

Abstract
Objectives
Cannabidiol (CBD) is hypothesized as a potential treatment for opioid addiction, with safety studies an important first step for medication development. We determined CBD safety and pharmacokinetics when administered concomitantly with a high-potency opioid in healthy subjects.
Methods
This double-blind, placebo-controlled cross-over study of CBD co-administered with intravenous fentanyl, was conducted at the Clinical Research Center in Mount Sinai Hospital, a tertiary care medical center in New York City. Participants were healthy volunteers aged 21–65 years with prior opioid exposure, regardless of route. Blood samples were obtained before and after 400 or 800 mg CBD pretreatment, followed by a single 0.5 (Session 1) or 1.0mcg/Kg (Session 2) intravenous fentanyl dose. The primary outcome was the Systematic Assessment for Treatment Emergent Events (SAFTEE) to assess safety and adverse effects. CBD peak plasma concentrations, time to reach peak plasma concentrations (tmax), and area under the curve (AUC) were measured.
Results
SAFTEE data were similar between groups without respiratory depression or cardiovascular complications during any test session. Following low dose CBD, tmax occurred at 3 and 1.5h (Sessions 1 and 2, respectively). Following high dose CBD, tmax occurred at 3 and 4h in Sessions 1 and 2, respectively. There were no significant differences in plasma CBD or cortisol (AUC p=NS) between sessions.
Conclusions
CBD does not exacerbate adverse effects associated with intravenous fentanyl administration. Co-administration of CBD and opioids was safe and well tolerated. These data provide the foundation for future studies examining CBD as a potential treatment for opioid abuse.
Keywords: Cannabidiol, pharmacokinetics, opioid dependence, cannabis
Introduction
Cannabidiol (CBD), a bioactive cannabinoid in marijuana (cannabis sativa), has received significant attention recently regarding its potential medicinal properties. A growing body of literature has reported therapeutic CBD effects for diverse disease processes including inflammation and cancer (Mechoulam et al., 2007; Mecha et al., 2013; Massi et al., 2013). Moreover, CBD has been shown to have anxiolytic properties (Fusar-Poli et al., 2009; Crippa et al., 2011) as well as therapeutic potential for psychosis (Borgwardt et al., 2008; Müller-Vahl and Emrich, 2008; Englund et al., 2013; Leweka et al., 2012). Importantly, CBD does not impair motor or cognitive performance (Crippa et al., 2004; Zuardi et al., 1982), which is important for medication development.
Recent research has also suggested CBD to have potential therapeutic effects for opioid abuse based on preclinical rat studies that demonstrated CBD to inhibit reinstatement of heroin-seeking behavior (Ren et al., 2009). Interestingly, the effects of CBD were observed to be specific to cue-induced relapse behavior and CBD continued to inhibit heroin seeking even two weeks after administration. Other animal studies have also reported CBD to reduce the rewarding effects of morphine (Katsidoni et al., 2013). These preclinical studies suggest CBD might be a potential treatment for opioid abuse. Identification of novel treatments for opioid dependence is important since there are growing public health concerns regarding the abuse, morbidity and mortality of opioids. Opioid dependence is a chronic, relapsing medical disorder characterized by drug craving, a persistent compulsion to use the drug with loss of control and profound drug-seeking behavior. The fact that heroin relapse vulnerability is linked to cue-induced craving response in opioid dependent patients highlights the critical need to develop therapeutic agents that diminish drug craving in this population (Fatseas et al., 2011).
No specific data presently exist regarding CBD tolerability and safety in regard to the interaction with opioids in humans, which is a critical first step for future clinical trials to examine CBD’s potential as a therapy for opioid craving and relapse in human abusers. As such, the objective of the current study was to first determine the safety and pharmacokinetics of CBD when administered concomitantly with opioid in normal human subjects. Based on previous investigations showing CBD to be safe (Crippa et al., 2004; Fusar-Poli et al., 2009; Zuardi et al., 1993; Zuardi et al., 2006), we hypothesized that CBD safety and pharmacokinetics would remain unchanged when co-administered at moderate doses (400–800mg) with intravenous fentanyl.
Methods
Study Design
Fentanyl was co-administered with oral CBD in healthy human volunteers at the Clinical Research Center in Mount Sinai Hospital in New York City based on the experimental design required by the Federal Drug Administration and IRB at Mount Sinai as the first phase to subsequently examine CBD as a potential medication in opioid abusers. The design was a double-blind placebo-controlled cross-over study. All volunteers signed a written informed consent document. A Data Safety and Monitoring Board (DSMB) reviewed and approved the protocol before enrollment and reviewed all study data.
Participants
Healthy volunteers were recruited through advertisement in the community and local newspapers and assigned a unique identification number. Inclusion Criteria: past exposure at least once to an opioid (i.e. codeine, morphine, fentanyl); and aged between 21 and 65 years. Exclusion Criteria: using any psychoactive drug at any time during the study; current diagnosis of drug dependence (except nicotine dependence) based on Structured Clinical Interview for DSM-IV (SCID-IV) interview; history of cardiac disease, arrhythmias, head trauma, seizures, or Axis I psychiatric conditions under DSM-IV examined with the Mini International Neuropsychiatric Interview-MINI; hypersensitivity to any opioid or cannabinoid; pregnant or breastfeeding; not using an appropriate method of contraception; or intoxication at the time of arrival on the site of the study or positive drug screen (screened for cocaine, cannabis, opiates, benzodiazepines, barbiturates, phencyclidine, amphetamines). Subjects were compensated $150 per session and $10 for screening; compensation was determined based on minimum wage, hours required for study participation and transportation fees. We enrolled 6 subjects per study group similar to CBD studies using the same dose range in healthy subjects (Crippa et al., 2004).
