are expressed at significant levels in several types of smooth muscle, including the intestinal tract,97,98,131,132 the urethra,101 the oviduct,133 airways,102,134,135 and vasculature.22,43,53-55,113,114,136-140 The role of CaCCs and anoctamins in determining pulmonary arterial tone is still to be evaluated. While ANO2 knockout mice develop and breed normally, exhibiting an apparently normal phenotype,141 ANO1 knockout mice die prematurely after birth because of malformation of their airways during embryonic development.142 This poses a serious challenge in attempting to determine the role of ANO1 in electromechanical coupling of blood vessels from immature animals, especially in resistance vessels, which will require the development of conditional and inducible genetically modified animal models. Very recently, Heinze et al.140 presented convincing data obtained with a novel conditional smooth muscle–specific and inducible ANO1 knockout mouse and suggested that the channel plays an important role in the regulation of peripheral resistance and systemic blood pressure in the angiotensin II–induced hypertensive model.
The few studies carried out thus far that have measured the level of expression of anoctamins in VSMCs, including PASMCs, have all reported much higher transcript levels of ANO1 than of ANO2.53-55 This is consistent with the fact that ICl(Ca) in vascular myocytes display a Ca2+ sensitivity that is similar to that of expressed ANO17,13-15,28 and is much higher than that reported for ANO2.143 Experiments with siRNA confirmed that ANO1 is the main pore-forming channel subunit composing the CaCC of rat pulmonary54 and cerebral55,144,145 artery myocytes and that it contributes significantly to agonist-induced tone in small rat mesenteric arteries.139 ANO1 was recently proposed to be a key component involved in the depolarization and contraction associated with the myogenic response of resistance cerebral arteries.144 In cerebral artery myocytes and HEK-293 (human embryonic kidney) cells overexpressing ANO1, exposure of the cell to hypo-osmotic medium to trigger the regulatory volume decrease response or stretching the cell membrane by the application of negative pressure activated a Cl− conductance that was inhibited by ANO1 antibodies or selective knockdown of ANO1 expression by siRNA.144 This idea stemmed from prior data showing that, under certain experimental conditions, ANO1 could be activated by cell swelling and act as a volume-regulated Cl− channel.146 The contribution of ANO1 to the myogenic tone of cerebral arteries may be unique to this blood vessel, as our group previously showed that NFA (100 μM) had no effect on the basal myogenic response of rabbit mesenteric small arteries, while it potently attenuated the vasoconstriction triggered by phenylephrine.42 The following sections discuss our current state of knowledge in regard to the contribution of various Ca2+ sources activating CaCCs in VSMCs and how these signaling interactions control the development of force and blood pressure.
Ca2+ sources triggering CaCCs in PASMCs
Activation of CaCCs by Ca2+ mobilization from inositol trisphosphate–sensitive stores
It was recognized very early that Ca2+ release from the sarcoplasmic reticulum (SR) represented an important source of Ca2+ stimulating CaCCs during agonist-induced vasoconstriction (original contributions are summarized in several reviews1,4,7). Vasoconstricting neurotransmitters and hormones binding to receptors coupled to Gq and other G-coupled receptors activate PLC, a membrane-bound enzyme that cleaves PIP2 into the second-messenger signaling molecules diacylglycerol (DAG) and inositol trisphosphate (InsP3; Fig. 5A). DAG stimulates different classes of protein kinase C isoforms, which phosphorylate a large number of targets that stimulate smooth muscle contraction and promote cell proliferation. InsP3 diffuses from the plasma membrane to the nearby SR, where it binds to one of three classes of InsP3 receptors identified in VSMCs (InsP3R1–R3), which are ligand-activated Ca2+ release channels. The binding of InsP3 to one of these receptors triggers the opening of the channel and passive Ca2+ diffusion caused by the large electrochemical gradient between the inside of the SR and the cytoplasm (∼5,000–10,000-fold). The large transient buildup of Ca2+ in the cytoplasm evoked by InsP3 is generally sufficient to elicit a contraction. The transient nature of the elevation in Ca2+ concentration is dictated by several factors, including the rate of biochemical synthesis and breakdown of InsP3, the contribution from Ca2+ extrusion and reuptake pathways, and the complex mechanisms controlling the gating of InsP3 receptors, which involve the dynamic interplay between InsP3, cytoplasmic and intraluminar Ca2+, and ATP.147-149
Figure 5.

Various sources of Ca2+ activating the Ca2+-activated Cl− channel (CaCC) ANO1 in vascular smooth muscle cells. A, The classical mode of activation of CaCCs by stimulation of a Gq-coupled receptor (GqPCR) with a constricting agonist (e.g., 5-hydroxytryptamine, angiotensin II, endothelin), leading to an elevation of intracellular Ca2+ concentration and contraction, is illustrated in this panel by the circled numbers: (1) binding of an agonist to a GqPCR; (2) breakdown of membrane-bound phosphatidyl-inositol by Gq-stimulated phospholipase C (PLC), leading to the production of the second messengers inositol trisphosphate (InsP3) and diacylglycerol (DAG); (3) while DAG stimulates protein kinase C (PKC), which targets many proteins involved in contraction and cell proliferation, InsP3 diffuses toward the sarcoplasmic reticulum (SR), where it binds to and activates a Ca2+-permeable receptor-operated channel (InsP3R), triggering Ca2+ release in the cytoplasm; (4) Ca2+ diffuses in the cytoplasm and activates contraction and ANO1; (5) opening of ANO1 leads to Cl− efflux and membrane depolarization due to an outwardly directed electrochemical gradient for Cl−, which stimulates voltage-gated L-type Ca2+ channels encoded by the gene CaV1.2; (6) Ca2+ entry through CaV1.2 would in turn promote actomyosin bridge cycling and further stimulate ANO1, establishing a positive-feedback loop sustaining membrane depolarization and Ca2+ entry; (7) there is evidence for stimulation of ANO1 in some vascular myocytes by Ca2+ entry through CaV1.2 triggering SR Ca2+ release from ryanodine receptors (RyR) in a process called Ca2+-induced Ca2+ release (CICR), which can provide an additional stimulus for ANO1 activation by CaV1.2; (8) spontaneous Ca2+ release by RyR in the SR, giving rise to “spontaneous transient inward currents,” or STICs, produced by transient openings of ANO1. B, Activation of ANO1 by voltage-independent Ca2+ entry pathways: (1) Ca2+ unbinding from the SR Ca2+ sensor protein STIM1 (stromal interacting molecule 1) by a decrease in the concentration of Ca2+ in the SR due to spontaneous Ca2+ leakage or mediated by an agonist leads to clustering of portions of the SR with the plasma membrane; (2) STIM1 then physically interacts with a complex composed of one or more members of the canonical transient receptor potential (TRPC) and/or Orai families of cation channels to trigger store-operated Ca2+ entry (SOCE); (3) Ca2+ entry via SOCE can then stimulate ANO1; (4) ANO1 can also be stimulated by Ca2+ influx through a receptor-operated channel stimulated by an agonist (ROCE); (5) although hypothetical, it is possible that ANO1 could be stimulated locally by Ca2+ influx through TRPV4 (vanilloid transient receptor potential) channels physically interacting with the anion channel.
Evidence from many laboratories has suggested that CaCCs may be an important and prime target in several types of VSMCs, since the inward current elicited by InsP3 signaling is mainly carried by Cl−, displays kinetics of activation that are consistent with those of CaCCs, and is sensitive to putative CaCC blockers such as NFA, A9C, NPPB, and DIDS.1,4,7 In PASMCs, constricting agonists binding to receptors that lead to activation of PLC, such as phenylephrine, an α1-adrenergic receptor agonist, and angiotensin II, have been shown to induce intracellular Ca2+ oscillations.150,151 Oscillatory membrane ICl(Ca) in the physiological range of membrane potentials were similarly triggered in PASMCs from several species by histamine,152 norepinephrine,152 ET-1,153 ATP,154 and angiotensin II.154 In some cases, the inward Cl− current cohabited with a transient outward Ca2+-activated K+ current (BKCa).152,153 Oscillatory Ca2+ transients and ICl(Ca) were abolished by caffeine or thapsigargin, which depleted Ca2+ stores and inhibited PLC. These transients were also abolished by an intracellular application of heparin, which inhibits InsP3 receptors. These studies confirmed the important role played by InsP3 signaling in activating CaCCs and driving membrane potential in PA myocytes; however, several questions remain unanswered. Is activation of CaCCs in PASMCs by InsP3-mediated Ca2+ release essential for eliciting the sustained depolarization triggered by agonists, or does it act only as a depolarizing “primer”? In addition to CaCCs, Ca2+ release from the SR may also influence directly or indirectly (e.g., store depletion, phosphorylation by Ca2+-dependent kinases and phosphatases) several types of plasma membrane ion channels (BKCa, voltage-dependent K+ channels, store- and receptor-operated channels, transient receptor potential [TRP] channels) that control membrane potential and Ca2+ entry into the cell. What is the relative contribution of these ion channels in the transient depolarization that is generally ascribed to InsP3 signaling? How does oscillatory Ca2+ signaling triggered by constricting agonists and the subsequent activation of plasma membrane channels, including CaCCs, interplay in determining force development in the intact pressurized pulmonary arterial tissue (e.g., intercellular coupling, synchronous vs. asynchronous events)?
Activation of CaCCs by ryanodine-sensitive Ca2+ stores
Benham and Bolton155 were the first to speculate about the existence of focal Ca2+-release events leading to the activation of “spontaneous transient outward currents,” or STOCs, in several types of VSMCs and visceral SMCs, which were manifest of the stimulation of clusters of BKCa. Nelson et al.156 later confirmed the existence of local Ca2+-release events, or “Ca2+ sparks,” produced by the opening of clusters of ryanodine receptors located in portions of the SR making close contact with the plasma membrane (∼20 nm). For a comprehensive review of the properties and regulation of Ca2+ sparks and their role in triggering BKCa and STOCs and regulating resting membrane potential (Em) and tone in intact arteries, the reader is invited to peruse excellent reviews on the topic.157-159
By analogy to STOCs, a similar type of electrical event called “spontaneous transient inward currents,” or STICs, was discovered a few years later in airway160-162 and vascular33,36,163-167 SMCs. Ion-replacement experiments confirmed that STICs originate from the coordinated opening of a group of Cl− channels triggered by a transient elevation of Ca2+ on the cytoplasmic side of the membrane that arises from ryanodine-sensitive Ca2+ stores (Fig. 5A). STOCs and STICs were shown to coexist in some SMCs, producing events called “STOICs.”168,169 STOICs were generally the product of an initial faster BKCa-mediated STOC followed by the slower CaCC-induced STIC. In a detailed analysis of the spatiotemporal relationship between Ca2+ sparks and STICs in mouse airway SMCs, Bao et al.170 suggested that the majority of CaCCs are arranged in clusters optimally located in the vicinity of groups of ryanodine receptors producing Ca2+ sparks to provide a functional unit tuning local Ca2+ signaling and cell excitability.
In the pulmonary circulation, STICs were first described in rabbit PASMCs.164 STICs in these cells were recorded simultaneously with STOCs, with a sequence of activation similar to that of STOICs described above, with the outward BKCa current generally preceding the activation of CaCCs. Consistent with the existence of STOCs and STICs was the later demonstration of the existence of ryanodine-dependent Ca2+ sparks in rat intralobar PA myocytes.171 Interestingly, STICs were more frequently observed than STOCs near the Em of PASMCs (approx. −50 mV).164 All 3 ryanodine receptor subtypes (RyR1–RyR3) are expressed in PASMCs,172-174 but they are differentially distributed within the cell, with RyR1 and RyR2 predominantly expressed near the plasma membrane and RyR1 and RyR3 exhibiting enhanced staining in the perinuclear region.172 Ca2+ sparks measured in these two regions displayed very distinct characteristics, possibly conferring unique profiles of Ca2+ regulation tuned for specific cellular functions. Thus, similar to those in airway SMCs,175 Ca2+ sparks in PASMCs appear to be generated primarily by the spontaneous opening of RyR1 and RyR2 Ca2+-release channels.172
Compared to Ca2+ sparks in cardiac myocytes, those recorded in PASMCs occurred less frequently and displayed lower amplitude but a similar time course and spatial spread. Interestingly, a low concentration of caffeine (0.5 mM) was shown to activate Ca2+ sparks and cause membrane depolarization in PASMCs, whereas the same treatment hyperpolarized SMCs from systemic arteries.171 This suggests that Ca2+ sparks preferentially activate STICs in adult PA myocytes, while they primarily target BKCa and STOCs in cells from systemic arteries. It is also consistent with the developmental downregulation of BKCa in SMCs from distal pulmonary arteries.176,177 Consistent with this profile, the Em of PASMCs178-180 and the basal tone of intact pulmonary arteries181 were unaffected by BKCa blockers such as tetraethylammonium chloride or charybdotoxin, again emphasizing that the role of BKCa in PASMCs, relative to that of CaCCs, is at best a minor one.
