

Abstract Abstract
Our previous work has shown that the increased lung endothelial permeability response to 14,15-epoxyeicosatrienoic acid (14,15-EET) in rat lung requires Ca2+ entry via vanilloid type-4 transient receptor potential (TRPV4) channels. Recent studies suggest that activation of TRPV4 channels in systemic vascular endothelium prolongs agonist-induced hyperpolarization and amplifies Ca2+ entry by activating Ca2+-activated K+ (KCa) channels, resulting in vessel relaxation. Activation of endothelial KCa channels thus has potential to increase the electrochemical driving force for Ca2+ influx via TRPV4 channels and to amplify permeability responses to TRPV4 activation in lung. To examine this hypothesis, we used Western blot analysis, electrophysiological recordings, and isolated-lung permeability measurements to document expression of TRPV4 and KCa channels and the potential for functional coupling. The results show that rat pulmonary microvascular endothelial cells express TRPV4 and 3 KCa channels of different conductances: large (BK), intermediate (IK), and small (SK3). However, TRPV4 channel activity modulates the IK and SK3, but not the BK, channel current density. Furthermore, the TRPV4-mediated permeability response to 14,15-EET in mouse lung is significantly attenuated by pharmacologic blockade of IK and SK3, but not BK, channels. Collectively, this functional coupling suggests that endothelial TRPV4 channels in rodent lung likely form signaling microdomains with IK and SK3 channels and that the integrated response dictates the extent of lung endothelial injury caused by 14,15-EET.
Keywords: KCa, BK, IK1, SK3, lung injury
Our previous work links activation of pulmonary vanilloid transient receptor potential subtype-4 (TRPV4) cation channels to Ca2+ influx into alveolar septal endothelium and a resultant increase in endothelial permeability.1-3 TRPV4-mediated impact of mechanical stress on lung endothelial permeability requires release of arachidonic acid from membrane stores. Subsequently, arachidonic acid–derived epoxyeicosatrienoic acids (EETs)2 elicit Ca2+ entry via endothelial TRPV4 channels.4,5 Our in vivo studies have shown that the impact of mechanical stress on lung endothelial permeability requires EET synthesis and can be mimicked by direct challenge with EETs.1,6
In addition to the Ca2+ influx via TRPV4 channels, EETs are known to activate Ca2+-activated potassium (KCa) channels both in vitro and in vivo.7-10 There are 5 members in the KCa channel family, characterized by their unitary conductances: large (BK), intermediate (IK), and 3 subtypes of small (SK): SK1, SK2, and SK3. SK3 and IK are thought to be the predominant KCa channels expressed in systemic vascular endothelia. Endothelial BK channel expression has been reported in some, but not all, studies.11-15 In genetically modified mice lacking both SK3 and IK expression, carotid artery endothelium exhibits minimal KCa currents, and mean arterial blood pressure is increased.16
Irrespective of the specific KCa channel involved, K+ efflux resulting from KCa channel activation can hyperpolarize the membrane potential. While, theoretically, hyperpolarization should increase the driving force for Ca2+ entry in endothelium,17 KCa activation has been reported to either enhance7 or have no impact on18 agonist-induced Ca2+ entry in systemic endothelium. Recent studies showed that endothelial TRPV4 channels modulate vasodilatation via their functional coupling with IK and SK3 channels in mesenteric arteries or with IK channels in cremaster arterioles.19-22 While little is known about functional coupling of TRPV4 and KCa channels in pulmonary endothelium, it is reasonable to propose that concomitant activation of TRPV4 and KCa channels might amplify the lung endothelial permeability response to EETs and, by extrapolation, the permeability response to mechanical stress. Such interplay would require the expression of KCa channels in the lung microvasculature, i.e., in alveolar septal endothelium. KCa channel expression has been documented in airway endothelium, epithelium, alveolar type II epithelial cells, and pulmonary arteries,13,23-25 although expression in the alveolar septal wall has not been extensively explored.26 Similarly, the few reports suggesting the involvement of KCa channels in regulation of endothelial permeability have been limited to the blood-brain tumor barrier.27-29
Taken together, these findings prompted us to examine the expression of TRPV4 and KCa channels and to test the hypothesis that these channels are functionally coupled in lung microvascular endothelium. Specifically, this study had 3 goals: (1) to document the expression levels of these channels in pulmonary microvascular endothelial cells (PMVECs) versus those in pulmonary arterial endothelial cells (PAECs),30,31 (2) to determine whether TRPV4-mediated Ca2+ influx modulates KCa channel currents in PMVECs, and (3) to investigate whether the TRPV4-induced increase in endothelial permeability is attenuated upon inhibition of KCa channels. Our results demonstrate that PMVECs and PAECs express TRPV4, BK, IK, and SK3 channels; that Ca2+ entry via TRPV4 channels selectively activates IK and SK, but not BK, channels; and that inhibition of IK and SK3 channel activity attenuates TRPV4-induced lung permeability.
Methods
Animals
For studies in isolated lung, male and female TRPV KO (TRPV4−/−) mice or wild-type littermates were used at 8–10 weeks of age. Experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of South Alabama and performed in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals.
Molecular biology and cell culture
Constructs used to express proteins in transfected cells were subcloned into a pCMV cytomegalovirus-based vector. HEK (human embryonic kidney 293) and COS-7 (monkey kidney fibroblast) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. Cells were transfected with pCMV expression plasmids encoding TRPV4, BK, IK, or SK3, using lipofectamine 2000 (Invitrogen; Carlsbad, CA). Cell lysates were collected 24–48 hours after transfection for Western blot analysis. Both cultured PMVECs and PAECs, passages 12–16, were maintained in DMEM complete growth medium (Invitrogen) containing high glucose (4.5 g/L) and sodium pyruvate (100 mg/L) supplemented with 10% fetal bovine serum.32-34
Western blot
Cell lysates from PMVECs (150 μg) and PAECs (150 μg), as well as HEK (for BK; 20 μg) or COS-7 (for TRPV4, IK, and SK3; 10–30 μg) cells with and without pCMV plasmid transfection, were used for protein analysis. Cells were homogenized in ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1% NP-40, 0.5% Na-deoxycholate (pH 8.0), and 1× protease inhibitor cocktail (Pierce, Rockford, IL). The samples were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. The primary antibodies used were all purchased from Alomone Labs (Jerusalem): TRPV4 (ACC-034, lot AN1550), BK (APC-021, lot AN1250), IK (ALM-051, lot AN03100), and SK3 (APC-025, lot AN0802). Horseradish peroxidase (HRP)–conjugated secondary antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX), and fluorescence-labeled (Dylight) secondary antibodies were from Thermo Scientific (Pittsburgh, PA). Blots were developed with a Kodak automated film-processing system and imaged with a digital Chemilluminescence imager (Fuji) or visualized with an Odyssey Infrared Imaging System (LI-COR Biosciences) according to manufacturer’s instructions.