Study Protocol
During this 2-session, double-blind design, participants were administered either placebo (Group 1), 400mg oral CBD (Group 2), or 800mg oral CBD (Group 3) prior to both 0.5mcg/Kg (Session 1) and 1.0mcg/Kg (Session 2) of IV fentanyl. Sessions were separated by at least 1 week to ensure a sufficient drug washout period. All participants were administered both doses of fentanyl with the low dose in the first session and high dose in the second (Table 1).
Table 1.
A blinded randomized experimental design assessed the potential safety of oral CBD administration prior to fentanyl administration (N=17)
| Groups: | Session 1 (Week 1): Fentanyl First Dose | Session 2 (Week 2): Fentanyl Second Dose |
|---|---|---|
|
| ||
| Group 1: placebo | CBD placebo + Fentanyl Dose 1 | CBD placebo + Fentanyl Dose 2 |
| Group 2: CBD 400 mg | CBD 400 mg + Fentanyl Dose 1 | CBD 400 mg + Fentanyl Dose 2 |
| Group 3: CBD 800 mg | CBD 800 mg + Fentanyl Dose 1 | CBD 800 mg + Fentanyl Dose 2 |
All participants received a low (week 1) and high (week 2) dose of fentanyl; fentanyl low and high doses were 0.5 and 1.0mcg/Kg. Participants in Group 1 received placebo, participants in Group 2 received oral CBD 400 mg and participants in Group 3 received oral CBD 800 mg. The two sessions were separated with a 1-week interval to ensure sufficient drug washout period.
Drugs
CBD, 99.9% pure CBD in corn oil, was encapsulated in gelatin capsules and supplied by GW Pharmaceutical. Placebo was corn oil in gelatin capsules identical to CBD capsules.
Intravenous administration was selected because it is the route of administration for many opioid users and minimizes inter-individual variation in bioavailability noted with oral opioid administration (Glare and Walsh, 1991). Fentanyl was safely administered in this dose-range in clinical settings and in previous studies conducted among healthy subjects and opioid-dependent subjects (Zacny et al., 1992).
Selection of 400 and 800mg CBD doses was based on safety and toxicology data regarding CBD in humans and animals, and also on previous CBD studies conducted in humans to assess other therapeutic properties. Our preliminary study showed a significant effect of CBD on heroin reinstatement behavior at 5mg/Kg in rodents (Ren et al., 2009). In humans, a similar CBD dose (300–400mg) decreased cortisol (Zuardi et al., 1993), a biomarker related to stress, and significantly altered cerebral blood flow in limbic and paralimbic brain structures, such as the amygdala, that are highly relevant to drug craving (Crippa et al., 2004). From a tolerability standpoint, 600 and 1280mg CBD has been administered to humans without toxicity or serious adverse events (Borgwardt et al., 2008; Zuardi et al., 1993; Zuardi et al., 2006; Consroe et al., 1991; Zuardi et al., 2009).
Laboratory session
The timeline for blood sampling and outcomes measures is summarized in Figure 1. Subjects were asked to fast for at least 8 h prior to the session. Upon arrival at approximately 9am, subjects were administered a light, standardized breakfast. CBD was administered 2h later. Another standardized meal was administered 4h after CBD. 200mL water was allowed prior to capsule intake, but no alcohol or caffeine was allowed; subjects were not allowed to engage in intense physical activity. One hour before CBD, a breathalyzer test was administered and a urine sample (rapid urine drug screen by immunoassay) collected to ensure that the subjects were not actively using alcohol, or other psychoactive substances (e.g. cocaine, amphetamines, opioids, sedatives, cannabis, and hallucinogens). Blood samples (each 10mL) were obtained through an indwelling cannula from the forearm. We allowed a 1h delay before giving a single 0.5 or 1.0mcg/Kg intravenous fentanyl citrate dose. Since the time to peak respiratory effects of IV fentanyl is ~5–15 min and peak CBD clinical effects were expected to be 1–2h (Crippa et al., 2004), with peak plasma concentrations at ~3h (Agurell et al., 1981), the delay between CBD and fentanyl administration ensured that CBD reached a significant plasma concentration before fentanyl possibly induced respiratory changes. The study timeline ensured that participants were under medical surveillance for the period during which expected adverse fentanyl effects would occur (up to 4h).
Figure 1. Experimental flowchart per study day for each subject.
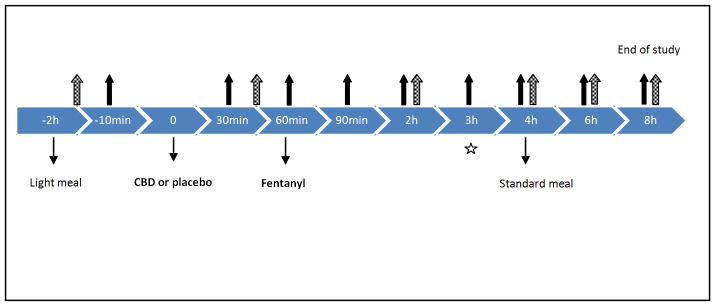
This flowchart outlines the session timeline to assess safety of cannabidiol (CBD) and fentanyl co-administration. Participants were provided a light meal 2 hours prior to CBD administration. Fentanyl was administered 60 minutes after administration of the CBD/placebo. A standard meal was provided 4 hours after CBD/placebo administration. Solid arrows indicate the time points when blood (10 mL) samples were collected. Vital signs, SAFTEE, VAS, PANAS and OVAS were also assessed at the times indicated by the solid arrows except for the 3h time point (denoted by a star). Dashed arrows indicate times for urine collection (45 min, 2,4,6, and 8h).