Do Ca2+ sparks and STICs influence pulmonary arterial membrane potential and tone under physiological conditions? It seems unlikely that STICs would depolarize Em under basal conditions, as both the probability of observing Ca2+ sparks and their frequency when detected (∼0.3 s−1) are low.171 However, their contribution could become significant during stimulation by certain agonists. ET-1, but not norepinephrine, enhanced Ca2+ spark frequency ∼3–4-fold,171 conceivably activating STICs and CaCCs sufficiently to depolarize Em and promote Ca2+ entry through voltage-gated Ca2+ channels. A subsequent study from the same group showed that the ET-1-mediated increase in Ca2+ spark frequency, amplitude, and duration involved the coordination of both InsP3 and ryanodine receptors.182 Crosstalk between ryanodine- and InsP3-sensitive Ca2+ stores in PA myocytes was confirmed by several groups.173,183 In summary, spontaneous and transient activation of CaCCs by Ca2+ sparks elicited by ryanodine-dependent and, in some cases, InsP3-dependent Ca2+ releasable pools may serve as an excitatory mechanism in PASMCs. The membrane depolarization induced by STICs, especially in the presence of agonists, may in turn enhance the open probability of voltage-gated Ca2+ channels, leading to increased Ca2+ entry and vasoconstriction (see next section).
Activation of CaCCs by Ca2+ entry through voltage-dependent Ca2+ channels
It was recognized early that Ca2+ entry through dihydropyridine-sensitive voltage-gated CaL represented an important source of Ca2+ triggering CaCCs in SMCs and other cell types (Fig. 5A). Evidence for a tight coupling between Ca2+ influx through CaL (encoded by CaV1.2) and CaCCs was reported in SMCs isolated from rat,44,45,184,185 rabbit,18,36,186 and mouse187 portal vein, rabbit esophagus,188 rabbit coronary artery,34 canine162 and guinea pig189 trachea, and rat41 and rabbit27 PA.
Figure 6 shows the results of typical whole-cell patch-clamp experiments, carried out in rat PASMCs by Yuan,41 illustrating the tight relationship between the two channels. In Figure 6A, step depolarizations from a holding potential of −70 mV to potentials ranging from −60 to +60 mV elicited a family of currents consisting, for most voltage steps, of a small early transient inward Ca2+ current (ICa(L)) followed by a large secondary inward or outward ICl(Ca), depending on membrane potential, that significantly overlapped the time course of inactivation of ICa(L). Because of the superimposed ICl(Ca) (in these experiments, the equilibrium potential for Cl− [Ecl] = 0 mV), ICa(L) reversed near +25 mV (Fig. 6B, left [Itr]), whereas the current measured at the end of the step reversed at a potential below 0 mV (Fig. 6B, middle [Itd]). Figure 6A also shows that upon return to the holding potential, a large, persisting inward tail current is apparent and reflects slow ICl(Ca) closure due to a mixture of voltage-dependent channel deactivation and Ca2+ reuptake and extrusion mechanisms. The classical slow ICl(Ca) tail current was virtually uncontaminated by ICa(L) because the latter deactivates very quickly after repolarization and has thus been used as a useful index of ICl(Ca) conductance. Analysis of the voltage dependence of the tail current shows that activation of ICl(Ca) occurred near the threshold for activation of ICa(L), peaked around +20 mV, and progressively declined with stronger membrane depolarizations, consistent with the reduced driving force for Ca2+ (Fig. 6B, right [Itail]). This and many other studies in SMCs confirmed that the inward tail current was carried by Cl− as it reversed near ECl and was not influenced by omitting permeable cations in the buffer. Whereas only the delayed current during the step and the inward tail current following repolarization were attenuated by the CaCC blockers NFA, DIDS, SITS, and A9C or by inclusion of a Ca2+ chelator such as EGTA or BAPTA in the pipette solution, both the early inward ICa(L) and delayed ICl(Ca) and the tail current were abolished by Ca2+ channel blockers such as nifedipine, nicardipine, Co2+, and Cd2+. Predictably, enhancing ICa(L) with the Ca2+ channel agonist Bay K 8644 enhanced ICa(L)-induced delayed ICl(Ca) and tail current. Finally, external Ca2+ replacement with Ba2+, which readily permeates CaL, abolished ICl(Ca), indicating that Ba2+ is unable to substitute for Ca2+ in activating CaCCs.
Figure 6.

Ca2+-activated Cl− currents triggered by Ca2+ entry through voltage-gated Ca2+ channels in rat pulmonary artery smooth muscle cells (PASMCs). A, Typical family of whole-cell currents (top traces) recorded from a single PASMC with the voltage-clamp protocol shown below the traces. The cell was bathed in a solution containing 1.8 mM Ca2+. B, Mean current-voltage relationships for currents measured as indicated in A and labeled as the early transient current (Itr) measured between 50 and 90 ms, the time-dependent current (Itd) measured at the end of the depolarizing step (between 750 and 790 ms), and the tail current (Itail) measured immediately after repolarization to the holding potential of −70 mV (between 900 and 940 ms). Each data point represents a mean ± SEM (n = 13). Reproduced from Yuan41 with permission from the American Physiological Society.
In some SMCs and other cell types exhibiting CaCC activity, activation of ICl(Ca) in response to CaL opening appears to involve Ca2+-induced Ca2+ release (CICR) and implicates mostly ryanodine (Fig. 5A), but in some cases InsP3 receptors, as perturbation of Ca2+ release or reuptake with agents such as caffeine, ryanodine, cyclopiazonic acid (CPA), 2-aminoethoxydiphenyl borate (2-APB), or tetracaine reduced the magnitude of ICa(L)-induced ICl(Ca).34,187,190 The possible contribution of CICR in stimulating ICl(Ca) in PASMCs is unknown.
Regardless of whether CaCCs are activated locally by Ca2+ sparks or by a rise in global [Ca2+]i, it is clear that ECl in VSMCs is physiologically set to optimally stimulate sustained Ca2+ influx through CaL. In smooth muscle, Cl− is actively accumulated inside the cell by the complex interplay between the HCO3-Cl− exchanger, the Na+-K+-2Cl− cotransporter, and an obscure transporter called “Pump III.”191 In rabbit PASMCs, ion flux experiments established an intracellular Cl− concentration of 51 mM, yielding a calculated equilibrium potential for Cl− of −26 mV,192 well above the Em measured in these cells41,192,193 and similar to that measured in other smooth muscles (for a review, see Wilson and Leblanc194). Thus, because ECl lies near the peak of the window Ca2+ current (approx. −20 mV), the depolarization evoked by activation of CaCCs favors sustained Ca2+ entry into the cell and contraction. This establishes a powerful positive-feedback loop, which is possibly counteracted by CaMKII-dependent inactivation of CaCC activity by phosphorylation. Support for the positive-feedback-loop hypothesis (Fig. 5A) came from indirect experiments examining the effects of Cl− channel blockers such as NFA, DIDS, and A9C on the vasoconstriction elicited by constricting agonists. Most physiological studies with arterial smooth muscles reported a significant attenuation of agonist-induced tone by CaCC blockers that was generally similar to or smaller than that produced by direct inhibition of CaL (e.g., nifedipine).1,4,7,38-42 The main issue that has hampered any definitive conclusion in regard to this proposed mechanism concerns the relatively low efficacy of the blockers to detect vasorelaxation and the fact that they have possible confounding effects on other processes affecting excitation-contraction coupling. Detailed reviews on the potentially detrimental side effects of CaCC blockers are available.1,4,7,30 For the majority of the studies supporting a role for CaCCs in electromechanical coupling, the key supporting argument was based on the observation that the blockers generally did not affect the contraction evoked by KCl-induced membrane depolarization. This implied that the vasorelaxation of precontracted arteries elicited by such inhibitors was not caused by a direct effect on the contractile machinery or by inhibition of Ca2+ entry through CaL. NFA (10–50 μM) caused relaxation of rat PAs precontracted with 5-hydroxytryptamine (5-HT) or phenylephrine and reversed the depolarization induced by 5-HT, but it produced no effect on intracellular Ca2+ release elicited by 5-HT.41 The recent discovery of TMEM16A permitted the development of high-throughput screening of small molecules targeting CaCCs with greater affinity and potency.57-59,195 One of these molecules, labeled T16AInh-A01, was shown to cause relaxation of several types of rat and mouse blood vessels precontracted with various agonists.22,43,113 Again, because ANO1 knockout mice die prematurely after birth, unequivocal testing of this hypothesis in pulmonary arteries, in particular in small-intrapulmonary-resistance vessels, will require the use of transgenic mice, whereby ANO1 can be downregulated or overexpressed in adult animals using inducible and conditional genetic strategies, as recently shown in a study examining the role of ANO1 in systemic hypertension.140
Activation of CaCCs by store- and receptor-operated Ca2+ entry
In many cell types, including VSMCs, depletion of intracellular Ca2+ stores leads to activation of a plasma membrane voltage-independent Ca2+ entry pathway, a process called “store-operated Ca2+ entry” (SOCE) or “capacitative Ca2+ entry.”196 SOCE appears to be prominent in the pulmonary circulation, having been detected in both PASMCs136,138,197-201 and venous SMCs.201 The major physiological functions of this mechanism are to replenish the stores after their emptying by agonist-mediated receptor signaling, leading to intracellular Ca2+ release, and to serve as a significant Ca2+ influx pathway during agonist-induced vasoconstriction.202-204 Experimentally, SOCE can be evoked by blocking Ca2+ reuptake into the SR with agents such as thapsigargin or CPA, both specific inhibitors of the SR Ca2+-ATPase (SERCA), in a cell exposed to a Ca2+-free buffer, which then leads to SR Ca2+ depletion. SOCE is unmasked by the reintroduction of external Ca2+ in the presence of the SERCA inhibitor and an L-type Ca2+ channel blocker such as nifedipine. Another method is to buffer intracellular Ca2+ with a chelating agent such as BAPTA or EGTA, which also causes SR Ca2+ depletion and activation of SOCE.
The molecular architecture of SOCE in smooth muscle is still unclear but has recently been the subject of more attention, with the discovery of two novel gene families directly involved in SOCE in T-lymphocytes, where “Ca2+ release–activated channels” (CRAC or CRACM channels) had been first characterized:205 (1) stromal interacting molecules 1 and 2 (STIM1 and STIM2)206-210 and (2) Orai1, Orai2, and Orai3.211-215 Strong evidence supports STIM1 and STIM2 as Ca2+-binding proteins acting as the sensors of Ca2+ depletion in the ER, which in turn interact with plasma membrane CRACs encoded by Orai genes. This process involves a substantial reorganization of the cytoskeleton, leading to translocation and clustering of STIMs, where it is believed to make close contact with the areas of the plasma membrane where SOCE is taking place. Significant progress has been made in the past few years, providing a conceptual framework defining the structure and protein domains involved in ER Ca2+ sensing, the interaction of STIMs with Orai, and the role of caveolae and molecular regulation of this process; the interested reader should consult excellent reviews available on these topics.216-221
In VSMCs, the situation is hazier, because even though store depletion clearly stimulates SOCE, the identity of the plasma membrane ion channels is unclear. Whereas the pore of CRAC and Orai channels is highly selective for Ca2+, the cation current activated by store depletion in VSMCs has been consistently shown to be a nonselective cation conduction pathway. Before the discovery of STIM and Orai proteins, investigators speculated that members of the TRP superfamily of ion channel genes, in particular those of the canonical subclass (TRPC), could be valid candidates, as many are nonselective channels displaying variable permeability to Ca2+.203,204,222,223 Studies have provided evidence for TRPC1, TRPC4, TRPC5, and/or TRPC6 as a part of the complex responsible for SOCE in VSMCs.203,222-226 More recent reports indicate that STIM1 and Orai1 are expressed in airway SMCs227 and VSMCs,200,227-231 where they were shown to play a significant role in SOCE, vascular tone, and cell proliferation. In addition to stimulating Orai1, STIM1 also interacts with TRPC1.232-234 Moreover, it has been speculated that after store depletion, STIM1, Orai1, and TRPC1 may form a ternary complex mediating voltage-independent SOCE (Fig. 5B).235-238
Although the molecular composition of the SOCE machinery and the nature of the divalent cation conduction pathway in PASMCs still are to be determined, it is clear that this Ca2+ entry pathway may represent an important additional trigger for activation of CaCCs in vascular myocytes. Yuan41 provided the first indirect evidence of coupling between SOCE and ICl(Ca) in rat PA myocytes. In the absence of external Ca2+, application of CPA to block the SERCA pump elicited transient ICl(Ca), consistent with Ca2+ first leaking from the SR stores and then being extruded, presumably through the plasma membrane Ca2+-ATPase and the Na+/Ca2+ exchanger. Readmission of external Ca2+ in the presence of CPA stimulated a transient ICl(Ca), although it is unclear whether a store-operated nonselective cation current was superimposed on the Cl− current. More recently, our group specifically addressed this question in rabbit PASMCs. In the presence of nifedipine to inhibit CaL, blocking the SERCA pump with thapsigargin136 or CPA136,138 activated a nonselective cation conductance displaying slight inward rectification, a response typical of that previously described in VSMCs by other investigators. A double-pulse protocol was used to analyze the relationship between SOCE and ICl(Ca). The traces in Figure 7Ab (Fig. 7 was reproduced from Angermann et al.138 with permission) show that in the presence of nifedipine, CPA elicited a membrane current from a holding potential of 0 mV that was mostly time independent during an initial step (1 s) ranging from −100 to +60 mV. A second pulse to +90 mV revealed the appearance of a slow, time-dependent outward current of which the magnitude grew with the level of hyperpolarization achieved during the first step. A slow inward tail current relaxing toward baseline was apparent upon return to −80 mV. The time-dependent currents at both +90 and −80 mV exhibited properties consistent with ICl(Ca) in PASMCs. Application of the SOCE blocker SKF-96365 abolished both the time-independent and the time-dependent currents evoked by CPA (Fig. 7Ac), whose magnitudes were similar to those recorded in the presence of nifedipine alone (Fig. 7Aa). Figure 7B, from the same study, showed that the voltage dependence of the current recorded at +90 mV with CPA and nifedipine displayed an inverse relationship to the voltage during the initial step, which was flattened by SKF-96365. Other series of experiments confirmed that the time-independent conductance is a nonselective cation current mainly permeable to Na+ and is blocked by SKF-96365 but is insensitive to NFA, whereas the time-dependent current components were abolished by the CaCC blocker. On the basis of these results, we hypothesized that CPA activated a store-operated nonselective cation conductance permeable to Ca2+ or indirectly permitting Ca2+ entry via reverse-mode Na+-Ca2+ exchange (NCX). Stronger hyperpolarizing steps would enhance Ca2+ entry through this pathway by increasing the driving force for Ca2+ and resulting in greater activation of ICl(Ca) during the subsequent step to +90 mV. Figure 8 (also from Angermann et al.138) shows the results of dual patch-clamp and Ca2+ fluorescence (Fluo-4) experiments confirming that in the presence of CPA and nifedipine, stronger and longer (3 s instead of 1 s) hyperpolarizing steps evoked larger, slowly developing Ca2+ transients that induced slow inward ICl(Ca) at very negative positive potentials and robust outward ICl(Ca) during the subsequent step to +90 mV. We concluded that in these recording conditions, reverse-mode NCX was not required for the SOCE-CaCC interaction, since (1) stepping to a positive potential (+90 mV) did not elicit a Ca2+ transient, as would be expected if sufficient Na+ had entered the cell via the store-operated nonselective cation current (ISOC; Fig. 8A, 8B), and (2) the activation of ICl(Ca) stimulated by CPA was unaffected by the NCX inhibitor KB-R7943 (Fig. 8D). These experiments highlighted the tight relationship between SOCE and CaCC activity in PASMCs (Fig. 8C). Although an indirect mode of activation of CaCCs by reverse-mode NCX was not demonstrated, our experiments did not rule out a potential role of the exchanger in this interaction under more physiological conditions, as the cells were dialyzed with a Na-free pipette solution.
