

Abstract Abstract
Although there are many studies focusing on the molecular pathways underlying lung vascular morphogenesis, the extracellular matrix (ECM)–dependent regulation of mesenchymal cell differentiation in vascular smooth muscle development needs better understanding. In this study, we demonstrate that the paired related homeobox gene transcription factor Prx1 maintains the elastic ECM properties, which are essential for vascular smooth muscle precursor cell differentiation. We have found that Prx1null mouse lungs exhibit defective vascular smooth muscle development, downregulated elastic ECM expression, and compromised transforming growth factor (TGF)–β localization and signaling. Further characterization of ECM properties using decellularized lung ECM scaffolds derived from Prx1 mice demonstrated that Prx1 is required to maintain lung ECM stiffness. The results of cell culture using stiffness-controlled 2-D and 3-D synthetic substrates confirmed that Prx1-dependent ECM stiffness is essential for promotion of smooth muscle precursor differentiation for effective TGF-β stimulation. Supporting these results, both decellularized Prx1null lung ECM and Prx1WT (wild type) ECM scaffolds with blocked TGF-β failed to support mesenchymal cell to 3-D smooth muscle cell differentiation. These results suggest a novel ECM-dependent regulatory pathway of lung vascular development wherein Prx1 regulates lung vascular smooth muscle precursor development by coordinating the ECM biophysical and biochemical properties.
Keywords: extracellular matrix properties, smooth muscle cell differentiation, 3-D culture, vascular morphogenesis, transcription factor
Proper vascular morphogenesis is critical for the development and regeneration of functional lung tissue and is required for the lungs’ main function of oxygenation. Recently, studies in stem cell differentiation exploring new pathways to morphogenesis have shown more dynamic interactions between microenvironmental cues and cells in this process. However, while intense research has been performed focusing on molecular networks regulating vascular morphogenesis, the mechanisms underlying extracellular matrix (ECM)–dependent vascular cell differentiation are not well understood.
The ECM is composed of a large collection of biochemically distinct components with different physical and biochemical properties.1 When constructed in an orderly manner, the ECM components possess unique physical, biochemical, and biomechanical properties that are essential for regulating cell behavior beyond providing structural support.
Growing evidence demonstrates that specific biophysical properties of ECM stiffness play an essential role in guiding stem/progenitor cell differentiation.2-5 For instance, the differentiation of human mesenchymal stem cells into an osteogenic phenotype was shown to be dependent on ECM stiffness.2,4 In contrast, biochemical properties of the ECM pertaining to its indirect or direct signaling capabilities allow cells to respond to microenvironmental cues using various signaling cascades. For example, the ECM can bind to myriad growth factors,6,7 including transforming growth factor (TGF)–β,8 and by doing so the ECM regulates growth factor distribution, activation, and presentation to cells in a spatially organized and regulated manner.9 During embryonic lung development, the biochemical properties of the ECM is essential for lung morphogenesis, which requires temporal and spatial gradients of growth factors surrounding the developing airway and vessels.8,10 These findings indicate that the signals from the environment surrounding the developing tissue play a pivotal role in guiding stem/progenitor cell fate determination and differentiation, while the role played by the ECM in cell signaling has not yet been well explained.
In the lungs, elastin and its associated microfibrils (i.e., fibrillins) are the major components of the lung ECM, and they play a pivotal role in lung homeostasis. Elastic ECM not only provides structural support for lung tissue,11,12 it appears to play a regulatory role in smooth muscle cell (SMC) proliferation and differentiation.13,14 Defects in both elastin and fibrillins result in vascular SMC growth abnormalities in human disease and animal models.8,13-16 Furthermore, TGF-β, a known regulator of stem/progenitor cell to vascular SMC differentiation,17,18 requires a level of ECM stiffness for latent form activation.19,20 In addition, fibrillin (Fbn) 1 modulates local TGF-β activation in the lungs by binding to the latent form of TGF-β.8 While this evidence highlights the pivotal role played by the ECM in lung vascular progenitor cell differentiation and functional maturation, the mechanism whereby elastic ECM regulates embryonic vascular cell differentiation is obscure.
To elucidate the mechanisms of elastic ECM-dependent lung vascular smooth muscle development, we focused on the homeobox gene transcription factor Prx1. In humans, several reports have suggested that agnathia-otocephaly complex (AGOTC) could be caused by a heterozygous mutation in the PRRX1 gene (167420) on chromosome 1q24.21-24 AGOTC is a rare, often lethal malformation characterized by absence or hypoplasia of the mandible, microstomia, hypoglossia/aglossia, and variable anterior midline fusion of the ears (melotia, synotia) that exhibits many similarities to the hypoplastic features of Prx1null homozygote mice,25 which exhibit skeletal defects affecting mandible, limbs, and vertebrae as well as vascular abnormalities and neonatal lethality. In addition, it has been shown that Prx1 colocalized with fibrillins within the developing elastic vascular wall during embryonic development.26,27 This suggests a tight association between Prx1 and fibrillins during vascular development, as a fibrillin mutation leads to abnormal morphogenesis in Marfan syndrome.28,29 Taken together, these lines of evidence suggest that Prx1 may regulate the composition of the elastic ECM and thereby have an effect on vascular development. Therefore, we hypothesized that Prx1 controls lung vascular smooth muscle development through its effects on the properties of the ECM. To investigate this hypothesis, we characterized the biochemical and biophysical ECM properties of Prx1WT (wild type) and Prx1null lungs and examined the effects of Prx1-dependent ECM on lung vascular smooth muscle development. The outcome of this study should contribute greatly to a better understanding of ECM-dependent embryonic lung tissue development and support the development of more effective therapies for tissue regeneration in pathological conditions of both newborns and adults.
Methods
Animals
Prx1 gene knockout mice were generated as described elsewhere.30 Polymerase chain reaction (PCR) was used to identify the three different expected genotypes: Prx1WT (wild type), Prx1 heterozygous, and Prx1null (homozygous-null). Genomic DNA from mouse tail was used in PCRs with the combination of primer pairs for the Prx1WT allele and the Prx1lacZ allele. All animal experiments were conducted in accordance with current National Institutes of Health (Bethesda, MD) guidelines.
Cell culture
C3H10T1/2 (10T1/2) cells (ATCC) were used as mesenchymal SMC precursor cells.17 Except in the case of TGF-β stimulation experiments, cells were maintained in 10% fetal bovine serum containing Dulbecco’s modified Eagle medium. Cells were cultured at 37°C in a humidified atmosphere with 5% CO2 until used for cellular experiments.
Transient transfection
Cytomegalovirus plasmid containing Prx1 construct30 and control vector were used to examine the downstream genes regulated by Prx1.
Decellularization of lungs
Intact fetal mouse lungs from E15.5 to D0 from Prx1WT and Prx1null mice were treated with hypotonic Tris buffer (pH 8) followed by hypertonic Tris buffer containing 1% Triton X-100 (pH 8).31 After decellularization, lung scaffolds were processed for characterization or left unfixed for biomechanical testing. Intact lungs were used as controls for decellularization experiments.
Polyacrylamide (PAA) gels
PAA gels of variable stiffness were prepared on glass coverslips using an established method.32 After being treated with sulfo-SANPAH (Thermo Fisher Scientific, Waltham, MA), collagen I (0.2 mg/mL; Advanced Biomatrix, Carlsbad, CA) was crosslinked to the gels before cells were plated.
Synthetic 3-D scaffolds (electrospun hyaluronic acid scaffolds)
Methacrylated hyaluronic acid (MeHA) was synthesized as described elsewhere.33,34 Briefly, MeHA was cospun with the carrier polymer PEO (molecular weight, 900 kDa; Sigma-Aldrich, St. Louis, MO) as described elsewhere, 34,35 and fiber size and view morphology were quantified by scanning electron microscopy (SEM; 7500F HRSEM; Jeol, Glen Ellyn, IL). After polymerization was accomplished by exposing ∼10 mW/cm2 365-nm light (Omnicure S1000 UV Spot Cure System; Exfo, Quebec City, QC), scaffolds were immersed in deionized H2O and washed to reach equilibrium swelling. The mechanical properties of MeHA gels were controlled by percent methacrylation or concentration of polymer used, and their stiffness was determined by atomic force microscopy (AFM).
Immunohistochemistry
The following primary antibodies were used: anti-Fbn1 and anti-Fbn2 antibodies (gift from R. Mecham, Washington University, St. Louis, MO), anti-tropoelastin antibody (Abcam, Cambridge, UK), anti-α–smooth muscle actin (SMA) antibody (Sigma-Aldrich), anti-γ-SMA antibody (Santa Cruz Biotechnology, Dallas, TX), anti-SM22α antibody (Abcam), anti–smooth muscle myosin heavy chain (SMMHC) antibody (Abcam), anti-TGF-β1 antibody (Promega, Madison, WI), and anti-TGF-β2 antibody (R&D Systems, Minneapolis, MN). For staining controls, equivalent concentrations of isotype-matched immunoglobulin (Ig) G were used. Sections were blocked for endogenous peroxidase activity, treated for antigen retrieval, and blocked for nonspecific antibody binding before being incubated with primary antibodies. The following dilutions were used for primary antibodies: Fbn1 and Fbn2 (1∶100), tropoelastin (1∶150), α-SMA (1∶400), γ-SMA (1∶50), SM22α (1∶100), and SMMHC (1∶100). Antigen detection was performed using species-specific secondary antibodies in conjunction with a Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA), followed by the 3,3′-diaminobenzidine reaction (Vector Laboratories), counterstaining with hematoxyline (Vector Laboratories), and processing for bright-field imaging (90i microscope; Nikon, Tokyo).
Immunofluorescence staining
Cells or scaffolds were fixed in 4% paraformaldehyde and then permeabilized with 0.2% Triton X-100–containing phosphate‐buffered saline. Next, materials were treated with blocking buffer and incubated with the following primary antibodies. To examine SMC differentiation, antibodies specific for α-SMA, γ-SMA, SM22α, and SMMHC were used. For detection of ECM components, antibodies specific for Fbn1, Fbn2, tropoelastin, and TGF-β1 were used. Species-specific fluorescent-conjugated antibodies (Molecular Probes, Thermo Fisher Scientific) were used for antigen detection. Stained materials were examined using a 90i fluorescence microscope (Nikon) or a LSM 710 two-photon confocal microscope (Zeiss, Jena; CDB Imaging Core at the University of Pennsylvania, Philadelphia, PA).