Abbreviations: OVAS, opioid specific visual analog scales, PANAS = positive and negative affect schedule, SAFTEE = systematic assessment for treatment emergent events, VAS = visual analog scale.
Sample Collection and Storage
CBD blood samples were collected in 6 mL lithium heparin Vacutainers, centrifuged at 4°C (3500 rpm for 10 min) within 2h of collection. Plasma was separated and stored in cryotubes at −80°C. All samples were labeled with a unique barcode and study number for each subject, test session number, date and time. Nine blood samples per session for cortisol testing (5mL) were sent directly to the hospital laboratory for processing. Urine samples also were obtained to evaluate CBD urinary excretion. Urine samples were collected in sterile mid stream collection cups, stored at −80°C.
Cannabidiol Assays
Plasma CBD was quantified by two-dimensional gas chromatography-mass spectrometry (GC-GC/MS) as previously described (Karschner et al., 2010). Briefly, 3mL of ice-cold acetonitrile was added to 1mL plasma to precipitate proteins. After mixing, samples were centrifuged and supernatant added to preconditioned solid phase extraction (SPE) columns. (United Chemical Technologies, Styre Screen SSTHC06Z). Analytes of interest (CBD, THC, 11-OH-THC, THCCOOH) were eluted with 3mL hexane: ethyl acetate: acetic acid (49:49:2, v/v), and dried under nitrogen. Samples were reconstituted with 25μL N,O-bis(trimethylsilyl) trifluoroacetamide with 1% trimethylchlorosilane (BSTFA) and derivatized for 0.5h at 70°C. Samples were centrifuged and analyzed by electron ionization GC-GC/MS. Limits of quantification (LOQ) were 0.125 μg/L for 11-OH-THC, and 0.25 μg/mL for CBD, THC and THCCOOH. Upper limits of linearity were 25 μg/L for CBD, and 100 μg/L for THC, 11-OH-THC and THCCOOH. Inter- and intra-assay imprecision (%CV) were ≤6.4 and ≤7.8% for all analytes, and quality control samples quantified within ±9.2% of target concentrations across the linear range.
For urine, CBD was analyzed by gas chromatography-mass spectrometry (GC/MS) according to a previously published method (Bergamaschi et al., 2013). CBD was linear from 2.5 – 500 μg/L. CBD intra- and inter-assay imprecision (%CV) was ≤3.2 and ≤3.4%, respectively. While intra- and inter-assay bias (%CV) was ≤11.3%.
Fentanyl assay
Highly sensitive enzyme linked immunosorbent assay (ELISA) kits (Immunalysis Corporation, Pomona, CA) were employed for fentanyl quantification from plasma samples. Briefly, 20μL of a diluted sample and 100μL aliquot of enzyme conjugate (drug labeled with horseradish peroxidase) were added to 96 well microplates coated with fentanyl antibodies and incubated for 60min. Assay limit of quantification was 0.5 μg/L.
Primary outcomes
We assessed safety and adverse effects with the Systematic Assessment for Treatment Emergent Events (SAFTEE) (Levine and Schooler, 1986). The SAFTEE has two forms, a General Inquiry (GI) and a Specific Inquiry (SI), which take approximately 20 min to administer. The GI is an open-ended inquiry about any physical or health problems and its impact on functioning. The SI is a detailed and systematic inquiry regarding 78 adverse effects divided into 23 categories corresponding to organ systems or body parts (Watson et al., 1988).
Secondary outcomes and Subjective Measures
Peak plasma concentration (Cmax), time to peak plasma concentration (tmax) and area under the curve (AUC) were calculated from CBD plasma concentrations. Urine sampling allowed estimation of clearance, calculated as percent of dose excreted over 8h. Cortisol plasma levels and AUC differences between groups also were examined. Plasma and urine AUC were calculated by the following equation: , where C1 = concentration at time 1 (t1), and C2 = concentration at time 2 (t2). Interaction between fentanyl dose and both (i) plasma CBD concentrations as well as (ii) urinary CBD clearance was assessed using mixed linear models.
Anxiety was assessed with a visual analog scale (VAS). The PANAS (Positive and Negative Affect Schedule) recorded positive and negative affect subscores as previously described (Watson et al., 1988). Specific Opiate VAS (OVAS) was administered to assess potential variations in fentanyl subjective effects. All scales were administered in less than 10 min.
Vital signs (blood pressure, heart rate, respiratory rate, oxygen saturation, and body temperature), respiratory function (inspired and expired oxygen and carbon dioxide concentrations, arterial hemoglobin–oxygen saturation and respiratory rate) and EKG were continuously monitored throughout the session. Vital signs were recorded as in Figure 1. Patients were monitored for respiratory depression (defined as diminished oxygen saturation below 90% or respiratory rate <12) and cardiovascular compromise (defined as abnormal heart rate or blood pressure outside of standard normal parameters) during the study.
Statistical Analysis
Continuous variables were summarized with means ± standard errors (SE). Student’s t-test, Kruskal-Wallis, and ANOVA (post-hoc testing with Tukey test) compared co-administration of CBD and fentanyl with fentanyl alone in terms of adverse events, physiological parameters variation and pharmacokinetic properties. CBD pharmacokinetics and subjective measures were first analyzed for skew and kurtosis, then analyzed for repeated measures analysis using either general or mixed linear models. Significance was set at 5% alpha and analyses were calculated by SPSS version 21 software (IBM, Chicago, IL).
