

Summary
In animals, Hox transcription factors define regional identity in distinct anatomical domains. How Hox genes encode this specificity is a paradox, because different Hox proteins bind with high affinity in vitro to similar DNA sequences. Here, we demonstrate that the Hox protein Ultrabithorax (Ubx) in complex with its cofactor Extradenticle (Exd) bound specifically to clusters of very low affinity sites in enhancers of the shavenbaby gene of Drosophila. These low affinity sites conferred specificity for Ubx binding in vivo, but multiple clustered sites were required for robust expression when embryos developed in variable environments. Although most individual Ubx binding sites are not evolutionarily conserved, the overall enhancer architecture—clusters of low affinity binding sites—is maintained and required for enhancer function. Natural selection therefore works at the level of the enhancer, requiring a particular density of low affinity Ubx sites to confer both specific and robust expression.
Introduction
Diversity along the anterior-posterior axis of animals results from differential expression of Hox transcription factors, which regulate different sets of target genes to determine the features specific to each anatomical region (McGinnis and Krumlauf, 1992). For example, in Drosophila, Sex combs reduced (Scr) determines anterior thoracic segments (Struhl, 1982; Wakimoto and Kaufman, 1981), whereas Ultrabithorax (Ubx) and abdominalA (abdA) specify thoracic and abdominal segments (Lewis, 1978; Sánchez-Herrero et al., 1985).
Hox protein specificity is paradoxical, because all Hox proteins have similar DNA binding domains (the homeodomain), particularly for residues that contact DNA directly (Akam, 1989; McGinnis and Krumlauf, 1992). As a result, all Hox proteins bind similar DNA sequences with high affinity (Berger et al., 2008; Mann et al., 2009; Noyes et al., 2008). In principle, one solution to this paradox is that sequences outside of the homeodomain, which have diverged among Hox proteins, allow interactions with a diversity of cofactors to confer specificity. However, only two cofactors, the homeodomain proteins Extra-denticle/Pbx (Exd) and Homothorax/MEIS (Hth) (Moens and Selleri, 2006), are known to interact with Hox proteins (Chan et al., 1994; Chang et al., 1995; Mann et al., 2009). Exd dimerizes with Hox proteins and Hth facilitates nuclear localization and DNA binding of Exd (Pai et al., 1998; Rieckhof et al., 1997; Ryoo et al., 1999). Thus, Hox specificity is unlikely to arise from interactions with a diversity of cofactors.
However, Hox protein structure is altered when bound to DNA with Exd, resulting in increased binding site specificity of Hox-Hth-Exd complexes in comparison with Hox monomers (Joshi et al., 2007; Slattery et al., 2011). In vivo support for this latent specificity model came from studies of artificial enhancers containing multimerized Hox-Exd binding sites (Ryoo and Mann, 1999). Therefore, it is not clear whether this mechanism is sufficient to account for the high degree of regulatory specificity exhibited by Hox proteins on native enhancers.
One clue that may inform the Hox specificity paradox is that many enhancers, including Hox-regulated enhancers, contain multiple binding sites for the same transcription factor (Arnone and Davidson, 1997; Gotea et al., 2010; Lifanov et al., 2003; Ochoa-Espinosa et al., 2005; Papatsenko et al., 2002; Stanojevic et al., 1991). These so-called homotypic binding site clusters are widespread, but the functions of clustered binding sites are understood in only a few cases. For example, homotypic clusters can fine-tune the response to graded transcription factors levels (Driever et al., 1989; Gaudet and Mango, 2002; Jiang and Levine, 1993; Rowan et al., 2010; Struhl et al., 1989), control the timing of enhancer activation (Gaudet and Mango, 2002), or determine whether binding results in repression or activation (Ramos and Barolo, 2013). However, elimination of individual binding sites in homotypic clusters often has little or no effect on enhancer activity (Doniger et al., 2005; Driever and Nüsslein-Volhard, 1989; Hersh and Carroll, 2005; Sarama¨ki et al., 2006; Stanojevic et al., 1991), suggesting that there may be additional reasons for the widespread existence of homotypic binding site clusters.
To gain insight into the Hox specificity paradox, we asked how Hox factors regulate native enhancers to achieve a specific pattern of epidermal trichomes along the anterior-posterior axis of Drosophila larvae. Trichome patterns display strong differences between adjacent segments in a Hox-dependent manner (Lewis, 1978; Sánchez-Herrero et al., 1985). Because shavenbaby (svb) is the master control gene for trichome development (Chanut-Delalande et al., 2006; Delon et al., 2003; Payre et al., 1999), we examined whether and how Hox factors regulate svb. We found that svb enhancers are directly regulated by Ubx and that they solve the Hox specificity paradox by employing clusters of low affinity Ubx-Exd binding sites. Specificity is encoded by low affinity sites and homotypic clusters of these sites provide regulatory robustness. This overall architecture—homotypic clusters of low affinity binding sites—is evolutionarily conserved and may provide a general mechanism to reconcile the need for both enhancer specificity and robustness.
Results
Ubx Positively Regulates svb Expression
In wild-type embryos of Drosophila melanogaster, cells of the ventral first abdominal segment (A1) differentiate a row of stout trichomes (Figure 1B). These trichomes were lost in the absence of Ubx (Figure 1D). Reciprocally, ectopic expression of Ubx using a heat shock inducible promoter (HS:Ubx) caused production of ectopic trichomes in thoracic segments (Figure 1F) (González-Reyes and Morata, 1990; Mann and Hogness, 1990). Because svb controls trichome development (Chanut-Delalande et al., 2006), we tested whether Ubx regulates svb expression. In wild-type embryos, svb was expressed strongly in cells of A1 and other abdominal segments that generate ventral trichomes and only weakly in the third thoracic segment (Figure 1A). In the absence of Ubx, svb expression was reduced in segment A1 (Figure 1C), consistent with the loss of the A1 trichomes in these larvae (Figure 1D). When we expressed Ubx ubiquitously, svb was upregulated in thoracic segments in a pattern similar to svb expression in segment A1 (Figure 1E). These results indicate that Ubx is required for expression of svb in the cells that generate A1 trichomes and that Ubx is sufficient to induce ectopic expression of svb when misexpressed in thoracic segments.
Figure 1. Ubx Is Necessary and Sufficient for svb Expression.
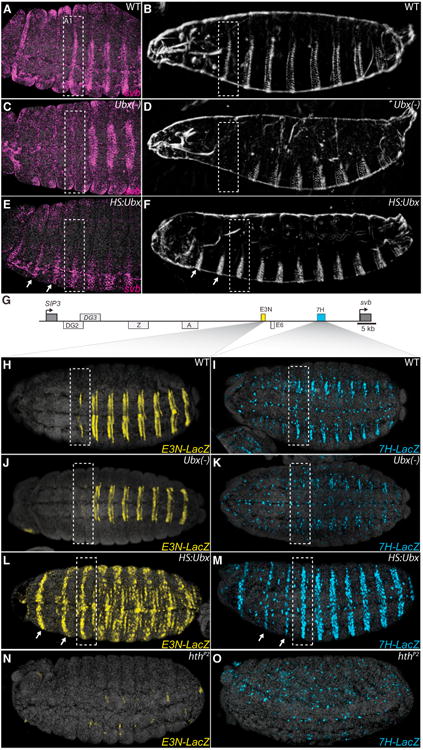
(A–F) Embryos stained with fluorescent svb mRNA probe and larval cuticle preps (B, D, and F) of the indicated genotypes. Loss of Ubx function transformed segment A1 into a thoracic segment that lacks svb expression (C) and larval trichomes (D), highlighted with bounding boxes. Ubiquitous expression of Ubx protein resulted in homeotic transformations of thoracic segments (arrows) into segments resembling segment A1 (E and F).
(G) Schematic of the svb upstream cis-regulatory region, indicating embryonic enhancers. The ventral enhancers E3N and 7H are highlighted in yellow and blue boxes, respectively. See also Figure S1.
(H–O) Expression of E3N∷lacZ or 7H∷lacZ reporter constructs (I, K, M, and O). Ubx was necessary for E3N and 7H reporter expression in segment A1 (J and K) and sufficient for their expression in thoracic segments when expressed ubiquitously (L and M).
(N and O) In hthP2 mutant embryos, activity of both the E3N and 7H enhancers was lost.
See also Table S1.
Ubx Controls Multiple svb Enhancers
To determine how Ubx regulates svb expression, we examined the effects of altered Ubx expression on two svb enhancers, called E and 7, that drive ventral stripes of expression (Figure 1G) (Frankel et al., 2010, 2011; McGregor et al., 2007). Through systematic functional dissection, we identified a 292 bp region of E, called E3N, and a 1,056 bp region of 7, called 7H (Figure S1 available online; Table S1), that each drove expression that accurately recapitulated the ventral patterns generated by the larger regions from which they were derived.
In wild-type embryos, E3N and 7H reporter genes were expressed in ventral rows of segments A1–A8 (Figures 1H and 1I). In embryos that lacked Ubx, E3N and 7H reporter gene expression was lost in the A1 segment (Figures 1J and 1K) and reduced in A2–A8 segments (Figures 1H–1K), consistent with the reduction in trichome numbers caused by loss of Ubx function (Lewis, 1978). Ectopic Ubx caused ectopic expression of E3N and 7H in thoracic segments and increased expression in abdominal segments (Figures 1L and 1M). In response to all manipulations of Ubx function, the expression patterns driven by E3N and 7H were similar to endogenous svb expression (Figures 1A–1F and 1H–1M). Therefore, these two enhancers respond to Ubx and, at least in part, capture the regulatory inputs of Hox genes to establish the anterior-posterior pattern of svb expression and trichomes.