Electrophysiology
For patch-clamp studies, cells were washed with 1× phosphate-buffered saline (PBS), trypsinized, and centrifuged. The cell pellet was resuspended in DMEM and seeded in a 35-mm cell culture dish on glass coverslips at 37°C for 2–4 days before experimentation. Single isolated cells without physical contact with neighboring cells were used for electrophysiological recording. Whole-cell patch-clamp recordings were performed on cultured PMVECs with an Axopatch 200B amplifier and a Digidata 1322A, and data were acquired with PClamp 8 software (all from Molecular Devices, Sunnyvale, CA). Cells were voltage clamped at resting membrane potential, and whole-cell currents were evoked every 20 seconds by applying a voltage protocol consisting of 3 segments: a 100-ms negative step to −80 mV, a 200-ms voltage ramp of −80 to +60 mV, and another 100-ms voltage step to +30 mV.35 Currents were sampled at 2 kHz and filtered at 1 kHz. Patch pipettes (3–5 MΩ) were filled with different pipette solutions, depending on the specific experimental requirements (see “Chemicals and solutions”). Series resistance and membrane capacitance were compensated for up to 80% to minimize errors. For each cell, the contributions of KCa channels were calculated from the digitally isolated steady-state currents (in the presence of selective inhibitors), normalized to the baseline whole-cell current density, before any pharmacological manipulation.
Chemicals and solutions
For patch-clamp experiments, the external bath solution contained (in mM) N-methyl-d-glucamine (NMDG) 145, KCl 6, glucose 5, HEPES 10, MgCl2 1, and CaCl2 2 (pH 7.35, 300 mOsm). The whole-cell internal pipette solution contained (in mM) K+ gluconate 130, KCl 4, NaCl 4, EDTA 2, HEPES 10, MgCl2 1, CaCl2 0.7, MgATP (adenosine triphosphate) 4, GTP (guanosine triphosphate) 0.3, and phosphocreatine 10 (pH 7.2, 308 mOsm). The concentrations of free Mg2+ (150 μM) and Ca2+ (0.7 μM) were calculated with Patcher’s Power Tools (Department of Membrane Biophysics at the Max Planck Institute for Biophysical Chemistry, Göttingen, Germany). The cesium (Cs)-based internal pipette solution contained (in mM) CsCl 95, CsOH 25, aspartic acid 50, NaCl 4, MgCl2 1, and HEPES 10 (pH 7.2, 302 mOsm). Osmolarity for all solutions was measured with Osmette III osmometer (Precision Systems, Natick, MA) and was adjusted with d-mannitol when necessary. Channel agonists and antagonists were perfused into recording chambers. GSK1016790A (GSK; 50 nM) and HC067047 (HC; 500 nM) were used as TRPV4 channel agonist and antagonist, respectively. Paxilline (1 μM), TRAM-34 (1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole; 1 μM), and apamin (200 nM) were used to block BK, IK, and SK channels, respectively. GSK was obtained from Sigma-Aldrich (St. Louis, MO). HC was obtained from Tocris (Minneapolis, MN). Paxilline was obtained from MP Biomedicals (Santa Ana, CA). Apamin and phosphocreatine were obtained from EMD Millipore (Philadelphia, PA). TRAM-34 was from Alomone Labs. All other chemicals were obtained from Fisher Scientific (Pittsburgh, PA).
Isolated lung and assessment of endothelial permeability
Lungs isolated from anesthetized mice (sodium pentobarbital, 50 mg/kg intraperitoneally) were perfused with 4% bovine serum albumin in Earle’s buffer containing physiological (2.2 mM) or low (0.02 mM) Ca2+. We have previously documented that lungs are stable in this low-Ca2+ perfusate and that this decrement in perfusate [Ca2+] is sufficient to prevent the permeability response to activation of TRPV4 channels.1,36 The filtration coefficient (Kf) was measured via the rate of lung weight gain 13–15 minutes after a step increase in venous pressure of 7–10 cm H2O.1,2,37 In each experimental group, the TRPV4 channel agonist 14,15-EET was added to the perfusate after baseline measurements were completed. Inhibitors were added before the baseline Kf measurement. Lung endothelial permeability was assessed with the following protocols.
1. We confirmed that the 14,15-EET-induced permeability response is TRPV4 dependent in lungs perfused with physiologic buffer. The permeability response to 14,15-EET (3 μM) was compared in lungs from wild-type mice in the absence (n = 6) or presence (n = 5) of the TRPV antagonist ruthenium red (RR, 3 μM) and in lungs from TRPV KO mice (n = 5). The Kf was measured at baseline and again 30 minutes after 14,15-EET administration.
2. To determine whether Ca2+ entry and KCa channel activation are required for the permeability response to 14,15-EET in wild- type lungs, we used the low-Ca2+/Ca2+ add-back protocol. The baseline Kf and a second Kf (30 min after addition of 14,15-EET) were measured in low-Ca2+ (0.02 mM) perfusate. The final Kf was measured 15 minutes after Ca2+ add-back to restore the physiological Ca2+ concentration (2.2 mM). The first group was challenged with 14,15-EET alone (3 μM; n = 5). In the next groups, before 14,15-EET administration lungs were pretreated with RR (3 μM, n = 4) to block TRPV channels or the combination of charybdotoxin (100 nM) and apamin (300 nM, n = 5) to block all KCa channels. We clarified the specific KCa channel isoforms involved, using iberiotoxin (100 nM, n = 5) to block BK channels, apamin (300 nM, n = 5) to block SK channels, and TRAM-34 (1 μM, n = 5) to block IK channels.
Data analysis and statistics
Data are expressed as mean ± standard error of mean. Electrophysiological data in cultured cells were analyzed and plotted in IgorPro (WaveMetrics, Lake Oswego, OR), and nonparametric Wilcoxon signed-rank tests were used to determine significance between groups of data. A paired t test was used for data within the same experimental group in this series. For functional studies in mouse lung, statistical comparisons between groups were performed via analysis of variance (ANOVA) with repeated measures and the Tukey post hoc test. A P value less than 0.05 was considered significant.
Results
TRPV4 and KCa channel expression in PMVECs
We first examined the expression of TRPV4, BK, IK, and SK3 channels in PMVECs and PAECs. These cells have been shown to retain expression of unique endothelial markers as well as to display stimulus-induced permeability increases in a pattern similar to that of their in vivo counterparts (for review see Stevens34). Figure 1A shows representative Western blots for TRPV4, BK, IK, and SK3 channel proteins as well as for β-actin as a loading control. Both PMVECs (MV [lane 3]) and PAECs (PA [lane 4]) express all of the probed proteins. To test the specificity of our antibodies against each ion channel, we used heterologous COS-7 and HEK293 expression systems without (Fig. 1A, minus sign [lane 1]) and with (Fig. 1A; plus sign [lane 2]) transfection of TRPV4, BK, IK, and SK3 channels; these data supplied negative and positive controls, respectively. Expression levels for each channel protein were obtained from band densitometry normalized to that of β-actin. The relative PMVEC/PAEC expression ratios are shown in Figure 1B. Expression of TRPV4, BK, and SK3 channels in PMVECs exceeded that in PAECs (ratios for TRPV4: 3.3 ± 1.0; BK: 1.8 ± 0.7; SK3: 1.9 ± 0.4). In contrast, the expression of IK channels was higher in PAECs (ratio for IK: 0.88 ± 0.1). These expression profiles are consistent with those from previous biochemical studies as well as some in vivo pharmacological studies.1,13,31,34
Figure 1.