Figure 7.

Store depletion activates Ca2+-activated Cl− currents in rabbit pulmonary artery smooth muscle cells. A, Three families of membrane currents recorded from the same smooth muscle cell and elicited by the voltage-clamp protocol displayed below the traces. The traces in a were recorded in the presence of 1 μM nifedipine (Nif). In the continued presence of Nif, the cell was then superfused with 30 μM cyclopiazonic acid (CPA) for 5 minutes (b) to deplete the stores. Subpanel c shows a family of traces recorded after application with the nonselective cation channel inhibitor SKF-96365 (SKF; 50 μM), with CPA and Nif still present. B, Mean current-voltage relationships for current density (pA/pF), measured at the end of the second step to +90 mV, as a function of the voltage during the preceding step. Each data point represents the mean ± SEM current recorded in the presence of 1 μM Nif alone (open circles; n = 5), with 30 μM CPA and Nif (filled circles; n = 12), or with Nif, CPA, and 50 μM SKF (filled squares; n = 4). There were no significant differences between the Nif-alone and Nif + CPA + SKF groups. All data points in the Nif + CPA group were significantly different from those of the other two groups (with a maximum P value of <0.05). The 3 lines passing through the data points represent linear regression fits. Reproduced from Angermann et al.138 with permission from the National Research Council of Canada.
Figure 8.

Voltage dependence of Ca2+ transients and Ca2+-activated Cl− currents evoked by store-operated Ca2+ entry (SOCE) in rabbit pulmonary artery smooth muscle cells. A, Family of membrane currents and intracellular Ca2+ transients measured simultaneously with the Ca2+ indicator Fluo-4 (expressed as F/F0) elicited by the protocol shown below the traces. The traces were recorded after 5 minutes of incubation with 1 μM nifedipine (Nif) and 30 μM cyclopiazonic acid (CPA) in the presence of 1.8 mM Ca2+ to induce SOCE. B, Mean current-voltage relationships for the instantaneous (filled circles) and late (open circles) membrane currents recorded in the presence of 1.8 mM Ca2+ with Nif and CPA for experiments similar to the one described in A. Each data point represents the mean ± SEM (n = 3). C, Dependence of normalized peak current at +90 mV during P2 (see protocol in A) on the magnitude of the Fluo-4 signal measured at the end of P1 (see protocol in A). Each data point represents the mean ± SEM of normalized peak current during P2 and Fluo-4 fluorescence at the end of P1 (n = 3). The solid line represents a second-degree polynomial fit. D, Peak current at +90 mV during P2 (see protocol in A) as a function of voltage during P1. Currents were recorded in the presence of Nif and CPA (filled circles) before the addition of 1 μM KB-R7943 (open circles), a Na+/Ca2+ exchange blocker. Each data point represents the mean ± SEM (n = 9). The two solid lines represent least squares exponential fits. Reproduced from Angermann et al.138 with permission from the National Research Council of Canada.
Further evidence in support of this interaction was also provided by Yamamura et al.,239 who showed that CaCC blockers such as NFA, flufenamic acid, and DIDS inhibited the transient and sustained phases of Ca2+ transients induced by angiotensin II and by store depletion in human PASMCs, which suggests that CaCCs and TMEM16A can both be activated by SOCE and receptor-operated Ca2+ entry (ROCE; Fig. 5B). Similarly to SOCE, TRPC channels are also thought to be the pore-forming subunits of ROCE in VSMCs.240-242
One conceptual problem in associating SOCE/ROCE with CaCCs is that ECl is more positive than Em. After stimulation with 5-HT in rat PA, inhibition of CaCCs by NFA caused membrane hyperpolarization,41 presumably by shifting Em toward EK (the equilibrium potential for K). Since Ca2+ influx during SOCE or ROCE would be expected to be increased by hyperpolarization due to an increased driving force for Ca2+, the hyperpolarization observed after the blocking of CaCCs by NFA should then enhance, rather than decrease, Ca2+ influx and intracellular Ca2+ levels. Consistent with the effects of NFA on SOCE and ROCE in human PA myocytes,239 we also observed a significant relaxation by the TMEM16A inhibitor T16AInh-A01 of small intralobar rat pulmonary arteries exposed to CPA and nifedipine. This then leads to the following questions: Is ECl stable and lying where expected (approx. −25 mV), or is it somehow changing because of the experimental conditions? What is the level of Em when CPA or thapsigargin is used to elicit SOCE, as opposed to a natural vasoconstrictor? Is the SOCE molecular complex physically and functionally linked to TMEM16A or to one of its regulatory subunits? More comprehensive research is required to answer these questions; intracellular membrane potential–recording experiments under very well controlled experimental conditions should help in elucidating this puzzle.
Potential role of CaCCs and ANO1 in pulmonary hypertension
Pulmonary hypertension (PH) is a rare chronic human disease exhibiting high morbidity and mortality rates. An elevation in pulmonary vascular resistance (PVR) is the direct consequence of PH, which enhances right ventricular (RV) afterload and in time leads to RV failure.243-247 The World Health Organization has recently reclassified the various forms of the disease into 5 categories: (1) pulmonary arterial hypertension (PAH), a progressive and severe syndrome displaying a low prognosis for survival, (2) PH associated with left heart disease, such as congenital heart disease, (3) PH associated with lung disease and/or hypoxia (e.g., chronic obstructive pulmonary disease), (4) thromboembolic PH, and (5) miscellaneous.244-247 PAH is defined by a pulmonary artery pressure (PAP) exceeding 25 mmHg at rest and 30 mmHg during exercise, a capillary wedge pressure ≤ 15 mmHg, and PVR ≥ 2–3 Wood units.244-247 Three major factors contribute to elevating PAP in PAH patients: (1) enhanced vasoconstriction, (2) partial or complete luminal obstruction due to extensive and complex remodeling of the intimal, medial, and adventitial layers of proximal and distal pulmonary arteries, characterized by enhanced proliferation, reduced apoptosis, and infiltration of inflammatory and progenitor cells, and (3) a propensity for in situ thrombosis.243-247
There is convincing evidence that PASMCs from animal models of PAH and humans with PAH are more depolarized and display higher [Ca2+]i than cells from healthy subjects and that this, at least initially, contributes to the observed changes in responsiveness to endogenous vasoconstrictors and remodeling.247-249 Consistent with this observation was the demonstration of a decreased voltage-dependent K+ current (IK) accompanied by a reduced expression of several members of the Kv superfamily of voltage-dependent K+ channels, including Kv1.5, Kv2.1, Kv4.3, and Kv9.3.250-259 It is thought that the depolarization caused by the reduced expression of K+ channel genes increases intracellular Ca2+ levels by activating CaL, which then triggers gene transcription, leading to reduced apoptosis and cell proliferation.248,256,260-263 One recent study revealed an upregulation of CaL activity and protein levels of the α1C pore-forming subunit of the channel in the chronic hypoxic neonatal piglet model.264 There is also evidence supporting the idea that other Ca2+ entry pathways might be altered in PAH.248,249,258,265 Several groups reported augmented ROCE and SOCE and in some cases increased expression of several members of the TRPC gene family, namely, TRPC1, TRPC3, TRPC4, and/or TRPC6, in pulmonary arteries from animal models of PH and in patients diagnosed with idiopathic PAH.198,225,265-269 Such changes may be important, because TRPC channels have been linked to PA cell proliferation.225,270 Recent studies also reported increased expression of one member of the “vallinoid” subfamily of TRP channels, TRPV4, in PASMCs from chronic hypoxic rats and suggested that the osmo-mechanosensitive channel could participate in the increase in myogenic and agonist-mediated tone of small pulmonary arteries in PH.271-273 Interestingly, TRPV4 and ANO1 were recently shown to physically interact and regulate water efflux through aquaporin channels in choroid plexus epithelial cells.274 It is thus tantalizing to speculate that a similar interaction could exist in PASMCs, whereby Ca2+ entry through TRPV4 could activate ANO1 and regulate tone in health and disease (Fig. 5B). Finally, the CRAC channel Orai2 and the SOCE Ca2+ sensor STIM2 (but not STIM1) were shown to be upregulated and were suggested to account for the enhancement of SOCE in PASMCs from patients with idiopathic PAH and chronic hypoxic rats.268
Very recently, two independent studies provided evidence for increased CaCC activity and expression of ANO1 in PA from two models of PH in the rat, monocrotaline-induced PH113 and PH caused by chronic hypoxia.43 In both animal models, CaCC current density in PASMCs from pulmonary hypertensive rats was elevated by ∼2-fold relative to that in control animals, and this observation was correlated by similar increases in ANO1 expression at the mRNA and protein levels, which is similar to that observed in a chronic mouse model of asthma135 and some forms of cancer.76,275-279 Consistent with these observations, the CaCC blockers NFA and T16AInh-A01 exerted more potent vasorelaxation of pulmonary arteries precontracted with 5-HT in pulmonary hypertensive than in control animals. Taken together, these studies suggest that CaCCs and ANO1 might be another important ionic conductance influencing pulmonary arterial resistance and blood flow in PH. This is further reinforced by the fact that crosstalk exists between CaCCs and SOCE/ROCE, and the proteins involved in these processes are also augmented in conditions favoring SMC proliferation and remodeling of the pulmonary arterial wall in PH. Indeed, ANO1 and ANO6 have been respectively shown to influence the direction and the speed of migration in Ehrlich-Lettre ascites cells, a cell model commonly used in cancer studies.280 Specific knockdown of ANO1 by siRNA transfection of rat mesenteric arteries was recently demonstrated to abrogate both “classical” ICl(Ca) and ICl.cGMP and to inhibit spontaneous cyclical vasomotion as well as agonist- and KCl-induced contractions of small arteries.139 Surprisingly, downregulation of ANO1 also reduced the expression at the mRNA level of bestrophins, previously shown to be involved in the generation of ICl.cGMP and the elicitation of rhythmic vasoconstrictions in this vascular bed,125,129 and the expression of CaV1.2 mRNA. The above study suggested that ANO1 plays an important function in regulating peripheral systemic vascular resistance. An earlier report by Wang et al.145 showed that in the 2-kidney, 2-clip systemic hypertensive rat model, ANO1 was downregulated and that this effect was responsible for the increased proliferation of basilar arterial SMCs. In this model, ANO1 was proposed to negatively regulate cell proliferation by altering the expression of cell cycle proteins and to essentially exert an antihypertensive effect. However, a more recent report showed that knockdown of ANO1 in a conditional smooth muscle–specific and inducible mouse model lowered systemic blood pressure in both untreated animals and mice rendered hypertensive by a chronic infusion of angiotensin II but had no effect on blood pressure of mice made hypertensive by a high-salt diet.140 Whether CaCCs and ANO1 play a similar role in the remodeling of the pulmonary arterial wall is unknown and will require investigation using a similar model.
Summary and concluding remarks
Progress made in the past 2 decades has shed new light on the functional and molecular properties of CaCCs in VSMCs. It is now known that CaCCs in PASMCs are small-conductance Ca2+-dependent anion channels modulated by voltage that are subjected to tight regulation by phosphorylation involving CaMKII, CaN, PP1, and PIP2 and that the pore-forming subunit is most likely encoded by ANO1. In pulmonary arterial smooth muscle, CaCCs can be activated by (1) mobilization of intracellular Ca2+ stores involving both InsP3- and ryanodine-sensitive Ca2+ stores triggered spontaneously by localized Ca2+ sparks or by G-protein coupled receptor stimulation and (2) Ca2+ entry via voltage-gated Ca2+ channels, SOCE, or ROCE.