Results
Baseline Characteristics
Seventeen subjects (N=6 high-dose CBD group, N=6 low-dose CBD group, N=5 placebo group) were recruited. Mean age 38.5 ±2.2, 47% female and 53% White/47% Black (no difference in age, gender or race among the three groups, p=NS). All subjects had a prior history of opioid exposure (the majority had received either hydrocodone, oxycodone or codeine in association with post-surgical care) to minimize any potential negative opioid response and were prescreened to assure absence of opioids via toxicology screens.
Primary Safety Endpoint
Each subject (n=17) had two sessions (thus N=34 sessions), each of which was analyzed independently. There were no serious adverse events such as respiratory depression or cardiovascular compromise in any subject. Minor adverse events reported by subjects during (and immediately after) sessions were recorded as dichotomous variables (in descending number of occurrences): dizziness/drowsiness (n=5), itching or rash (n=3), headache (n=2), abdominal discomfort (n=2), nausea/vomiting (n=2), diarrhea (n=2). There were no significant associations between CBD Cmax (peak of maximum concentration) and occurrence of adverse events (t-test p=NS). The two episodes of vomiting occurred in subjects with significantly lower CBD Cmax (t-test p<0.05). One subject developed itching and a rash that resolved after treatment with diphenhydramine. SAFTEE results are summarized in Table 2.
Table 2.
Systematic Assessment for Treatment Emergent Events (SAFTEE) Data
| Fentanyl Session | Group 1 | Group 2 | Group 3 |
|---|---|---|---|
|
| |||
| Session 1 | Itching | Dizzy (n=2) | Blurry vision |
| Drowsy | Drowsy (n=2) | Diarrhea* | |
| Bruxism | Headache | ||
| Session 2 | Itching | Drowsy (n=3) | Abdominal pain (n=2)* |
| Nausea | Anxiety | Diaphoresis | |
| Dizzy | Dizzy | Dizzy | |
| Dry Mouth | Nausea | Diarrhea* | |
| Vomit | Itching | ||
| Vomit* | |||
Fentanyl Dosing: Session 1 = 0.5mcg/Kg, Session 2 = 1.0mcg/Kg
CBD Dosing: Low = 400 mg, High = 800 mg
Indicates same individual
Clinical monitoring of vital signs (temperature, heart rate, respiratory rate, blood pressure, oxygen saturation) occurred continuously and was analyzed at time points according to Figure 1 using AUC between groups. There were no occurrences of respiratory depression (breaths/min <12) or cardiovascular compromise (mean arterial pressure <60mmHg, heart rate >100) in any subject at any time point. Higher fentanyl dose (1.0 vs. 0.5 mcg/kg) was associated with slightly lower respiratory rate and temperature (mean AUC t-test p<0.05), however, cardiovascular parameters were similar between sessions (p=NS). CBD dose (0 vs. 400 vs. 800mg) was not associated with any significant differences in vital sign parameters throughout the study (mean AUC ANOVA p=NS for all).
Plasma CBD Concentrations
Plasma CBD concentrations were measured at baseline and at selected time points according to the protocol outlined in Figure 1. Group 1 (placebo) measurements served as the negative control. In Group 2 (400mg CBD), time to peak plasma concentrations (tmax) occurred at 3h (mean Cmax 181.2±39.8μg/L) and 1.5h (Cmax 114.2±19.5μg/L) in Sessions 1 (0.5 mcg/kg fentanyl) and 2 (1.0 mcg/kg fentanyl), respectively (Fig. 2A). In Group 3 (800mg CBD), tmax occurred at 3h (mean Cmax 221.1±35.6μg/L) and 4h (mean Cmax 157.1±49.0μg/L) in Sessions 1 and 2, respectively (Fig. 2B). In Group 2, AUC (mcg*hr/dL) for Sessions 1 and 2 were 704±283 and 482±314, while Group 3 with the highest CBD dose tested had AUCs of 867± 304 and 722±443, respectively (F[2,14] = 55.34; p < 0.0001). There was no significant interaction between plasma CBD concentrations across fentanyl sessions as demonstrated in Figure 2C (mixed linear model p=NS); 800 mg CBD tended to have higher plasma concentrations irrespective of the fentanyl dose.
Figure 2. Plasma CBD Concentrations.
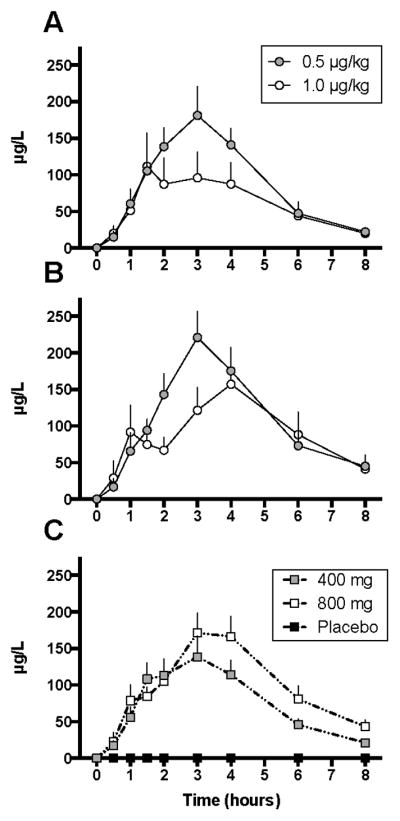
Mean (± SEM) plasma CBD concentrations across time (in hrs) were not significantly affected by Fentanyl dose (AUC Wilcoxon p=NS). Mean (± SEM) plasma CBD concentrations for Low-Dose (400 mg) CBD are in Panel A and High-Dose (800 mg) CBD is in Panel B. Panel C represents the combined curves for all Groups (0, 400mg and 800 mg CBD) across the two Fentanyl sessions.