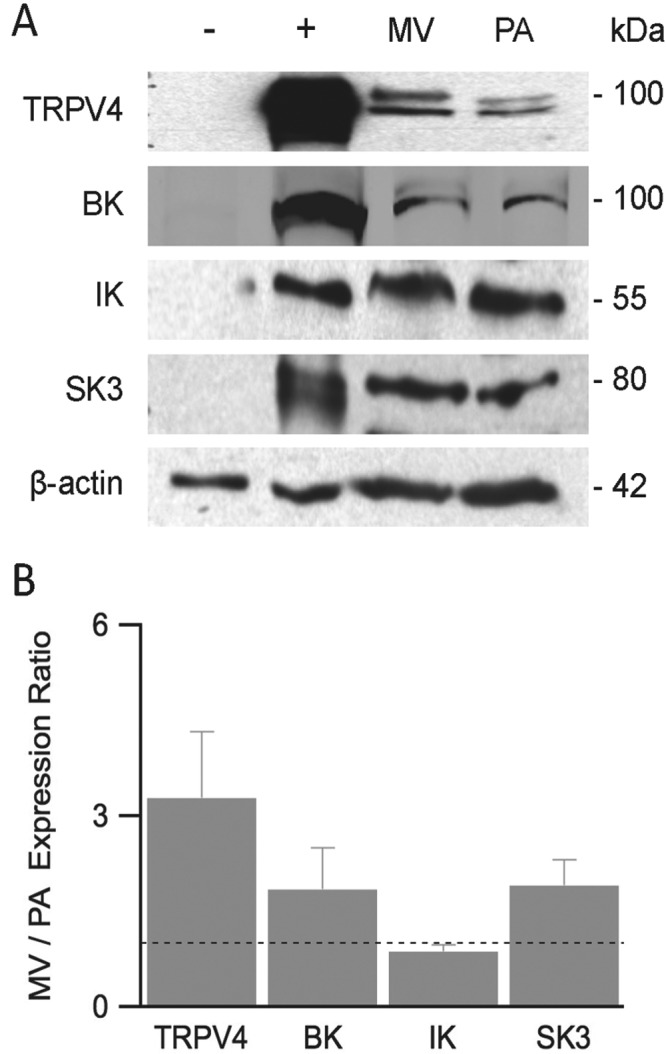
Ion channel expression in PMVECs and PAECs. Western blot analysis was performed to examine TRPV4, BK, IK, and SK3 channels and β-actin expression in heterologous expression systems, as well as in PMVECs and PAECs. A, Representative Western blot analysis of cell lysates of nontransfected (lane 1) and transfected (lane 2) HEK or COS-7 cells, used as negative and positive controls, respectively, PMVECs (lane 3, MV), and PAECs (lane 4, PA). B, After densitometry, values for TRPV4, BK, IK, and SK3 were normalized to β-actin, then expression in MV was reported relative to that for PA. A ratio of 1, shown as the dotted line, would indicate equal protein expression in PMVECs and PAECs. BK, IK, SK3: large-, intermediate-, and small-conductance (subtype 3) KCa (Ca2+-activated potassium) channels, respectively; COS-7: monkey kidney fibroblast; HEK: human embryonic kidney; PAECs: pulmonary arterial endothelial cells; PMVECs: pulmonary microvascular endothelial cells; TRPV4: transient receptor potential vanilloid 4.
Characterization of KCa channel currents in PMVECs
To directly study functional interplay between TRPV4 and KCa channels, patch-clamp recordings were obtained in cultured PMVECs. Whole-cell currents were elicited in the presence of ∼0.7 μM free internal Ca2+ to restrain full activation of KCa channels with a voltage-clamp protocol delivered every 20 seconds. The voltage protocol consisted of 3 segments: a 100-ms voltage step to −80 mV from resting potential, a 200-ms voltage ramp from −80 to +60 mV, and a 100-ms voltage step to +30 mV (Fig. 2A). The initial hyperpolarizing step to −80 mV was used to calculate cell membrane capacitance and resistance, the voltage ramp from −80 to +60 mV was used to reveal the profile of whole-cell currents, and the voltage step to +30 mV was used to quantify steady-state currents, as shown in Figure 2F.
Figure 2.

Whole-cell voltage-clamp recordings in PMVECs. A, Voltage-clamp recording protocol. Cells were held at their resting potential and stepped to −80 mV for 100 ms before a voltage ramp of −80 to +60 mV (200 ms), a voltage step to +30 mV (100 ms), and return to resting potential. B, Representative whole-cell current densities elicited by the voltage-clamp protocol shown in A. After stable baseline recordings (control), sequential additions of paxilline (1 μM), TRAM-34 (1 μM), and apamin (200 nM) selectively blocked BK, IK, and SK channels, respectively. C–E, BK (C), IK (D), and SK (E) currents were isolated from digital subtraction of the voltage-ramp portion of traces in B. F, Normalized BK, IK, and SK channel current density to whole-cell current density, calculated from the steady-state current density recordings at +30 mV. BK, IK, SK: large-, intermediate-, and small-conductance KCa (Ca2+-activated potassium) channels, respectively; PMVECs: pulmonary microvascular endothelial cells; TRAM-34: 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole.
Following stable baseline recordings (Fig. 2B, control), KCa channel blockers were applied. Paxilline (1 μM), TRAM-34 (1 μM), and apamin (200 nM) were sequentially added to selectively block BK, IK, and SK channels, respectively (Fig. 2B). Bath application of paxilline reduced the whole-cell current density by 15%; BK current density was then isolated by digital subtraction (Fig. 2C). Subsequent addition of TRAM-34 and then apamin further reduced current density by 21% and 11%, respectively; IK and SK current densities were then isolated by digital subtraction (Fig. 2D, 2E). Figure 2F shows normalized contribution of KCa channels to steady-state whole-cell current density at +30 mV. The average contributions for BK, IK, and SK were 20.7% ± 2.8%, 11.1% ± 1.9%, and 13.5% ± 2.3%, respectively (Fig. 2F), with an averaged membrane capacitance of 22.5 ± 2.7 pF (n = 6). Results from these studies concur with our Western blot data, indicating the presence of functional BK, IK, and SK3 channels in PMVECs. Under these recording conditions, BK channels conducted twice as much steady-state current as IK or SK channels at +30 mV.