Electron microscopy
Transmission electron microscopy (TEM)
Intact or decellularized embryo lungs were processed for TEM using a modification of a protocol described elsewhere.30 Ultrathin sections of Epon 812–embedded (EMS, Hatfield, PA) specimens were stained with ulanyl acetate and lead citrate before being examined under a 1010 electron microscope (Jeol).
SEM
Lung materials were fixed, dehydrated, and then chemically dried using hexamethyldisilazane. Dried specimens were Palladium sputter coated before being examined under an XL20 ESEM (Philips, Amsterdam).
Western immunoblotting
Protein was extracted from lung tissue and concentrations were determined using the Pierce Protein Assay Reagent (Thermo Fisher Scientific). Ten micrograms of protein was separated on an 8%–16% Tris-HCl sodium dodecyl sulfate–PAA gel and then transferred to polyvinylidene fluoride membranes. After being incubated with blocking buffer, membranes were incubated with primary antibodies using the following: Fbn1 and Fbn2 (gift of R. Mecham), tropoelastin (Abcam), TGF-β1 (Promega), phosphorylated Smad2 (Cell Signaling Technology, Danvers, MA), and total Smad2/3 (Cell Signaling Technology). After incubation with species-specific secondary antibodies conjugated with horseradish peroxidase, protein was visualized using an enhanced chemiluminescence kit and examined using a Fuji imager (Fujifilm, Tokyo/GE Healthcare, Chalfont St Giles, UK). β-Actin (Sigma-Aldrich) was used as a loading control.
Real-time quantitative PCR
Total RNA was extracted from lung tissue or cells, and complementary DNA (cDNA) was then synthesized and processed for SYBR green–based real-time quantitative PCR analysis using the LightCycler 480 instrument (Roche, Basel). All target gene expressions were normalized to a housekeeping gene.
Stiffness measurements
Indentation testing
The stiffness of decellularized lung scaffolds was measured using a custom-made microindenter.36 Briefly, lung scaffolds were brought into contact with the probe (blunt-ended tungsten wire, 550 μm in diameter), and the magnitudes of indentation and force were measured. The average indentation and force on the sample were derived and used to quantify sample stiffness by the method of Hayes et al.37
Mechanical testing by AFM
To measure the stiffness of HA scaffolds at the tissue level, synthesized HA scaffolds were rehydrated, and the mechanical force was measured by AFM. The mechanical force was calculated using a cantilever spring constant determined by the vendor’s thermal method.
TGF-β stimulation experiments
Cells were incubated with serum-free medium overnight before being treated with recombinant TGF-β1 (Cell Signaling Technology).17 Either cells were fixed for immunostaining or total RNA was harvested for real-time PCR.
TGF-β function-blocking experiments
TGF-β blocking
Ten micrograms of neutralizing antibody per milliliter against TGF-β1 (R&D Systems) was added to the medium when 10T1/2 cells were seeded in the decellularized scaffolds. As a control, an identical concentration of chicken IgY (R&D Systems) was added to the medium. 10T1/2 cell differentiation was examined by immunostaining, and intensity was quantified using ImageJ software (National Institutes of Health).
TGF-βR1 signaling blocking
10T1/2 cells were serum starved overnight before being incubated with 10 mM SB431542 (Cell Signaling Technology) or 10 mM LY364947 (Sigma-Aldrich) with or without TGF-β1 stimulation and then processed for immunostaining or real-time PCR to examine canonical TGF-β signaling pathways. As a control, vehicle alone was added to the medium.
Statistics
All experiments were performed in triplicate unless otherwise stated. Statistical analysis was performed using Prism software (GraphPad, San Diego, CA) to compare the groups. One-way analysis of variance with the Newman-Keuls posttest was used to compare more than two groups, and the unpaired Student t test was used to compare two groups. A P value of less than 0.05 was considered significant. All values are shown as means ± SD unless otherwise specified.
Detailed methods are available in the supplement.
Results
Prx1 regulates expression of elastic ECM components during lung development
We first examined expression of elastic ECM components in Prx1WT and Prx1null lungs to determine whether Prx1 regulates expression of the major elastic ECM components within developing lungs. We focused on E15.5 to newborn (D0) stages since the expression level of lung ECM components is higher during the late gestational period. The expression of tropoelastin, Fbn1, and Fbn2 was significantly lower in Prx1null lungs than in Prx1WT lungs at both the protein (Figs. 1A, 1B, S1) and the messenger RNA (mRNA; Fig. 1C) level. Immunohistochemical studies also suggested that tropoelastin protein expression was significantly reduced around the developing vessels and in the adjacent mesenchyme (Fig. 1A, bottom) as well as around airways. Collectively, these data suggest that Prx1 controls the composition of the elastic ECM in developing lung vessels. To investigate further, we examined the ultrastructure of vessels in E17.5 embryo lungs using TEM. Strikingly, the results showed that, whereas Prx1WT lungs possessed a dense, solid elastic lamina, Prx1null lungs displayed a thinner, patchy, discontinuous elastic lamina (Fig. 1D). To examine whether Prx1 directly affects the expression of these elastic ECM components, a full-length Prx1 cDNA-containing plasmid30 was transiently transfected to 10T1/2 cells, a mesenchymal precursor cell line, and mRNA expression of these elastic ECM components was examined (Fig. S1B–S1D). Overexpression of the Prx1 gene (Fig. S1B) was followed by increased mRNA expression of tropoelastin (Fig. S1C; 2.968e6 ± 1.175e7 vs. 0.9973e7 ± 0.9e9) and Fbn1 (Fig. S1D; 4.961e4 ± 1.566e4 vs. 3.234e4 ± 0.150e4). In support of the above results, because the promoter regions of tropoelastin, Fbn1, and Fbn2 contain conservative Prx1-binding sequences, it is plausible that Prx1 directly binds to and regulates expression of these elastic ECM components.
Figure 1.

Prx1 controls expression of tropoelastin (TE), fibrillin (Fbn) 1, and Fbn2 during lung vascular development. A, Immunohistochemical staining for TE, Fbn1, and Fbn2 in lung sections derived from E15.5 Prx1WT (left) and Prx1null (right) mice. Arrows indicate developing vessels, in which TE expression is observed in Prx1WT mice but not in Prx1null mice. Brown color indicates positive staining; hematoxylin staining in blue is nuclear counterstaining. B, Western immunoblotting for Fbn1, Fbn2, and TE protein in Prx1WT and Prx1null lungs isolated at E17.5. β-Actin was used as a loading control. C, Real-time polymerase chain reaction for TE and Fbn2 messenger RNA expression in Prx1WT and Prx1null lungs isolated at E17.5. Target gene expression was normalized to GAPDH expression. One asterisk indicates P < 0.05, and two asterisks indicate P < 0.01. D, Semithin section images (top) and transmission electron microscope images (bottom) of pulmonary arteries of E17.5 Prx1WT (left) and Prx1null (right) mouse lungs. Arrows indicate the internal elastic lamina. Scale bars = 5 μm. Western immunoblotting was performed using at least three different litters. WT: wild type; EC: endothelial cell; SMC: smooth muscle cell.
Prx1null lungs have defective vascular SMC differentiation
Since elastic ECM properties are essential for vascular SMC differentiation,13,14 we examined vascular SMC differentiation in Prx1WT and Prx1null lungs. Immunostaining of E15.5 and D0 lungs for α-SMA (Fig. 2A, left; Fig. 2B), γ-SMA (Fig. 2A, middle), and SM22α (Fig. 2A, right) showed that SMCs within developing pulmonary vessels in Prx1WT mice strongly expressed SMC markers (i.e., α-SMA, γ-SMA, and SM22α) and possessed a spindle-shaped morphology (Fig. 2B). In contrast, Prx1null lungs expressed reduced levels of the SMC markers examined, and vascular wall cells appeared to possess a rounded, more primitive morphology (Fig. 2B). Furthermore, whole-mount immunostaining of E17.5 lungs with α-SMA clearly showed that 3-D development and differentiation of vascular SMCs was deficient in Prx1null lungs compared with Prx1WT lungs (Fig. 2C). Consistent with the staining results, mRNA expression of α-SMA (Fig. 2D), SM22α (Fig. 2E), and SMMHC (Fig. 2F) in Prx1null lungs at this stage was significantly reduced compared with that in Prx1WT lungs.
Figure 2.

Prx1 promotes vascular smooth muscle cell differentiation. A, Immunohistochemistry for α–smooth muscle actin (SMA; left), γ-SMA (middle), and SM22α (right) in lungs of E15.5 (top) and D0 (bottom) Prx1WT and Prx1null mice. Positive staining by 3,3′-diaminobenzidine (DAB) reaction is brown; nuclear counterstaining is blue. Arrows point to vessel walls. Scale bars = 20 μm. B, Higher magnification of α-SMA immunohistochemistry on D0 Prx1WT and Prx1null mice lung sections. Positive staining by DAB reaction is brown; nuclear counterstaining is blue. Arrows point to the smooth muscle layer of vessels. Scale bar = 20 μm. C, 3-D projection images of whole-mount lung immunofluorescence staining for α-SMA in E17.5 lungs from Prx1WT (left) and Prx1null (right) mice. Scale bar = 20 μm. D–F, Real-time polymerase chain reaction for α-SMA (D), SM22α (E), and smooth muscle myosin heavy chain (SMMHC; F) messenger RNA expression in E17.5 lungs isolated from Prx1WT and Prx1null mice. Target gene expression was normalized to GAPDH expression. Three asterisks indicate P < 0.001. WT: wild type.
TGF-β signaling was downregulated in Prx1null lungs
It was recently reported that ECM properties regulate the activity of TGF-β1 signaling,19,38 a potent inducer of vascular SMC differentiation.8 Since Prx1-deficient mice demonstrated compromised ECM composition and deficient SMC differentiation, we examined TGF-β expression and signaling in Prx1WT and Prx1null lungs. Immunohistochemistry on Prx1WT and Prx1null lung sections showed that both TGF-β1 and TGF-β2 strongly localize in and around the developing vessels in Prx1WT lungs (Fig. 3A), while in Prx1null lungs both TGF-β1 and TGF-β2 are less expressed in the vicinity of vessels and appear frequently in parenchyma (Fig. 3A). Interestingly, Western blotting and real-time PCR showed that total TGF-β1 expression in Prx1WT and Prx1null lungs was not significantly different at the protein and mRNA levels (Fig. 3B, 3C); however, phosphorylation of the TGF-β downstream signaling mediator Smad2 (Fig. 3D) and expression of the TGF-β-regulated protein plasminogen activator inhibitor type 1 (PAI-1; Fig. 3E) were reduced in Prx1null lungs compared with Prx1WT lungs. These results indicate that Prx1 regulates TGF-β signaling, potentially because of its effects on lung matrix.