Hox proteins bind DNA with Exd and Hth (Mann et al., 2009) and embryos lacking either hth or exd display homeotic transformations of trichome patterns (Jürgens et al., 1984; Peifer and Wieschaus, 1990; Rieckhof et al., 1997). To test if the Exd-Hth complex contributes to Ubx regulation of svb expression, we assayed expression of the E3N and 7H enhancers in embryos homozygous for a strongly hypomorphic hth allele, hthP2, which cannot facilitate nuclear localization of Exd (Noro et al., 2006; Rieckhof et al., 1997). E3N and 7H expression was abrogated in hthP2 embryos (Figures 1N and 1O), suggesting that Ubx requires Exd and Hth for activation of these svb enhancers.
The loss of E3N and 7H activity in abdominal segments in hthP2 embryos suggests that multiple Hox genes activate these enhancers. While Ubx specifies the trichomes in A1, it acts together with abdA to specify trichomes in more posterior segments (Lewis, 1978). Ubx and AbdA have similar DNA-binding specificities in complex with Exd (Karch et al., 1990; Slattery et al., 2011) and either Ubx or abdA is sufficient to drive svb expression in ventral abdominal stripes (Coiffier et al., 2008). Accordingly, we found that embryos deficient for Ubx and abdA expressed neither E3N nor 7H in abdominal stripes (see below), indicating that both Ubx and AbdA activate the E3N and 7H svb enhancers.
Ubx Regulates the E3N and 7H Enhancers Directly through Multiple Low Affinity Binding Sites
To determine whether Ubx binds to svb enhancers, first we examined genome-wide Ubx and Hth chromatin immunoprecipitation data (Choo et al., 2011). These data revealed in vivo binding of Ubx and Hth at the E3N and 7H regions, as well as at other svb enhancer regions (Figure S1). These results suggest that Ubx may regulate the E3N and 7H svb enhancers directly, which we tested further below.
Surprisingly, the DNA sequences of E3N and 7H contained no Hox-Exd sites that match those previously identified by systematic evolution of ligands by exponential enrichment sequencing (SELEX-seq) (Slattery et al., 2011). Therefore, we systematically searched for Ubx binding sites in E3N using electrophoretic mobility shift assays (EMSAs) of overlapping DNA fragments (Figures 2A and S2; Table S2). Ubx showed concentration-dependent binding to E3N1 and E3N2 fragments, but only when in complex with both Hth and Exd (Figures 2C, S2B, and S2C). These fragments bound Ubx-Exd in complex with either full-length Hth (HthFL) or with HthHM, similar to a naturally occurring isoform of Hth that lacks a homeodomain but that can translocate Exd to the nucleus (Ryoo et al., 1999). Neither HthHM-Exd nor HthFL-Exd bound to these E3N subfragments in the absence of Ubx (Figures S2B and S2C). Thus, despite the absence of predicted Ubx binding sites in E3, these data revealed binding of Ubx-Exd-Hth trimers— hereafter abbreviated as Ubx-Exd for simplicity—to several regions of this enhancer.
Figure 2. The svb E3N Enhancer Contains a Cluster of Ubx-Exd Binding Sites.
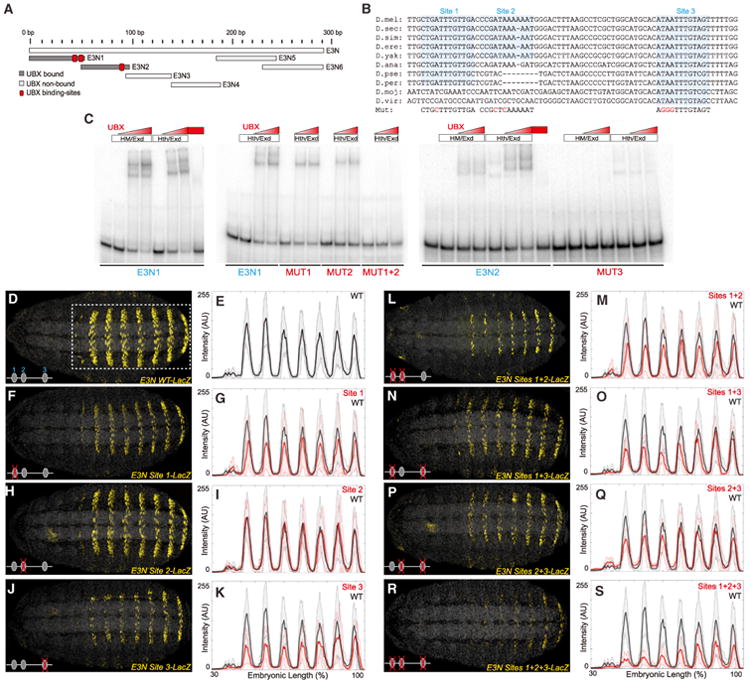
(A) A schematic of the regions tested for their ability to bind Ubx-Exd, assayed via EMSAs. See also Figures S2 and S3.
(B) Sequence alignment for the region of the E3N enhancer containing the three Ubx-Exd sites, labeled and highlighted. Dashes indicate gaps in the aligned sequence. Mutations of the Ubx-Exd binding sites are shown (Mut).
(C) Ubx-Hth-Exd bound specifically to each of the three sites, as demonstrated with EMSAs. In this and the following figures, Hth and HM refer to the full-length (HthFL) and homeodomainless (HthHM) isoforms of Hth, respectively.
(D–S) Expression of E3N∷lacZ reporter constructs with Ubx-Exd sites altered as indicated (B), juxtaposed with plots of average expression in the region outlined in (D) (n = 10 for each genotype). In all plots, the black and red lines denote expression driven by the wild-type and modified enhancers, respectively. Shaded areas indicate ±1 SD. AU, arbitrary units of fluorescence intensity.
See also Figures S4 and S5 and Tables S1 and S2.
To identify the Ubx-Exd binding sites in the E3N subregions, we systematically tested binding of Ubx-Exd to oligonucleotides mutated at each 5′ -AT dinucleotide pair (Figure S3) and found that most of the Ubx-Exd binding activity came from three sites (Figure 2C). Mutation of each of two binding sites in E3N1 reduced Ubx-Exd binding and mutation of both together abolished Ubx-Exd binding (Figure 2C, see also Figure S3). In the E3N2 fragment, we found a third site that, when mutated, abolished Ubx-Exd binding (Figures S3P and S3Q). Mutation of an additional site located near the 5′ end of E3N1 also reduced Ubx-Exd binding, suggesting that this region may contain another low affinity Ubx-Exd binding site (Figures S2 and S3D). The Ubx-Exd binding sites in E3N show variable levels of evolutionary conservation and only site 3 is conserved across all sequenced Drosophila species.
We next tested, in vivo, the role of Ubx-Exd sites identified in vitro by generating transgenic constructs with all possible combinations of the three sites mutated (Figure 2). Mutation of either site 1 or site 3 reduced the expression levels driven by E3N (Figures 2F, 2G, 2J, and 2K). Mutation of site 2 had no detectable effect on E3N expression (Figures 2H and 2I), including when combined with either site 1 or site 3 mutations (Figures 2L, 2M, 2P, and 2Q). However, when both site 1 and site 3 were mutated, the E3N enhancer still drove weak expression, which was reduced further upon knockout of site 2 (Figures 2N, 2O, 2R, and 2S). Thus, all three Ubx-Exd sites in the E3N enhancer are functional in vivo.
We obtained very similar results for Ubx binding to the 7H enhancer. In vitro assays identified three low affinity Ubx-Exd binding sites in 7H (Figures S4A and S4B). Individual mutation of each of the three Ubx-Exd binding sites did not modify the activity of 7H in embryos, either qualitatively (Figures S4C, S4G, S4K, and S4O) or quantitatively (Figures S4D, S4H, S4L, and S4P). In contrast, simultaneous mutation of sites 1 and 2, or sites 2 and 3, decreased 7H activity (Figures S4E–S4N) and mutation of all three sites almost completely abrogated 7H expression (Figures S4Q and S4R). Collectively, these results indicate that, as observed for E3N, the 7H svb enhancer uses at least three low affinity Ubx-Exd sites to drive expression along the AP axis of embryos. The absence of Ubx-Exd sites in E3N or 7H that match those detected by SELEX-seq (Slattery et al., 2011) implies that these sites have very low affinity for Ubx-Exd (see below).
In addition to Ubx, the E3N and 7H enhancers are regulated in vivo by abdA (Figure S5). Therefore, we tested whether the Ubx-Exd sites we identified could also bind AbdA-Hth-Exd (AbdA-Exd). In vitro, AbdA-Exd bound to the same E3N and 7H binding sites as Ubx-Exd did, and binding was abrogated when these sites were mutated (Figure S5). Thus, Ubx-Exd and AbdA-Exd directly regulate the E3N and 7H enhancers through the same binding sites.
Taken together, these data show that both the E3N and 7H svb enhancers contain clusters of low affinity Ubx/AbdA-Exd binding sites that are required to drive svb expression in ventral abdominal stripes. They further indicate that these sites mediate the action of Ubx in segment A1 and Ubx plus AbdA in segments A2–A8.
Proper Regulation of a svb Enhancer Requires Low Affinity Ubx Binding Sites
While our in vivo assays demonstrated that all of the Hox-Exd sites in E3N and 7H are required for proper function, it is not clear why these enhancers employ low affinity rather than higher affinity binding sites. We hypothesized that the low affinity of these binding sites may be part of the solution to the Hox specificity paradox. To explore this idea, we analyzed previously published data in which the DNA sequence preferences of all Drosophila Hox-Exd complexes were measured using SELEX-seq, resulting in relative affinity scores from 0.03 to 1 (Slattery et al., 2011). Using these data, we asked if there was any correlation between affinity and specificity. For example, do sequences with low affinity versus high affinity for Ubx-Exd display preference for Ubx-Exd compared to other Hox-Exd complexes? The results of this analysis were striking; only sequences with a relative affinity lower than 0.3 bound Ubx/AbdA-Exd specifically compared to the other Hox-Exd complexes (Figure 3). Moreover, as the relative affinity for Ubx/AbdA-Exd decreased, the number of sequences that bound specifically to Ubx/AbdA-Exd increased (Figure 3). These data imply that Hox-Exd complexes display, at least in vitro, a tradeoff between binding affinity and specificity.