There remain many intriguing and pressing questions about the properties of CaCCs and their interaction with other signaling molecules. Are CaCCs encoded only by ANO1, or is there a role for other anoctamins, perhaps ANO2 and/or ANO6, or bestrophins in forming the native channel in PASMCs? How are native CaCCs regulated by phosphorylation at the single-channel level, and is ANO1 similarly and directly regulated, or is one of its unidentified accessory subunits targeted by kinase activity? How does the regulation of CaCCs by phosphorylation influence membrane potential, intracellular Ca2+ dynamics, and arterial tone? Under physiological conditions, activation of CaCCs is hypothesized to increase SMC excitability by promoting membrane depolarization, activation of voltage-dependent Ca2+ channels, increased Ca2+ influx, and vasomotor tone. Support for this hypothesis is based on the ability of pharmacological agents to reverse the depolarization and contraction induced by vasoconstrictors. Unequivocal testing of this hypothesis will require a demonstration that the depolarization and contraction to vasoconstrictors are impaired in intact arteries from ANO1 conditional-knockout mice or following ANO1 knockdown by RNA silencing in vitro, as was recently done in cerebral144 and mesenteric139 arteries. Moreover, is ANO1 (and its spliced variants) differentially expressed in the pulmonary arterial tree, as recently shown in systemic arteries?140 What is the functional impact of the interaction of CaCCs with SOCE and ROCE under physiological conditions, as well as in PH, where these signaling pathways are upregulated? Is there a role for CaCCs and anoctamins in hypoxic pulmonary vasoconstriction? Is the increased expression of the CaCC/ANO1 channel seen in animal models of PH also present in human PH? In addition to promoting vasoconstriction, does enhanced expression of the channel influence remodeling of the pulmonary arterial wall in PH, and if so, by what mechanism? How is ANO1 expression genetically regulated in health and pulmonary arterial diseases? There is no doubt that the discovery of anoctamins has resurrected an interest in this field and identified a new therapeutic target in the battle against the deleterious consequences of PAH, which will lead to exciting research in the near future.
Source of Support: This study was supported by grants to NL from the National Institutes of Health (NIH; R01 HL075477 and R01 HL091238), an NIH grant to FB (R01 HL091238), and a grant to IAG from the British Heart Foundation (PG/05/038). The publication was also made possible by a grant to NL (NCRR 5 P20 RR15581) from the National Center for Research Resources (NCRR), a defunct branch of the NIH that supported a Center of Biomedical Research Excellence at the University of Nevada School of Medicine, Reno (2000–2010). The contents of the article are solely the responsibility of the authors and do not necessarily represent the official views of the NCRR or the NIH. RJA was supported by an NIH Ruth L. Kirschstein National Research Service Predoctoral Fellowship (F31 HL090023).
Conflict of Interest: None declared.
References
- 1.Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. Am J Physiol Cell Physiol 1996;271(2):C435–C454. [DOI] [PubMed]
- 2.Frings S, Reuter D, Kleene SJ. Neuronal Ca2+-activated Cl− channels—homing in on an elusive channel species. Prog Neurobiol 2000;60(3):247–289. [DOI] [PubMed]
- 3.Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiol Rev 2000;80(1):31–81. [DOI] [PubMed]
- 4.Kitamura K, Yamazaki J. Chloride channels and their functional roles in smooth muscle tone in the vasculature. Jpn J Pharmacol 2001;85(4):351–357. [DOI] [PubMed]
- 5.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev 2002;82(2):503–568. [DOI] [PubMed]
- 6.Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol 2005;67:719–758. [DOI] [PubMed]
- 7.Leblanc N, Ledoux J, Saleh S, Sanguinetti A, Angermann J, O’Driscoll K, Britton F, Perrino BA, Greenwood IA. Regulation of calcium-activated chloride channels in smooth muscle cells: a complex picture is emerging. Can J Physiol Pharmacol 2005;83(7):541–556. [DOI] [PubMed]
- 8.Berg J, Yang H, Jan LY. Ca2+-activated Cl− channels at a glance. J Cell Sci 2012;125(6):1367–1371. [DOI] [PMC free article] [PubMed]
- 9.Huang F, Wong X, Jan LY. International Union of Basic and Clinical Pharmacology. LXXXV: Calcium-activated chloride channels. Pharmacol Rev 2012;64(1):1–15. [DOI] [PMC free article] [PubMed]
- 10.Kunzelmann K, Schreiber R, Kmit A, Jantarajit W, Martins JR, Faria D, Kongsuphol P, Ousingsawat J, Tian Y. Expression and function of epithelial anoctamins. Exp Physiol 2012;97(2):184–192. [DOI] [PubMed]
- 11.Tian Y, Schreiber R, Kunzelmann K. Anoctamins are a family of Ca2+-activated Cl− channels. J Cell Sci 2012;125(21):4991–4998. [DOI] [PubMed]
- 12.Byrne NG, Large WA. Membrane mechanism associated with muscarinic receptor activation in single cells freshly dispersed from the rat anococcygeus muscle. Br J Pharmacol 1987;92(2):371–379. [DOI] [PMC free article] [PubMed]
- 13.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008;322(5901):590–594. [DOI] [PubMed]
- 14.Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008;134(6):1019–1029. [DOI] [PMC free article] [PubMed]
- 15.Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008;455(7217):1210–1215. [DOI] [PubMed]
- 16.Arreola J, Melvin JE, Begenisich T. Activation of calcium-dependent chloride channels in rat parotid acinar cells. J Gen Physiol 1996;108(1):35–47. [DOI] [PMC free article] [PubMed]
- 17.Kuruma A, Hartzell HC. Bimodal control of a Ca2+-activated Cl− channel by different Ca2+ signals. J Gen Physiol 2000;115(1):59–80. [DOI] [PMC free article] [PubMed]
- 18.Greenwood IA, Ledoux J, Leblanc N. Differential regulation of Ca2+-activated Cl− currents in rabbit arterial and portal vein smooth muscle cells by Ca2+-calmodulin-dependent kinase. J Physiol 2001;534(2):395–408. [DOI] [PMC free article] [PubMed]
- 19.Klöckner U. Intracellular calcium ions activate a low-conductance chloride channel in smooth-muscle cells isolated from human mesenteric artery. Pfluegers Arch 1993;424(3):231–237. [DOI] [PubMed]
- 20.Van Renterghem C, Lazdunski M. Endothelin and vasopressin activate low conductance chloride channels in aortic smooth muscle cells. Pfluegers Arch 1993;425(1–2):156–163. [DOI] [PubMed]
- 21.Hirakawa Y, Gericke M, Cohen RA, Bolotina VM. Ca2+-dependent Cl− channels in mouse and rabbit aortic smooth muscle cells: regulation by intracellular Ca2+ and NO. Am J Physiol Heart Circ Physiol 1999;277(5):H1732–H1744. [DOI] [PubMed]
- 22.Davis AJ, Shi J, Pritchard HA, Chadha PS, Leblanc N, Vasilikostas G, Yao Z, Verkman AS, Albert AP, Greenwood IA. Potent vasorelaxant activity of the TMEM16A inhibitor T16Ainh-A01. Br J Pharmacol 2013;168(3):773–784. [DOI] [PMC free article] [PubMed]
- 23.Piper AS, Large WA. Multiple conductance states of single Ca2+-activated Cl− channels in rabbit pulmonary artery smooth muscle cells. J Physiol 2003;547(1):181–196. [DOI] [PMC free article] [PubMed]
- 24.Saleh SN, Angermann JE, Sones WR, Leblanc N, Greenwood IA. Stimulation of Ca2+-gated Cl− currents by the calcium-dependent K+ channel modulators NS1619 [1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-2H-benzimidazol-2-one] and isopimaric acid. J Pharmacol Exp Ther 2007;321(3):1075–1084. [DOI] [PubMed]
- 25.Amédée T, Large WA, Wang Q. Characteristics of chloride currents activated by noradrenaline in rabbit ear artery cells. J Physiol 1990;428(1):501–516. [DOI] [PMC free article] [PubMed]
- 26.Greenwood IA, Large WA. Modulation of the decay of Ca2+-activated Cl− currents in rabbit portal vein smooth muscle cells by external anions. J Physiol 1999;516(2):365–376. [DOI] [PMC free article] [PubMed]
- 27.Greenwood IA, Ledoux J, Sanguinetti A, Perrino BA, Leblanc N. Calcineurin Aα but not Aβ augments ICl(Ca) in rabbit pulmonary artery smooth muscle cells. J Biol Chem 2004;279(37):38830–38837. [DOI] [PubMed]
- 28.Angermann JE, Sanguinetti AR, Kenyon JL, Leblanc N, Greenwood IA. Mechanism of the inhibition of Ca2+-activated Cl− currents by phosphorylation in pulmonary arterial smooth muscle cells. J Gen Physiol 2006;128(1):73–87. [DOI] [PMC free article] [PubMed]
- 29.Piper AS, Greenwood IA. Anomalous effect of anthracene-9-carboxylic acid on calcium-activated chloride currents in rabbit pulmonary artery smooth muscle cells. Br J Pharmacol 2003;138(1):31–38. [DOI] [PMC free article] [PubMed]
- 30.Ledoux J, Greenwood IA, Leblanc N. Dynamics of Ca2+-dependent Cl− channel modulation by niflumic acid in rabbit coronary arterial myocytes. Mol Pharmacol 2005;67(1):163–173. [DOI] [PubMed]
- 31.Greenwood IA, Leblanc N. Overlapping pharmacology of Ca2+-activated Cl− and K+ channels. Trends Pharmacol Sci 2007;28(1):1–5. [DOI] [PubMed]
- 32.Greenwood IA, Large WA. Properties of a Cl− current activated by cell swelling in rabbit portal vein vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 1998;275(5):H1524–H1532. [DOI] [PubMed]
- 33.Hogg RC, Wang Q, Large WA. Action of niflumic acid on evoked and spontaneous calcium-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. Br J Pharmacol 1994;112(3):977–984. [DOI] [PMC free article] [PubMed]
- 34.Lamb FS, Volk KA, Shibata EF. Calcium-activated chloride current in rabbit coronary artery myocytes. Circ Res 1994;75(4):742–750. [DOI] [PubMed]
- 35.Greenwood IA, Large WA. Comparison of the effects of fenamates on Ca-activated chloride and potassium currents in rabbit portal vein smooth muscle cells. Br J Pharmacol 1995;116(7):2939–2948. [DOI] [PMC free article] [PubMed]
- 36.Greenwood IA, Large WA. Analysis of the time course of calcium-activated chloride “tail” currents in rabbit portal vein smooth muscle cells. Pfluegers Arch 1996;432(6):970–979. [DOI] [PubMed]
- 37.Piper AS, Greenwood IA, Large WA. Dual effect of blocking agents on Ca2+-activated Cl− currents in rabbit pulmonary artery smooth muscle cells. J Physiol 2002;539(1):119–131. [DOI] [PMC free article] [PubMed]
- 38.Lamb FS, Barna TJ. Chloride ion currents contribute functionally to norepinephrine-induced vascular contraction. Am J Physiol Heart Circ Physiol 1998;275(1):H151–H160. [DOI] [PubMed]
- 39.Criddle DN, de Moura RS, Greenwood IA, Large WA. Effect of niflumic acid on noradrenaline-induced contractions of the rat aorta. Br J Pharmacol 1996;118(4):1065–1071. [DOI] [PMC free article] [PubMed]
- 40.Criddle DN, de Moura RS, Greenwood IA, Large WA. Inhibitory action of niflumic acid on noradrenaline- and 5-hydroxytryptamine-induced pressor responses in the isolated mesenteric vascular bed of the rat. Br J Pharmacol 1997;120(5):813–818. [DOI] [PMC free article] [PubMed]
- 41.Yuan XJ. Role of calcium-activated chloride current in regulating pulmonary vasomotor tone. Am J Physiol Lung Cell Mol Physiol 1997;272(5):L959–L968. [DOI] [PubMed]