Figure 3.

Prx1 regulates transforming growth factor (TGF)–β signaling in embryonic lungs. A, Immunohistochemical staining for TGF-β1 and TGF-β2 in lung sections derived from E15.5 (top) and D0 (bottom) Prx1WT (left) and Prx1null (right) mice. Brown indicates positive staining of antibodies. While TGF-β localization is restricted to the developing vessel area (arrows) in Prx1WT lungs, in Prx1null lungs TGF-β expression is observed in areas other than the vessel wall vicinity (arrowheads). Hematoxylin staining (blue) is used for nuclear counterstaining. Scale bar = 40 μm. B, Western blotting for TGF-β1 protein expression in E17.5 lungs from Prx1WT and Prx1null mice. C, Real-time polymerase chain reaction (PCR) for TGF-β1 messenger RNA (mRNA) expression in E17.5 lungs from Prx1WT and Prx1null mice. D, Western blotting for phosphorylated Smad2 (P-Smad2; top) and total Smad 2/3 (middle) in E17.5 lungs from Prx1WT and Prx1null mice. E, Real-time PCR for plasminogen activator inhibitor type 1 (PAI-1) mRNA expression in E17.5 lungs from Prx1WT and Prx1null mice. For Western immunoblotting (B, D), β-actin was used as a loading control. For mRNA expression by real-time PCR (C, E), target gene expression was normalized to GAPDH. Two asterisks indicate P < 0.01. WT: wild type.
Prx1 controls the biomechanical properties of lung ECM
Since our data suggested a strong link between Prx1-dependent formation of elastic ECM and development of the lung vasculature, we further examined the ECM in Prx1WT and Prx1null lungs. To characterize the ECM derived from Prx1WT and Prx1null lungs and to elucidate the role played by Prx1-dependent ECM properties in vascular SMC differentiation, we developed a system to decellularize fetal mouse lungs in which proper removal of cells from the tissue leaves behind a complex ECM and associated proteins, including growth factors.39 Decellularized scaffolds have proven to be useful tools for studying cell signaling in an appropriate 3-D ECM environment as well as for tissue engineering studies designed for clinical applications.40 Therefore, we used the decellularized scaffolds to examine the architectural, biomechanical, and biochemical properties of the ECM in Prx1null lungs.
First, we used hematoxylin nuclear staining on intact and decellularized frozen lung sections and confirmed that cells were removed successfully by the decellularization process (Fig. 4A). Next, we immunostained intact and decellularized Prx1WT lungs to determine whether decellularized scaffolds retain ECM components. The results showed that the decellularized scaffolds clearly retained Fbn2, tropoelastin, and TGF-β1 (Fig. 4B) as well as basement component laminin (Fig. S2). We also examined the stiffness of intact and decellularized Prx1WT lung scaffolds (Fig. 4C). As other groups have observed,41 decellularization leads to reduced stiffness (1,486 ± 1,029 Pa) compared with that in intact lungs (2,577 ± 670 Pa), likely due to loss of cellular tension. These results confirmed that cells were successfully removed while ECM components were retained.
Figure 4.

Fetal lung extracellular matrix (ECM) is well preserved after decellularization. A, Hematoxylin staining for cell nuclei on frozen sections derived from intact (left) and decellularized (right) E17.5 Prx1WT lungs to confirm the removal of cells from tissue. Scale bar = 20 μm. B, Immunofluorescence staining for fibrillin (Fbn) 2 (top), tropoelastin (TE; middle), and transforming growth factor (TGF)–β (bottom) within intact (left) and decellularized (right) Prx1WT lungs to examine the preservation of elastic ECM components in decellularized ECM scaffolds. Scale bar = 20 μm. C, Stiffness (Young’s modulus) of intact and decellularized E17.5 Prx1WT lungs, determined by indentation testing. WT: wild type.
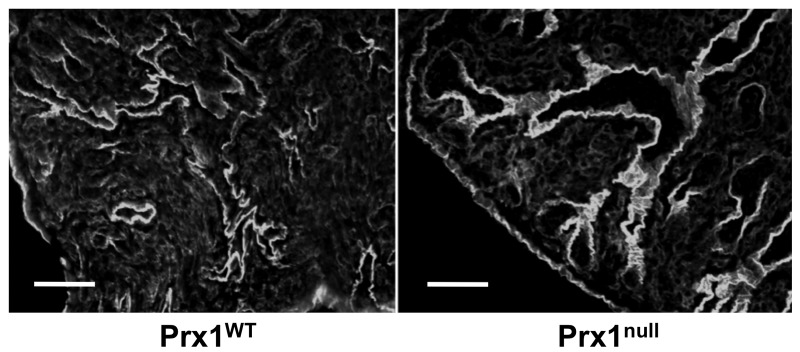
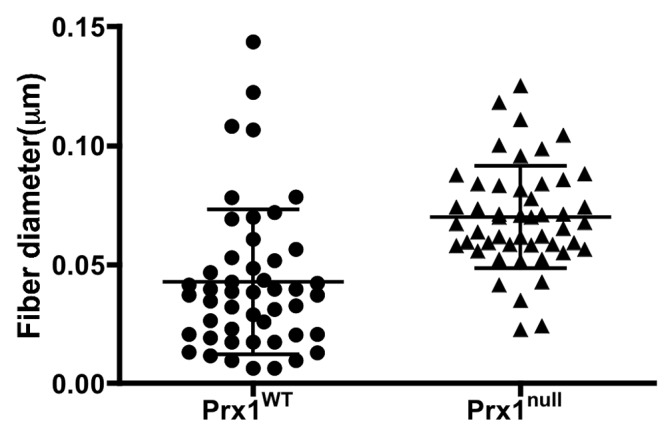
We used these lung scaffolds to characterize Prx1-dependent properties of the lung ECM. Consistent with the staining of intact tissue (Fig. 1), immunostaining showed that Fbn2 and tropoelastin were decreased in Prx1null scaffolds compared with Prx1WT lung scaffolds (Fig. 5A). Furthermore, the localization of TGF-β1 in the ECM was diffuse in Prx1null scaffolds, whereas it was strongly deposited in the vicinity of the airway and vessels in Prx1WT lung scaffolds (Fig. 5A). We also examined the stiffness of E17.5 lung scaffolds by indentation testing with a 550-μm-diameter probe (Fig. 5B). Prx1null lung scaffolds (398 ± 209 Pa) were significantly softer than Prx1WT lung scaffolds (1486 ± 1029 Pa; Fig. 5B). We then used SEM and TEM studies to examine the ultrastructural architecture of the ECM. SEM images showed that Prx1WT scaffolds possessed a well-assembled ECM, bearing a variety of sizes of interlinked fibrillar networks (Fig. 5C, left). In contrast, Prx1null lung scaffolds possessed a less fibrillar and more disorganized structure (Fig. 5C, right). TEM images were consistent with SEM level analysis, showing that E17.5 Prx1null lung scaffolds possessed a less developed fibrillar network (Fig. 5D, right) compared with that of the Prx1WT scaffolds (Fig. 5D, left). Fiber sizes constructing lung ECM scaffolds were measured using XL20 ESEM imaging software (Philips) and ImageJ. We found that fiber sizes of Prx1null ECM were significantly different from those of Prx1WT ECM. Prx1WT ECM fibrils (42.7 ± 30.5 nm) were on average of smaller diameter than Prx1null ECM fibrils (69.8 ± 21.5 nm) but had a wider range of sizes (Fig. S3), which is consistent with the ultrastructural observations (Fig. 5C, 5D). These results clearly indicate that Prx1 controls ECM architecture and physical properties by regulating ECM composition.
Figure 5.

Prx1 controls the biophysical properties of fetal lung extracellular matrix (ECM). A, Immunofluorescence staining for fibrillin (Fbn) 2 (top), tropoelastin (TE; middle), and transforming growth factor (TGF)–β (bottom) in Prx1WT (left) and Prx1null (right) lung scaffolds at E17.5. Arrows indicate decellularized vessels. Scale bar = 20 μm. B, Stiffness (Young’s modulus) of decellularized E17.5 lung scaffolds from Prx1WT and Prx1null mice, determined by indentation testing. Two asterisks indicate P < 0.01. C, Scanning electron photomicrographs of decellularized lung scaffolds from Prx1WT and Prx1null mice demonstrating the ultrastructure and fine architecture of lung ECM. D, Transmission electron micrographs of decellularized lung scaffolds from Prx1WT and Prx1null mice demonstrating the 2-D ultrastructure and ECM fiber formation of lung ECM. WT: wild type.
ECM stiffness and TGF-β signaling tightly cooperate in vascular SMC differentiation in 2-D and 3-D synthetic substrates
To determine whether Prx1-dependent ECM stiffness regulates vascular SMC differentiation and whether TGF-β signaling is involved in this process, we first confirmed that 10T1/2 mesenchymal precursor cells differentiate into SMC-like cells in response to TGF-β1.17,42 An immunocytochemical study showed that 10T1/2 cells respond to the TGF-β1 stimulation and induce SMC differentiation markers, such as α-SMA, γ-SMA, SM22α, and SMMHC (Fig. 6A), while cells not stimulated or cotreated with TGF-β and LY364947, an inhibitor of the TGF-β type I receptor, exhibited low-level expression (Fig. 6A). Activation of TGF-β signaling was confirmed by demonstrating increased expression of PAI-1, a well-recognized target of the canonical TGF-β pathway (Fig. 6B, bottom). Conversely, cotreatment of 10T1/2 cells with TGF-β1 and LY364947 resulted in PAI-1 expression at basal levels (Fig. 6B, bottom). In parallel, mRNA expression levels of α-SMA and γ-SMA were significantly increased in response to TGF-β stimulation but remained at basal levels with inclusion of the inhibitor (Fig. 6B, top, middle). Western immunoblotting (Fig. 6C) for α- and γ-SMA together with phosphorylated Smad2 showed the same response at the protein level as at the mRNA level. These studies confirm that 10T1/2 cells have the capacity to adopt a SMC-like phenotype in response to TGF-β signaling via the canonical pathway.
Figure 6.