Figure 3. Inverse Correlation between Sequence Affinity and Specificity.

The proportion of 12mer sequences bound by various Hox-Exd complexes versus relative affinity of these 12mers for Ubx/AbdA-Exd is shown as colored bars (specificity groups). The number of 12mers in each affinity bin is plotted as a gray line. Average relative affinities of 12mers were calculated for four pairs of Hox-Exd complexes with similar binding profiles: (1) Labial and Pb, (2) Dfd and Scr, (3) AbdB and Antp, and (4) Ubx and AbdA. Sequences specific for Ubx/AbdA-Exd (green bars) are more prevalent in lower affinity bins than in higher affinity bins.
To test whether this affinity-specificity tradeoff holds in vivo, we generated E3N transgenic variants in which we varied the affinity of the Ubx-Exd binding sites according to the relative affinities predicted by SELEX-seq (Figure 4A) (Slattery et al., 2011). Although none of the binding sites found in the native svb enhancer were identified by SELEX-seq, we estimate (based on the core 8-mer) that they have relative affinities <0.03 (Slattery et al., 2011). Every mutation that increased the affinity of Ubx-Exd sites resulted in qualitative or quantitative changes in E3N enhancer expression (Figure 4). For example, converting either native sites 1 or 2 to high-affinity sites (scores of 0.87 or 0.79, respectively) resulted in increased expression in the normal domain of E3N and ectopic expression anteriorly and dorsally (Figures 4C and 4D). Replacing site 3 in E3N with the highest affinity site (score of 1.0) also resulted in ectopic expression in anterior segments and in the intestine (Figure 4B). We further explored the functional consequences of gradually increasing the affinity of a Hox-Exd binding site by replacing site 3 with sites that have a range of relative affinities, from 0.06 to 0.72. A small increase in affinity to 0.06 resulted in higher levels of E3N expression within its normal expression domain (Figures 4H and 4I). Increasing the affinity to 0.25, 0.65, and 0.72 altered levels of expression in the normal domains of E3N and induced ectopic expression in anterior segments (white arrows in Figures 4E–4G).
Figure 4. Conversion of Low Affinity Ubx-Exd Binding Sites to Higher Affinity Sites Results in Ectopic Expression.
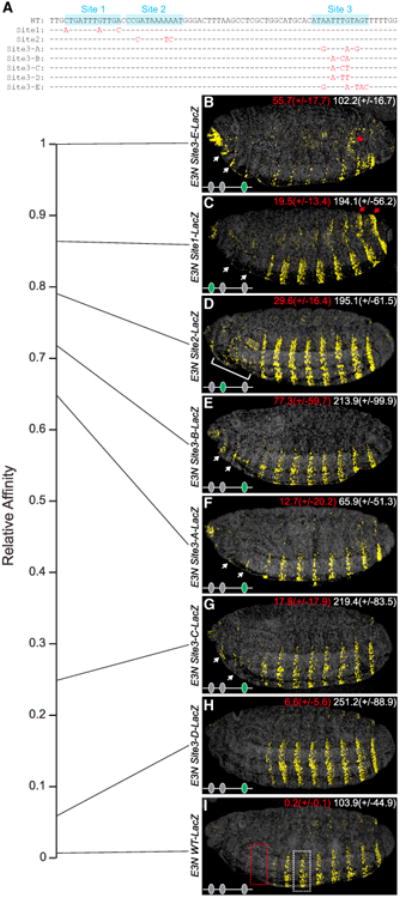
(A) Aligned E3N sequences from wild-type and mutated sequences. Dashes and red letters indicate unaltered and modified sequence, respectively.
(B–I) Embryos carrying E3N∷lacZ constructs, with Ubx-Exd sites altered as indicated in (A). The numbers in the top right of each panel indicate the average levels of expression in the regions outlined in (I) (n = 10 for each genotype), measured in arbitrary units of fluorescence intensity. Numbers in parantheses indicate ±1 SD. White arrows and brackets denote expression in domains anterior to segment A1 (B–G). The red asterisk marks ectopic staining in the intestine; red arrows indicate ectopic dorsal and lateral expression (C).
See also Figure S6 and Table S1.
We also observed strong position effects of a high affinity site, similar to observations in a previous study (Swanson et al., 2010). Placing the highest affinity site 5′ of the E3N enhancer resulted in ectopic expression in anterior segments, but decreased expression in the normal domain (Figure S6B). In contrast, placing this site inside the enhancer increased expression in the normal domain and generated ectopic expression in multiple regions (Figure S6C). We cannot rule out the possibility, however, that these position-dependent effects resulted from the creation or destruction of binding sites for additional factors.
Taken together, these results indicate that Hox-Exd sites with higher affinity than the native sites alter the specificity of the svb enhancer, demonstrating that the affinity-specificity tradeoff that was inferred from in vitro data also pertains in vivo.
High Affinity Hox Binding Sites Decreased the Specificity of Enhancer Function
Replacement of native sites with high affinity sites caused ectopic expression mostly outside of the domains of Ubx and abdA expression (Figure S5), suggesting that these high affinity sites bound transcription factors other than Ubx and AbdA. Indeed, in embryos deficient for Ubx, the E3N enhancers with high affinity binding sites showed the expected reduction of expression in A1 (where Ubx is the only Hox gene active), but they also continued to drive ectopic expression in anterior segments (Figures 5A–5D).
Figure 5. Low Affinity Ubx-Exd Binding Sites Provide High Ubx-Exd Specificity.
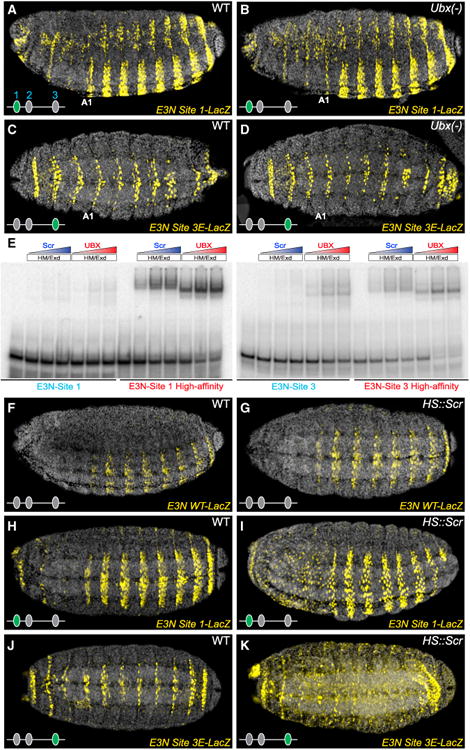
(A–D) Embryos carrying E3N∷lacZ constructs, with Ubx-Exd sites altered as indicated in (Figure 4A). In embryos deficient for Ubx, E3N∷lacZ with high affinity sites drove extensive ectopic expression (B and D).
(E) Scr-Exd did not bind to wild-type E3N Ubx-Exd sites in vitro, as demonstrated with EMSAs. However, both Scr-Exd and Ubx-Exd bound to high-affinity Ubx-Exd sites.
(F–K) Ubiquitous expression of Scr (hs∷Scr) did not alter expression of the wild-type E3N∷lacZ (G), but caused ectopic expression of E3N∷lacZ carrying high-affinity Ubx-Exd sites (I and K).
See also Tables S1 and S2.
Sex comb reduced (Scr) was an attractive candidate for driving some of the ectopic anterior expression of svb enhancers carrying high-affinity sites. Scr is expressed in anterior segments (Kuroiwa et al., 1985) and SELEX-seq data indicated that Scr-Exd can bind to high-affinity Ubx/Exd binding sites (Slattery et al., 2011). When assayed on the E3N svb enhancer, Scr-Exd showed little or no in vitro binding to the native E3N sites, but it bound to the high affinity sites even more strongly than Ubx-Exd bound to the native sites (Figure 5E). In vivo, uniform expression of Scr produced no obvious changes in the expression of wild-type E3N (Figures 5F and 5G), but drove ectopic expression of E3N variants that carried one high-affinity site (Figures 5H–5K). Thus, replacing low affinity Ubx/AbdA-Exd sites with high-affinity sites enabled the E3N enhancer to respond to Scr. In addition to Scr, it is likely that other homeodomain transcription factors bind and activate the E3N enhancers carrying high-affinity sites to generate their broad domains of ectopic expression.
Together, our results indicate that the native low affinity Ubx/AbdA-Exd binding sites in the E3N enhancer confer specificity for Ubx-Exd and AbdA-Exd over other Hox proteins, such as Scr, and probably over additional homeodomain factors.
Clusters of HOX Binding Sites Confer Robustness to Genetic and Environmental Variability
As discussed earlier, some of the Ubx/AbdA-Exd binding sites in the E3N and 7H enhancers can be mutated with minimal effects on reporter gene expression (Figures 2 and S4). It is not clear, therefore, why these enhancers contain multiple Hox binding sites. We wondered if the multiple, apparently redundant, Ubx-Exd binding sites within individual svb enhancers contribute to transcriptional robustness, in the same way that multiple enhancers of svb confer robustness in the face of environmental and genetic variation (Frankel et al., 2010).