- 42.Remillard CV, Lupien MA, Crépeau V, Leblanc N. Role of Ca2+- and swelling-activated Cl− channels in α1-adrenoceptor-mediated tone in pressurized rabbit mesenteric arterioles. Cardiovasc Res 2000;46(3):557–568. [DOI] [PubMed]
- 43.Sun H, Xia Y, Paudel O, Yang XR, Sham JS. Chronic hypoxia-induced upregulation of Ca2+-activated Cl− channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol 2012;590(15):3507–3521. [DOI] [PMC free article] [PubMed]
- 44.Pacaud P, Loirand G, Lavie JL, Mironneau C, Mironneau J. Calcium-activated chloride current in rat vascular smooth muscle cells in short-term primary culture. Pfluegers Arch 1989;413(6):629–636. [DOI] [PubMed]
- 45.Baron A, Pacaud P, Loirand G, Mironneau C, Mironneau J. Pharmacological block of Ca2+-activated Cl− current in rat vascular smooth muscle cells in short-term primary culture. Pfluegers Arch 1991;419(6):553–558. [DOI] [PubMed]
- 46.Busch AE, Herzer T, Wagner CA, Schmidt F, Raber G, Waldegger S, Lang F. Positive regulation by chloride channel blockers of IsK channels expressed in Xenopus oocytes. Mol Pharmacol 1994;46(4):750–753. [PubMed]
- 47.Farrugia G, Rae JL, Szurszewski JH. Characterization of an outward potassium current in canine jejunal circular smooth muscle and its activation by fenamates. J Physiol 1993;468(1):297–310. [DOI] [PMC free article] [PubMed]
- 48.Ottolia M, Toro L. Potentiation of large conductance KCa channels by niflumic, flufenamic, and mefenamic acids. Biophys J 1994;67(6):2272–2279. [DOI] [PMC free article] [PubMed]
- 49.Gögelein H, Dahlem D, Englert HC, Lang HJ. Flufenamic acid, mefenamic acid and niflumic acid inhibit single nonselective cation channels in the rat exocrine pancreas. FEBS Lett 1990;268(1):79–82. [DOI] [PubMed]
- 50.Cruickshank SF, Baxter LM, Drummond RM. The Cl− channel blocker niflumic acid releases Ca2+ from an intracellular store in rat pulmonary artery smooth muscle cells. Br J Pharmacol 2003;140(8):1442–1450. [DOI] [PMC free article] [PubMed]
- 51.Kato K, Evans AM, Kozlowski RZ. Relaxation of endothelin-1-induced pulmonary arterial constriction by niflumic acid and NPPB: mechanism(s) independent of chloride channel block. J Pharmacol Exp Ther 1999;288(3):1242–1250. [PubMed]
- 52.Bradley E, Fedigan S, Webb T, Hollywood MA, Thornbury KD, McHale NG, Sergeant GP. Pharmacological characterization of TMEM16A currents. Channels 2014;8(4):308–320. [DOI] [PMC free article] [PubMed]
- 53.Davis AJ, Forrest AS, Jepps TA, Valencik ML, Wiwchar M, Singer CA, Sones WR, Greenwood IA, Leblanc N. Expression profile and protein translation of TMEM16A in murine smooth muscle. Am J Physiol Cell Physiol 2010;299(5):C948–C959. [DOI] [PMC free article] [PubMed]
- 54.Manoury B, Tamuleviciute A, Tammaro P. TMEM16A/Anoctamin1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol 2010;588(13):2305–2314. [DOI] [PMC free article] [PubMed]
- 55.Thomas-Gatewood C, Neeb ZP, Bulley S, Adebiyi A, Bannister JP, Leo MD, Jaggar JH. TMEM16A channels generate Ca2+-activated Cl currents in cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol 2011;301(5):H1819–H1827. [DOI] [PMC free article] [PubMed]
- 56.De La Fuente R, Namkung W, Mills A, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol 2008;73(3):758–768. [DOI] [PubMed]
- 57.Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem 2011;286(3):2365–2374. [DOI] [PMC free article] [PubMed]
- 58.Namkung W, Yao Z, Finkbeiner WE, Verkman AS. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J 2011;25(11):4048–4062. [DOI] [PMC free article] [PubMed]
- 59.Namkung W, Thiagarajah JR, Phuan PW, Verkman AS. Inhibition of Ca2+-activated Cl− channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J 2010;24(11):4178–4186. [DOI] [PMC free article] [PubMed]
- 60.Wang YX, Kotlikoff MI. Inactivation of calcium-activated chloride channels in smooth muscle by calcium/calmodulin-dependent protein kinase. Proc Natl Acad Sci USA 1997;94(26):14918–14923. [DOI] [PMC free article] [PubMed]
- 61.Ledoux J, Greenwood I, Villeneuve LR, Leblanc N. Modulation of Ca2+-dependent Cl− channels by calcineurin in rabbit coronary arterial myocytes. J Physiol 2003;552(3):701–714. [DOI] [PMC free article] [PubMed]
- 62.Ayon R, Sones W, Forrest AS, Wiwchar M, Valencik ML, Sanguinetti AR, Perrino BA, Greenwood IA, Leblanc N. Complex phosphatase regulation of Ca2+-activated Cl− currents in pulmonary arterial smooth muscle cells. J Biol Chem 2009;284(47):32507–32521. [DOI] [PMC free article] [PubMed]
- 63.Wiwchar M, Ayon R, Greenwood IA, Leblanc N. Phosphorylation alters the pharmacology of Ca2+-activated Cl− channels in rabbit pulmonary arterial smooth muscle cells. Br J Pharmacol 2009;158(5):1356–1365. [DOI] [PMC free article] [PubMed]
- 64.Pritchard HA, LeBlanc N, Albert AP, Greenwood IA. Inhibitory role of phosphatidylinositol 4,5-bisphosphate on TMEM16A encoded calcium-activated chloride channels in rat pulmonary artery. Br J Pharmacol 2014;171(18):4311–4321. [DOI] [PMC free article] [PubMed]
- 65.Cunningham SA, Awayda MS, Bubien JK, Ismailov II, Arrate MP, Berdiev BK, Benos DJ, Fuller CM. Cloning of an epithelial chloride channel from bovine trachea. J Biol Chem 1995;270(52):31016–31026. [DOI] [PubMed]
- 66.Fuller CM, Benos DJ. Electrophysiological characteristics of the Ca2+-activated Cl− channel family of anion transport proteins. Clin Exp Pharmacol Physiol 2000;27(11):906–910. [DOI] [PubMed]
- 67.Pauli BU, AbdelGhany M, Cheng HC, Gruber AD, Archibald HA, Elble RC. Molecular characteristics and functional diversity of CLCA family members. Clin Exp Pharmacol Physiol 2000;27(11):901–905. [DOI] [PubMed]
- 68.Fuller CM, Ji HL, Tousson A, Elble RC, Pauli BU, Benos DJ. Ca2+-activated Cl− channels: a newly emerging anion transport family. Pfluegers Arch 2001;443(1 suppl.):S107–S110. [DOI] [PubMed]
- 69.Elble RC, Ji G, Nehrke K, DeBiasio J, Kingsley PD, Kotlikoff MI, Pauli BU. Molecular and functional characterization of a murine calcium-activated chloride channel expressed in smooth muscle. J Biol Chem 2002;277(21):18586–18591. [DOI] [PubMed]
- 70.Huang P, Liu J, Di AK, Robinson NC, Musch MW, Kaetzel MA, Nelson DJ. Regulation of human CLC-3 channels by multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem 2001;276(23):20093–20100. [DOI] [PubMed]
- 71.Robinson NC, Huang P, Kaetzel MA, Lamb FS, Nelson DJ. Identification of an N-terminal amino acid of the CLC-3 chloride channel critical in phosphorylation-dependent activation of a CaMKII-activated chloride current. J Physiol 2004;556(2):353–368. [DOI] [PMC free article] [PubMed]
- 72.Suzuki M, Mizuno A. A novel human Cl− channel family related to Drosophila flightless locus. J Biol Chem 2004;279(21):22461–22468. [DOI] [PubMed]
- 73.Suzuki M. The Drosophila tweety family: molecular candidates for large-conductance Ca2+-activated Cl− channels. Exp Physiol 2006;91(1):141–147. [DOI] [PubMed]
- 74.Sun H, Tsunenari T, Yau KW, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci USA 2002;99(6):4008–4013. [DOI] [PMC free article] [PubMed]
- 75.Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol Rev 2008;88(2):639–672. [DOI] [PubMed]
- 76.Hartzell HC, Yu K, Xiao Q, Chien LT, Qu Z. Anoctamin/TMEM16 family members are Ca2+-activated Cl− channels. J Physiol 2009;587(10):2127–2139. [DOI] [PMC free article] [PubMed]
- 77.Stöhr H, Marquardt A, Nanda I, Schmid M, Weber BH. Three novel human VMD2-like genes are members of the evolutionary highly conserved RFP-TM family. Eur J Hum Genet 2002;10(4):281–284. [DOI] [PubMed]
- 78.Krämer F, Stöhr H, Weber BH. Cloning and characterization of the murine Vmd2 RFP-TM gene family. Cytogenet Genome Res 2004;105(1):107–114. [DOI] [PubMed]
- 79.Qu Z, Wei RW, Mann W, Hartzell HC. Two bestrophins cloned from Xenopus laevis oocytes express Ca2+-activated Cl− currents. J Biol Chem 2003;278(49):49563–49572. [DOI] [PubMed]
- 80.Bakall B, Marmorstein LY, Hoppe G, Peachey NS, Wadelius C, Marmorstein AD. Expression and localization of bestrophin during normal mouse development. Invest Ophthalmol Vis Sci 2003;44(8):3622–3628. [DOI] [PubMed]
- 81.Milenkovic VM, Rivera A, Horling F, Weber BH. Insertion and topology of normal and mutant bestrophin-1 in the endoplasmic reticulum membrane. J Biol Chem 2007;282(2):1313–1321. [DOI] [PubMed]
- 82.Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, Sandgren O, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet 1998;19(3):241–247. [DOI] [PubMed]
- 83.Tsunenari T, Sun H, Williams J, Cahill H, Smallwood P, Yau KW, Nathans J. Structure-function analysis of the bestrophin family of anion channels. J Biol Chem 2003;278(42):41114–41125. [DOI] [PMC free article] [PubMed]
- 84.Qu ZQ, Fischmeister R, Hartzell C. Mouse bestrophin-2 is a bona fide Cl− channel: identification of a residue important in anion binding and conduction. J Gen Physiol 2004;123(4):327–340. [DOI] [PMC free article] [PubMed]
- 85.O’Driscoll KE, Hatton WJ, Burkin HR, Leblanc N, Britton FC. Expression, localization, and functional properties of Bestrophin 3 channel isolated from mouse heart. Am J Physiol Cell Physiol 2008;295(6):C1610–C1624. [DOI] [PMC free article] [PubMed]
- 86.O’Driscoll KE, Leblanc N, Hatton WJ, Britton FC. Functional properties of murine bestrophin 1 channel. Biochem Biophys Res Commun 2009;384(4):476–481. [DOI] [PMC free article] [PubMed]
- 87.Xiao Q, Prussia A, Yu K, Cui YY, Hartzell HC. Regulation of bestrophin Cl channels by calcium: role of the C terminus. J Gen Physiol 2008;132(6):681–692. [DOI] [PMC free article] [PubMed]
- 88.Chien LT, Zhang ZR, Hartzell HC. Single Cl− channels activated by Ca2+ in Drosophila S2 cells are mediated by bestrophins. J Gen Physiol 2006;128(3):247–259. [DOI] [PMC free article] [PubMed]
- 89.Pifferi S, Pascarella G, Boccaccio A, Mazzatenta A, Gustincich S, Menini A, Zucchelli S. Bestrophin-2 is a candidate calcium-activated chloride channel involved in olfactory transduction. Proc Natl Acad Sci USA 2006;103(34):12929–12934. [DOI] [PMC free article] [PubMed]
- 90.Scudieri P, Sondo E, Ferrera L, Galietta LJ. The anoctamin family: TMEM16A and TMEM16B as calcium-activated chloride channels. Exp Physiol 2012;97(2):177–183. [DOI] [PubMed]
- 91.Grubb S, Poulsen KA, Juul CA, Kyed T, Klausen TK, Larsen EH, Hoffmann EK. TMEM16F (Anoctamin 6), an anion channel of delayed Ca2+ activation. J Gen Physiol 2013;141(5):585–600. [DOI] [PMC free article] [PubMed]
- 92.Shimizu T, Iehara T, Sato K, Fujii T, Sakai H, Okada Y. TMEM16F is a component of a Ca2+-activated Cl− channel but not a volume-sensitive outwardly rectifying Cl− channel. Am J Physiol Cell Physiol 2013;304(8):C748–C759. [DOI] [PubMed]
- 93.Yang H, Kim A, David T, Palmer D, Jin T, Tien J, Huang F, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012;151(1):111–122. [DOI] [PMC free article] [PubMed]
- 94.Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem 2013;288(19):13305–13316. [DOI] [PMC free article] [PubMed]