10T1/2 cell differentiation to a smooth muscle phenotype is transforming growth factor (TGF)–β dependent. A, Immunostaining for α–smooth muscle actin (SMA), γ-SMA, SM22α, and smooth muscle myosin heavy chain (SMMHC; all positive reactions are green) in 10T1/2 cells, with or without TGF-β1 treatment, in the presence or absence of a TGF-β type I receptor (TGF-βRI) inhibitor (LY364947). Scale bar = 10 μm. B, α- and γ-SMA messenger RNA expression in 10T1/2 cells after stimulation with TGF-β1, with or without LY364947. Plasminogen activator inhibitor type 1 (PAI-1) was used as a positive control for canonical TGF-β signaling activation. Signals were normalized to GAPDH. C, Western immunoblotting for α- and γ-SMA expression in 10T1/2 cells after stimulation with TGF-β1, with or without a TGF-βRI inhibitor (SB431542). Phosphorylation of Smad2 (P-Smad2) was examined as a marker of canonical TGF-β signaling. GAPDH was used as a loading control.
We used 10T1/2 cells cultured on stiffness-controlled PAA gels in 2-D culture (Fig. 7A) to determine whether Prx1-dependent biophysical properties of the ECM are critical factors in controlling mesenchymal cell differentiation and whether Prx1-dependent ECM stiffness alone is sufficient or whether TGF-β signaling needs to be involved in this process.32,38 By adjusting the concentration of bis-acrylamide within the gels, the bulk physical properties of these substrates were designed to match the stiffness of either Prx1WT (i.e., 1,500-Pa) or Prx1null (i.e., 500-Pa) lung scaffolds. Expression of the SMC differentiation markers α-SMA, SM22α, and SMMHC was examined following culture of 10T1/2 cells on these PAA gels. Fluorescence microscopy images showed that in the absence of TGF-β1, 10T1/2 cells cultured on the stiffer substrate developed a relatively elongated morphology and more marker expression (Fig. 7B); with TGF-β1 treatment, characteristics of the SMC-like phenotype, such as cell elongation and SMC marker expression, were further increased (Fig. 7B, top vs. bottom). In contrast, 10T1/2 cells cultivated on the softer Prx1null-stiffness substrate retained mesenchymal-like morphology and expressed low levels of SMC markers with or without TGF-β1 stimulation (Fig. 7B) compared with cells cultured on stiffer Prx1WT-stiffness substrates. To quantify the morphological change of cell shape, the aspect ratio was measured (Fig. 7C). Supporting the cell morphological observation of immunocytochemistry, 10T1/2 cells cultured on stiffer substrates had a higher aspect ratio (more elongated SMC-like morphology), and the effect of TGF-β on the aspect ratio was enhanced by a stiffer substrate. Real-time PCR for SMC differentiation markers (α-SMA, SM22α, and SMMHC) demonstrated that mRNA expression of the SMC differentiation markers was induced by stiffer substrates and enhanced by TGF-β signaling (Fig. 7D–7F), which is parallel to the results of immunostaining. We also noted that among the SMC differentiation markers we examined, expression of α-SMA, an initial SMC differentiation marker, showed the most significant differences between the culture settings (Fig. 7B, 7D) compared with the expression of SM22α (Fig. 7B, 7E) and SMMHC (Fig. 7B, 7F), which are more mature SMC markers and are significantly induced only in the cells seeded on the 1,500-Pa substrates with TGF-β stimulation. This indicates that the 24-hour biophysical and biochemical stimulation leads to an initial stage of SMC differentiation from embryonic precursor cells but is not sufficient to achieve full differentiation. Taken together, these results indicate that ECM stiffness is a key determinant in controlling 10T1/2 cell differentiation to SMCs in conjunction with TGF-β stimulation. Our results also indicate that Prx1-dependent ECM stiffness alone is not sufficient to support mesenchymal cells to differentiate to the smooth muscle phenotype and requires coordination with TGF-β signaling. For this event, Prx1 likely promotes SMC differentiation via its effect on both the biophysical and the biochemical properties of the developing lung ECM.
Figure 7.

Transforming growth factor (TGF)–β–dependent 10T1/2 cell differentiation tightly interacts with Prx1-dependent extracellular matrix (ECM) stiffness. A, Schema for the stiffness-controlled polyacrylamide (PAA) gel 2-D culture system. B, 10T1/2 cells were cultured on PAA gel 2-D substrates with a stiffness of either 500 (top) or 1,500 (bottom) Pa, which is parallel to the E17.5 Prx1-lung stiffness, in the presence or absence of TGF-β stimulation. Smooth muscle cell differentiation was evaluated by expression of α–smooth muscle actin (SMA; left, green), SM22α (middle, green), and smooth muscle myosin heavy chain (SMMHC; right, red). 4′,6‐Diamidino‐2‐phenylindole dihydrochloride (blue) was used for nuclear staining. Scale bar = 10 μm. C, Aspect ratio of cells cultured on 2-D PAA gels under the above-described conditions was examined to quantify cell morphology change. D–F, Real-time polymerase chain reaction results for 10T1/2 cells cultured on 2-D PAA gels of either 500- or 1,500-Pa stiffness with or without TGF-β stimulation. α-SMA (D), SM22α (E), and SMMHC (F) messenger RNA expression was examined and normalized by GAPDH or cyclophilin A (cyclo A) expression. One asterisk indicates P < 0.05, and two asterisks indicate P < 0.01. G, 3-D projection of confocal images of F-actin-stained 10T1/2 cells cultured in soft and stiff 3-D methacrylated hyaluronic acid synthetic scaffolds. F-actin is red, nucleus is blue. Scale bar = 20 μm.
To further examine the effect of ECM stiffness on cell differentiation under more physiological conditions, we cultured 10T1/2 cells within chemically engineered 3-D substrates of different stiffness. Electrospun, MeHA gels were coated with type 1 collagen before being seeded with cells.35 The stiffness of these synthetic scaffolds was measured by AFM after the scaffolds were rehydrated to determine the physiological stiffness. We used MeHA gels with stiffness of ≃1,000 Pa to represent a soft ECM and of ∼3,000–5,000 Pa to represent a stiffer ECM for cell culture.34 3-D projections of confocal microscope images outlined by cellular F-actin staining showed that on soft 3-D scaffolds 10T1/2 cells sporadically aggregated and failed to differentiate or assemble functional vascular SMC-like networks (Fig. 7G). Within the stiffer 3-D microenvironment, however, cells grew into the MeHA substrate and demonstrated prominent network formation with elongated cell morphologies (Fig. 7G), consistent with functional SMC differentiation. Although the synthetic scaffold stiffness is not identical to the Prx1WT and Prx1null lung scaffold stiffness because of technical limitations, these results suggest that 3-D stiffness cues have a stronger effect on 10T1/2 cell to SMC functional differentiation than 2-D cues.
Prx1-dependent ECM regulates SMC differentiation in a TGF-β-dependent manner
Finally, we asked whether acellular Prx1WT and Prx1null lung scaffolds differentially support SMC differentiation. We seeded and cultured 10T1/2 cells in decellularized lung scaffolds derived from Prx1WT and Prx1null mice. After 5 days in culture, cells had clearly migrated into both Prx1WT and Prx1null scaffolds, yet their organization and morphology greatly differed: recolonization of vascular structures was clearly evident for 10T1/2 cells in Prx1WT lung scaffolds (Fig. 8A, left) compared with the Prx1null scaffolds, within which cells appeared to be randomly distributed (Fig. 8A, right). Furthermore, expression of α-SMA of seeded cells in Prx1WT scaffolds was greater than that in Prx1null scaffolds (Fig. 8A, 8B). SEM images for ultrafine structures support the immunostaining results that 10T1/2 cells on Prx1WT scaffolds possess a more elongated SMC-like phenotype compared with those cultivated on Prx1null scaffolds (Fig. 8E). Consistent with the idea that Prx1-dependent ECM properties play a major role in mesenchymal cell to SMC differentiation, we tested whether 10T1/2 cell differentiation within Prx1WT scaffolds is simply dependent on the mechanical properties of the ECM or is also dependent on the biochemical properties of the ECM, TGF-β. We pretreated Prx1WT scaffolds with a function-blocking anti-TGF-β1 antibody and then seeded 10T1/2 cells. Whereas IgY-treated controls adopted the expected elongated and differentiated phenotype, including expression of α-SMA (Fig. 8C), cultures treated with anti-TGF-β1 IgY failed to colonize scaffolds appropriately, adopted a nondifferentiated morphology, and expressed reduced levels of α-SMA (Fig. 8C, 8D). Collectively, these results strongly suggest that Prx1 plays a critical role in lung vascular SMC differentiation during embryonic development by coordinating the biophysical and biochemical properties of the ECM.
Figure 8.

Prx1-dependent extracellular matrix regulates lung vascular smooth muscle cell differentiation. A, Immunofluorescence staining for α–smooth muscle actin (SMA; green) in 10T1/2 cells seeded within E17.5 Prx1WT (left) or Prx1null (right) lung scaffolds. Insets show enlarged 10T1/2 cell images. Nuclei stained blue by 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI) staining. Scale bar = 20 μm. B, Fluorescence intensity of α-SMA immunostaining on frozen sections of 10T1/2 cells cultured in Prx1WT and Prx1null scaffolds (A) was quantified using ImageJ. One asterisk indicates P < 0.05. C, Immunofluorescence staining for α-SMA (green) of 10T1/2 cells seeded in Prx1WT scaffolds, treated with either control immunoglobulin (Ig) Y (left) or transforming growth factor (TGF)–β function-blocking IgY (right). Nuclei stained blue by DAPI. Scale bar = 20 μm. D, Fluorescence intensity of α-SMA immunostaining on frozen sections of 10T1/2 cells cultured in Prx1WT scaffolds with or without function-blocking TGF-β antibody (C), quantified using ImageJ. Two asterisks indicate P < 0.01. E, Scanning electron photomicrographs showing the ultrastructure of 10T1/2 cells in Prx1WT (left) and Prx1null (right) lung scaffolds after 5 days in culture. Cells are highlighted in red. Scale bar = 5 μm. WT: wild type.
Discussion
Vascular morphogenesis is critical for the development and regeneration of functional lung tissues. In the past, research on developmental biomechanics was focused on developing theoretical models and using in vitro experimental approaches for model validation. However, recently it has been recognized that morphogenesis occurs through more dynamic interactions between cells and through biochemical and biomechanical microenvironmental cues that are not well understood from individual cell culture models. In this study, we demonstrated that the homeobox transcription factor Prx1 plays an essential role in embryonic lung vascular SMC differentiation by regulating the biophysical and biochemical properties of the ECM (Fig. 9).
Figure 9.