To test this hypothesis, first we examined the effects of altered levels of Ubx on the expression of E3N enhancers. The wild-type E3N enhancer drove normal expression in embryos heterozygous for an Ubx null mutation (Figures 6A, 6B, 6M, and 6N). In contrast, all E3N enhancers that contained single mutations in the Ubx-Exd binding sites drove dramatically lower levels of expression in Ubx heterozygotes, compared to wild-type embryos (Figures 6C–6T). Similar effects were observed for most combinations of the mutations (Figures 6I, 6J, 6U, and 6V). These results indicate that E3N requires multiple sites to confer robustness when Ubx dose is perturbed. The reduced activity of these E3N enhancers in Ubx heterozygotes also provides further evidence that the Ubx-Exd binding sites respond to Ubx in vivo.
Figure 6. The svb E3N Enhancer Contains a Cluster of Ubx-Exd Binding Sites that Confer Robustness against Environmental and Genetic Variation.
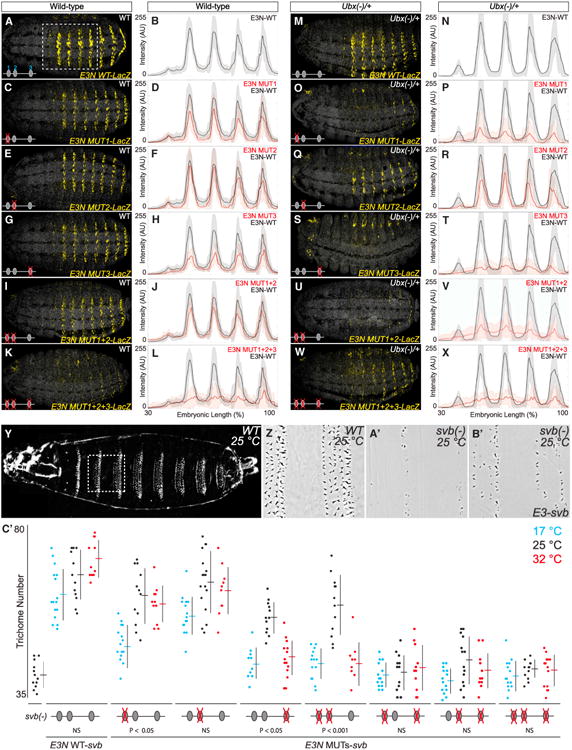
(A–X) Wild-type (A–L) and Ubx heterozygote (M–X) embryos carrying E3N∷lacZ constructs with Ubx-Exd sites altered as indicated in Figure 2B, juxtaposed with plots of average expression in the region outlined in (D) (n = 10 for each genotype). Shaded bounding areas indicate ±1 SD. AU, arbitrary units of fluorescence intensity.
(Y–B′) Cuticle preps showing that the E3N∷svb transgene (B′) in a svb null mutant background rescued a subset of the wild-type trichome pattern (cf. Y–A′).
(C′) The number of trichomes in the larval A2 segment for the corresponding genotypes. The error bars indicate ±1 SD. Significance values are sequential Bonferroni test p values, to control the type I error rate, from separate ANOVA tests for each genotype.
See also Table S1.
Next, we assayed the effects of environmental variation on enhancer activity by exploiting the fact that svb enhancers driving a svb cDNA in svb null embryos provide a sensitive and quantitative readout of enhancer function (Frankel et al., 2010, 2011). We reared embryos at 17°C and 32°C, temperature extremes that are still compatible with normal development (Powsner, 1935). In embryos carrying the wild-type E3N∷svb rescue construct, trichome numbers were relatively invariant to temperature extremes (Figure 6C). In contrast, enhancers containing a single mutated Ubx/Exd binding site showed reduced rescue of trichomes at extreme temperatures (Figure 6C). Furthermore, the simultaneous mutation of site 1 and 2 abrogated trichome rescue at extreme temperatures, while other double or triple combinations led to no rescue (Figure 6C′).
These results indicate that multiple Ubx-Exd binding sites are required for normal enhancer function and to cope with variable genetic backgrounds and environments, similar to conditions faced by flies in the wild.
Ubx Binds a Cluster of Low Affinity Binding Sites in the Orthologous E3N Enhancer from a Distantly Related Species
We wondered whether the enhancer architecture discovered for E3N and 7H, with homotypic clusters of low affinity Hox-Exd sites, is an evolutionarily conserved feature of svb enhancers. Because the large-scale cis-regulatory landscape of svb has been well conserved in Drosophila virilis (Frankel et al., 2012), we examined this question by focusing on the E3N region of D. virilis.
Drosophila melanogaster and D. virilis last shared a common ancestor ∼40 mya and the E3N region displays little sequence conservation between these species (Figure 7A). We thus employed EMSAs to identify, in an unbiased manner, all of the Ubx-Exd binding sites putatively present in the D. virilis E3N orthologous region (VE3N). We found that four fragments— VE3N1, VE3N2, VE3N5, and VE3N9—bound Ubx-Exd in vitro (Figures 7B–7D). Comprehensive mutagenesis of these fragments revealed five Ubx-Exd binding sites (Figures 7B, 7C, and S7). One of these sites is evolutionarily conserved and four sites display no sequence conservation to D. melanogaster and only weak conservation to closely related species (Figure 7A). As observed for D. melanogaster, none of the Ubx-Exd binding sites of VE3N were detected by SELEX-seq, indicating that they are low affinity sites.
Figure 7. Multiple Low Affinity Poorly Conserved Ubx-Exd Binding Sites Regulate the Drosophila virilis E3N Enhancer.
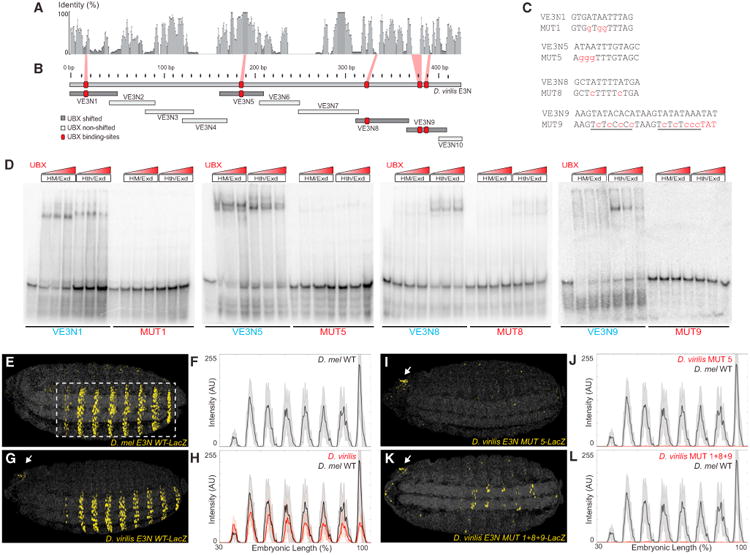
(A) Sequence conservation over a 10 bp sliding window for a sequence alignment of the E3N region from ten Drosophila species.
(B) Regions tested for the ability to bind Ubx-Exd, assayed via EMSAs (see also Figure S7). The positions of the Ubx-Exd sites are indicated with red boxes.
(C) E3N Ubx-Exd binding-site sequences aligned with site-specific mutations indicated in lowercase, red letters.
(D) Ubx-HthFL-Exd and Ubx-HthHM-Exd bound five sites in the D. virilis E3N enhancer, as demonstrated with EMSAs (see also Figure S7). This binding was reduced when the sites were mutated (MUT).
(E–L) Embryos carrying E3N∷LacZ constructs, with Ubx-Exd sites altered as indicated in (C), juxtaposed with plots of average expression (n = 10 for each genotype). Black lines denote expression driven by the D. melanogaster and D. virilis enhancers, respectively.
See also Tables S1 and S2.
We next tested whether these Ubx-Exd sites function in vivo by generating transgenic D. melanogaster lines that contained either the wild-type or mutated D. virilis VE3N enhancers. The wild-type VE3N enhancer drove lower levels of expression than did its D. melanogaster E3N counterpart (Figures 7E–7H), a result observed often in tests of orthologous enhancers (Crocker et al., 2008; Ludwig et al., 1998). Mutation of the conserved site present in VE3N resulted in the loss of reporter expression (Figures 7I and 7J). To test if only this site was required for VE3N expression, we mutated the four nonconserved sites, leaving the conserved site intact. This reporter also displayed very little VE3N activity (Figures 7K and 7L). Therefore, multiple Ubx-Exd binding sites, at least some of which are poorly conserved, contribute to the proper regulation of the D. virilis VE3N enhancer.
Taken together, these results indicate that clustering of low affinity Ubx-Exd sites is an evolutionarily conserved strategy used by svb enhancers, although many of the individual binding sites are not conserved across species.
Discussion
We have demonstrated that the Hox protein Ubx regulates separate enhancers of the svb gene by binding, with its cofactors Exd and Hth, to clusters of low affinity binding sites. Combining in vitro and in vivo assays, we provided experimental demonstration of an affinity-specificity tradeoff for Hox proteins, such that enhancers that integrate Hox inputs to drive regionalized expression are unlikely to utilize high affinity Hox binding sites. Forced to utilize low affinity sites, enhancers have evolved to contain multiple binding sites to ensure regulatory robustness to genetic and environmental variations. Most individual Ubx-Exd sites have evolved rapidly, but evolution has conserved overall enhancer architecture, with clusters of low affinity sites.