- 95.Ferrera L, Caputo A, Ubby I, Bussani E, Zegarra-Moran O, Ravazzolo R, Pagani F, Galietta LJ. Regulation of TMEM16A chloride channel properties by alternative splicing. J Biol Chem 2009;284(48):33360–33368. [DOI] [PMC free article] [PubMed]
- 96.Rock JR, Harfe BD. Expression of TMEM16 paralogs during murine embryogenesis. Dev Dyn 2008;237(9):2566–2574. [DOI] [PubMed]
- 97.Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, Jan LY. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci USA 2009;106(50):21413–21418. [DOI] [PMC free article] [PubMed]
- 98.Hwang SJ, Blair PJ, Britton FC, O’Driscoll KE, Hennig G, Bayguinov YR, Rock JR, Harfe BD, Sanders KM, Ward SM. Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J Physiol 2009;587(20):4887–4904. [DOI] [PMC free article] [PubMed]
- 99.Schreiber R, Uliyakina I, Kongsuphol P, Warth R, Mirza M, Martins JR, Kunzelmann K. Expression and function of epithelial anoctamins. J Biol Chem 2010;285(10):7838–7845. [DOI] [PMC free article] [PubMed]
- 100.Orta G, Ferreira G, José O, Treviño CL, Beltrán C, Darszon A. Human spermatozoa possess a calcium-dependent chloride channel that may participate in the acrosomal reaction. J Physiol 2012;590(11):2659–2675. [DOI] [PMC free article] [PubMed]
- 101.Sancho M, García-Pascual A, Triguero D. Presence of the Ca2+-activated chloride channel anoctamin 1 in the urethra and its role in excitatory neurotransmission. Am J Physiol Renal Physiol 2012;302(3):F390–F400. [DOI] [PubMed]
- 102.Huang F, Zhang H, Wu M, Yang H, Kudo M, Peters CJ, Woodruff, PG, et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc Natl Acad Sci USA 2012;109(40):16354–16359. [DOI] [PMC free article] [PubMed]
- 103.Yu K, Duran C, Qu Z, Cui YY, Hartzell HC. Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ Res 2012;110(7):990–999. [DOI] [PMC free article] [PubMed]
- 104.Tian Y, Kongsuphol P, Hug M, Ousingsawat J, Witzgall R, Schreiber R, Kuntzelmann K. Calmodulin-dependent activation of the epithelial calcium-dependent chloride channel TMEM16A. FASEB J 2011;25(3):1058–1068. [DOI] [PubMed]
- 105.Jung J, Nam JH, Park HW, Oh U, Yoon JH, Lee MG. Dynamic modulation of ANO1/TMEM16A HCO3− permeability by Ca2+/calmodulin. Proc Natl Acad Sci USA 2013;110(1):360–365. [DOI] [PMC free article] [PubMed]
- 106.Vocke K, Dauner K, Hahn A, Ulbrich A, Broecker J, Keller S, Frings S, Möhren F. Calmodulin-dependent activation and inactivation of anoctamin calcium-gated chloride channels. J Gen Physiol 2013;142(4):381–404. [DOI] [PMC free article] [PubMed]
- 107.Xiao Q, Yu K, Perez-Cornejo P, Cui Y, Arreola J, Hartzell HC. Voltage- and calcium-dependent gating of TMEM16A/Ano1 chloride channels are physically coupled by the first intracellular loop. Proc Natl Acad Sci USA 2011;108(21):8891–8896. [DOI] [PMC free article] [PubMed]
- 108.Fallah G, Romer T, Detro-Dassen S, Braam U, Markwardt F, Schmalzing G. TMEM16A(a)/anoctamin-1 shares a homodimeric architecture with CLC chloride channels. Mol Cell Proteomics 2011;10(2):M110.004697. doi:10.1074/mcp.M110.004697. [DOI] [PMC free article] [PubMed]
- 109.Sheridan JT, Worthington EN, Yu K, Gabriel SE, Hartzell HC, Tarran R. Characterization of the oligomeric structure of the Ca2+-activated Cl− channel Ano1/TMEM16A. J Biol Chem 2011;286(2):1381–1388. [DOI] [PMC free article] [PubMed]
- 110.Tien J, Lee HY, Minor DL Jr., Jan YN, Jan LY. Identification of a dimerization domain in the TMEM16A calcium-activated chloride channel (CaCC). Proc Natl Acad Sci USA 2013;110(16):6352–6357. [DOI] [PMC free article] [PubMed]
- 111.Adomaviciene A, Smith KJ, Garnett H, Tammaro P. Putative pore-loops of TMEM16/anoctamin channels affect channel density in cell membranes. J Physiol 2013;591(14):3487–3505. [DOI] [PMC free article] [PubMed]
- 112.O’Driscoll KE, Pipe RA, Britton FC. Increased complexity of Tmem16a/Anoctamin 1 transcript alternative splicing. BMC Mol Biol 2011;12:35. doi:10.1186/1471-2199-12-35. [DOI] [PMC free article] [PubMed]
- 113.Forrest AS, Joyce TC, Huebner ML, Ayon RJ, Wiwchar M, Joyce J, Freitas N, et al. Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol 2012;303(12):C1229–C1243. [DOI] [PMC free article] [PubMed]
- 114.Ohshiro J, Yamamura H, Saeki T, Suzuki Y, Imaizumi Y. The multiple expression of Ca2+-activated Cl− channels via homo- and hetero-dimer formation of TMEM16A splicing variants in murine portal vein. Biochem Biophys Res Commun 2014;443(2):518–523. [DOI] [PubMed]
- 115.Ferrera L, Scudieri P, Sondo E, Caputo A, Caci E, Zegarra-Moran O, Ravazzolo R, Galietta LJ. A minimal isoform of the TMEM16A protein associated with chloride channel activity. Biochim Biophys Acta Biomembr 2011;1808(9):2214–2223. doi:10.1016/j.bbamem.2011.05.017. [DOI] [PMC free article] [PubMed]
- 116.Sondo E, Scudieri P, Tomati V, Caci E, Mazzone A, Farrugia G, Ravazzolo R, Galietta LJ. Non-canonical translation start sites in the TMEM16A chloride channel. Biochim Biophys Acta Biomembr 2014;1838(1B):89–97. doi:10.1016/j.bbamem.2013.08.010. [DOI] [PMC free article] [PubMed]
- 117.Mazzone A, Bernard CE, Strege PR, Beyder A, Galietta LJ, Pasricha PJ, Rae JL, et al. Altered expression of Ano1 variants in human diabetic gastroparesis. J Biol Chem 2011;286(15):13393–13403. [DOI] [PMC free article] [PubMed]
- 118.Ni YL, Kuan AS, Chen TY. Activation and inhibition of TMEM16A calcium-activated chloride channels. PLoS ONE 2014;9:e86734. doi:10.1371/journal.pone.0086734. [DOI] [PMC free article] [PubMed]
- 119.Yuan H, Gao C, Chen Y, Jia M, Geng J, Zhang H, Zhan Y, Boland LM, An H. Divalent cations modulate TMEM16A calcium-activated chloride channels by a common mechanism. J Membr Biol 2013;246(12):893–902. [DOI] [PubMed]
- 120.Berkefeld H, Fakler B, Schulte U. Ca2+-activated K+ channels: from protein complexes to function. Physiol Rev 2010;90(4):1437–1459. [DOI] [PubMed]
- 121.Terashima H, Picollo A, Accardi A. Purified TMEM16A is sufficient to form Ca2+-activated Cl− channels. Proc Natl Acad Sci USA 2013;110(48):19354–19359. [DOI] [PMC free article] [PubMed]
- 122.Yu K, Zhu J, Qu Z, Cui YY, Hartzell HC. Activation of the Ano1 (TMEM16A) chloride channel by calcium is not mediated by calmodulin. J Gen Physiol 2014;143(2):253–267. [DOI] [PMC free article] [PubMed]
- 123.Yu Y, Kuan A-S, Chen T-Y. Calcium-calmodulin does not alter the anion permeability of the TMEM16A calcium-activated chloride channel. Biophys J 2014;106(suppl. 1):P144a. [DOI] [PMC free article] [PubMed]
- 124.Chao SH, Suzuki Y, Zysk JR, Cheung WY. Activation of calmodulin by various metal cations as a function of ionic radius. Mol Pharmacol 1984;26(1):75–82. [PubMed]
- 125.Matchkov VV, Larsen P, Bouzinova EV, Rojek A, Boedtkjer DM, Golubinskaya V, Pedersen FS, Aalkjær C, Nilsson H. Bestrophin-3 (vitelliform macular dystrophy 2–like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circ Res 2008;103(8):864–872. [DOI] [PubMed]
- 126.Matchkov VV, Aalkjaer C, Nilsson H. A cyclic GMP-dependent calcium-activated chloride current in smooth-muscle cells from rat mesenteric resistance arteries. J Gen Physiol 2004;123(2):121–134. [DOI] [PMC free article] [PubMed]
- 127.Piper AS, Large WA. Direct effect of Ca2+-calmodulin on cGMP-activated Ca2+-dependent Cl− channels in rat mesenteric artery myocytes. J Physiol 2004;559(2):449–457. [DOI] [PMC free article] [PubMed]
- 128.Piper AS, Large WA. Single cGMP-activated Ca2+-dependent Cl− channels in rat mesenteric artery smooth muscle cells. J Physiol 2004;555(2):397–408. [DOI] [PMC free article] [PubMed]
- 129.Broegger T, Jacobsen JC, Dam VS, Boedtkjer DM, Kold-Petersen H, Pedersen FS, Aalkjaer C, Matchkov VV. Bestrophin is important for the rhythmic but not the tonic contraction in rat mesenteric small arteries. Cardiovasc Res 2011;91(4):685–693. [DOI] [PubMed]
- 130.Duran C, Qu Z, Osunkoya AO, Cui Y, Hartzell HC. ANOs 3–7 in the anoctamin/Tmem16 Cl− channel family are intracellular proteins. Am J Physiol Cell Physiol 2012;302(3):C482–C493. [DOI] [PMC free article] [PubMed]
- 131.Gomez-Pinilla PJ, Gibbons SJ, Bardsley MR, Lorincz A, Pozo MJ, Pasricha PJ, Van de Rijn M, et al. Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol 2009;296(6):G1370–G1381. [DOI] [PMC free article] [PubMed]
- 132.Sanders KM, Zhu MH, Britton F, Koh SD, Ward SM. Anoctamins and gastrointestinal smooth muscle excitability. Exp Physiol 2012;97(2):200–206. [DOI] [PMC free article] [PubMed]
- 133.Dixon RE, Hennig GW, Baker SA, Britton FC, Harfe BD, Rock JR, Sanders KM, Ward SM. Electrical slow waves in the mouse oviduct are dependent upon a calcium activated chloride conductance encoded by Tmem16a. Biol Reprod 2012;86(1):1–7. [DOI] [PMC free article] [PubMed]
- 134.Gallos G, Remy KE, Danielsson J, Funayama H, Fu XW, Chang HY, Yim P, Xu DB, Emala CW. Functional expression of the TMEM16 family of calcium-activated chloride channels in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 2013;305(9):L625–L634. [DOI] [PMC free article] [PubMed]
- 135.Zhang CH, Li Y, Zhao W, Lifshitz LM, Li H, Harfe BD, Zhu MS, ZhuGe R. The transmembrane protein 16A Ca2+-activated Cl− channel in airway smooth muscle contributes to airway hyperresponsiveness. Am J Respir Crit Care Med 2013;187(4):374–381. [DOI] [PMC free article] [PubMed]
- 136.Forrest AS, Angermann JE, Raghunathan R, Lachendro C, Greenwood IA, Leblanc N. Intricate interaction between store-operated calcium entry and calcium-activated chloride channels in pulmonary artery smooth muscle cells. In: Yuan JXJ, Ward JPT, eds. Membrane receptors, channels and transporters in pulmonary circulation. Advances in Experimental Medicine and Biology. Vol. 661. Dordrecht: Humana, 2010:31–55. [DOI] [PubMed]
- 137.Sones WR, Davis AJ, Leblanc N, Greenwood IA. Cholesterol depletion alters amplitude and pharmacology of vascular calcium-activated chloride channels. Cardiovasc Res 2010;87(3):476–484. [DOI] [PMC free article] [PubMed]
- 138.Angermann JE, Forrest AS, Greenwood IA, Leblanc N. Activation of Ca2+-activated Cl− channels by store-operated Ca2+ entry in arterial smooth muscle cells does not require reverse-mode Na+/Ca2+ exchange. Can J Physiol Pharmacol 2012;90(7):903–921. [DOI] [PubMed]
- 139.Dam VS, Boedtkjer DM, Nyvad J, Aalkjaer C, Matchkov V. TMEM16A knockdown abrogates two different Ca2+-activated Cl− currents and contractility of smooth muscle in rat mesenteric small arteries. Pfluegers Arch 2014;466(7):1391–1409. [DOI] [PMC free article] [PubMed]
- 140.Heinze C, Seniuk A, Sokolov MV, Huebner AK, Klementowicz AE, Szijártó IA, Schleifenbaum J, et al. Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J Clin Invest 2014;124(2):675–686. [DOI] [PMC free article] [PubMed]
- 141.Billig GM, Pál B, Fidzinski P, Jentsch TJ. Ca2+-activated Cl− currents are dispensable for olfaction. Nat Neurosci 2011;14(6):763–769. [DOI] [PubMed]
- 142.Rock JR, Futtner CR, Harfe BD. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev Biol 2008;321(1):141–149. [DOI] [PubMed]