Basal TRPV4 channel activity in PMVECs
To reveal whole-cell TRPV4 current density in PMVECs, a Cs-based internal pipette solution was used to minimize K+ currents. Whole-cell TRPV4 channel currents were elicited with the same voltage-clamp protocol shown in Figure 2A. Voltage ramp from −80 to +60 mV for 200 ms elicited a small whole-cell current density (Fig. 3A). Application of GSK (50 nM), a selective TRPV4 agonist,19,38,39 increased the current density by 72%; digital subtraction isolated the GSK-sensitive TRPV4 channel activation (Fig. 3C). Subsequent application of the selective TRPV4 channel antagonist HC (500 nM)40 reduced current density by 60% (Fig. 3B); the HC-sensitive TRPV4 channel current density is shown in Figure 3D. Normalized results showed that activation of TRPV4 channels by GSK increased whole-cell current density by 34.8% ± 5.8%, from 19.9 ± 2.7 to 27.3 ± 4.6 pA/pF (n = 6). Subsequent blocking by HC decreased current density by 43.3% ± 7.3%, to 13.9 ± 1.1 pA/pF (Fig. 3B). On average, blocking TRPV4 channels resulted in a greater reduction in current density than did activating them (8.5% ± 2.0%, P < 0.05, n = 6; paired t test), suggesting the existence of basal activity of TRPV4 channels in PMVECs in our recording conditions.
Figure 3.

TRPV4 channel current density recording using Cs-based internal solution. A, Representative whole-cell current densities elicited by a −80 to +60 mV voltage ramp for 200 ms in control conditions or following subsequent application of GSK or HC. B, The effect of GSK or HC on TRPV4 channel current density normalized to control. Normalized changes in current densities were calculated from the steady-state current density recordings at +30 mV. Asterisks indicate statistical significance using paired t test (P < 0.05). C, D, The effect of GSK (C) and HC (D) on TRPV4 channel current density isolated from digital subtraction of the current densities shown in A. Averaged membrane capacitance was 18.5 ± 2.3 pF. GSK: GSK1016790A; HC: HC067047; TRPV4: transient receptor potential vanilloid 4.
Activating TRPV4 channels significantly increases IK and SK, but not BK, channel current densities
To test the hypothesis that TRPV4 channels are functionally coupled with KCa channels in PMVECs, we pharmacologically modulated TRPV4 channel activity while recording whole-cell KCa currents. Since TRPV4 channels nonselectively conduct both inward Na+ and Ca2+ currents, we replaced external Na+ ions with nonpermeant NMDG ions to minimize Na+-induced inward currents and consequently reveal outward K+ conductance.4 Our results showed that GSK-dependent activation of TRPV4 channels consistently increased whole-cell current density, with a reversal potential close to the K+ reversal potential. Current increases were abolished in the absence of external Ca2+ (data not shown), consistent with TRPV4-dependent Ca2+ activation of KCa channels (Fig. 4A). As shown by the representative recordings in Figure 4A, following stable baseline recordings (control), 50 nM GSK increased the whole-cell current density by 48% (GSK). Subsequent sequential additions of paxilline, TRAM-34, and apamin reduced current densities, and the reductions were attributable to the blockade of BK, IK, and SK channels, respectively (Fig. 4A). The effects of GSK and each of the KCa channel antagonists were digitally isolated (Fig. 4C–4F). KCa channels’ contributions to whole-cell currents were calculated from their respective isolated current densities, normalized to the baseline whole-cell current density obtained before the addition of GSK. Summarized results showed that the contributions of IK and SK channels increased in the presence of GSK (Fig. 4B; IK: 33.0% ± 4.1%, SK: 28.2% ± 3.8%, n = 6), as compared to those in the absence of GSK (dotted lines; P < 0.05). In contrast, following TRPV4 activation BK channels contributed 26.6% ± 5.4% (Fig. 4B; n = 6) to the steady-state whole-cell current density, an effect not significantly different from that in controls (dotted line; P = 0.67). Together, these results indicate that while TRPV4 channel activation selectively increases IK and SK channel activity, it has little effect on BK channel activity in PMVECs.
Figure 4.

TRPV4 channel activation increases IK and SK channel current densities. A, Representative whole-cell current densities elicited by the 200-ms −80 to +60 mV voltage-ramp protocol. Following stable baseline recordings, GSK (50 nM) was used to activate TRPV4 channels. Sequential applications of paxilline (500 nM), TRAM-34 (1 μM), and apamin (200 nM) were used to selectively inhibit BK, IK, and SK channels, respectively. B, Bar graph showing BK, IK, and SK channel current densities, in the presence of GSK, normalized to baseline whole-cell current density (control in A). Normalized results were calculated from the steady-state current density recordings at +30 mV. Dashed lines indicate normalized averages of each channel current density in the absence of GSK, as shown in Figure 2F. Asterisks indicate statistical significance (P < 0.05). C–F, The effect of GSK (C), paxilline (D), TRAM-34 (E), and apamin (F) on whole-cell current density isolated from digital subtraction of the traces shown in A. Averaged membrane capacitance was 23.8 ± 3.2 pF. BK, IK, SK: large-, intermediate-, and small-conductance KCa (Ca2+-activated potassium) channels, respectively; GSK: GSK1016790A; TRAM-34: 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole; TRPV4: transient receptor potential vanilloid 4.
Inhibiting TRPV4 channels selectively reduces IK and SK, but not BK, channel current densities
The results presented above suggest that Ca2+ influx via TRPV4 channels on PMVECs selectively activates IK and SK, but not BK, channels. This outcome predicts that blocking endogenous TRPV4 channel activity will reduce IK and SK channel currents and thus their contribution to whole-cell current density. We found that blocking TRPV4 channels with 500 nM HC reduced whole-cell current density by 28% (Fig. 5A, from control to HC). The HC-dependent reduction was abolished in the absence of external Ca2+ (data not shown), suggesting that blockade of Ca2+ influx via TRPV4 reduces KCa current density. Moreover, each subsequent addition of the KCa channel blockers paxilline, TRAM-34, and apamin further decreased current density by 12%, 7%, and 6%, respectively (Fig. 5C–5F).
Figure 5.

TRPV4 channel inhibition reduces IK and SK channel current densities. A, Representative whole-cell current density elicited by the 200-ms −80 to +60 mV voltage-ramp protocol. Following stable baseline recordings (control), HC (500 nM) was used to inhibit TRPV4 channels. Sequential applications of paxilline (500 nM), TRAM-34 (1 μM), and apamin (200 nM) were used to selectively inhibit BK, IK, and SK channels, respectively. B, Bar graph showing BK, IK, and SK channel current densities in the presence of HC, normalized to baseline whole-cell current density (control in A). Normalized results were calculated from the steady-state current density recordings at +30 mV. Dashed lines indicate normalized averages of each channel current density in the absence of HC, as shown in Figure 2F. Asterisks indicate statistical significance (P < 0.05). C–F, The effect of HC (C), paxilline (D), TRAM-34 (E), and apamin (F) on whole-cell current density isolated from digital subtraction of the traces shown in A. Averaged membrane capacitance was 22.3 ± 4.5 pF. BK, IK, SK: large-, intermediate-, and small-conductance KCa (Ca2+-activated potassium) channels, respectively; HC: HC067047; TRAM-34: 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole; TRPV4: transient receptor potential vanilloid 4.