Abbreviations: NS = not significant; SEM = standard error of the mean; * indicates significant difference between groups at one given time point (t-test p<0.5).
Urinary CBD Excretion
Urinary excretion of unchanged CBD (<5% total) and CBD conjugates (metabolite, majority of excretion) were measured in subjects’ urine at time points according to the protocol outlined in Figure 1. After the low dose CBD, mean peak urinary concentrations (Umax) of CBD conjugates occurred at 6 h (Umax = 4.6μg/L) and 2h (Umax = 2.9μg/L) in Sessions 1 and 2, respectively. In the high dose CBD group, mean peak urinary concentrations of CBD conjugates occurred at 4h (Umax = 3.7μg/L) and 6h (Umax = 2.8μg/L) in Sessions 1 and 2, respectively. Higher fentanyl dose in Session 2 was associated with significantly decreased mean CBD clearance (F[2,12] = 5.42; p = 0.02).
Plasma Fentanyl concentrations
None of the subjects in either the placebo or CBD groups showed detectable plasma fentanyl concentrations.
Plasma Cortisol
Each subject had plasma cortisol concentrations (μg/dL) analyzed at each time point and AUC (mcg*h/dL) calculated over 8h. Mean plasma cortisol concentrations in Group 1 (placebo) in Sessions 1 and 2 were 46.9±12.7 and 56.3±20.9, respectively. Mean plasma cortisol concentrations in Group 2 (low dose CBD) in Sessions 1 and 2 were 55.9±24.1 and 69.7±27.2, respectively, and in Group 3 (high dose CBD) 46.2±23.9 and 69.3±40.4, respectively. There were no significant between-group or within-group correlations between cortisol concentration AUCs using ANOVA (p=NS), or when analyzed by CBD dose at any study time point (Pearson R ranged from −0.134 to +0.21, p=NS for all). When plasma cortisol from all study time points were combined, there was no correlation between CBD dose and cortisol (R=0.065, p=NS).
Subjective Measures
Anxiety VAS, PANAS (positive and negative subscores), and OVAS were administered across 8 time points for each Session as per Figure 1. There were no significant main effects for CBD for any of the subjective measures (Table 3). A significant time x CBD interaction was evident for the positive PANAS (F[14,105]=4.96; p<0.0001) and total OVAS (F[14,105]=3.8; p<0.0001) measures, but no significant group differences at individual times were observed following correction for multiple comparisons (data not shown).
Table 3.
Subjective Measures Analysis by Treatment Group
| Treatment Group | PANAS | OVAS | |
|---|---|---|---|
| Positive* | Negative | ||
| CBD 0mg- 0.5 Fentanyl | 33.56 | 10.04 | 19.17 |
| CBD 0mg- 1.0 Fentanyl | 31.50 | 10.04 | 19.61 |
| CBD 400mg-0.5 Fentanyl | 23.81 | 10.08 | 18.44 |
| CBD 400mg-1.0 Fentanyl | 23.10 | 10.25 | 17.52 |
| CBD 800mg-0.5 Fentanyl | 33.85 | 10.81 | 24.56 |
| CBD 800mg-1.0 Fentanyl | 33.06 | 10.40 | 23.92 |
Mean scores for the subjective measures (PANAS, OVAS) and the results of the repeated measures statistical analysis.
Mixed linear model with repeated measures.
Abbreviations: OVAS=Opiate Visual Analog Scales; PANAS=Positive and Negative Affect Scales; S=session number.
Discussion
The main findings of this study were that CBD was well tolerated at doses up to 800mg (approximately 10–15 mg/kg), with no significant pharmacokinetics changes with opioid co-administration. Plasma CBD Cmax and tmax were not significantly altered by fentanyl co-administration. Similarly, no effect was evident for urinary CBD and metabolite excretion except at the higher fentanyl dose in which CBD clearance was reduced. Importantly, fentanyl co-administration did not produce respiratory depression or cardiovascular complications during the test sessions and CBD did not potentiate fentanyl effects. Furthermore, CBD clearance did not differ when co-administered with opioids. Our results add to the growing body of literature evaluating the safety of CBD administration in humans. Together with prior safety studies (Consroe et al., 1991; Zuardi et al., 1995; Tomida et al., 2006; Rosenkrantz et al., 1981), these data show that CBD is well tolerated and safe, which is critical for the development of CBD as a treatment intervention, even in relation to opioid abuse.
To assess the effects of CBD on subjects’ affect, we performed an exploratory analysis of validated subjective measures scales (PANAS and OVAS) (Watson et al., 1988) as well as anxiety VAS. The results suggest that CBD at the doses examined do not markedly alter affect, consistent with other reports (Fusar-Poli et al., 2009), or exacerbate fentanyl’s subjective effects. Further studies are necessary in larger populations to fully discern CBD effects on mood across a large dose range.