Homotypic clusters of transcription factor binding sites are pervasive in animal genomes (Arnone and Davidson, 1997; Gotea et al., 2010; Lifanov et al., 2003; Ochoa-Espinosa et al., 2005; Papatsenko et al., 2002; Stanojevic et al., 1991) and several models have been proposed to explain their existence (Doniger et al., 2005; Giorgetti et al., 2010; He et al., 2012; Segal et al., 2008). Our results provide experimental evidence that homotypic clusters of Hox binding sites can confer robustness to enhancers. This may reflect a more widespread phenomenon. Although many enhancers contain homotypic clusters with low affinity sites, previous studies have rarely detected changes in expression by deleting individual binding sites (Doniger and Fay, 2007; Driever and Nüsslein-Volhard, 1989; Estella et al., 2008; Giorgianni and Mann, 2011; Hersh and Carroll, 2005; Saramäki et al., 2006; Stanojevic et al., 1991). However, these mutated enhancers have not been tested in variable environments. It is possible that many of these clustered sites confer regulatory robustness.
It is useful to compare our results with previous studies that have demonstrated specific regulatory functions for homotypic clusters. For example, clustered binding sites in an enhancer of the Drosophila hunchback gene mediate cooperative DNA binding by Bicoid, which provides threshold-dependent enhancer activity (Driever et al., 1989; Lebrecht et al., 2005; Struhl et al., 1989). In other cases, clusters of homotypic binding sites act in a noncooperative manner to allow enhancers to respond in a graded fashion (Giorgetti et al., 2010), for example to determine expression levels in response to transcription factor concentrations (Driever et al., 1989; Gaudet and Mango, 2002; Rowan et al., 2010). It is worth noting that in these cases, where homotypic clusters mediate specific linear or nonlinear outputs, enhancers are bound by transcription factors that belong to small paralogous families: e.g., two paralogs for Msn2 (Hasan et al., 2002); three for p53 (Belyi et al., 2010); two for Dorsal (Silverman and Maniatis, 2001); and five for NFkB (Silverman and Maniatis, 2001). In contrast, there are 84 homeodomain-containing proteins encoded in the Drosophila genome, many with overlapping specificities (Berger et al., 2008; Noyes et al., 2008). Therefore, in previously described examples of homotypic clusters, binding affinity may not be a strong constraint on specificity.
For the Hox regulated svb enhancers, low affinity Ubx/AbdA-Exd binding sites enable specificity, while the clustering of low affinity sites confers phenotypic robustness. This is a fundamentally different constraint on clustered binding sites than observed in all previous examples. The affinity-specificity tradeoff, initially supported by our computational analysis of in vitro data, was confirmed in vivo by progressively increasing the affinity of the Ubx-Exd binding sites. While replacement of low affinity sites with higher affinity sites always quantitatively altered enhancer activity, either positively or negatively, most higher affinity sites generated strong ectopic expression. As we show, this ectopic expression is driven, at least in part, by gaining the binding of additional Hox proteins, which are normally not involved in the regulation of these enhancers. Other studies have performed replacement of low affinity sites with higher affinity sites and, in some cases, they have also observed ectopic expression (Busser et al., 2012; Driever et al., 1989; Gaudet and Mango, 2002; Jiang and Levine, 1993; Peterson et al., 2012; Ramos and Barolo, 2013; Scardigli et al., 2003; Stewart-Ornstein et al., 2013; Struhl et al., 1989). These altered patterns of expression may reflect increased sensitivity of enhancers to the same transcription factor that binds to the wild-type enhancer (Jiang and Levine, 1993). We observed a similar effect for Ubx and AbdA-dependent upregulation of svb enhancers in the cells in which they are normally expressed. In addition, however, we found that sites with higher affinity resulted in a reduced specificity, due to the binding of additional homeodomain proteins, such as Scr, to svb enhancers. Our computational analyses suggest that this affinity-specificity tradeoff is a fundamental property of Hox proteins and would therefore influence the architecture of enhancers that must generate specific outputs in response to Hox factors. We suggest that transcription factors that belong to other large paralog groups may exhibit a similar affinity-specificity tradeoff and that enhancers regulated by these factors may also exploit clusters of low affinity sites.
Our results help to explain previous difficulties with bioinformatic prediction of functional Hox binding sites, because low affinity sites are difficult to detect reliably. Indeed, the low affinity sites that implement Hox regulation within svb enhancers share little similarity with canonical Hox or Hox-Exd binding sites. Consequently, a very large number of seemingly disparate DNA sequences can confer low affinity binding for Hox proteins. If Hox-Exd sites are often clustered in the genome, then signals from genome-wide chromatin immunoprecipitation sequencing (ChIP-seq) will reflect binding to the entire cluster (as we observed) and the signals associated with individual low affinity sites may be difficult to discern from noise. Identification of important low affinity sites will require a change in computational approaches to analyzing genome-wide data. Currently, it is de rigueur to apply an arbitrary threshold to genome-wide data and then to analyze only signals above this threshold. This approach is likely to bias detection toward high affinity sites, whose functions may be distinct from those of clusters of low affinity sites.
Our findings provide insight into how different Hox proteins regulate specific target genes to generate phenotypic diversity across the anterior-posterior axis. One unanswered question is how the many low affinity DNA sequences, which appear to share little in common, are bound by the same Hox-Exd complex with apparently similar affinity. It is possible that variations in DNA shape (deviations from the structure of canonical B-DNA) influence Hox-Exd binding to low affinity sites (Dror et al., 2014; Joshi et al., 2007; Rohs et al., 2009). It remains unclear if very different sequences can adopt similar shapes, or whether instead the Hox-Exd complex can recognize a range of shapes. Resolution of this question will require structural studies of Hox-Exd complexes bound to a range of low affinity DNA sequences and quantitative analysis of their binding dynamics in vivo.
Experimental Procedures
Fly Strains and Transgenic Constructs
DNA fragments were cloned into the reporter constructs placZattB and pHSPattB GFP and the pRSQsvb rescue construct (Frankel et al., 2011) (see Table S1). Mutations were introduced using site-directed mutagenesis (Genescript). Plasmids were integrated into the attP2 landing site by Rainbow Transgenic Flies. Additional strains used were: svbR9/FM7c twi∷GFP (Delon et al., 2003); HS∷Ubx-1; Ubx1; hthP2 (Noro et al., 2006); and Ubx1abdA-D24AbdBD18 (Bloomington stock 1108).
Embryo Staining and Cuticles
Stage 15/16 embryos were collected, fixed, and stained using standard protocols with mouse anti-bGal (1:1,000, Promega) and anti-mouse AlexaFluor (1:500, Invitrogen) antibodies. Cuticles were prepared following standard protocols, imaged with phase-contrast microscopy, and ventral trichomes in larval A2 segments were counted.
Image Analysis
Embryos carrying reporter constructs were imaged on a Leica SPE Confocal Microscope. Sum projections of confocal stacks were assembled, images were scaled, background was subtracted using a 50-pixel rolling-ball radius and plot profiles of fluorescence intensity were analyzed using ImageJ software (http://rsb.info.nih.gov/ij). Data from the plot profiles were analyzed further in MATLAB (http://www.mathworks.com) (Crocker and Stern, 2013).
In Vitro Affinity-Specificity Tradeoff Calculations
Average relative affinities of 12mers from SELEX-seq data (Slattery et al., 2011) were calculated for four pairs of Hox-Exd complexes that share similar binding preferences: (1) Labial and Pb, (2) Dfd and Scr, (3) Antp and AbdB, and (4) Ubx and AbdA. Specificity groups (colored bars in Figure 3) were defined as having an average relative affinity ≥0.05 for bound complexes and <0.03 for unbound complexes. The proportion and total number of sequences in each specificity group were calculated for ten bins based on their Ubx/AbdA-Exd relative affinities.
DNA Alignments
Multiple sequence alignments were performed using Geneious (http://www.geneious.com) with MUSCLE alignment algorithms (anchor optimized).
Protein Purification and EMSAs
Ubx (isoform IVa), abdA, HthHM-Exd, and HthFL-Exd constructs, protein purification, and EMSA conditions were described previously (Lelli et al., 2011). Further experimental details are provided in Table S2.
Supplementary Material
Figure S1. Defining the Minimal Ventral Embryonic svb Enhancers, Related to Figure 1 and Table S1
(A) Schematic of the svb upstream cis-regulatory region, indicating embryonic enhancer elements. The ventral embryonic enhancers E3N and 7H are highlighted in yellow and blue, respectively.
(B–D) Early stage 15 embryo carrying both E∷lacZ and 7∷GFP reporter constructs, co-stained with antibodies against β -Galactosidase and GFP (B), demonstrate partially overlapping patterns of expression.
(E) Schematic of a subset of the E-derived enhancer fragments tested with transgenic reporter constructs (also see Figure 1). Enhancers indicated with filled boxes drove expression.
(F–H) Early stage 15 embryos carrying three of the E-derived enhancer reporter constructs—E, E3, and E3N—stained with an antibody against β -Galactosidase.
(I) Schematic of a subset of the 7 enhancer fragments tested with transgenic reporter constructs. Enhancers indicated with filled boxes drove expression and those with unfilled boxes showed no expression.
(J–O) Early stage 15 embryos carrying six of the 7-derived enhancer reporter constructs—7, 7B, 7D, 7H, 7I, and 7J—stained with an antibody against b-Galactosidase. Further reduction of 7H to 7J led to additional anterior expression (O).
(P) Log2 ratio enrichment profiles for Ubx and Hth across the svb upstream cis-regulatory region, with the minimal E3N and 7H enhancers indicated in yellow and blue, respectively.
Figure S2. Identification of Regions of the E3N Enhancer that Bind Ubx-Exd In Vitro, Related to Figure 2 and Table S1
(A) A schematic of the regions tested for the ability to bind Ubx-Exd assayed with EMSAs.