The contributions of IK and SK to whole-cell currents were calculated by normalizing their respective isolated current to the baseline whole-cell currents obtained before the application of HC. In the presence of HC to block TRPV4 channels, both IK and SK channel contributions (Fig. 5B, n = 7) were significantly reduced, as compared to those in the absence of HC (dotted lines in Fig. 5B; P < 0.05). Furthermore, consistent with results from TRPV4 channel activation, the normalized BK channel contribution was not affected in the presence of HC (Fig. 5B, n = 7; P = 0.29). Taken together, our electrophysiological results indicate that TRPV4 channels are functionally coupled with both IK and SK, but not with BK, channels. Thus, Ca2+ influx via TRPV4 channels selectively activates IK and SK channels in PMVECs.
14,15-EET-dependent activation of TRPV4 channels increases endothelial permeability in isolated lung
To assess whether 14,15-EET affects lung endothelial permeability in a TRPV4-dependent fashion, we measured Kf in lungs isolated from wild-type and TRPV4 KO mice. While Kf was stable in lungs treated with vehicle alone, addition of 3 μM 14,15-EET, an indirect activator of TRPV4 channels, to the recirculating perfusate increased Kf 2.9-fold in wild-type mouse lungs (Fig. 6). This 14,15-EET-induced increase in permeability was completely abolished by pretreatment of lung with RR, which unidirectionally blocks Ca2+ influx via TRPV channels, or by using lungs from TRPV4 KO mice. Therefore, the endothelial permeability increase induced by 14,15-EET is dependent on TRPV4 channel activation, consistent with our previous studies using rat lung.1,6 Collectively, these results support the notion that Ca2+ entry via TRPV4 channels is required for EET-induced increases in endothelial permeability in mouse lung.
Figure 6.
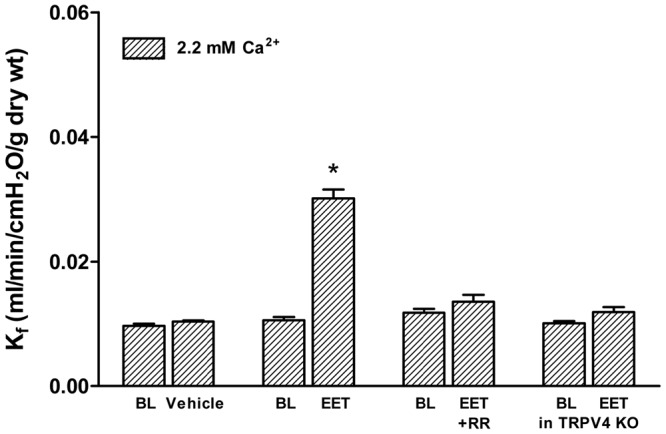
The permeability response to 14,15-EET requires TRPV4 channels. To document the requirement for TRPV4 channels, paired measurements of the filtration coefficient Kf were made in the isolated mouse lung at baseline (BL) and after treatment with vehicle (100% ethanol, n = 3) or 14,15-EET (3 μM, n = 6). The permeability response to 14,15-EET was attenuated after blockade of TRPV channels in wild-type (WT) lungs with ruthenium red (RR, 3 μM, n = 5) or in lungs from TRPV4−/− mice (TRPV4 KO, n = 5). Asterisk indicates P < 0.05 versus baseline. 14,15-EET: 14,15-epoxyeicosatrienoic acid; TRPV4: transient receptor potential vanilloid 4.
Inhibiting IK or SK, but not BK, channels attenuates 14,15-EET-induced endothelial permeability increase in isolated lung
We next asked whether 14,15-EET-induced Ca2+ influx via TRPV4 channels selectively activates IK and SK channels in intact lungs and whether activation of these KCa channels contributes to the increase in permeability. To address these questions, we utilized a low-Ca2+/Ca2+ add-back paradigm1 to identify Ca2+ entry–specific contributions to modulation of lung endothelial permeability (Fig. 7). In lungs perfused with low-Ca2+ buffer (0.02 mM), baseline Kf was no different from that measured in physiologic buffer. Further, in low-Ca2+ buffer the 14,15-EET-induced increase in Kf was markedly attenuated. The addition of Ca2+ to achieve a physiologic Ca2+ concentration of (2.2 mM) restored the 14,15-EET-induced increase in Kf. Permeability studies were repeated in the presence of KCa channel blockers, using the same paradigm. Blockade of all 3 KCa channels with the combination of charybdotoxin and apamin and pretreatment with apamin or TRAM-34 alone to block SK or IK channels, respectively, inhibited the 14,15-EET-induced increase in Kf to a similar extent. In contrast, pretreatment with the selective BK channel inhibitor iberiotoxin had no impact on the 14,15-EET-induced permeability response. These data are consistent with our electrophysiological recordings and support the notion that activation of either IK or SK amplifies the permeability response to TRPV4 channel activation.
Figure 7.

Role of KCa channels in the 14,15-EET-induced permeability response. Permeability increased significantly with 14,15-EET (3 μM, n = 5) in lungs from wild-type mice. This response was attenuated in low-Ca2+ buffer and was restored by Ca2+ add-back. Pretreatment of lungs with iberiotoxin (IbTx, 100 nM, n = 5) had no effect on the Kf response to 14,15-EET. In contrast, pretreatment of lungs with the combination of charybdotoxin (ChTx, 100 nM) and apamin (300 nM) to block all KCa channels (n = 4), with apamin alone to block SK channels (300 nM, n = 5), or with TRAM-34 (1 μM, n = 5) to block IK channels significantly attenuated this response. Note that 14,15-EET had no effect in any group in low-Ca2+ buffer. Asterisks indicate P < 0.05; only groups with 14,15-EET alone or 14,15-EET in presence of IbTx showed significant increases in permeability (response in low vs. normal Ca2+ or baseline [BL] vs. normal Ca2+), 2-way ANOVA. BK, IK, SK: large-, intermediate-, and small-conductance KCa (Ca2+-activated potassium) channels, respectively; 14,15-EET: 14,15-epoxyeicosatrienoic acid; KCa: Ca2+-activated potassium; TRAM-34: 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole.
Discussion
This study (1) compares the expression patterns of TRPV4 and KCa channels in PMVECs and PAECs, (2) provides direct evidence linking TRPV4 channel activation and the resultant Ca2+-dependent modulation of selective KCa channel activity in pulmonary endothelium, and (3) documents amplification of TRPV4-dependent increases in lung endothelial permeability by concomitant activation of IK or SK3 channels. Both PMVECs and PAECs derived from rat lung express TRPV4, BK, IK, and SK3 channel protein on Western blots, consistent with electrophysiological recordings showing the presence of their functional currents. Importantly, we determined that calcium influx via TRPV4 channels selectively activates IK and SK channels in these cells. Similarly, our functional studies in isolated mouse lung show that inhibition of IK or SK channels, but not that of BK channels, blocks TRPV4-induced increases in lung endothelial permeability.