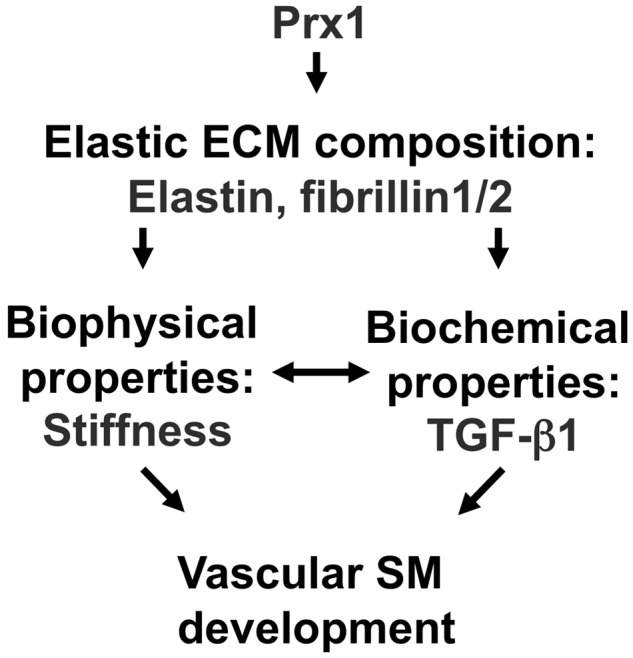
Model of the Prx1-dependent regulation of smooth muscle cell (SMC) differentiation. Prx1 regulates expression of elastic extracellular matrix (ECM) components, which regulates biophysical and biochemical properties in developing lungs. Proper biochemical and biophysical ECM properties are required for vascular SMC differentiation. TGF: transforming growth factor; SM: smooth muscle.
In this study, the results comparing Prx1WT and Prx1null mice demonstrated that Prx1 regulates expression of elastic ECM components, such as tropoelastin, Fbn1, and Fbn2, in the lungs. In Prx1null lungs, the expression of these elastic ECM components is downregulated around the developing vessels, and vascular SMC differentiation is compromised. Elastic ECM is a major component of the lungs, consisting of elastin and associated microfibrils, such as fibrillins.11,12,43-47 In animal models, defects of a single gene of these elastic ECM components have been shown to contribute to various vascular diseases. For example, mutations in tropoelastin cause occlusive arterial disease in humans,15,48 and elastin-deficient mice die shortly after birth due to aberrant subendothelial cell proliferation and reorganization of the SMC layer.13,49 In addition, elastin-associated microfibril proteins in fibrillin50,51 and fibulin45,52,53 families also regulate vascular SMC differentiation. However, the upstream regulator orchestrating these elastic ECMs during embryonic development is unknown. While previous studies have shown that Prx1 regulates vascular SMC differentiation in the adult by direct transcriptional regulation of α-SMA transactivation54,55 acting as a cofactor for serum response factor,55,56 this mechanism has not been found to regulate initial SMC marker gene expression in early embryonic stages.57 Therefore, the differences in elastic ECM composition between Prx1WT and Prx1null lungs suggest a new indirect role for the transcription factor Prx1 in embryonic lung vascular morphogenesis by organizing elastic ECM during embryonic development.
Decellularized lung ECM scaffolds enabled us to demonstrate that differences in elastic ECM composition between Prx1WT and Prx1null strongly affect the architecture and ultrastructural fiber formation of ECM in embryonic lungs. For instance, SEM images and fiber measurement showed that the fiber diameter of Prx1WT has wide variations, while Prx1null has a more uniform intermediate size, which supports previous findings that the insufficiency of elastin and fibrillins prevents the proper maturation of fibers.11,58 The variation in the fiber size of the ECM in Prx1WT indicates more maturation of ECM fibers to construct the proper microenvironment for vascular development, while Prx1null ECM with a uniform size of fibers results in a lack of a specific microstructure to support functional vascular development. Therefore, the fiber size of the ECM could be a key indicator of the biophysical properties of the ECM. In this study, this structural maturation in the ECM by Prx1 was reflected in the biophysical property of stiffness, resulting in a softer Prx1null ECM (398 ± 209 Pa) and a more rigid Prx1WT ECM (1,486 ± 1,029 Pa). Similar to the recent studies that show that the stiffness of the ECM has a profound influence on stem/progenitor cell fate determination and differentiation in adult tissues in both normal and pathological settings,2-4,59,60 our results suggest that embryonic lung vascular morphogenesis is regulated by ECM stiffness and that Prx1 plays a key role in this regulation, a notion supported by the cell culture results using stiffness-controlled 2-D and 3-D synthetic substrates.
In addition to biophysical properties, biochemical properties, such as growth factor stimulation of the ECM, play an essential role in regulating cell growth. During embryonic lung development, it has been well established that temporal spatial gradients of growth factors surrounding the developing airway and vessels are essential for lung morphogenesis.6-8,10,61 However, many studies using transgenic animal models and/or conventional cell culture systems could not elucidate the dynamics of the microenvironment in tissue and organ development. In this study, our results show that while the expression of TGF-β was not significantly different between Prx1WT and Prx1null lungs, downstream canonical signaling activity was decreased in Prx1null lungs. Furthermore, the immunohistochemistry of intact and ECM scaffold lungs showed that the localization of TGF-β is diffusely spread out in Prx1null lungs, while the localization is condensed around the growing vessels and airways in Prx1WT lungs. Since an elastic matrix has been considered to play an essential role in vascular cell differentiation and functional maturation by acting as a reservoir for multiple growth factors40 and since fibrillins tether TGF-β,8,62 which controls its bioavailability, these differences in TGF-β localization are regulated by the Prx1-dependent elastic ECM components. Supported by previous studies using fibrillin-deficient mice, which present hypoplasia of airway and vascular systems due to compromised TGF-β tempospatial availability during development,63-65 our results suggest that Prx1 maintains a precise bioavailability of TGF-β during lung development by regulating elastic ECM properties.
We further examined the interaction between TGF-β activation and stiffness of Prx1-dependent ECM in lung development since a previous study showed that ECM stiffness can regulate activation of TGF-β and induce cell differentiation.19 Since our results showed that canonical TGF-β signaling was downregulated in Prx1null lungs accompanied by low stiffness in the ECM, stiffness of Prx1-dependent ECM plays a role in regulating local TGF-β activation and subsequent SMC differentiation. In addition, the results using stiffness-controlled 2-D and 3-D culture substrates and decellularized lung scaffolds demonstrated that Prx1-dependent ECM stiffness has a substantial effect on SMC differentiation even without TGF-β stimulation and that TGF-β alone could not fully rescue SMC differentiation on soft Prx1null scaffolds. On the other hand, our results also showed that matrix stiffness alone is not sufficient to control vascular SMC differentiation, while blocking TGF-β dramatically reduced SMC differentiation and functional maturation. Since stiffness of the ECM also regulates noncanonical TGF-β activation in different settings and regulates cell differentiation and fate determinations,60,66 it is plausible that Prx1-dependent ECM also regulates TGF-β signaling via a noncanonical pathway; this requires future investigation.
Our results strongly suggest that Prx1 regulates a unique pathway in lung vascular smooth muscle development by coordinating biophysical and biochemical properties of the ECM during embryonic development. In addition to TGF-β, there is also a possibility that Prx1-dependent ECM coordinates other growth factor pathways in lung vascular morphogenesis. For example, Wnt, a key regulator of lung morphogenesis, responds to ECM stiffness;67 we have reported that Wnt7b signaling is regulated by tenascin C, which is downstream of Prx1.68 In addition, the roles played by ECM characteristics other than stiffness, such as composition and architecture, in SMC differentiation require further investigation. It is plausible that Prx1 maintains temporal and spatial stiffness and growth factor bioavailability by precisely controlling the ECM properties around the developing embryonic lung tissue; this requires future investigation. Once technical limitations for regional stiffness measurements can be overcome, we will have the ability to generate more precise information about the temporal and spatial coordination of biophysical and biochemical signals controlled by Prx1. These future studies will enable better understanding of the regulation of embryonic lung cell differentiation, tissue development, and functional maturation controlled by ECM properties, which would create a fundamental understanding of ontogeny of the cardiovascular system and the development of cell-ECM-based therapies.
In conclusion, we report here that Prx1 indirectly regulates lung vascular SMC differentiation by controlling elastic ECM properties. By focusing on Prx1, this work provides essential information on the role played by the transcription factor in association with the control of ECM properties in specifying the final shape and architecture of lung vascular morphogenesis. We believe that our findings contribute to advancing knowledge of the biophysical and biochemical processes that occur during normal and abnormal embryonic lung vascular development and have implications for the design of regenerative therapies by combining stem/progenitor cells and ECM applications.
Acknowledgments
We gratefully thank Ms. Dipal Patel, Ms. Amanda Romney, Ms. Cathryn Herbst, and Mr. Ja On Kim for technical assistance. We gratefully acknowledge the assistance of the University of Pennsylvania Cell and Developmental Biology Microscopy Core Facility for confocal and scanning electron microscopy (SEM) imaging, the Path BioResource Electron Microscopy Resource for transmission electron microscopy imaging, and the Penn Regional Nanotechnology Facility for SEM imaging of polymers.
Supplement.
Methods
Animals
Prx1 gene knockout mice were generated as described elsewhere.30 Briefly, the Prx1lacZ allele was generated by introducing an in-frame LacZ gene into the Prx1 locus to disrupt Prx1 functions. Polymerase chain reaction (PCR) was used to identify the three different expected genotypes: Prx1WT (wild type), Prx1 heterozygous, and Prx1null (homozygous-null). Genomic DNA from mouse tails was used for PCRs for genotyping with the following primer pairs: Prx1WT allele, 5′-GACCAGTTGAACTCTGAGGAGAAGAAGAAGAGAAA-3′ (forward) and 5′-CCCTCAGTGGATAGTAGTATATCGAACACAATATA-3′ (reverse); and Prx1lacZ allele, 5′-GCATCGAGCTGGGTAATAAGGGTTGGCAAT-3′ (forward) and 5′-GACACCAGACCAACTGGTAATGGTAGCCAC-3′ (reverse). The following conditions were used for PCR: 94°C for 1 minute, 62°C for 1 minute, and 72°C for 1 minute, for 40 cycles. Mouse lungs isolated from Prx1WT and Prx1null mice of gestational ages E15.5, E17.5, and newborn (D0) were used for the experiments. All animal experiments were conducted in accordance with current National Institutes of Health guidelines.