(B–G) Ubx-HthFL-Exd and Ubx-HthHM-Exd bound fragments E3N1 (B) and E3N2 (C), as demonstrated with EMSAs. There was also very weak binding of Ubx-HthFL-Exd and Ubx-HthHM-Exd to fragment E3N5 (F).
Figure S3. E3N Contains a Cluster of Three Ubx-Exd Binding Sites, Related to Figure 2 and Table S2
(A) A schematic of the regions tested for the ability to bind Ubx-Exd assayed with EMSAs.
(B and C) The aligned E3N1 and E3N2 sequences from wild-type and mutated sequences. Dashes indicate non-altered sequence. Red letters denote mutated sites. Blue boxes highlight Ubx-Exd binding sites.
(D–Q) Ubx-HthFL-Exd and Ubx-HthHM-Exd EMSAs for each enhancer fragment outlined in (B) and (C). Mutation of the putative Ubx-Exd binding sites shown in (B) and (C) led to decreased binding of Ubx-Exd.
Figure S4. The svb 7H Enhancer Contains a Cluster of Ubx-Exd Binding Sites, Related to Figure 2 and Tables S1 and S2
(A) A multiple sequence alignment for selected regions of the 7H-enhancer, with the three Ubx-Exd sites labeled and highlighted. Dashes indicate gaps in the aligned sequence. Bracketed numbers indicate the number of base pairs between the aligned sequences shown. Mutations of the Ubx-Exd binding-sites are shown (MUTS).
(B) Ubx-HthFL-Exd and Ubx-HthHM-Exd bound each of the three sites, as demonstrated with EMSAs. This binding was reduced when the sites were mutated (MUT).
(C, G, K, O, E, I, M, and Q) Early stage 15 embryos carrying 7H∷lacZ reporter constructs stained with an antibody against β-Galactosidase, with Ubx sites mutated as indicated in (A).
(D, H, L, P, F, J, N, and R) Plots showing profiles of average expression in the region outlined in (C) for the corresponding genotype (n = 10 for each genotype). In all plots, the black lines denote WT embryos and the red lines denote the MUT line indicated. Shaded bounding areas indicate ± 1 s.d. AU, arbitrary units of fluorescence intensity.
Figure S5. AbdominalA Binds to the Ubx Binding Sites in the 7H and E3N Enhancers, Related to Figure 2 and Tables S1 and S2
(A) Early stage 15 embryos stained with an antibody against AbdA and Ubx.
(B) Larval cuticle prep of the indicated genotype.
(C) AbdA-HthFL-Exd and AbdA-HthHM-Exd bound each of the Ubx-Exd sites in the E3N enhancer (see Figure 2), as demonstrated with EMSAs. This binding was decreased when the sites were mutated (as per Figure 2).
(D and E) Early stage 15 embryos carrying E3N∷lacz reporter constructs, stained with an antibody against β-Galactosidase, with genotypes indicated. (F) AbdA-HthFL-Exd and AbdA-HthHM-Exd bound each of the Ubx-Exd sites in the 7H enhancer (see Figure S4), as demonstrated with EMSAs. This binding was decreased when the sites were mutated (as per Figure S4).
(G and H) Early stage 15 embryos carrying 7H∷lacZ reporter constructs, stained with an antibody against β-Galactosidase, with genotypes indicated.
Figure S6. Addition of a High Affinity Site Results in Ectopic Expression, Related to Figure 4 and Table S1
(A) A sequence alignment of the wild-type and mutated versions of the E3N region, showing the modified sequences in red.
(B and C) Early stage 16 embryos carrying E3N∷lacZ reporter constructs stained with an antibody against β-Galactosidase, with an ectopic highest-affinity Ubx-Exd site added either 5′ (B) or internally (C) to the E3N enhancer. The numbers shown in the top right of each panel indicate the average level of expression in the region outlined in Figure 6I for the corresponding genotypes (n = 10 for each genotype), measured in arbitrary units of fluorescence intensity. Numbers in parantheses indicate ± 1 s.d. White arrows denote expression in domains anterior to segment A1 (A,B). Red arrows indicate ectopic dorsal and lateral expression (B).
Figure S7. Identification of Regions of the D. virilis E3N Enhancer that Bind Ubx-Exd In Vitro, Related to Figure 7 and Table S2
(A) A schematic of the regions tested for the ability to bind Ubx-Exd, assayed via EMSAs. Filled boxes indicate regions that bound Ubx-Exd.
(B–G) Ubx-HthFL-Exd and Ubx-HthHM-Exd bound fragments VE3N1 (B), VE3N5 (F), VE3NI (I), and VE3N9 (J), as demonstrated with EMSAs.
(L) A sequence alignment of the wild-type and mutated versions of the VE3N9 region, showing the mutated putative Ubx-Exd binding sites in red.
(M) EMSAs demonstrated that both putative Ubx-Exd binding sites must be mutated to eliminate binding of Ubx-HthHM-Exd or Ubx-HthFL-Exd to VE3N9.
(N) DNA sequence alignment of the E3N enhancer region. Shaded boxes indicate regions that bound Ubx-Exd in D. melanogaster (green) and D. virilis (purple).
Acknowledgments
We thank A. Sterling for technical assistance with EMSA experiments, A. Lemire and V. FitzPatrick for bioinformatics assistance, E. Preger for use of her 7∷GFP reporter construct, and J.Cande, Y. Ding, A. Lemire, J.Mast, E.Preger, and T. Shirangi for helpful discussions. This work was supported in part by NIH grant GM054510 to R.S.M. and ANR grant (ChronoNet) to F.P.
Footnotes
Supplemental Information: Supplemental Information includes seven figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2014.11.041.
Author Contributions: The experimental plan was conceived by J.C., N.A., R.S.M., and D.L.S. Most in vivo assays were performed by J.C. All in vitro assays and computational analysis were performed by N.A., assisted by L.R. and R.S.M. A.P.M., A.A., P.V., S.P., F.P., and D.L.S. defined the minimal enhancer elements. N.F. and S.W. performed some in vivo assays. J.C. assembled all figures. J.C., R.S.M., and D.L.S. wrote the paper, with input from N.A., A.P.M., N.F., S.W., S.P., and, especially, from F.P.
References
- Akam M. Hox and HOM: homologous gene clusters in insects and vertebrates. Cell. 1989;57:347–349. doi: 10.1016/0092-8674(89)90909-4. [DOI] [PubMed] [Google Scholar]
- Arnone MI, Davidson EH. The hardwiring of development: organization and function of genomic regulatory systems. Development. 1997;124:1851–1864. doi: 10.1242/dev.124.10.1851. [DOI] [PubMed] [Google Scholar]
- Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, Levine AJ. The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol. 2010;2:a001198. doi: 10.1101/cshperspect.a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Peña-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser BW, Shokri L, Jaeger SA, Gisselbrecht SS, Singhania A, Berger MF, Zhou B, Bulyk ML, Michelson AM. Molecular mechanism underlying the regulatory specificity of a Drosophila homeodomain protein that specifies myoblast identity. Development. 2012;139:1164–1174. doi: 10.1242/dev.077362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SKK, Jaffe L, Capovilla M, Botas J, Mann RS. The DNA binding specificity of Ultrabithorax is modulated by cooperative interactions with extradenticle, another homeoprotein. Cell. 1994;78:603–615. doi: 10.1016/0092-8674(94)90525-8. [DOI] [PubMed] [Google Scholar]
- Chang CP, Shen WF, Rozenfeld S, Lawrence HJ, Largman C, Cleary ML. Pbx proteins display hexapeptide-dependent cooperative DNA binding with a subset of Hox proteins. Genes Dev. 1995;9:663–674. doi: 10.1101/gad.9.6.663. [DOI] [PubMed] [Google Scholar]
- Chanut-Delalande H, Fernandes I, Roch F, Payre F, Plaza S. Shavenbaby couples patterning to epidermal cell shape control. PLoS Biol. 2006;4:e290. doi: 10.1371/journal.pbio.0040290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo SW, White R, Russell S. Genome-wide analysis of the binding of the Hox protein Ultrabithorax and the Hox cofactor Homothorax in Drosophila. PLoS ONE. 2011;6:e14778. doi: 10.1371/journal.pone.0014778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coiffier D, Charroux B, Kerridge S. Common functions of central and posterior Hox genes for the repression of head in the trunk of Drosophila. Development. 2008;135:291–300. doi: 10.1242/dev.009662. [DOI] [PubMed] [Google Scholar]
- Crocker J, Stern DL. TALE-mediated modulation of transcriptional enhancers in vivo. Nat Methods. 2013;10:762–767. doi: 10.1038/nmeth.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker J, Tamori Y, Erives A. Evolution acts on enhancer organization to fine-tune gradient threshold readouts. PLoS Biol. 2008;6:e263. doi: 10.1371/journal.pbio.0060263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delon I, Chanut-Delalande H, Payre F. The Ovo/Shavenbaby transcription factor specifies actin remodelling during epidermal differentiation in Drosophila. Mech Dev. 2003;120:747–758. doi: 10.1016/s0925-4773(03)00081-9. [DOI] [PubMed] [Google Scholar]
- Doniger SW, Fay JC. Frequent gain and loss of functional transcription factor binding sites. PLoS Comput Biol. 2007;3:e99. doi: 10.1371/journal.pcbi.0030099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doniger SW, Huh J, Fay JC. Identification of functional transcription factor binding sites using closely related Saccharomyces species. Genome Res. 2005;15:701–709. doi: 10.1101/gr.3578205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driever W, Nüsslein-Volhard C. The bicoid protein is a positive regulator of hunchback transcription in the early Drosophila embryo. Nature. 1989;337:138–143. doi: 10.1038/337138a0. [DOI] [PubMed] [Google Scholar]