Several methodological considerations are critical to interpretation of our in vitro electrophysiology results. First, our recording pipette solution contained 0.7 μM free [Ca2+], i.e., only enough to activate approximately half of IK and SK channels.41,42 This empirical concentration was used so that the changes in Ca2+ influx through TRPV4 channels could be reflected on whole-cell KCa current density. Second, the relatively low selectivity of TRPV4 channels for Ca2+ versus Na+ (a ratio of 6–10)4 affected electrophysiological recordings. When we used a more physiological extracellular solution containing Na+, activation of TRPV4 channels did not consistently increase whole-cell current density. We concluded that this was likely due to TPRV4-dependent Na+ influx opposing KCa-dependent K+ efflux. Consequently, to study the functional coupling of TRPV4 and KCa channels, NMDG was used to replace Na+ in the extracellular solution to minimize Na+ influx and unmask changes in whole-cell K+ currents due to activation or inhibition of TRPV4 channels. Furthermore, the exclusion of Na+ currents allowed us to identify an unexpected basal TRPV4 channel activity (Fig. 3). We speculate that the basal TRPV4 current may be due in part to the direct physical contact of patch glass on PMVECs as TRPV4 channels in endothelial cells are activated by mechanical stress.43 Third, paxilline and iberiotoxin were used to block BK channels in cell recordings and functional studies, respectively. Both of these BK inhibitors bind to the S5-S6 loop and block the pore of BK channels, albeit with different binding affinity—Kd (the distribution coefficient) for iberiotoxin is 1 nM, while that for paxilline is 10 nM. At the concentrations used in this study, they should selectively block BK channels without any off-target effects (iberiotoxin: 100 nM;44 paxilline: 1 μM45). Finally, the involvement of endothelial SK3 channels was inferred from prior studies; however, SK1 and SK2 channels are also apamin sensitive. To rule out involvement of SK1 and SK2, we quantified the expression of SK1, SK2, and SK3 messenger RNA (mRNA), normalized to that of GAPDH (glyceraldehyde 3-phosphate dehydrogenase), using real-time polymerase chain reaction. Compared to mouse brain, used as a positive control for SK1 and SK2, PMVECs expressed SK3 mRNA (PMVEC/brain = 5.8%) but negligible SK1 (0.2%) and SK2 (0.01%) mRNA levels (duplicates of 3 independent samples per group). These data, along with the protein expression profile in PMVECs, support the notion that SK3 channels predominately underlie the apamin-sensitive current in our recordings.
With respect to our ex vivo isolated-lung functional studies, one might question our use of 14,15-EET instead of GSK to activate TRPV4 channels, since EETs also activate KCa channels. We and others have documented the important role TRPV4 channels play in regulating pulmonary vascular endothelial integrity, particularly within the alveolar septal capillary network, where TRPV4-mediated loss of barrier integrity leads to pulmonary edema and alveolar flooding.1,2 In isolated dog, rat, and mouse lungs, TRPV4 channels can be activated by mechanical stress due to high pulmonary venous pressure or high airway pressure, EETs, and pharmacological channel activators.1,2,36,46,47 These effects are specific to TRPV4-mediated Ca2+ entry, as permeability does not increase under these conditions in lungs from TRPV4 KO mice1,46 or when the availability of extracellular Ca2+ is limited.1 Thus, the regulation of TRPV4 in lung microvascular endothelium appears to be conserved across these species. As a result, we would argue that 14,15-EET is physiologically relevant, since the impact of mechanical stress on lung endothelial permeability requires EET synthesis.2,36 Our results in cultured rat PMVECs suggest that Ca2+ entry via TRPV4 can directly activate IK and SK currents, with little impact on BK currents. In intact mouse lung, the EET-induced permeability response is attenuated with IK or SK channel blockers, but not with a BK channel blocker. We tested the permeability response to direct TRPV4 activation with 4α-phorbol-12,13-didecanoate (4αPDD, 10 μM) alone or 4αPDD with apamin (300 nM) in isolated mouse lungs (n = 4 per group). As a result, while Kf increased (P = 0.02), on average, by 2.7- and 1.8-fold, respectively, the impact of apamin alone did not achieve statistical significance (P = 0.07). We have not assessed the impact of concomitant SK3/IK blockade in this setting. Thus, our data collectively document activation of KCa channels secondary to TRPV4 activation in cultured rat PMVECs and amplification of the TRPV4-mediated permeability response to EETs via involvement of IK and SK3 channels in isolated mouse lung. Similar regulation of IK and SK3 due to direct TRPV4 activation has been observed in endothelial cells isolated from mouse mesenteric arteries.19 Nonetheless, given the limited impact of apamin alone on the permeability response to 4αPDD, we cannot definitively invoke a requisite causal link between Ca2+ entry via TRPV4 and subsequent KCa activation in intact mouse lung.
We recently suggested that Ca2+ signals in lung microvascular endothelium are constrained within local microdomains, on the basis of our observation that Ca2+ entry via TRPV4, but not via T-type Ca2+ channel expressed in the same cells, increases lung endothelial permeability.3 The current study provides additional evidence supporting the notion that TRPV4, SK3, and IK channels in lung microvascular endothelium operate within a functional microdomain that excludes BK channels. Recent studies have indicated subcellular functional coupling between TRPV4, SK3, and IK channels in several systemic vascular beds, while their coupling of TRPV4 with BK channels is not fully characterized.19,20,31,48-50 In PMVECs, KCa channel expression profiles, as well as their functional interplay with TRPV4, had not been previously reported. In previous work, KCa activation has been reported to either enhance7 or have no impact on18 agonist-induced Ca2+ entry in systemic endothelium. In systemic endothelium, both TRPV4 and SK3 channels interact with caveolin-1, a major structural determinant of caveolar formation.51,52 However, functional studies have shown inconsistent results.14,18-20,53 Variation in the agonists used and the expression pattern of functionally linked KCa channels may underlie such divergent responses. Fleming and colleagues7 showed that KCa channel activation by EETs hyperpolarizes the membrane potential and increases the driving force for Ca2+ entry following bradykinin stimulation in rat mesenteric endothelial cells. While EET-induced hyperpolarization may plausibly be dependent on activation of IK and SK3 channels10 or BK channels,53 our results show that only IK and SK3 channels functionally connect to TRPV4 channels in rat lung microvascular endothelial cells and in intact mouse lung. The observation that GSK, a direct activator of TRPV4 channels, elicits IK and SK3 channel currents in vitro provides additional evidence that TRPV4, IK, and SK3 channels must be closely arrayed in a signaling microdomain. In contrast, functional coupling between TRPV4 and BK and SK3 channels was recently shown in distal tubules of the kidney.54 However, despite their expression in PMVECs, BK channels are neither functionally coupled to TRPV4 channels nor involved in 14,15-EET-induced lung injury. As BK channels are sensitive to both voltage and Ca2+ activation, small changes in intracellular [Ca2+] should be detected by our voltage-clamp protocol. In fact, mibefradil, a selective blocker for T-type Ca2+ channels, reduced BK current density, indicating the capability of BK channels to sense Ca2+ influx via these voltage-gated Ca2+ channels (data not shown). Notably, T-type Ca2+ channels are expressed in lung microvascular endothelium, though not coupled to regulation of endothelial permeability.3
In conclusion, our work provides support for functional coupling between TRPV4-IK and TRPV4-SK3 channels in vitro, such that the Ca2+ entry elicited by activation of TRPV4 channels is amplified by subsequent activation of both IK and SK3 channels. This would be most relevant in the setting of lung mechanical stress. Breaking this functional link limits TRPV4-mediated lung damage. This functional coupling of TRPV4 to distinct KCa channels is reminiscent of our earlier work showing distinct outcomes for the Ca2+ transients elicited by activation of TRPV4 and T-type Ca2+ channels in mouse lung alveolar septal capillaries: Ca2+ entry via TRPV4 increased lung endothelial permeability, whereas Ca2+ entry via the T-type Ca2+ channels increased P-selectin expression.3 Collectively, these studies provide strong support for discrete Ca2+ microdomains in lung alveolar septal endothelium, whereby Ca2+ entry via TRPV4 and T-type Ca2+ channels is constrained to modulate distinct downstream functional targets.