Cell culture
We used mouse embryo origin C3H10T1/2 (10T1/2) cells (ATCC) as mesenchymal SMC precursor cells.17,69 Except in the case of transforming growth factor (TGF)–β stimulation experiments, cells were maintained in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific), 2 mM l-glutamine, 0.1 mM nonessential amino acids (Gibco), and antibiotics-antimicotics (Gibco). Cells were cultured at 37°C in a humidified atmosphere with 5% CO2 until used for cellular experiments.
Transient transfection
Cytomegalovirus plasmids with or without the Prx1 construct described elsewhere30 were used for transfection experiments to examine the downstream genes regulated by Prx1. These plasmids were transiently transfected into 10T1/2 cells using Lipofectamine (Invitrogen, Thermo Fisher Scientific) and cultured for 48 hours, and total RNA was harvested to examine messenger RNA (mRNA) expression.
Decellularization of lungs
Intact fetal mouse lungs with gestational ages from E15.5 to newborn (D0) were removed from Prx1WT and Prx1null mice, treated with 10 mM Tris buffer containing 5 mM ethylenediaminetetraacetic acid (EDTA; pH 8), and then incubated in 50 mM Tris buffer containing 1% Triton X-100, 1.5 M KCl, and 5 mM EDTA (pH 8)31 at 4°C. Following decellularization, lung scaffolds were either fixed for biochemical and morphological characterization or left unfixed for biomechanical testing. Intact lungs were used as controls for decellularization experiments.
Polyacrylamide (PAA) gels
PAA gels of variable stiffness were prepared on glass coverslips using an established method.32 Briefly, gels were a mixture of 7.5% acrylamide and 0.01%–0.3% bis-acrylamide impregnated with 0.5 mg/mL sulfosuccinimidyl-6-(4′-azido-2′-nitrophenyl-amino) hexanoate (Sulfo-SANPAH; Pierce). As an extracellular matrix (ECM) protein, a thin layer of collagen (0.2 mg/mL; Advanced Biomatrix) was crosslinked to the PAA gels at 4°C overnight, and gels were then soaked in serum-free culture medium for at least 2 hours at 37°C before cells were plated. It has been demonstrated that the layer of matrix protein does not alter the stiffness of the system,32 and the final thickness of the gels was ∼100–150 μm.32
Synthetic 3-D scaffolds (electrospun hyaluronic acid scaffolds)
Methacrylated hyaluronic acid (MeHA) was synthesized by reacting methacrylic anhydride (20-fold excess; Sigma-Aldrich) with a 1% w/v sodium hyaluronate (molecular weight, 74 kDa; Lifecore Biomedical, Chaska, MN) in deionized H2O solution adjusted to pH 8 with 5 N NaOH on ice for 24 h, as described elsewhere.33,34 After the reaction, the polymer was purified by dialysis against water for 48 h and then lyophilized to produce the final product. The percent methacrylation (percentage of HA repeat units with a methacrylate group) was adjusted on the basis of the amount of methacrylic anhydride added to the reaction and was determined with 1H NMR (Advance 360 MHz; Bruker Optics, Billerica, MA). MeHA was cospun with the carrier polymer polyethylene oxide (PEO; molecular weight, 900 kDa; Sigma-Aldrich) at 2% and 3% w/v, respectively, as described elsewhere.34,35 Briefly, the polymer solution (HA, PEO, and 0.05% w/v of the photoinitiator I2959 [2-methyl-1-(4-[hydroxyethoxy]phenyl)-2-methyl-1-propanone; Ciba Geigy, Tarrytown, NY]) in water was ejected at 1.2 mL/h using a syringe pump (KD Scientific, Holliston, MA) through a 12-cm-long 18-gauge needle charged to 22 kV onto a grounded flat plate at a distance of 15 cm. To quantify fiber size and view morphology, scaffolds were examined using scanning electron microscopy (SEM; 7500F HRSEM; Jeol). Polymerization was accomplished by exposing as-spun MeHA mats in a purged nitrogen chamber to ∼10 mW/cm2 365-nm light (Omnicure S1000 UV Spot Cure System; Exfo) with a collimating adapter for 5 minutes. Subsequently, scaffolds were immersed in deionized H2O to remove unreacted MeHA and PEO and were washed for 24 hours to encourage removal of PEO and to reach equilibrium swelling. The mechanical properties of MeHA gels were controlled by percent methacrylation or concentration of polymer used, and their stiffness was determined by atomic force microscopy (AFM; Bioscope DAFMLN-AM head [Veeco Instruments, Plainview, NY] mounted on an Axiovert 100 microscope [Zeiss]).
Immunohistochemistry
The following primary antibodies were used for section staining: anti-fibrillin (Fbn) 1 and Fbn2 rabbit polyclonal antibodies (gift of R. Mecham, University of Washington), anti-tropoelastin polyclonal antibody (Abcam), anti-α-smooth muscle actin (SMA) monoclonal antibody (clone 1A4; Sigma-Aldrich), anti-γ-SMA monoclonal antibody (clone B4; Santa Cruz Biotech), anti-SM22α polyclonal antibody (Abcam), anti–smooth muscle myosin heavy chain (SMMHC) monoclonal antibody (Abcam), anti-TGF-β1 polyclonal antibody (Promega), and anti-TGF-β2 polyclonal antibody (R&D Systems). For controls, tissue sections were incubated with equivalent concentrations of isotype-matched immunoglobulin (Ig) G. Unless otherwise indicated, all procedures were conducted at room temperature. Sections were deparaffinized, rehydrated, and then incubated with 3% H2O2 in methanol for 20 minutes to block endogenous peroxidase activity. After washing in tap water and phosphate‐buffered saline (PBS), an antigen retrieval step was performed. For Fbn1, Fbn2, and tropoelastin staining, sections were exposed to a solution containing 6 M guanidine-HCl and 50 mM dithiothreitol for 15 minutes, followed by treatment with 100 mM iodoacetamide in the dark for 15 minutes. After antigen retrieval, sections were washed in PBS and then blocked for 1 hour in blocking buffer, which was 1% bovine serum albumin (PBSA) containing 10% serum and 0.1% Tween-20. Each primary antibody was diluted in blocking buffer (Fbn1 and Fbn2, 1∶100; tropoelastin, 1∶150; α-SMA, 1∶400; γ-SMA, 1∶50; SM22α, 1∶100; SMMHC, 1∶100), applied to sections, and then incubated overnight at 4°C. Antigen detection was performed using biotinylated species-specific secondary antibodies followed by avidin-biotin complex from the Vectastain Elite ABC Kit (Vector Laboratories), and then the 3,3′-diaminobenzidine reaction (Vector Laboratories) was performed for final visualization. After being washed in tap water, sections were counterstained with hematoxyline (Vector Laboratories), dehydrated in serial concentrations of ethanol, cleared in Citrisolv (Fisher Scientific, Thermo Fisher Scientific), and mounted using Cytoseal 60 medium (Richard-Allan Scientific, Thermo Fisher Scientific) before being subjected to bright-field imaging under a microscope (90i; Nikon).
Immunofluorescence staining
Cells or scaffolds were fixed in 4% paraformaldehyde for 15 minutes, washed with PBS, and then permeabilized with 0.2% Triton X-100–containing PBS for 15 minutes. After brief washing in PBS, materials were treated with blocking buffer (10% serum and 0.2% Triton X-100–containing PBSA) and then incubated with primary antibodies overnight at 4°C. To examine mesenchymal precursor to SMC differentiation, SMC marker antibodies specific for α-SMA (1∶200), γ-SMA (1∶100), SM22α (1∶100), and SMMHC (1∶100; the same antibodies used for immunohistochemistry) were used. For detection of ECM components, antibodies specific for Fbn1 (1∶50), Fbn2 (1∶50), tropoelastin (1∶100), laminin (Abcam; 1∶100), and TGF-β1 (1∶50) were used. Species-specific fluorescent-conjugated antibodies (Molecular Probes; all 1∶100) were used for primary antigen detection. 4′,6‐Diamidino‐2‐phenylindole dihydrochloride (KPL, Gaithersburg, MD) was used for nuclear staining, and specimens were mounted in mounting medium (KPL). Stained materials were examined using a Nikon 90i fluorescence microscope or a Zeiss LSM 710 two-photon confocal microscope (CDB Imaging Core at the University of Pennsylvania, Philadelphia, PA), and some were further processed for quantitative analysis. Quantification of the staining intensity was performed using ImageJ software (National Institutes of Health).
Electron microscopy
Transmission electron microscopy (TEM)
Intact or decellularized embryo lungs were processed using a modification of a protocol described elsewhere.30 Briefly, materials were fixed with 2.5% glutaraldehyde containing 0.1 M cacodylate buffer at 4°C overnight followed by postfixation with 2% OsO4 (EMS) containing 0.1 M cacodylate buffer for 1 hour. Specimens were then dehydrated in graded ethanols before being embedded in Epon 812 (EMS). Semithin sections were stained with toluidine blue to determine the orientation of the ultrathin sections. Ultrathin sections of embedded specimens were stained with ulanyl acetate (EMS) and lead citrate (EMS) before being examined under a Jeol 1010 electron microscope.
Scanning electron microscopy (SEM)
Decellularized lung scaffolds were fixed in 2% glutaraldehyde containing 0.1 M cacodylate buffer, dehydrated with a graded series of ethanol, and then chemically dried using hexamethyldisilazane. Dried specimens were sputter coated with palladium before being examined using Philips XL20 ESEM imaging software (CDB Imaging Core at the University of Pennsylvania, Philadelphia, PA).
Western immunoblotting
Protein was extracted from Prx1 mouse lungs in ice-cold radioimmunoprecipitation assay buffer (Millipore, Billerica, MA) with protease and phosphatase inhibitors (Pierce), and protein concentrations were determined using the Pierce Protein Assay Reagent (Pierce). Equal amounts of lysate were separated on a 8%–16% Tris-HCl sodium dodecyl sulfate–PAA gel (Pierce) and transferred to a polyvinylidene fluoride membrane. Membranes were blocked with 5% nonfat dry milk, and analysis was performed with primary antibodies against Fbn1 and Fbn2 (polyclonal antibodies; gift of R. Mecham), tropoelastin (polyclonal antibody; Abcam), TGF-β1 (polyclonal antibody; Promega), phosphorylated Smad2 (polyclonal antibody; Cell Signaling Technology), and total Smad2/3 (polyclonal antibody; Cell Signaling Technology) and β-actin (Sigma-Aldrich). Horseradish peroxidase–conjugated species-specific secondary antibodies (Chemi-Con, Tokyo) and enhanced chemiluminescence kit (Amersham, Amersham, UK) were used for detection. Blots were imaged using a Fuji imager (Fujifilm/GE Healthcare). Densitometric analysis was performed using ImageJ.