- Driever W, Thoma G, Nüsslein-Volhard C. Determination of spatial domains of zygotic gene expression in the Drosophila embryo by the affinity of binding sites for the bicoid morphogen. Nature. 1989;340:363–367. doi: 10.1038/340363a0. [DOI] [PubMed] [Google Scholar]
- Dror I, Zhou T, Mandel-Gutfreund Y, Rohs R. Covariation between homeodomain transcription factors and the shape of their DNA binding sites. Nucleic Acids Res. 2014;42:430–441. doi: 10.1093/nar/gkt862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estella C, McKay DJ, Mann RS. Molecular integration of wingless, decapentaplegic, and autoregulatory inputs into Distalless during Drosophila leg development. Dev Cell. 2008;14:86–96. doi: 10.1016/j.devcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel N, Davis GK, Vargas D, Wang S, Payre F, Stern DL. Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature. 2010;466:490–493. doi: 10.1038/nature09158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel N, Erezyilmaz DF, McGregor AP, Wang S, Payre F, Stern DL. Morphological evolution caused by many subtle-effect substitutions in regulatory DNA. Nature. 2011;474:598–603. doi: 10.1038/nature10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel N, Wang S, Stern DL. Conserved regulatory architecture underlies parallel genetic changes and convergent phenotypic evolution. Proc Natl Acad Sci USA. 2012;109:20975–20979. doi: 10.1073/pnas.1207715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet J, Mango SE. Regulation of organogenesis by the Caenorhabditis elegans FoxA protein PHA-4. Science. 2002;295:821–825. doi: 10.1126/science.1065175. [DOI] [PubMed] [Google Scholar]
- Giorgetti L, Siggers T, Tiana G, Caprara G, Notarbartolo S, Corona T, Pasparakis M, Milani P, Bulyk ML, Natoli G. Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol Cell. 2010;37:418–428. doi: 10.1016/j.molcel.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Giorgianni MW, Mann RS. Establishment of medial fates along the proximodistal axis of the Drosophila leg through direct activation of dachshund by Distalless. Dev Cell. 2011;20:455–468. doi: 10.1016/j.devcel.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Reyes A, Morata G. The developmental effect of over-expressing a Ubx product in Drosophila embryos is dependent on its interactions with other homeotic products. Cell. 1990;61:515–522. doi: 10.1016/0092-8674(90)90533-k. [DOI] [PubMed] [Google Scholar]
- Gotea V, Visel A, Westlund JM, Nobrega MA, Pennacchio LA, Ovcharenko I. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res. 2010;20:565–577. doi: 10.1101/gr.104471.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan R, Leroy C, Isnard AD, Labarre J, Boy-Marcotte E, Toledano MB. The control of the yeast H2O2 response by the Msn2/4 transcription factors. Mol Microbiol. 2002;45:233–241. doi: 10.1046/j.1365-2958.2002.03011.x. [DOI] [PubMed] [Google Scholar]
- He X, Duque TSPC, Sinha S. Evolutionary origins of transcription factor binding site clusters. Mol Biol Evol. 2012;29:1059–1070. doi: 10.1093/molbev/msr277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersh BM, Carroll SB. Direct regulation of knot gene expression by Ultrabithorax and the evolution of cis-regulatory elements in Drosophila. Development. 2005;132:1567–1577. doi: 10.1242/dev.01737. [DOI] [PubMed] [Google Scholar]
- Jiang J, Levine M. Binding affinities and cooperative interactions with bHLH activators delimit threshold responses to the dorsal gradient morphogen. Cell. 1993;72:741–752. doi: 10.1016/0092-8674(93)90402-c. [DOI] [PubMed] [Google Scholar]
- Joshi R, Passner JM, Rohs R, Jain R, Sosinsky A, Crickmore MA, Jacob V, Aggarwal AK, Honig B, Mann RS. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell. 2007;131:530–543. doi: 10.1016/j.cell.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgens G, Wieschaus E, Nüsselin-Volhard C, Kluding H. Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster II Zygotic loci on the third chromosome. Rouxs Arch Dev Biol. 1984;193:283–295. doi: 10.1007/BF00848157. [DOI] [PubMed] [Google Scholar]
- Karch F, Bender W, Weiffenbach B. abdA expression in Drosophila embryos. Genes Dev. 1990;4:1573–1587. doi: 10.1101/gad.4.9.1573. [DOI] [PubMed] [Google Scholar]
- Kuroiwa A, Kloter U, Baumgartner P, Gehring WJ. Cloning of the homeotic Sex combs reduced gene in Drosophila and in situ localization of its transcripts. EMBO J. 1985;4(13B):3757–3764. doi: 10.1002/j.1460-2075.1985.tb04145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrecht D, Foehr M, Smith E, Lopes FJP, Vanario-Alonso CE, Reinitz J, Burz DS, Hanes SD. Bicoid cooperative DNA binding is critical for embryonic patterning in Drosophila. Proc Natl Acad Sci USA. 2005;102:13176–13181. doi: 10.1073/pnas.0506462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli KM, Noro B, Mann RS. Variable motif utilization in homeotic selector (Hox)-cofactor complex formation controls specificity. Proc Natl Acad Sci USA. 2011;108:21122–21127. doi: 10.1073/pnas.1114118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EB. A gene complex controlling segmentation in Drosophila. Nature. 1978;276:565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- Lifanov AP, Makeev VJ, Nazina AG, Papatsenko DA. Homotypic regulatory clusters in Drosophila. Genome Res. 2003;13:579–588. doi: 10.1101/gr.668403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig MZ, Patel NH, Kreitman M. Functional analysis of eve stripe 2 enhancer evolution in Drosophila: rules governing conservation and change. Development. 1998;125:949–958. doi: 10.1242/dev.125.5.949. [DOI] [PubMed] [Google Scholar]
- Mann RS, Hogness DS. Functional dissection of Ultrabithorax proteins in D. melanogaster. Cell. 1990;60:597–610. doi: 10.1016/0092-8674(90)90663-y. [DOI] [PubMed] [Google Scholar]
- Mann RS, Lelli KM, Joshi R. Hox specificity unique roles for cofactors and collaborators. Curr Top Dev Biol. 2009;88:63–101. doi: 10.1016/S0070-2153(09)88003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis W, Krumlauf R. Homeobox genes and axial patterning. Cell. 1992;68:283–302. doi: 10.1016/0092-8674(92)90471-n. [DOI] [PubMed] [Google Scholar]
- McGregor AP, Orgogozo V, Delon I, Zanet J, Srinivasan DG, Payre F, Stern DL. Morphological evolution through multiple cis-regulatory mutations at a single gene. Nature. 2007;448:587–590. doi: 10.1038/nature05988. [DOI] [PubMed] [Google Scholar]
- Moens CB, Selleri L. Hox cofactors in vertebrate development. Dev Biol. 2006;291:193–206. doi: 10.1016/j.ydbio.2005.10.032. [DOI] [PubMed] [Google Scholar]
- Noro B, Culi J, McKay DJ, Zhang W, Mann RS. Distinct functions of homeodomain-containing and homeodomain-less isoforms encoded by homothorax. Genes Dev. 2006;20:1636–1650. doi: 10.1101/gad.1412606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, Wolfe SA. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell. 2008;133:1277–1289. doi: 10.1016/j.cell.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa-Espinosa A, Yucel G, Kaplan L, Pare A, Pura N, Oberstein A, Papatsenko D, Small S. The role of binding site cluster strength in Bicoid-dependent patterning in Drosophila. Proc Natl Acad Sci USA. 2005;102:4960–4965. doi: 10.1073/pnas.0500373102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai CY, Kuo TS, Jaw TJ, Kurant E, Chen CT, Bessarab DA, Salzberg A, Sun YH. The Homothorax homeoprotein activates the nuclear localization of another homeoprotein, extradenticle and suppresses eye development in Drosophila. Genes Dev. 1998;12:435–446. doi: 10.1101/gad.12.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatsenko DA, Makeev VJ, Lifanov AP, Régnier M, Nazina AG, Desplan C. Extraction of functional binding sites from unique regulatory regions the Drosophila early developmental enhancers. Genome Res. 2002;12:470–481. doi: 10.1101/gr.212502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payre F, Vincent A, Carreno S. ovo/svb integrates Wingless and DER pathways to control epidermis differentiation. Nature. 1999;400:271–275. doi: 10.1038/22330. [DOI] [PubMed] [Google Scholar]
- Peifer M, Wieschaus E. Mutations in the Drosophila gene extra-denticle affect the way specific homeo domain proteins regulate segmental identity. Genes Dev. 1990;4:1209–1223. doi: 10.1101/gad.4.7.1209. [DOI] [PubMed] [Google Scholar]
- Peterson KA, Nishi Y, Ma W, Vedenko A, Shokri L, Zhang X, McFarlane M, Baizabal JM, Junker JP, van Oudenaarden A, et al. Neural-specific Sox2 input and differential Gli-binding affinity provide context and positional information in Shh-directed neural patterning. Genes Dev. 2012;26:2802–2816. doi: 10.1101/gad.207142.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powsner L. The effects of temperature on the durations of the developmental stages of Drosophila melanogaster. Physiol Zool. 1935;8:474–520. [Google Scholar]