Acknowledgment
We thank the Center for Lung Biology for providing cultured pulmonary endothelial cells.
Source of Support: This work was supported by grants HL066299 (MIT) and HL102056 (MTL) from the National Institutes of Health.
Conflict of Interest: None declared.
References
- 1.Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res 2006;99(9):988–995. [DOI] [PMC free article] [PubMed]
- 2.Jian MY, King JA, Al-Mehdi AB, Liedtke W, Townsley MI. High vascular pressure–induced lung injury requires P450 epoxygenase–dependent activation of TRPV4. Am J Respir Cell Mol Biol 2008;38(4):386–392. [DOI] [PMC free article] [PubMed]
- 3.Wu S, Jian MY, Xu YC, Zhou C, Al-Mehdi AB, Liedtke W, Shin HS, Townsley MI. Ca2+ entry via α1G and TRPV4 channels differentially regulates surface expression of P-selectin and barrier integrity in pulmonary capillary endothelium. Am J Physiol Lung Cell Mol Physiol 2009;297(4):L650–L657. [DOI] [PMC free article] [PubMed]
- 4.Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol 2004;286(2):C195–C205. [DOI] [PubMed]
- 5.Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, et al. Modulation of the Ca2+ permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res 2005;97(9):908–915. [DOI] [PubMed]
- 6.Alvarez DF, Gjerde EA, Townsley MI. Role of EETs in regulation of endothelial permeability in rat lung. Am J Physiol Lung Cell Mol Physiol 2004;286(2):L445–L451. [DOI] [PubMed]
- 7.Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, Haendeler J, et al. Epoxyeicosatrienoic acids regulate Trp channel–dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler Thromb Vasc Biol 2007;27(12):2612–2618. [DOI] [PubMed]
- 8.Campbell WB, Deeter C, Gauthier KM, Ingraham RH, Falck JR, Li PL. 14,15-Dihydroxyeicosatrienoic acid relaxes bovine coronary arteries by activation of KCa channels. Am J Physiol Heart Circ Physiol 2002;282(5):H1656–H1664. [DOI] [PubMed]
- 9.Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 2005;97(12):1270–1279. [DOI] [PubMed]
- 10.Weston AH, Félétou M, Vanhoutte PM, Falck JR, Campbell WB, Edwards G. Bradykinin-induced, endothelium-dependent responses in porcine coronary arteries: involvement of potassium channel activation and epoxyeicosatrienoic acids. Br J Pharmacol 2005;145(6):775–784. [DOI] [PMC free article] [PubMed]
- 11.Wang XL, Ye D, Peterson TE, Cao S, Shah VH, Katusic ZS, Sieck GC, Lee HC. Caveolae targeting and regulation of large conductance Ca2+-activated K+ channels in vascular endothelial cells. J Biol Chem 2005;280(12):11656–11664. [DOI] [PubMed]
- 12.Gauthier KM, Liu C, Popovic A, Albarwani S, Rusch NJ. Freshly isolated bovine coronary endothelial cells do not express the BKCa channel gene. J Physiol 2002;545(3):829–836. [DOI] [PMC free article] [PubMed]
- 13.Vang A, Mazer J, Casserly B, Choudhary G. Activation of endothelial BKCa channels causes pulmonary vasodilation. Vasc Pharmacol 2010;53(3–4):122–129. [DOI] [PubMed]
- 14.Grgic I, Kaistha BP, Hoyer J, Köhler R. Endothelial Ca2+-activated K+ channels in normal and impaired EDHF-dilator responses—relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol 2009;157(4):509–526. [DOI] [PMC free article] [PubMed]
- 15.Papassotiriou J, Köhler R, Prenen J, Krause H, Akbar M, Eggermont J, Paul M, Distler A, Nilius B, Hoyer J. Endothelial K+ channel lacks the Ca2+ sensitivity–regulating β subunit. FASEB J 2000;14(7):885–894. [PubMed]
- 16.Brähler S, Kaistha A, Schmidt VJ, Wölfle SE, Busch C, Kaistha BP, Kacik M, et al. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 2009;119(17):2323–2332. [DOI] [PubMed]
- 17.Hoebel BG, Kostner GM, Graier WF. Activation of microsomal cytochrome P450 mono-oxygenase by Ca2+ store depletion and its contribution to Ca2+ entry in porcine aortic endothelial cells. Br J Pharmacol 1997;121(8):1579–1588. [DOI] [PMC free article] [PubMed]
- 18.Cohen KD, Jackson WF. Membrane hyperpolarization is not required for sustained muscarinic agonist-induced increases in intracellular Ca2+ in arteriolar endothelial cells. Microcirculation 2005;12(2):169–182. [DOI] [PMC free article] [PubMed]
- 19.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012;336(6081):597–601. [DOI] [PMC free article] [PubMed]
- 20.Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ, Dora KA. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc Natl Acad Sci USA 2012;109(44):18174–18179. [DOI] [PMC free article] [PubMed]
- 21.Ma X, Du J, Zhang P, Deng J, Liu J, Lam FF, Li RA, Huang Y, Jin J, Yao X. Functional role of TRPV4-KCa2.3 signaling in vascular endothelial cells in normal and streptozotocin-induced diabetic rats. Hypertension 2013;62(1):134–139. [DOI] [PubMed]
- 22.Yap FC, Taylor MS, Lin MT. Ovariectomy-induced reductions in endothelial SK3 channel activity and endothelium-dependent vasorelaxation in murine mesenteric arteries. PLoS ONE 2014;9:e104686. doi:10.1371/journal.pone.0104686. [DOI] [PMC free article] [PubMed]
- 23.Bardou O, Trinh NT, Brochiero E. Molecular diversity and function of K+ channels in airway and alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 2009;296(2):L145–L155. [DOI] [PubMed]
- 24.Bonnet S, Archer SL. Potassium channel diversity in the pulmonary arteries and pulmonary veins: implications for regulation of the pulmonary vasculature in health and during pulmonary hypertension. Pharmacol Ther 2007;115(1):56–69. [DOI] [PubMed]
- 25.Leroy C, Privé A, Bourret JC, Berthiaume Y, Ferraro P, Brochiero E. Regulation of ENaC and CFTR expression with K+ channel modulators and effect on fluid absorption across alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 2006;291(6):L1207–L1219. [DOI] [PubMed]