The potential to examine CBD in human opiate abusers is intriguing based on results from our preclinical animal data (Ren et al., 2009). It is predicted that CBD would have a significant effect on inhibiting heroin-seeking behavior. However, there are still large gaps of knowledge regarding CBD actions in the brain, as this phytocannabinoid is much less studied than THC. It was previously demonstrated that CBD did not affect brain morphine and methadone concentrations in mice (Reid and Bornheim, 2001). However, it was suggested that that although CBD has weak activity at CB1 and CB2 receptors, it may act as an inverse agonist at these receptors (Pertwee et al., 2002; Thomas et al., 2007). In addition, CBD stimulates the transient receptor potential V2 (TRPV2) protein (Qin et al., 2008), also known as vanilloid receptor-like 1 (VRL-1), a member of the TRP superfamily of nonselective, ligand-gated cation channels that appear to serve as so-called ionotropic cannabinoid receptors. CBD also was reported to alter the hydrolysis and cellular uptake of the endocannabinoid anandamide (Bisogno et al., 2001). CBD modulates allosterically μ and delta opioid receptors, but only at high concentrations (Kathmann et al., 2006). μ opioid receptors are co-localized with type 1 cannabinoid receptor (CB1) in striatal output projection neurons of the nucleus accumbens and dorsal striatum that modulates reward, goal-directed behavior, and habit formation relevant to addiction (Rodriguez et al., 2001). CB1 and μ opioid receptors are not only physically associated, but also share Gi-alpha-mediated intracellular signaling and interact to modulate neurotransmitter release in the nucleus accumbens (Schoffelmeer et al., 2006). Given the growing clinical interest in CBD, and in relation to addictive disorders, future studies are needed to further explore the molecular mechanisms by which CBD may inhibit opioid-seeking behaviors and endogenous opioid mediated reward pathways.
There are limitations that must be considered with these results. Our experimental design was subject to some degree of selection bias due to not studying pharmacokinetic properties across all ages, gender and ethnic backgrounds; however, the pharmacokinetic properties of CBD have already been examined and the main objective of this study was to assess CBD safety in combination with an opioid agonist. Moreover, whether these effects generalize to opioid dependent individuals and other relevant clinical populations are important to assess in the next phase of investigation. Self-report may have led to bias via distortion of information either intentionally or unintentionally, but our combination of self-report (e.g. questionnaire) and objective measures (e.g. vital signs, urine testing, blood sampling) to examine eligibility criteria and the safety outcomes decreased such risk. Finally, we studied relatively low to moderate doses of CBD and fentanyl and only included participants who were not opioid dependent out of concerns associated with development of drug abuse behaviors following administration in large doses. While we cannot generalize our results to opioid dependent patients, the tolerance to opiates that characterize such individuals may actually decrease the risk associated with opioid intoxication compared to subjects with limited opioid exposure such as those included in this study.
In conclusion, capsules of 400 and 800mg CBD did not exacerbate adverse effects associated with IV fentanyl administration. Co-administration of CBD and opioid is safe and well tolerated. The results of this study pave the way for future studies to examine the effect of CBD on opioid craving in association with opioid dependence in humans.
Acknowledgments
The study was funded by a research grant from the National Institutes of Health grant DA027781 (YLH) and CTSA (UL1RR029887). This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health. We would like to thank the members of the Hurd Laboratory and the Clinical Research Center at the Icahn School of Medicine at Mount Sinai for excellent technical support, administrative guidance, and study conduct.
Footnotes
Declarations of Interest: None.
References
- Agurell S, Carlsson S, Lindgren JE, et al. Interactions of delta 1-tetrahydrocannabinol with cannabinol and cannabidiol following oral administration in man. Assay of cannabinol and cannabidiol by mass fragmentography. Experientia. 1981;37:1090–2. doi: 10.1007/BF02085029. [DOI] [PubMed] [Google Scholar]
- Agurell S, Halldin M, Lindgren JE, et al. Pharmacokinetics and metabolism of delta 1-tetrahydrocannabinol and other cannabinoids with emphasis on man. Pharmacol Rev. 1986;38:21–43. [PubMed] [Google Scholar]
- Barta WD, Kurth ME, Stein MD, et al. Craving and self-efficacy in the first five weeks of methadone maintenance therapy: a daily process study. J Stud Alcohol Drugs. 2009;70:735–40. doi: 10.15288/jsad.2009.70.735. [DOI] [PubMed] [Google Scholar]