- 143.Pifferi S, Dibattista M, Menini A. TMEM16B induces chloride currents activated by calcium in mammalian cells. Pfluegers Arch 2009;458(6):1023–1038. [DOI] [PubMed]
- 144.Bulley S, Neeb ZP, Burris SK, Bannister JP, Thomas-Gatewood CM, Jangsangthong W, Jaggar JH. TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res 2012;111(8):1027–1036. [DOI] [PMC free article] [PubMed]
- 145.Wang M, Yang H, Zheng LY, Zhang Z, Tang YB, Wang GL, Du YH, et al. Downregulation of TMEM16A calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension through promoting basilar smooth muscle cell proliferation. Circulation 2012;125(5):697–707. [DOI] [PubMed]
- 146.Almaça J, Tian Y, Aldehni F, Ousingsawat J, Kongsuphol P, Rock JR, Harfe BD, Schreiber R, Kunzelmann K. TMEM16 proteins produce volume regulated chloride currents that are reduced in mice lacking TMEM16A. J Biol Chem 2009;284(42):28571–28578. [DOI] [PMC free article] [PubMed]
- 147.Patterson RL, Boehning D, Snyder SH. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem 2004;73:437–465. [DOI] [PubMed]
- 148.Bezprozvanny I. The inositol 1,4,5-trisphosphate receptors. Cell Calcium 2005;38(3–4):261–272. [DOI] [PubMed]
- 149.Wagner LE II, Yule DI. Differential regulation of the InsP3 receptor type-1 and -2 single channel properties by InsP3, Ca2+ and ATP. J Physiol 2012;590(14):3245–3259. [DOI] [PMC free article] [PubMed]
- 150.Guibert C, Pacaud P, Loirand G, Marthan R, Savineau JP. Effect of extracellular ATP on cytosolic Ca2+ concentration in rat pulmonary artery myocytes. Am J Physiol Lung Cell Mol Physiol 1996;271(3):L450–L458. [DOI] [PubMed]
- 151.Hamada H, Damron DS, Hong SJ, Van Wagoner DR, Murray PA. Phenylephrine-induced Ca2+ oscillations in canine pulmonary artery smooth muscle cells. Circ Res 1997;81(5):812–823. [DOI] [PubMed]
- 152.Wang Q, Large WA. Action of histamine on single smooth muscle cells dispersed from the rabbit pulmonary artery. J Physiol 1993;468(1):125–139. [DOI] [PMC free article] [PubMed]
- 153.Salter KJ, Turner JL, Albarwani S, Clapp LH, Kozlowski RZ. Ca2+-activated Cl− and K+ channels and their modulation by endothelin-1 in rat pulmonary arterial smooth muscle cells. Exp Physiol 1995;80(5):815–824. [DOI] [PubMed]
- 154.Guibert C, Marthan R, Savineau JP. Angiotensin II-induced Ca2+-oscillations in vascular myocytes from the rat pulmonary artery. Am J Physiol Lung Cell Mol Physiol 1996;270(4):L637–L642. [DOI] [PubMed]
- 155.Benham CD, Bolton TB. Spontaneous transient outward currents in single visceral and vascular smooth muscle cells. J Physiol 1986;381(1):385–406. [DOI] [PMC free article] [PubMed]
- 156.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science 1995;270(5236):633–637. [DOI] [PubMed]
- 157.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol 1995;268(4):C799–C822. [DOI] [PubMed]
- 158.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 2000;278(2):C235–C256. [DOI] [PubMed]
- 159.Wellman GC, Nelson MT. Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium 2003;34(3):211–229. [DOI] [PubMed]
- 160.Janssen LJ, Sims SM. Acetylcholine activates non-selective cation and chloride conductances in canine and guinea-pig tracheal myocytes. J Physiol 1992;453(1):197–218. [DOI] [PMC free article] [PubMed]
- 161.Janssen J, Sims SM. Spontaneous transient inward currents and rhythmicity in canine and guinea-pig tracheal smooth muscle cells. Pfluegers Arch 1994;427(5–6):473–480. [DOI] [PubMed]
- 162.Janssen LJ, Sims SM. Ca2+-dependent Cl− current in canine tracheal smooth muscle cells. Am J Physiol Cell Physiol 1995;269(1):C163–C169. [DOI] [PubMed]
- 163.Wang Q, Hogg RC, Large WA. Properties of spontaneous inward currents recorded in smooth muscle cells isolated from the rabbit portal vein. J Physiol 1992;451(1):525–537. [DOI] [PMC free article] [PubMed]
- 164.Hogg RC, Wang Q, Helliwell RM, Large WA. Properties of spontaneous inward currents in rabbit pulmonary artery smooth muscle cells. Pfluegers Arch 1993;425(3–4):233–240. [DOI] [PubMed]
- 165.Hogg RC, Wang Q, Large WA. Time course of spontaneous calcium-activated chloride currents in smooth muscle cells from the rabbit portal vein. J Physiol 1993;464(1):15–31. [DOI] [PMC free article] [PubMed]
- 166.Hogg RC, Wang Q, Large WA. Effects of Cl channel blockers on Ca-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. Br J Pharmacol 1994;111(4):1333–1341. [DOI] [PMC free article] [PubMed]
- 167.Greenwood IA, Hogg RC, Large WA. Effect of frusemide, ethacrynic acid and indanyloxyacetic acid on spontaneous Ca-activated currents in rabbit portal vein smooth muscle cells. Br J Pharmacol 1995;115(5):733–738. [DOI] [PMC free article] [PubMed]
- 168.ZhuGe RH, Sims SM, Tuft RA, Fogarty KE, Walsh JV. Ca2+ sparks activate K+ and Cl− channels, resulting in spontaneous transient currents in guinea-pig tracheal myocytes. J Physiol 1998;513(3):711–718. [DOI] [PMC free article] [PubMed]
- 169.Williams BA, Sims SM. Calcium sparks activate calcium-dependent Cl− current in rat corpus cavernosum smooth muscle cells. Am J Physiol Cell Physiol 2007;293(4):C1239–C1251. [DOI] [PubMed]
- 170.Bao R, Lifshitz LM, Tuft RA, Bellvé K, Fogarty KE, ZhuGe R. A close association of RyRs with highly dense clusters of Ca2+-activated Cl− channels underlies the activation of STICs by Ca2+ sparks in mouse airway smooth muscle. J Gen Physiol 2008;132(1):145–160. [DOI] [PMC free article] [PubMed]
- 171.Remillard CV, Zhang WM, Shimoda LA, Sham JS. Physiological properties and functions of Ca2+ sparks in rat intrapulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2002;283(2):L433–L444. [DOI] [PubMed]
- 172.Yang XR, Lin MJ, Yip KP, Jeyakumar LH, Fleischer S, Leung GP, Sham JS. Multiple ryanodine receptor subtypes and heterogeneous ryanodine receptor-gated Ca2+ stores in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2005;289(2):L338–L348. [DOI] [PubMed]
- 173.Zheng YM, Wang QS, Rathore R, Zhang WH, Mazurkiewicz JE, Sorrentino V, Singer HA, Kotlikoff MI, Wang YX. Type-3 ryanodine receptors mediate hypoxia-, but not neurotransmitter-induced calcium release and contraction in pulmonary artery smooth muscle cells. J Gen Physiol 2005;125(4):427–440. [DOI] [PMC free article] [PubMed]
- 174.Zheng YM, Wang QS, Liu QH, Rathore R, Yadav V, Wang YX. Heterogeneous gene expression and functional activity of ryanodine receptors in resistance and conduit pulmonary as well as mesenteric artery smooth muscle cells. J Vasc Res 2008;45(6):469–479. [DOI] [PMC free article] [PubMed]
- 175.Lifshitz LM, Carmichael JD, Lai FA, Sorrentino V, Bellvé K, Fogarty KE, ZhuGe R. Spatial organization of RYRs and BK channels underlying the activation of STOCs by Ca2+ sparks in airway myocytes. J Gen Physiol 2011;138(2):195–209. [DOI] [PMC free article] [PubMed]
- 176.Porter VA, Rhodes MT, Reeve HL, Cornfield DN. Oxygen-induced fetal pulmonary vasodilation is mediated by intracellular calcium activation of KCa channels. Am J Physiol Lung Cell Mol Physiol 2001;281(6):L1379–L1385. [DOI] [PubMed]
- 177.Rhodes MT, Porter VA, Saqueton CB, Herron JM, Resnik ER, Cornfield DN. Pulmonary vascular response to normoxia and KCa channel activity is developmentally regulated. Am J Physiol Lung Cell Mol Physiol 2001;280(6):L1250–L1257. [DOI] [PubMed]
- 178.Yuan XJ. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res 1995;77(2):370–378. [DOI] [PubMed]
- 179.Archer SL, Huang JM, Reeve HL, Hampl V, Tolarová S, Michelakis E, Weir EK. Differential distribution of electrophysiologically distinct myocytes in conduit and resistance arteries determines their response to nitric oxide and hypoxia. Circ Res 1996;78(3):431–442. [DOI] [PubMed]
- 180.Shimoda L, Sylvester JT, Sham JS. Inhibition of voltage-gated K+ current in rat intrapulmonary arterial myocytes by endothelin-1. Am J Physiol Lung Cell Mol Physiol 1998;274(5):L842–L853. [DOI] [PubMed]
- 181.Wiener CM, Banta MR, Dowless MS, Flavahan NA, Sylvester JT. Mechanisms of hypoxic vasodilation in ferret pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 1995;269(3):L351–L357. [DOI] [PubMed]
- 182.Zhang WM, Yip KP, Lin MJ, Shimoda LA, Li WH, Sham JS. ET-1 activates Ca2+ sparks in PASMC: local Ca2+ signaling between inositol trisphosphate and ryanodine receptors. Am J Physiol Lung Cell Mol Physiol 2003;285(3):L680–L690. [DOI] [PubMed]
- 183.Ng LC, Wilson SM, McAllister CE, Hume JR. Role of InsP3 and ryanodine receptors in the activation of capacitative Ca2+ entry by store depletion or hypoxia in canine pulmonary arterial smooth muscle cells. Br J Pharmacol 2007;152(1):101–111. [DOI] [PMC free article] [PubMed]
- 184.Pacaud P, Loirand G, Mironneau C, Mironneau J. Noradrenaline activates a calcium-activated chloride conductance and increases the voltage-dependent calcium current in cultured single cells of rat portal vein. Br J Pharmacol 1989;97(1):139–146. [DOI] [PMC free article] [PubMed]
- 185.Pacaud P, Loirand G, Baron A, Mironneau C, Mironneau J. Ca2+ channel activation and membrane depolarization mediated by Cl− channels in response to noradrenaline in vascular myocytes. Br J Pharmacol 1991;104(4):1000–1006. [DOI] [PMC free article] [PubMed]
- 186.Greenwood IA, Leblanc N, Gordienko DV, Large WA. Modulation of ICl(Ca) in vascular smooth muscle cells by oxidizing and cysteine-reactive reagents. Pfluegers Arch 2002;443(3):473–482. [DOI] [PubMed]
- 187.Saleh SN, Greenwood IA. Activation of chloride currents in murine portal vein smooth muscle cells by membrane depolarization involves intracellular calcium release. Am J Physiol Cell Physiol 2005;288(1):C122–C131. [DOI] [PubMed]
- 188.Akbarali HI, Giles WR. Ca2+ and Ca2+-activated Cl− currents in rabbit oesophageal smooth muscle. J Physiol 1993;460(1):117–133. [DOI] [PMC free article] [PubMed]
- 189.Hazama H, Nakajima T, Hamada E, Omata M, Kurachi Y. Neurokinin A and Ca2+ current induce Ca2+-activated Cl− currents in guinea-pig tracheal myocytes. J Physiol 1996;492(2):377–393. [DOI] [PMC free article] [PubMed]
- 190.Hollywood MA, Sergeant GP, McHale NG, Thornbury KD. Activation of Ca2+-activated Cl− current by depolarizing steps in rabbit urethral interstitial cells. Am J Physiol Cell Physiol 2003;285(2):C327–C333. [DOI] [PubMed]
- 191.Chipperfield AR, Harper AA. Chloride in smooth muscle. Prog Biophys Mol Biol 2000;74(3–5):175–221. [DOI] [PubMed]
- 192.Casteels R, Kitamura K, Kuriyama H, Suzuki H. The membrane properties of the smooth muscle cells of the rabbit main pulmonary artery. J Physiol 1977;271(1):41–61. [DOI] [PMC free article] [PubMed]
- 193.Clapp LH, Gurney AM. ATP-sensitive K+ channels regulate resting potential of pulmonary arterial smooth muscle cells. Am J Physiol Heart Circ Physiol 1992;262(3):H916–H920. [DOI] [PubMed]
- 194.Wilson SM, Leblanc N. Membrane electrical properties of vascular smooth muscle cells of the pulmonary circulation. In: Yuan J X-J, ed. Ion channels in the pulmonary vasculature. New York: Taylor & Francis, 2005:1–24.