Real-time quantitative PCR (qPCR)
Total RNA was isolated from Prx1 mouse lungs or cultured cells using an RNeasy Kit (Qiagen, Venlo, Netherlands) followed by complementary DNA (cDNA) synthesis using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific). Transcript levels were measured by SYBR Green–based real-time PCR using LightCycler 480 SYBR Green I Master Mix (Roche) and the LightCycler 480 instrument (Roche). Each mRNA expression value was normalized to the expression of a housekeeping gene (GAPDH or cyclophilin A). Primer sequences were as follows: mouse plasminogen activator inhibitor type 1, 5′-CCACAAAGGTCTCATGGACCAT-3′ and 5′-TGAAAGTGTTGTGCCCTCCAC-3′; mouse TGF-β1, 5′-TGACGTCACTGGAGTTGTACGG-3′ and 5′-GGTTCATGTCATGGATGGTGC-3′; mouse α-SMA, 5′-AGCGTGAGATTGTCCGTGACAT-3′ and 5′-GCGTTCGTTTCCAATGGTGA-3′; mouse γ-SMA, 5′-GCCCTGGATTTCGAGAATGA-3′ and 5′-AAGCGCTCGTTGCCAATAGT-3′; mouse SM22α, 5′-GCGCCTGGGCTTCCA-3′ and 5′-CAGGCTGTTCACCAATTTGCT-3′; mouse SMMHC, 5′-GGTCGTGGAGTTGGTGGA-3′ and 5′-CTGCCATGTCCTCCACCTTAGAA-3′; mouse tropoelastin, 5′-AACCCACAGGACAAGGAAATCAGAC-3′ and 5′-TCACCAAATCCATCGTCAAGGC-3′; mouse Fbn2, 5′-AGGATGACGAAAACGCTCTGTCCC-3′ and 5′-TACGCTCTCCAGGCTGATTTGCTC-3′; mouse GAPDH, 5′-ATGTGTCCGTCGTGGATCTGA-3′ and 5′-TTGAAGTCGCAGGAGACAACCT-3′; and mouse cyclophilin A, 5′-ATGGTCAACCCCACCGTGT-3′ and 5′-TCCTGCTGTCTTTGGAACTTTGTC-3′.
Stiffness measurements
Indentation testing
The stiffness of decellularized lung scaffolds was measured using a custom-made microindenter.36 Briefly, lung scaffolds were brought into contact with the probe (blunt-ended tungsten wire, 550 μm in diameter), and the magnitude of indentation and force was measured for every 10 μm of upward displacement of the sample toward the probe. The average indentation and force on the sample were derived and used to quantify sample stiffness by the method of Hayes et al.37
Mechanical testing by AFM
To measure the stiffness of HA scaffolds at the tissue level, synthesized HA scaffolds were rehydrated, and the mechanical force was measured by AFM. The mechanical force was calculated using a cantilever spring constant determined by the vendor’s thermal method.
TGF-β stimulation experiments
Cells at 80% confluence were incubated with serum-free medium overnight and then treated with 5 ng/mL recombinant TGF-β1 (Cell Signaling Technology) at a concentration of 1 ng/μL3 in DMEM/2% FBS for 24 hours. The same concentration of bovine serum albumin in identical medium was used as a control. Cells were either fixed for immunostaining or used to harvest total RNA for real-time qPCR.
TGF-β function-blocking experiments
TGF-β blocking
10T1/2 cells were seeded within Prx1WT scaffolds, with or without 10 mg/mL neutralizing antibody against TGF-β1 (R&D Systems) in F12/DMEM plus 2% FBS for 1–7 days. As a control, an identical concentration of chicken IgY was added to the medium. During culture, antibody was added to the medium every other day. 10T1/2 cell differentiation was examined by immunostaining for α-SMA on the frozen sections of lung scaffolds cultured with cells and examined by fluorescence microscopy. Intensity was quantified using ImageJ.
TGF-βR1 signaling blocking
When 10T1/2 cells reached more than 90% confluence, cells were serum starved overnight and then incubated with 10 mM SB431542 (Cell Signaling Technology) or 10 mM LY364947 (Sigma-Aldrich) compounds with or without TGF-β1 stimulation for 48 hours and then processed for immunostaining or real-time qPCR to examine canonical TGF-β signaling pathways. As a control, vehicle alone was added to the medium.
Statistics
All experiments were performed in triplicate unless otherwise stated. Statistical analysis was performed using Prism software (GraphPad) to compare the groups. One-way analysis of variance with the Newman-Keuls posttest was used to compare more than two groups, and the two-tailed unpaired t test was used to compare two groups. A P value of less than 0.05 was considered significant. All values are shown as means ± SD unless otherwise specified.
Figure S1.

Prx1 controls expression of elastic extracellular matrix (ECM) components. A, Quantification of elastic ECM protein expression by densitometry Western blotting analysis in ImageJ. All target protein values were standardized by β-actin intensity. B–D, Real-time polymerase chain reaction for Prx1 (B), tropoelastin (ELN; C), and fibrillin (Fbn) 1 (D) messenger RNA expression in 10T1/2 cells transiently transfected with empty or Prx1-containing construct. Target gene expression was normalized to cyclophilin A (Cyclo A) expression. One asterisk indicates P < 0.05, and two asterisks indicate P < 0.001. WT: wild type.
Figure S2.
Laminin immunostaining on decellularized lung scaffolds derived from E17.5 Prx1WT and Prx1null mice. Scale bar = 40 μm. WT: wild type.
Figure S3.
Measurement of the diameter of fibers consisting of the extracellular matrix scaffolds in Prx1WT and Prx1null lungs using XL20 ESEM imaging software (Philips) and ImageJ (National Institutes of Health).
Reference Cited Only in the Supplement
- 69.Pinney DF, Emerson CP Jr. 10T1/2 cells: an in vitro model for molecular genetic analysis of mesodermal determination and differentiation. Environ Health Perspect 1989;80:221–227. [DOI] [PMC free article] [PubMed]
Source of Support: This work was supported by a grant from the American Heart Association (09SDG2080312), the Penn Institute for Regenerative Medicine Technology and Tool Funding (KI-S), National Institutes of Health (NIH) R01 (HL079196; PLJ), a Fellowship in Science and Engineering from the David and Lucile Packard Foundation (JAB), and NIH R01 (DK058123; RGW).
Conflict of Interest: None declared.
Supplements
SupplementPulmCirc-005-382.s001.pdf (777.8KB, pdf)
References
- 1.Ozbek S, Balasubramanian PG, Chiquet-Ehrismann R, Tucker RP, Adams JC. The evolution of extracellular matrix. Mol Biol Cell 2010;21:4300–4305. [DOI] [PMC free article] [PubMed]
- 2.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell 2006;126:677–689. [DOI] [PubMed]
- 3.Wells RG, Discher DE. Matrix elasticity, cytoskeletal tension, and TGF-β: the insoluble and soluble meet. Sci Signal 2008;1:pe13. [DOI] [PMC free article] [PubMed]
- 4.Janmey PA, Winer JP, Murray ME, Wen Q. The hard life of soft cells. Cell Motil Cytoskeleton 2009;66:597–605. [DOI] [PMC free article] [PubMed]
- 5.Fernandes H, Mentink A, Bank R, Stoop R, van Blitterswijk C, de Boer J. Endogenous collagen influences differentiation of human multipotent mesenchymal stromal cells. Tissue Eng Part A 2010;16:1693–1702. [DOI] [PubMed]
- 6.Park Y, Rangel C, Reynolds MM, Caldwell MC, Johns M, Nayak M, Welsh CJ, McDermott S, Datta S. Drosophila perlecan modulates FGF and hedgehog signals to activate neural stem cell division. Dev Biol 2003;253:247–257. [DOI] [PubMed]
- 7.Giros A, Morante J, Gil-Sanz C, Fairen A, Costell M. Perlecan controls neurogenesis in the developing telencephalon. BMC Dev Biol 2007;7:29. [DOI] [PMC free article] [PubMed]
- 8.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-β activation contributes to pathogenesis in marfan syndrome. Nat Genet 2003;33:407–411. [DOI] [PubMed]
- 9.Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009;326:1216–1219. [DOI] [PMC free article] [PubMed]
- 10.Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell 2010;18:8–23. [DOI] [PMC free article] [PubMed]
- 11.Kielty CM, Sherratt MJ, Shuttleworth CA. Elastic fibres. J Cell Sci 2002;115:2817–2828. [DOI] [PubMed]
- 12.Trask TM, Trask BC, Ritty TM, Abrams WR, Rosenbloom J, Mecham RP. Interaction of tropoelastin with the amino-terminal domains of fibrillin-1 and fibrillin-2 suggests a role for the fibrillins in elastic fiber assembly. J Biol Chem 2000;275:24400–24406. [DOI] [PubMed]
- 13.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature 1998;393:276–280. [DOI] [PubMed]
- 14.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. The importance of elastin to aortic development in mice. Am J Physiol Heart Circ Physiol 2010;299:H257–H264. [DOI] [PMC free article] [PubMed]
- 15.Curran ME, Atkinson DL, Ewart AK, Morris CA, Leppert MF, Keating MT. The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell 1993;73:159–168. [DOI] [PubMed]
- 16.Mellody KT, Freeman LJ, Baldock C, Jowitt TA, Siegler V, Raynal BD, Cain SA, Wess TJ, Shuttleworth CA, Kielty CM. Marfan syndrome–causing mutations in fibrillin-1 result in gross morphological alterations and highlight the structural importance of the second hybrid domain. J Biol Chem 2006;281:31854–31862. [DOI] [PubMed]
- 17.Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF-β, and heterotypic cell-cell interactions mediate endothelial cell–induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol 1998;141:805–814. [DOI] [PMC free article] [PubMed]
- 18.Kurpinski K, Lam H, Chu J, Wang A, Kim A, Tsay E, Agrawal S, Schaffer DV, Li S. Transforming growth factor–β and notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells 2010;28:734–742. [DOI] [PubMed]
- 19.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J Cell Biol 2007;179:1311–1323. [DOI] [PMC free article] [PubMed]