- Bergamaschi MM, Barnes A, Queiroz RH, et al. Impact of enzymatic and alkaline hydrolysis on CBD concentration in urine. Anal Bioanal Chem. 2013;405:4679–89. doi: 10.1007/s00216-013-6837-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Hanus L, De Petrocellis L, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. 2001;134:845–52. doi: 10.1038/sj.bjp.0704327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgwardt SJ, Allen P, Bhattacharyya S, et al. Neural basis of Delta-9-tetrahydrocannabinol and cannabidiol: effects during response inhibition. Biol Psychiatry. 2008;64:966–73. doi: 10.1016/j.biopsych.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Broadbear JH, Winger G, Woods JH. Self-administration of fentanyl, cocaine and ketamine: effects on the pituitary-adrenal axis in rhesus monkeys. Psychopharmacology (Berl) 2004;176:398–406. doi: 10.1007/s00213-004-1891-x. [DOI] [PubMed] [Google Scholar]
- Crippa JA, Zuardi AW, Garrido GE, et al. Effects of cannabidiol (CBD) on regional cerebral blood flow. Neuropsychopharmacology. 2004;29:417–26. doi: 10.1038/sj.npp.1300340. [DOI] [PubMed] [Google Scholar]
- Crippa JA, Derenusson GN, Ferrari TB, et al. Neural basis of anxiolytic effects of cannabidiol (CBD) in generalized social anxiety disorder: a preliminary report. J Psychopharmacol. 2011;25:121–30. doi: 10.1177/0269881110379283. [DOI] [PubMed] [Google Scholar]
- Consroe P, Kennedy K, Schram K. Assay of plasma cannabidiol by capillary gas chromatography/ion trap mass spectroscopy following high-dose repeated daily oral administration in humans. Pharmacol Biochem Behav. 1991;40:517–22. doi: 10.1016/0091-3057(91)90357-8. [DOI] [PubMed] [Google Scholar]
- Consroe P, Laguna J, Allender J, et al. Controlled clinical trial of cannabidiol in Huntington’s disease. Pharmacol Biochem Behav. 1991;40:701–8. doi: 10.1016/0091-3057(91)90386-g. [DOI] [PubMed] [Google Scholar]
- Dijkstra BA, De Jong CA, Bluschke SM, et al. Does naltrexone affect craving in abstinent opioid-dependent patients? Addict Biol. 2007;12:176–82. doi: 10.1111/j.1369-1600.2007.00067.x. [DOI] [PubMed] [Google Scholar]
- Ellgren M, Spano SM, Hurd YL. Adolescent cannabis exposure alters opiate intake and opioid limbic neuronal populations in adult rats. Neuropsychopharmacology. 2007;32:607–615. doi: 10.1038/sj.npp.1301127. [DOI] [PubMed] [Google Scholar]
- Englund A, Morrison PD, Nottage J, et al. Cannabidiol inhibits THC-elicited paranoid symptoms and hippocampal-dependent memory impairment. J Psychopharmacol. 2013;27:19–27. doi: 10.1177/0269881112460109. [DOI] [PubMed] [Google Scholar]
- Fatseas M, Denis C, Massida Z, et al. Cue-induced reactivity, cortisol response and substance use outcome in treated heroin dependent individuals. Biol Psychiatry. 2011;70:720–7. doi: 10.1016/j.biopsych.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Fusar-Poli P, Crippa JA, Bhattacharyya S, et al. Distinct effects of {delta}9-tetrahydrocannabinol and cannabidiol on neural activation during emotional processing. Arch Gen Psychiatry. 2009;66:95–105. doi: 10.1001/archgenpsychiatry.2008.519. [DOI] [PubMed] [Google Scholar]
- Glare PA, Walsh TD. Clinical pharmacokinetics of morphine. Ther Drug Monit. 1991;13:1–23. doi: 10.1097/00007691-199101000-00001. [DOI] [PubMed] [Google Scholar]
- Harvey DJ, Samara E, Mechoulam R. Urinary metabolites of cannabidiol in dog, rat and man and their identification by gas chromatography-mass spectrometry. J Chromatogr. 1991;562:299–322. doi: 10.1016/0378-4347(91)80587-3. [DOI] [PubMed] [Google Scholar]
- Hyman SM, Fox H, Hong KI, et al. Stress and drug-cue-induced craving in opioid-dependent individuals in naltrexone treatment. Exp Clin Psychopharmacol. 2007;15:134–43. doi: 10.1037/1064-1297.15.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilgen M, Jain A, Kim HM, et al. The effect of stress on craving for methadone depends on the timing of last methadone dose. Behav Res Ther. 2008;46:1170–5. doi: 10.1016/j.brat.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Karschner EL, Barnes AJ, Lowe RH, et al. Validation of a two-dimensional gas chromatography mass spectrometry method for the simultaneous quantification of cannabidiol, Δ9-Tetrahydrocannabinol (THC), 11- Hydroxy-THC and 11-nor-9-Carboxy-THC in Plasma. Analytical and Bioanalytical Chemistry. 2010;397:603–11. doi: 10.1007/s00216-010-3599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathmann M, Flau K, Redmer A, et al. Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:354–61. doi: 10.1007/s00210-006-0033-x. [DOI] [PubMed] [Google Scholar]
- Katsidoni V, Anagnostou I, Panagis G. Cannabidiol inhibits the reward-facilitating effect of morphine: involvement of 5-HT1A receptors in the dorsal raphe nucleus. Addict Biol. 2013;18:286–96. doi: 10.1111/j.1369-1600.2012.00483.x. [DOI] [PubMed] [Google Scholar]
- Langleben DD, Ruparel K, Elman I, et al. Acute effect of methadone maintenance dose on brain FMRI response to heroin-related cues. Am J Psychiatry. 2008;165:390–4. doi: 10.1176/appi.ajp.2007.07010070. [DOI] [PubMed] [Google Scholar]
- Levine J, Schooler NR. SAFTEE: a technique for the systematic assessment of side effects in clinical trials. Psychopharmacol Bull. 1986;22:343–81. [PubMed] [Google Scholar]
- Leweke FM, Piomelli D, Pahlisch F, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. 2012;2:e94. doi: 10.1038/tp.2012.15. [Abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi P, Solinas M, Cinquina V, et al. Cannabidiol as potential anticancer drug. Br J Clin Pharmacol. 2013;75:303–12. doi: 10.1111/j.1365-2125.2012.04298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecha M, Feliú A, Iñigo PM, et al. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: A role for A2A receptors. Neurobiol Dis. 2013;59:141–50. doi: 10.1016/j.nbd.2013.06.016. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Peters M, Murillo-Rodriguez E, et al. Cannabidiol--recent advances. Chem Biodivers. 2007;4:1678–92. doi: 10.1002/cbdv.200790147. [DOI] [PubMed] [Google Scholar]