- Ramos AI, Barolo S. Low-affinity transcription factor binding sites shape morphogen responses and enhancer evolution. Philos Trans R Soc Lond B Biol Sci. 2013;368:20130018. doi: 10.1098/rstb.2013.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieckhof GE, Casares F, Ryoo HD, Abu-Shaar M, Mann RS. Nuclear translocation of extradenticle requires homothorax, which encodes an extradenticle-related homeodomain protein. Cell. 1997;91:171–183. doi: 10.1016/s0092-8674(00)80400-6. [DOI] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–1253. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S, Siggers T, Lachke SA, Yue Y, Bulyk ML, Maas RL. Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev. 2010;24:980–985. doi: 10.1101/gad.1890410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD, Mann RS. The control of trunk Hox specificity and activity by Extradenticle. Genes Dev. 1999;13:1704–1716. doi: 10.1101/gad.13.13.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD, Marty T, Casares F, Affolter M, Mann RS. Regulation of Hox target genes by a DNA bound Homothorax/Hox/Extradenticle complex. Development. 1999;126:5137–5148. doi: 10.1242/dev.126.22.5137. [DOI] [PubMed] [Google Scholar]
- Sánchez-Herrero E, Vernós I, Marco R, Morata G. Genetic organization of Drosophila bithorax complex. Nature. 1985;313:108–113. doi: 10.1038/313108a0. [DOI] [PubMed] [Google Scholar]
- Saramäki A, Banwell CM, Campbell MJ, Carlberg C. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006;34:543–554. doi: 10.1093/nar/gkj460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scardigli R, Bäumer N, Gruss P, Guillemot F, Le Roux I. Direct and concentration-dependent regulation of the proneural gene Neurogenin2 by Pax6. Development. 2003;130:3269–3281. doi: 10.1242/dev.00539. [DOI] [PubMed] [Google Scholar]
- Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- Silverman N, Maniatis T. NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes Dev. 2001;15:2321–2342. doi: 10.1101/gad.909001. [DOI] [PubMed] [Google Scholar]
- Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Zhou T, Rohs R, Honig B, Bussemaker HJ, Mann RS. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell. 2011;147:1270–1282. doi: 10.1016/j.cell.2011.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanojevic D, Small S, Levine M. Regulation of a segmentation stripe by overlapping activators and repressors in the Drosophila embryo. Science. 1991;254:1385–1387. doi: 10.1126/science.1683715. [DOI] [PubMed] [Google Scholar]
- Stewart-Ornstein J, Nelson C, DeRisi J, Weissman JS, El-Samad H. Msn2 coordinates a stoichiometric gene expression program. Curr Biol. 2013;23:2336–2345. doi: 10.1016/j.cub.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G. Genes controlling segmental specification in the Drosophila thorax. Proc Natl Acad Sci USA. 1982;79:7380–7384. doi: 10.1073/pnas.79.23.7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Struhl K, Macdonald PM. The gradient morphogen bicoid is a concentration-dependent transcriptional activator. Cell. 1989;57:1259–1273. doi: 10.1016/0092-8674(89)90062-7. [DOI] [PubMed] [Google Scholar]
- Swanson CI, Evans NC, Barolo S. Structural rules and complex regulatory circuitry constrain expression of a Notch- and EGFR-regulated eye enhancer. Dev Cell. 2010;18:359–370. doi: 10.1016/j.devcel.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto BT, Kaufman TC. Analysis of larval segmentation in lethal genotypes associated with the antennapedia gene complex in Drosophila melanogaster. Dev Biol. 1981;81:51–64. doi: 10.1016/0012-1606(81)90347-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Defining the Minimal Ventral Embryonic svb Enhancers, Related to Figure 1 and Table S1
(A) Schematic of the svb upstream cis-regulatory region, indicating embryonic enhancer elements. The ventral embryonic enhancers E3N and 7H are highlighted in yellow and blue, respectively.
(B–D) Early stage 15 embryo carrying both E∷lacZ and 7∷GFP reporter constructs, co-stained with antibodies against β -Galactosidase and GFP (B), demonstrate partially overlapping patterns of expression.
(E) Schematic of a subset of the E-derived enhancer fragments tested with transgenic reporter constructs (also see Figure 1). Enhancers indicated with filled boxes drove expression.
(F–H) Early stage 15 embryos carrying three of the E-derived enhancer reporter constructs—E, E3, and E3N—stained with an antibody against β -Galactosidase.
(I) Schematic of a subset of the 7 enhancer fragments tested with transgenic reporter constructs. Enhancers indicated with filled boxes drove expression and those with unfilled boxes showed no expression.
(J–O) Early stage 15 embryos carrying six of the 7-derived enhancer reporter constructs—7, 7B, 7D, 7H, 7I, and 7J—stained with an antibody against b-Galactosidase. Further reduction of 7H to 7J led to additional anterior expression (O).
(P) Log2 ratio enrichment profiles for Ubx and Hth across the svb upstream cis-regulatory region, with the minimal E3N and 7H enhancers indicated in yellow and blue, respectively.
Figure S2. Identification of Regions of the E3N Enhancer that Bind Ubx-Exd In Vitro, Related to Figure 2 and Table S1
(A) A schematic of the regions tested for the ability to bind Ubx-Exd assayed with EMSAs.
(B–G) Ubx-HthFL-Exd and Ubx-HthHM-Exd bound fragments E3N1 (B) and E3N2 (C), as demonstrated with EMSAs. There was also very weak binding of Ubx-HthFL-Exd and Ubx-HthHM-Exd to fragment E3N5 (F).
Figure S3. E3N Contains a Cluster of Three Ubx-Exd Binding Sites, Related to Figure 2 and Table S2
(A) A schematic of the regions tested for the ability to bind Ubx-Exd assayed with EMSAs.
(B and C) The aligned E3N1 and E3N2 sequences from wild-type and mutated sequences. Dashes indicate non-altered sequence. Red letters denote mutated sites. Blue boxes highlight Ubx-Exd binding sites.
(D–Q) Ubx-HthFL-Exd and Ubx-HthHM-Exd EMSAs for each enhancer fragment outlined in (B) and (C). Mutation of the putative Ubx-Exd binding sites shown in (B) and (C) led to decreased binding of Ubx-Exd.
Figure S4. The svb 7H Enhancer Contains a Cluster of Ubx-Exd Binding Sites, Related to Figure 2 and Tables S1 and S2
(A) A multiple sequence alignment for selected regions of the 7H-enhancer, with the three Ubx-Exd sites labeled and highlighted. Dashes indicate gaps in the aligned sequence. Bracketed numbers indicate the number of base pairs between the aligned sequences shown. Mutations of the Ubx-Exd binding-sites are shown (MUTS).
(B) Ubx-HthFL-Exd and Ubx-HthHM-Exd bound each of the three sites, as demonstrated with EMSAs. This binding was reduced when the sites were mutated (MUT).
(C, G, K, O, E, I, M, and Q) Early stage 15 embryos carrying 7H∷lacZ reporter constructs stained with an antibody against β-Galactosidase, with Ubx sites mutated as indicated in (A).
(D, H, L, P, F, J, N, and R) Plots showing profiles of average expression in the region outlined in (C) for the corresponding genotype (n = 10 for each genotype). In all plots, the black lines denote WT embryos and the red lines denote the MUT line indicated. Shaded bounding areas indicate ± 1 s.d. AU, arbitrary units of fluorescence intensity.
Figure S5. AbdominalA Binds to the Ubx Binding Sites in the 7H and E3N Enhancers, Related to Figure 2 and Tables S1 and S2
(A) Early stage 15 embryos stained with an antibody against AbdA and Ubx.
(B) Larval cuticle prep of the indicated genotype.
(C) AbdA-HthFL-Exd and AbdA-HthHM-Exd bound each of the Ubx-Exd sites in the E3N enhancer (see Figure 2), as demonstrated with EMSAs. This binding was decreased when the sites were mutated (as per Figure 2).
(D and E) Early stage 15 embryos carrying E3N∷lacz reporter constructs, stained with an antibody against β-Galactosidase, with genotypes indicated. (F) AbdA-HthFL-Exd and AbdA-HthHM-Exd bound each of the Ubx-Exd sites in the 7H enhancer (see Figure S4), as demonstrated with EMSAs. This binding was decreased when the sites were mutated (as per Figure S4).
(G and H) Early stage 15 embryos carrying 7H∷lacZ reporter constructs, stained with an antibody against β-Galactosidase, with genotypes indicated.
Figure S6. Addition of a High Affinity Site Results in Ectopic Expression, Related to Figure 4 and Table S1
(A) A sequence alignment of the wild-type and mutated versions of the E3N region, showing the modified sequences in red.
(B and C) Early stage 16 embryos carrying E3N∷lacZ reporter constructs stained with an antibody against β-Galactosidase, with an ectopic highest-affinity Ubx-Exd site added either 5′ (B) or internally (C) to the E3N enhancer. The numbers shown in the top right of each panel indicate the average level of expression in the region outlined in Figure 6I for the corresponding genotypes (n = 10 for each genotype), measured in arbitrary units of fluorescence intensity. Numbers in parantheses indicate ± 1 s.d. White arrows denote expression in domains anterior to segment A1 (A,B). Red arrows indicate ectopic dorsal and lateral expression (B).
Figure S7. Identification of Regions of the D. virilis E3N Enhancer that Bind Ubx-Exd In Vitro, Related to Figure 7 and Table S2
(A) A schematic of the regions tested for the ability to bind Ubx-Exd, assayed via EMSAs. Filled boxes indicate regions that bound Ubx-Exd.
(B–G) Ubx-HthFL-Exd and Ubx-HthHM-Exd bound fragments VE3N1 (B), VE3N5 (F), VE3NI (I), and VE3N9 (J), as demonstrated with EMSAs.
(L) A sequence alignment of the wild-type and mutated versions of the VE3N9 region, showing the mutated putative Ubx-Exd binding sites in red.
(M) EMSAs demonstrated that both putative Ubx-Exd binding sites must be mutated to eliminate binding of Ubx-HthHM-Exd or Ubx-HthFL-Exd to VE3N9.
(N) DNA sequence alignment of the E3N enhancer region. Shaded boxes indicate regions that bound Ubx-Exd in D. melanogaster (green) and D. virilis (purple).