- 20.Buscemi L, Ramonet D, Klingberg F, Formey A, Smith-Clerc J, Meister JJ, Hinz B. The single-molecule mechanics of the latent TGF-β1 complex. Curr Biol 2011;21:2046–2054. [DOI] [PubMed]
- 21.Schiffer C, Tariverdian G, Schiesser M, Thomas MC, Sergi C. Agnathia-otocephaly complex: report of three cases with involvement of two different Carnegie stages. Am J Med Genet 2002;112:203–208. [DOI] [PubMed]
- 22.Herman S, Delio M, Morrow B, Samanich J. Agnathia-otocephaly complex: a case report and examination of the OTX2 and PRRX1 genes. Gene 2012;494:124–129. [DOI] [PubMed]
- 23.Dasouki M, Andrews B, Parimi P, Kamnasaran D. Recurrent agnathia-otocephaly caused by DNA replication slippage in PRRX1. Am J Med Genet A 2013;161A:803–808. [DOI] [PubMed]
- 24.Celik T, Simsek PO, Sozen T, Ozyuncu O, Utine GE, Talim B, Yigit S, Boduroglu K, Kamnasaran D. PRRX1 is mutated in an otocephalic newborn infant conceived by consanguineous parents. Clin Genet 2012;81:294–297. [DOI] [PubMed]
- 25.Martin JF, Bradley A, Olson EN. The paired-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes Dev 1995;9:1237–1249. [DOI] [PubMed]
- 26.Bergwerff M, Gittenberger-de Groot AC, DeRuiter MC, van Iperen L, Meijlink F, Poelmann RE. Patterns of paired-related homeobox genes PRX1 and PRX2 suggest involvement in matrix modulation in the developing chick vascular system. Dev Dyn 1998;213:59–70. [DOI] [PubMed]
- 27.Bergwerff M, Gittenberger-de Groot AC, Wisse LJ, DeRuiter MC, Wessels A, Martin JF, Olson EN, Kern MJ. Loss of function of the Prx1 and Prx2 homeobox genes alters architecture of the great elastic arteries and ductus arteriosus. Virchows Arch 2000;436:12–19. [DOI] [PubMed]
- 28.Kainulainen K, Karttunen L, Puhakka L, Sakai L, Peltonen L. Mutations in the fibrillin gene responsible for dominant ectopia lentis and neonatal marfan syndrome. Nat Genet 1994;6:64–69. [DOI] [PubMed]
- 29.Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular understanding of marfan syndrome. Am J Med Genet C Semin Med Genet 2005;139C:4–9. [DOI] [PubMed]
- 30.Ihida-Stansbury K, McKean DM, Gebb SA, Martin JF, Stevens T, Nemenoff R, Akeson A, Vaughn J, Jones PL. Paired-related homeobox gene Prx1 is required for pulmonary vascular development. Circ Res 2004;94:1507–1514. [DOI] [PubMed]
- 31.Gilbert TW, Sellaro TL, Badylak SF. Decellularization of tissues and organs. Biomaterials 2006;27:3675–3683. [DOI] [PubMed]
- 32.Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton 2005;60:24–34. [DOI] [PubMed]
- 33.Burdick JA, Chung C, Jia X, Randolph MA, Langer R. Controlled degradation and mechanical behavior of photopolymerized hyaluronic acid networks. Biomacromolecules 2005;6:386–391. [DOI] [PMC free article] [PubMed]
- 34.Sundararaghavan HG, Metter RB, Burdick JA. Electrospun fibrous scaffolds with multiscale and photopatterned porosity. Macromol Biosci 2010;10:265–270. [DOI] [PMC free article] [PubMed]
- 35.Sundararaghavan HG, Burdick JA. Gradients with depth in electrospun fibrous scaffolds for directed cell behavior. Biomacromolecules 2011;12:2344–2350. [DOI] [PMC free article] [PubMed]
- 36.Levental I, Levental KR, Klein EA, Assoian R, Miller RT, Wells RG, Janmey PA. A simple indentation device for measuring micrometer-scale tissue stiffness. J Phys Condens Matter 2010;22:194120. [DOI] [PMC free article] [PubMed]
- 37.Hayes WC, Keer LM, Herrmann G, Mockros LF. A mathematical analysis for indentation tests of articular cartilage. J Biomech 1972;5:541–551. [DOI] [PubMed]
- 38.Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. Transforming growth factor–β and substrate stiffness regulate portal fibroblast activation in culture. Hepatology 2007;46:1246–1256. [DOI] [PubMed]
- 39.Charbonneau NL, Ono RN, Corson GM, Keene DR, Sakai LY. Fine tuning of growth factor signals depends on fibrillin microfibril networks. Birth Defects Res C Embryo Today 2004;72:37–50. [DOI] [PubMed]
- 40.Badylak SF. The extracellular matrix as a biologic scaffold material. Biomaterials 2007;28:3587–3593. [DOI] [PubMed]
- 41.Witzenburg C, Raghupathy R, Kren SM, Taylor DA, Barocas VH. Mechanical changes in the rat right ventricle with decellularization. J Biomech 2012;45:842–849. [DOI] [PMC free article] [PubMed]
- 42.Chen S, Kulik M, Lechleider RJ. Smad proteins regulate transcriptional induction of the SM22α gene by TGF-β. Nucleic Acids Res 2003;31:1302–1310. [DOI] [PMC free article] [PubMed]
- 43.Ushiki T. Collagen fibers, reticular fibers and elastic fibers: a comprehensive understanding from a morphological viewpoint. Arch Histol Cytol 2002;65:109–126. [DOI] [PubMed]
- 44.Kielty CM, Sherratt MJ, Marson A, Baldock C. Fibrillin microfibrils. Adv Protein Chem 2005;70:405–436. [DOI] [PubMed]
- 45.Huang J, Davis EC, Chapman SL, Budatha M, Marmorstein LY, Word RA, Yanagisawa H. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ Res 2010;106:583–592. [DOI] [PMC free article] [PubMed]
- 46.Chapman SL, Sicot FX, Davis EC, Huang J, Sasaki T, Chu ML, Yanagisawa H. Fibulin-2 and fibulin-5 cooperatively function to form the internal elastic lamina and protect from vascular injury. Arterioscler Thromb Vasc Biol 2010;30:68–74. [DOI] [PMC free article] [PubMed]
- 47.Wang X, LeMaire SA, Chen L, Carter SA, Shen YH, Gan Y, Bartsch H, et al. Decreased expression of fibulin-5 correlates with reduced elastin in thoracic aortic dissection. Surgery 2005;138:352–359. [DOI] [PubMed]
- 48.Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet 1993;5:11–16. [DOI] [PubMed]
- 49.Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY. Impaired distal airway development in mice lacking elastin. Am J Respir Cell Mol Biol 2000;23:320–326. [DOI] [PubMed]
- 50.Hubmacher D, Tiedemann K, Reinhardt DP. Fibrillins: from biogenesis of microfibrils to signaling functions. Curr Top Dev Biol 2006;75:93–123. [DOI] [PubMed]
- 51.Carta L, Pereira L, Arteaga-Solis E, Lee-Arteaga SY, Lenart B, Starcher B, Merkel CA, et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem 2006;281:8016–8023. [DOI] [PMC free article] [PubMed]
- 52.Wan W, Yanagisawa H, Gleason RL Jr. Biomechanical and microstructural properties of common carotid arteries from fibulin-5 null mice. Ann Biomed Eng 2010;38:3605–3617. [DOI] [PMC free article] [PubMed]
- 53.Nakamura T, Lozano PR, Ikeda Y, Iwanaga Y, Hinek A, Minamisawa S, Cheng CF, et al. Fibulin-5/dance is essential for elastogenesis in vivo. Nature 2002;415:171–175. [DOI] [PubMed]
- 54.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 2004;84:767–801. [DOI] [PubMed]
- 55.Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res 2005;96:280–291. [DOI] [PubMed]
- 56.Yoshida T, Hoofnagle MH, Owens GK. Myocardin and Prx1 contribute to angiotensin II–induced expression of smooth muscle α-actin. Circ Res 2004;94:1075–1082. [DOI] [PubMed]
- 57.Shang Y, Yoshida T, Amendt BA, Martin JF, Owens GK. Pitx2 is functionally important in the early stages of vascular smooth muscle cell differentiation. J Cell Biol 2008;181:461–473. [DOI] [PMC free article] [PubMed]
- 58.Wagenseil JE, Mecham RP. New insights into elastic fiber assembly. Birth Defects Res C Embryo Today 2007;81:229–240. [DOI] [PubMed]
- 59.Engler AJ, Carag-Krieger C, Johnson CP, Raab M, Tang HY, Speicher DW, Sanger JW, Sanger JM, Discher DE. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. J Cell Sci 2008;121:3794–3802. [DOI] [PMC free article] [PubMed]
- 60.Park JS, Chu JS, Tsou AD, Diop R, Tang Z, Wang A, Li S. The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-β. Biomaterials 2011;32:3921–3930. [DOI] [PMC free article] [PubMed]
- 61.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 1999;68:729–777. [DOI] [PubMed]
- 62.Taipale J, Keski-Oja J. Growth factors in the extracellular matrix. FASEB J 1997;11:51–59. [DOI] [PubMed]
- 63.Zeng X, Gray M, Stahlman MT, Whitsett JA. TGF-β1 perturbs vascular development and inhibits epithelial differentiation in fetal lung in vivo. Dev Dyn 2001;221:289–301. [DOI] [PubMed]
- 64.Lecart C, Cayabyab R, Buckley S, Morrison J, Kwong KY, Warburton D, Ramanathan R, Jones CA, Minoo P. Bioactive transforming growth factor–beta in the lungs of extremely low birthweight neonates predicts the need for home oxygen supplementation. Biol Neonate 2000;77:217–223. [DOI] [PubMed]
- 65.Sterner-Kock A, Thorey IS, Koli K, Wempe F, Otte J, Bangsow T, Kuhlmeier K, et al. Disruption of the gene encoding the latent transforming growth factor–β binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev 2002;16:2264–2273. [DOI] [PMC free article] [PubMed]
- 66.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 2012;196:395–406. [DOI] [PMC free article] [PubMed]
- 67.Chen JH, Chen WL, Sider KL, Yip CY, Simmons CA. β-Catenin mediates mechanically regulated, transforming growth factor–β1–induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler Thromb Vasc Biol 2010;31:590–597. [DOI] [PubMed]
- 68.Cohen ED, Ihida-Stansbury K, Lu MM, Panettieri RA, Jones PL, Morrisey EE. Wnt signaling regulates smooth muscle precursor development in the mouse lung via a tenascin C/PDGFR pathway. J Clin Invest 2009;119:2538–2549. [DOI] [PMC free article] [PubMed]