- Müller-Vahl KR, Emrich HM. Cannabis and schizophrenia: towards a cannabinoid hypothesis of schizophrenia. Expert Rev Neurother. 2008;8:1037–48. doi: 10.1586/14737175.8.7.1037. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA, Craib SJ, et al. Cannabidiol antagonizes cannabinoid receptor agonists and noradrenaline in the mouse vas deferens. Eur J Pharmacol. 2002;456:99–106. doi: 10.1016/s0014-2999(02)02624-9. [DOI] [PubMed] [Google Scholar]
- Qin N, Neeper MP, Liu Y, et al. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci. 2008;28:6231–8. doi: 10.1523/JNEUROSCI.0504-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MJ, Bornheim LM. Cannabinoid-induced alterations in brain disposition of drugs of abuse. Biochem Pharmacol. 2001;61:1357–67. doi: 10.1016/s0006-2952(01)00616-5. [DOI] [PubMed] [Google Scholar]
- Ren Y, Whittard J, Higuera-Matas A, et al. Cannabidiol, a nonpsychotropic component of cannabis, inhibits cue-induced heroin seeking and normalizes discrete mesolimbic neuronal disturbances. J Neurosci. 2009;29:14764–9. doi: 10.1523/JNEUROSCI.4291-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JJ, Mackie K, Pickel VM, et al. Ultrastructural localization of the CB1 cannabinoid receptor in mu-opioid receptor patches of the rat Caudate putamen nucleus. J Neurosci. 2001;21:823–33. doi: 10.1523/JNEUROSCI.21-03-00823.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkrantz H, Fleischman RW, Grant RJ. Toxicity of short-term administration of cannabinoids to rhesus monkeys. Toxicol Appl Pharmacol. 1981;58:118–31. doi: 10.1016/0041-008x(81)90122-8. [DOI] [PubMed] [Google Scholar]
- Scavone JL, Sterling RC, Van Bockstaele EJ. Cannabinoid and opioid interactions: Implications for opiate dependence and withdrawal. Neuroscience. 2013;248:637–54. doi: 10.1016/j.neuroscience.2013.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoffelmeer AN, Hogenboom F, Wardeh G, et al. Interactions between CB1 cannabinoid and mu opioid receptors mediating inhibition of neurotransmitter release in rat nucleus accumbens core. Neuropharmacology. 2013;51:773. doi: 10.1016/j.neuropharm.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Solinas M, Panlilio LV, Tanda G, et al. Cannabinoid agonists but not inhibitors of endogenous cannabinoid transport or metabolism enhance the reinforcing efficacy of heroin in rats. Neuropsychopharmacology. 2005;30:2046–57. doi: 10.1038/sj.npp.1300754. [DOI] [PubMed] [Google Scholar]
- Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. British Journal of Pharmacology. 2007;150:613–623. doi: 10.1038/sj.bjp.0707133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasiewicz HC, Jacobs MM, Wilkinson MB, et al. Proenkephalin Mediates the Enduring Effects of Adolescent Cannabis Exposure Associated with Adult Opiate Vulnerability. Biological Psychiatry. 2012;72:803–10. doi: 10.1016/j.biopsych.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida I, Azuara-Blanco A, House H, et al. Effect of sublingual application of cannabinoids on intraocular pressure: a pilot study. J Glaucoma. 2006;15:349–53. doi: 10.1097/01.ijg.0000212260.04488.60. [DOI] [PubMed] [Google Scholar]
- United Nations Office on Drugs and Crime. [Accessed 25 September 2014];World Drug Report. 2012 whqlibdoc.who.int/publications/2009/9789241547543_eng.pdf.
- Walter M, Wiesbeck GA, Bloch N, et al. Psychobiological responses to drug cues before and after methadone intake in heroin-dependent patients: a pilot study. Eur Neuropsychopharmacol. 2008;18:390–3. doi: 10.1016/j.euroneuro.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Watson D, Clark LA, Tellegen A. Development and validation of brief measures of positive and negative affect: the PANAS scales. J Pers Soc Psychol. 1988;54:1063–70. doi: 10.1037//0022-3514.54.6.1063. [DOI] [PubMed] [Google Scholar]
- Welch SP. Interaction of the cannabinoid and opioid systems in the modulation of nociception. Int Rev Psychiatry. 2009;21:143–51. doi: 10.1080/09540260902782794. [DOI] [PubMed] [Google Scholar]
- Zacny JP, Lichtor JL, Zaragoza JG, et al. Effects of fasting on responses to intravenous fentanyl in healthy volunteers. J Subst Abuse. 1992;4:197–207. doi: 10.1016/0899-3289(92)90019-t. [DOI] [PubMed] [Google Scholar]
- Zuardi AW, Shirakawa I, Finkelfarb E, et al. Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology (Berl) 1982;76:245–50. doi: 10.1007/BF00432554. [DOI] [PubMed] [Google Scholar]
- Zuardi AW, Morais SL, Guimarães FS, et al. Antipsychotic effect of cannabidiol. J Clin Psychiatry. 1995;56:485–6. [PubMed] [Google Scholar]
- Zuardi AW, Hallak JE, Dursun SM, et al. Cannabidiol monotherapy for treatment-resistant schizophrenia. J Psychopharmacol. 2006;20:683–6. doi: 10.1177/0269881106060967. [DOI] [PubMed] [Google Scholar]
- Zuardi AW, Crippa JA, Hallak JE, et al. Cannabidiol for the treatment of psychosis in Parkinson’s disease. J Psychopharmacol. 2009;23:979–83. doi: 10.1177/0269881108096519. [DOI] [PubMed] [Google Scholar]