- 26.Thompson-Vest N, Shimizu Y, Hunne B, Furness JB. The distribution of intermediate-conductance, calcium-activated, potassium (IK) channels in epithelial cells. J Anat 2006;208(2):219–229. [DOI] [PMC free article] [PubMed]
- 27.Gu YT, Qin LJ, Qin X, Xu F. The molecular mechanism of dexamethasone-mediated effect on the blood-brain tumor barrier permeability in a rat brain tumor model. Neurosci Lett 2009;452(2):114–118. [DOI] [PubMed]
- 28.Hu J, Yuan X, Ko MK, Yin D, Sacapano MR, Wang X, Konda BM, et al. Calcium-activated potassium channels mediated blood-brain tumor barrier opening in a rat metastatic brain tumor model. Mol Cancer 2007;6:22. [DOI] [PMC free article] [PubMed]
- 29.Ningaraj NS, Rao M, Hashizume K, Asotra K, Black KL. Regulation of blood-brain tumor barrier permeability by calcium-activated potassium channels. J Pharmacol Exp Ther 2002;301(3):838–851. [DOI] [PubMed]
- 30.Stevens T, Thompson WJ. Regulation of pulmonary microvascular endothelial cell cyclic adenosine monophosphate by adenylyl cyclase: implications for endothelial barrier function. Chest 1999;116(suppl. 1):32S–33S. [DOI] [PubMed]
- 31.Parker JC, Hashizumi M, Kelly SV, Francis M, Mouner M, Meyer AL, Townsley MI, Wu S, Cioffi DL, Taylor MS. TRPV4 calcium entry and surface expression attenuated by inhibition of myosin light chain kinase in rat pulmonary microvascular endothelial cells. Physiol Rep 2013;1:e00121. doi:10.1002/phy2.121. [DOI] [PMC free article] [PubMed]
- 32.Wu S, Haynes J Jr., Taylor JT, Obiako BO, Stubbs JR, Li M, Stevens T. Cav3.1 (α1G) T-type Ca2+ channels mediate vaso-occlusion of sickled erythrocytes in lung microcirculation. Circ Res 2003;93(4):346–353. [DOI] [PubMed]
- 33.King J, Hamil T, Creighton J, Wu S, Bhat P, McDonald F, Stevens T. Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc Res 2004;67(2):139–151. [DOI] [PubMed]
- 34.Stevens T. Functional and molecular heterogeneity of pulmonary endothelial cells. Proc Am Thorac Soc 2011;8(6):453–457. [DOI] [PubMed]
- 35.Lin MT, Adelman JP, Maylie J. Modulation of endothelial SK3 channel activity by Ca2+-dependent caveolar trafficking. Am J Physiol Cell Physiol 2012;303(3):C318–C327. [DOI] [PMC free article] [PubMed]
- 36.Hamanaka K, Jian MY, Weber DS, Alvarez DF, Townsley MI, Al-Mehdi AB, King JA, Liedtke W, Parker JC. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol 2007;293(4):L923–L932. [DOI] [PubMed]
- 37.Parker JC, Townsley MI. Evaluation of lung injury in rats and mice. Am J Physiol Lung Cell Mol Physiol 2004;286(2):L231–L246. [DOI] [PubMed]
- 38.Sullivan MN, Francis M, Pitts NL, Taylor MS, Earley S. Optical recording reveals novel properties of GSK1016790A-induced vanilloid transient receptor potential channel TRPV4 activity in primary human endothelial cells. Mol Pharmacol 2012;82(3):464–472. [DOI] [PMC free article] [PubMed]
- 39.Willette RN, Bao W, Nerurkar S, Yue T, Doe CP, Stankus G, Turner GH, et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: part 2. J Pharmacol Exp Ther 2008;326(2):443–452. [DOI] [PubMed]
- 40.Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, Hayward NJ, et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci USA 2010;107(44):19084–19089. [DOI] [PMC free article] [PubMed]
- 41.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 1998;395(6701):503–507. [DOI] [PubMed]
- 42.Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci USA 1997;94(21):11651–11656. [DOI] [PMC free article] [PubMed]
- 43.Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA 2003;100(23):13698–13703. [DOI] [PMC free article] [PubMed]
- 44.Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem 1990;265(19):11083–11090. [PubMed]
- 45.Sanchez M, McManus OB. Paxilline inhibition of the alpha-subunit of the high-conductance calcium-activated potassium channel. Neuropharmacology 1996;35(7):963–968. [DOI] [PubMed]
- 46.Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian M, Costell M, et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 2012;4:159ra148. doi:10.1126/scitranslmed.3004276. [DOI] [PubMed]
- 47.Yin J, Hoffmann J, Kaestle SM, Neye N, Wang L, Baeurle J, Liedtke W, et al. Negative-feedback loop attenuates hydrostatic lung edema via a cGMP-dependent regulation of transient receptor potential vanilloid 4. Circ Res 2008;102(8):966–974. [DOI] [PubMed]
- 48.Hannah RM, Dunn KM, Bonev AD, Nelson MT. Endothelial SKCa and IKCa channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab 2011;31(5):1175–1186. [DOI] [PMC free article] [PubMed]
- 49.Earley S. Endothelium-dependent cerebral artery dilation mediated by transient receptor potential and Ca2+-activated K+ channels. J Cardiovasc Pharmacol 2011;57(2):148–153. [DOI] [PubMed]
- 50.Sayner SL, Alexeyev M, Dessauer CW, Stevens T. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ Res 2006;98(5):675–681. [DOI] [PubMed]
- 51.Absi M, Burnham MP, Weston AH, Harno E, Rogers M, Edwards G. Effects of methyl β-cyclodextrin on EDHF responses in pig and rat arteries; association between SKCa channels and caveolin-rich domains. Br J Pharmacol 2007;151(3):332–340. [DOI] [PMC free article] [PubMed]
- 52.Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, Rodella LF, et al. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation 2008;117(8):1065–1074. [DOI] [PubMed]
- 53.Baron A, Frieden M, Bény JL. Epoxyeicosatrienoic acids activate a high-conductance, Ca2+-dependent K+ channel on pig coronary artery endothelial cells. J Physiol 1997;504(3):537–543. [DOI] [PMC free article] [PubMed]
- 54.Berrout J, Mamenko M, Zaika OL, Chen L, Zang W, Pochynyuk O, O’Neil RG. Emerging role of the calcium-activated, small conductance, SK3 K+ channel in distal tubule function: regulation by TRPV4. PLoS ONE 2014;9:e95149. doi:10.1371/journal.pone.0095149. [DOI] [PMC free article] [PubMed]