- 195.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov 2008;8(2):153–171. [DOI] [PMC free article] [PubMed]
- 196.Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev 1997;77(4):901–930. [DOI] [PubMed]
- 197.Ng LC, Gurney AM. Store-operated channels mediate Ca2+ influx and contraction in rat pulmonary artery. Circ Res 2001;89(10):923–929. [DOI] [PubMed]
- 198.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 2004;95(5):496–505. [DOI] [PubMed]
- 199.Zhang S, Yuan JX, Barrett KE, Dong H. Role of Na+/Ca2+ exchange in regulating cytosolic Ca2+ in cultured human pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 2005;288(2):C245–C252. [DOI] [PubMed]
- 200.Ng LC, McCormack MD, Airey JA, Singer CA, Keller PS, Shen XM, Hume JR. TRPC1 and STIM1 mediate capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. J Physiol 2009;587(11):2429–2442. [DOI] [PMC free article] [PubMed]
- 201.Peng G, Lu W, Li X, Chen Y, Zhong N, Ran P, Wang J. Expression of store-operated Ca2+ entry and transient receptor potential canonical and vanilloid-related proteins in rat distal pulmonary venous smooth muscle. Am J Physiol Lung Cell Mol Physiol 2010;299(5):L621–L630. [DOI] [PMC free article] [PubMed]
- 202.Remillard CV, Yuan JX. TRP channels, CCE, and the pulmonary vascular smooth muscle. Microcirculation 2006;13(8):671–692. [DOI] [PubMed]
- 203.Albert AP, Saleh SN, Peppiatt-Wildman CM, Large WA. Multiple activation mechanisms of store-operated TRPC channels in smooth muscle cells. J Physiol 2007;583(1):25–36. [DOI] [PMC free article] [PubMed]
- 204.Guibert C, Ducret T, Savineau JP. Voltage-independent calcium influx in smooth muscle. Prog Biophys Mol Biol 2008;98(1):10–23. [DOI] [PubMed]
- 205.Prakriya M, Lewis RS. CRAC channels: activation, permeation, and the search for a molecular identity. Cell Calcium 2003;33(5–6):311–321. [DOI] [PubMed]
- 206.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005;437(7060):902–905. [DOI] [PMC free article] [PubMed]
- 207.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 2005;15(13):1235–1241. [DOI] [PMC free article] [PubMed]
- 208.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 2005;169(3):435–445. [DOI] [PMC free article] [PubMed]
- 209.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER–plasma membrane junctions. J Cell Biol 2006;174(6):815–825. [DOI] [PMC free article] [PubMed]
- 210.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 2006;174(6):803–813. [DOI] [PMC free article] [PubMed]
- 211.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006;441(7090):179–185. [DOI] [PubMed]
- 212.Mercer JC, DeHaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW Jr. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem 2006;281(34):24979–24990. [DOI] [PMC free article] [PubMed]
- 213.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006;443(7108):230–233. [DOI] [PubMed]
- 214.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 2006;443(7108):226–229. [DOI] [PMC free article] [PubMed]
- 215.DeHaven WI, Smyth JT, Boyles RR, Putney JW Jr. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release–activated calcium channels. J Biol Chem 2007;282(24):17548–17556. [DOI] [PubMed]
- 216.Hewavitharana T, Deng X, Soboloff J, Gill DL. Role of STIM and Orai proteins in the store-operated calcium signaling pathway. Cell Calcium 2007;42(2):173–182. [DOI] [PubMed]
- 217.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature 2007;446(7133):284–287. [DOI] [PubMed]
- 218.Wu MM, Luik RM, Lewis RS. Some assembly required: constructing the elementary units of store-operated Ca2+ entry. Cell Calcium 2007;42(2):163–172. [DOI] [PMC free article] [PubMed]
- 219.Frischauf I, Schindl R, Derler I, Bergsmann J, Fahrner M, Romanin C. The STIM/Orai coupling machinery. Channels 2008;2(4):261–268. [DOI] [PubMed]
- 220.Lewis RS. Store-operated calcium channels: new perspectives on mechanism and function. Cold Spring Harb Perspect Biol 2011;3(12):a003970. doi:10.1101/cshperspect.a003970. [DOI] [PMC free article] [PubMed]
- 221.McNally BA, Prakriya M. Permeation, selectivity, and gating in store-operated CRAC channels. J Physiol 2012;590(17):4179–4191. [DOI] [PMC free article] [PubMed]
- 222.Beech DJ, Muraki K, Flemming R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J Physiol 2004;559(3):685–706. [DOI] [PMC free article] [PubMed]
- 223.Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW Jr. The mammalian TRPC cation channels. Biochim Biophys Acta Mol Cell Res 2004;1742(1–3):21–36. doi:10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed]
- 224.Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ Res 2001;88(1):84–87. [DOI] [PubMed]
- 225.Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 2002;283(1):L144–L155. [DOI] [PubMed]
- 226.Xu SZ, Boulay G, Flemming R, Beech DJ. E3-targeted anti-TRPC5 antibody inhibits store-operated calcium entry in freshly isolated pial arterioles. Am J Physiol Heart Circ Physiol 2006;291(6):H2653–H2659. [DOI] [PubMed]
- 227.Peel SE, Liu B, Hall IP. ORAI and store-operated calcium influx in human airway smooth muscle cells. Am J Respir Cell Mol Biol 2008;38(6):744–749. [DOI] [PMC free article] [PubMed]
- 228.Lu W, Wang J, Peng G, Shimoda LA, Sylvester JT. Knockdown of stromal interaction molecule 1 attenuates store-operated Ca2+ entry and Ca2+ responses to acute hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 2009;297(1):L17–L25. [DOI] [PMC free article] [PubMed]
- 229.Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, Trebak M. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J 2009;23(8):2425–2437. [DOI] [PMC free article] [PubMed]
- 230.Beech DJ. Orai1 calcium channels in the vasculature. Pfluegers Arch 2012;463(5):635–647. [DOI] [PMC free article] [PubMed]
- 231.Trebak M. STIM/Orai signaling complexes in vascular smooth muscle. J Physiol 2012;590(17):4201–4208. [DOI] [PMC free article] [PubMed]
- 232.Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, Worley PF. STIM1 carboxyl-terminus activates native SOC, Icrac and TRPC1 channels. Nat Cell Biol 2006;8(9):1003–1001. [DOI] [PubMed]
- 233.Alicia S, Angélica Z, Carlos S, Alfonso S, Vaca L. STIM1 converts TRPC1 from a receptor-operated to a store-operated channel: moving TRPC1 in and out of lipid rafts. Cell Calcium 2008;44(5):479–491. [DOI] [PubMed]
- 234.Li J, Sukumar P, Milligan CJ, Kumar B, Ma ZY, Munsch CM, Jiang LH, Porter KE, Beech DJ. Interactions, functions, and independence of plasma membrane STIM1 and TRPC1 in vascular smooth muscle cells. Circ Res 2008;103(8):e97–e104. [DOI] [PMC free article] [PubMed]
- 235.Ong HL, Cheng KT, Liu X, Bandyopadhyay BC, Paria BC, Soboloff J, Pani B, et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx: evidence for similarities in store-operated and calcium release—activated calcium channel components. J Biol Chem 2007;282(12):9105–9116. [DOI] [PMC free article] [PubMed]
- 236.Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem 2008;283(19):12935–12940. [DOI] [PMC free article] [PubMed]
- 237.Jardin I, Lopez JJ, Salido GM, Rosado JA. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J Biol Chem 2008;283(37):25296–25304. [DOI] [PubMed]
- 238.Jardin I, Salido GM, Rosado JA. Role of lipid rafts in the interaction between hTRPC1, Orai1 and STIM1. Channels 2008;2(6):401–403. [DOI] [PubMed]
- 239.Yamamura A, Yamamura H, Zeifman A, Yuan JX. Activity of Ca-activated Cl channels contributes to regulating receptor- and store-operated Ca entry in human pulmonary artery smooth muscle cells. Pulm Circ 2011;1(2):269–279. [DOI] [PMC free article] [PubMed]
- 240.Jung S, Strotmann R, Schultz N, Plant TD. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am J Physiol Cell Physiol 2002;282(2):C347–C359. [DOI] [PubMed]
- 241.Maruyama Y, Nakanishi Y, Walsh EJ, Wilson DP, Welsh DG, Cole WC. Heteromultimeric TRPC6-TRPC7 channels contribute to arginine vasopressin-induced cation current of A7r5 vascular smooth muscle cells. Circ Res 2006;98(12):1520–1527. [DOI] [PubMed]
- 242.Tai K, Hamaide MC, Debaix H, Gailly P, Wibo M, Morel N. Agonist-evoked calcium entry in vascular smooth muscle cells requires IP3 receptor-mediated activation of TRPC1. Eur J Pharmacol 2008;583(1):135–147. [DOI] [PubMed]
- 243.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009;135(3):794–804. [DOI] [PubMed]
- 244.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009;54(1 suppl.):S43–S54. [DOI] [PubMed]
- 245.Stenmark KR, Meyrick B, Galiè N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiologic discovery and pharmacologic cure. Am J Physiol Lung Cell Mol Physiol 2009;297(6):L1013–L1032. [DOI] [PubMed]
- 246.Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 2010;121(18):2045–2066. [DOI] [PMC free article] [PubMed]
- 247.Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol 2012;302(8):H1546–H1562. [DOI] [PMC free article] [PubMed]
- 248.Mandegar M, Remillard CV, Yuan JX. Ion channels in pulmonary arterial hypertension. Prog Cardiovasc Dis 2002;45(2):81–114. [DOI] [PubMed]
- 249.Shimoda LA, Wang J, Sylvester JT. Ca2+ channels and chronic hypoxia. Microcirculation 2006;13(8):657–670. [DOI] [PubMed]
- 250.Smirnov SV, Robertson TP, Ward JP, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. Am J Physiol Heart Circ Physiol 1994;266(1):H365–H370. [DOI] [PubMed]
- 251.Peng W, Hoidal JR, Karwande SV, Farrukh IS. Effect of chronic hypoxia on K+ channels: regulation in human pulmonary vascular smooth muscle cells. Am J Physiol Cell Physiol 1997;272(4):C1271–C1278. [DOI] [PubMed]
- 252.Osipenko ON, Alexander D, MacLean MR, Gurney AM. Influence of chronic hypoxia on the contributions of non-inactivating and delayed rectifier K currents to the resting potential and tone of rat pulmonary artery smooth muscle. Br J Pharmacol 1998;124(7):1335–1337. [DOI] [PMC free article] [PubMed]
- 253.Archer SL, Souil E, Dinh-Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen-Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 1998;101(11):2319–2330. [DOI] [PMC free article] [PubMed]
- 254.Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr., Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 1998;98(14):1400–1406. [DOI] [PubMed]
- 255.Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet 1998;351(9104):726–727. [DOI] [PubMed]
- 256.Michelakis ED, Dyck JR, McMurtry MS, Wang S, Wu XC, Moudgil R, Hashimoto K, Puttagunta L, Archer SL. Gene transfer and metabolic modulators as new therapies for pulmonary hypertension. Increasing expression and activity of potassium channels in rat and human models. In: Roach RC, Wagner PD, Hackett PH, eds. Hypoxia: from genes to the bedside. Advances in Experimental Medicine and Biology. Vol. 502. New York: Plenum, 2001:401–418. [DOI] [PubMed]
- 257.Mandegar M, Yuan JX. Role of K+ channels in pulmonary hypertension. Vasc Pharmacol 2002;38(1):25–33. [DOI] [PubMed]
- 258.Remillard CV, Yuan JX. High altitude pulmonary hypertension: role of K+ and Ca2+ channels. High Alt Med Biol 2005;6(2):133–146. [DOI] [PMC free article] [PubMed]
- 259.Moudgil R, Michelakis ED, Archer SL. The role of K+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 2006;13(8):615–632. [DOI] [PubMed]
- 260.Archer SL, Reeve HL, Michelakis E, Puttagunta L, Waite R, Nelson DP, Dinauer MC, Weir EK. O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc Natl Acad Sci USA 1999;96(14):7944–7949. [DOI] [PMC free article] [PubMed]
- 261.Reeve HL, Michelakis E, Nelson DP, Weir EK, Archer SL. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J Appl Physiol 2001;90(6):2249–2256. [DOI] [PubMed]
- 262.Archer S, Michelakis E. The mechanism(s) of hypoxic pulmonary vasoconstriction: potassium channels, redox O2 sensors, and controversies. News Physiol Sci 2002;17(4):131–137. [DOI] [PubMed]
- 263.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 2008;294(2):H570–H578. [DOI] [PubMed]
- 264.Hirenallur SD, Haworth ST, Leming JT, Chang J, Hernandez G, Gordon JB, Rusch NJ. Upregulation of vascular calcium channels in neonatal piglets with hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2008;295(5):L915–L924. [DOI] [PMC free article] [PubMed]
- 265.Firth AL, Remillard CV, Yuan JX. TRP channels in hypertension. Biochim Biophys Acta Mol Basis Dis 2007;1772(8):895–906. doi:10.1016/j.bbadis.2007.02.009. [DOI] [PMC free article] [PubMed]
- 266.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 2004;101(38):13861–13866. [DOI] [PMC free article] [PubMed]
- 267.Zhang S, Patel HH, Murray F, Remillard CV, Schach C, Thistlethwaite PA, Insel PA, Yuan JX. Pulmonary artery smooth muscle cells from normal subjects and IPAH patients show divergent cAMP-mediated effects on TRPC expression and capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 2007;292(5):L1202–L1210. [DOI] [PubMed]
- 268.Song MY, Makino A, Yuan JX. STIM2 contributes to enhanced store-operated Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Pulm Circ 2011;1(1):84–94. [DOI] [PMC free article] [PubMed]
- 269.Liu XR, Zhang MF, Yang N, Liu Q, Wang RX, Cao YN, Yang XR, Sham JS, Lin MJ. Enhanced store-operated Ca2+ entry and TRPC channel expression in pulmonary arteries of monocrotaline-induced pulmonary hypertensive rats. Am J Physiol Cell Physiol 2012;302(1):C77–C87. [DOI] [PMC free article] [PubMed]
- 270.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 2001;280(2):H746–H755. [DOI] [PubMed]
- 271.Yang XR, Lin AH, Hughes JM, Flavahan NA, Cao YN, Liedtke W, Sham JS. Upregulation of osmo-mechanosensitive TRPV4 channel facilitates chronic hypoxia-induced myogenic tone and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2012;302(6):L555–L568. [DOI] [PMC free article] [PubMed]
- 272.Earley S, Leblanc N. Serotonin receptors take the TRiPV4 highway in chronic hypoxic pulmonary hypertension. Focus on “TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension.” Am J Physiol Cell Physiol 2013;305(7):C690–C692. [DOI] [PMC free article] [PubMed]
- 273.Xia Y, Fu Z, Hu J, Huang C, Paudel O, Cai S, Liedtke W, Sham JS. TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension. Am J Physiol Cell Physiol 2013;305(7):C704–C715. [DOI] [PMC free article] [PubMed]
- 274.Takayama Y, Shibasaki K, Suzuki Y, Yamanaka A, Tominaga M. Modulation of water efflux through functional interaction between TRPV4 and TMEM16A/anoctamin 1. FASEB J 2014;28(5):2238–2248. [DOI] [PubMed]
- 275.Galindo BE, Vacquier VD. Phylogeny of the TMEM16 protein family: some members are overexpressed in cancer. Int J Mol Med 2005;16(5):919–924. [PubMed]
- 276.Duvvuri U, Shiwarski DJ, Xiao D, Bertrand C, Huang X, Edinger RS, Rock JR, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res 2012;72(13):3270–3281. [DOI] [PMC free article] [PubMed]
- 277.Atala A. Re: inhibition of Ca2+ -activated Cl− channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. J Urol 2013;189(6):2393. [DOI] [PubMed]
- 278.Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, Rebhan M, et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci USA 2013;110(11):E1026–E1034. [DOI] [PMC free article] [PubMed]
- 279.Ubby I, Bussani E, Colonna A, Stacul G, Locatelli M, Scudieri P, Galietta L, Pagani F. TMEM16A alternative splicing coordination in breast cancer. Mol Cancer 2013;12:75. [DOI] [PMC free article] [PubMed]
- 280.Jacobsen KS, Zeeberg K, Sauter DR, Poulsen KA, Hoffmann EK, Schwab A. The role of TMEM16A (ANO1) and TMEM16F (ANO6) in cell migration. Pfluegers Arch 2013;465(12):1753–1762. [DOI] [PMC free article] [PubMed]