

Abstract
We describe a patient with a severe juvenile polyposis phenotype, due to a de novo deletion of chromosome 10q22.3-q24.1. He was initially diagnosed with Juvenile polyposis syndrome (JPS) at age 4 after presenting with hematochezia due to multiple colonic juvenile polyps. He then represented at 23 years with recurrent hematochezia from juvenile polyps in his ileoanal pouch. He is one of the earliest reported cases of JPS associated with a large deletion of chromosome 10. Since his initial diagnosis of JPS further studies have confirmed an association between JPS and mutations in BMPR1A in chromosome band 10q23.2, which is in close proximity to PTEN. Mutations in PTEN cause Cowden syndrome (CS) and other PTEN hamartoma tumor syndromes. Due to the chromosome 10 deletion involving contiguous portions of BMPR1A and PTEN in our patient, he may be at risk for CS associated cancers and features, in addition to the polyps associated with JPS. This case presents new challenges in developing appropriate surveillance algorithms to account for the risks associated with each syndrome and highlights the importance of longitudinal follow-up and transitional care between pediatric and adult gastroenterology for patients with hereditary polyposis syndromes.
Keywords: Gastroenterology, Hamartoma syndrome, multiple, Intestinal polyposis, Pediatrics
INTRODUCTION
Colorectal cancer is the second leading cause of cancer death of men and women in the United States [Siegel et al., 2013]. Approximately 5% of colon cancers can be attributed to well-defined inherited syndromes, such as Lynch syndrome, Familial Adenomatous Polyposis, and hamartomatous polyposis syndromes – including Juvenile polyposis syndrome (JPS), Cowden syndrome (CS), and Peutz-Jeghers syndrome [Jasperson et al., 2010]. Inherited hamartomatous polyposis syndromes occur less frequently than adenomatous polyp syndromes and account for <1% of colorectal cancers [Schreibman et al., 2005]. JPS is a rare autosomal dominant syndrome characterized by histologically distinct juvenile polyps in the gastrointestinal tract and an increased risk of colorectal cancer. Juvenile polyps are hamartomas with a normal epithelium and dense stroma, an inflammatory infiltrate, and a smooth surface with dilated mucous-filled cystic glands in the lamina propria [Schreibman et al., 2005]. They are named for the specific histologic features and not for the age of the patient when the polyp arises; isolated juvenile polyps can be found in adults. JPS has been linked to mutations in both SMAD4 on chromosome band 18q21.2 and BMPR1A on chromosome band 10q23.2, both of which play a role in the BMP/TGF-beta signaling pathway [Dahdaleh et al., 2012; Howe et al., 2001; Howe et al., 1998b; Sayed et al., 2002]. JPS is defined by ≥5 juvenile polyps in the colon or rectum, juvenile polyps in other parts of the gastrointestinal tract, or any number of juvenile polyps in a patient with one or more relatives affected with JPS [Jass et al., 1988]. JPS can also be divided into three subtypes: juvenile polyposis coli and generalized juvenile polyposis based on the location of polyps; and juvenile polyposis of infancy which is characterized by early presentation and a more aggressive course with diarrhea and exudative enteropathy [Dahdaleh et al., 2012]. Location and extent of polyps can vary, even within the same family. JPS carries an estimated lifetime risk of colorectal cancer of up to 40%, with 17 to 21% of cases occurring by age 34 and up to 55% occurring by age 43 [Howe et al, 1998a; Jass et al., 1988]. There is also an increased risk of stomach, small intestine, and pancreatic cancers.
Cowden syndrome (CS) is a member of the PTEN hamartomatous polyp syndromes which also includes Bannayan-Riley-Ruvalcaba syndrome. CS is a rare, autosomal dominant syndrome, and is associated with the PTEN gene located on chromosome band 10q23.3, approximately 1 Mb telomeric to BMPR1A [Dahdaleh et al., 2012]. PTEN is a tumor suppressor gene and is also involved in regulating the cell cycle, apoptosis, and angiogenesis [Manfredi 2010]. CS has variable penetrance and common manifestations include thyroid abnormalities (>50%), gastrointestinal hamartomatous polyps (30–85%), macrocephaly (35%), mucocutaneous hamartomas, trichilemmomas, acral keratoses, papillomatous lesions, and a risk of developmental delay [Dahdaleh et al., 2012; Manfredi, 2010; Stanich et al., 2011]. A number of patients with CS will not exhibit the cutaneous findings and these patients may be clinically indistinguishable from JPS [Schreibman et al., 2005]. The association between colorectal cancer and CS is controversial. However, Kato et al. [ 2000] found a colon cancer incidence of 9% among CS patients in the Japanese national registry. Extra-intestinal malignancies associated with CS include breast (30–40%), thyroid (10%), endometrial, and renal cell carcinoma [Manfredi, 2010; Schreibman et al., 2005].
CLINICAL REPORT
We present a follow-up study of one of the earliest reported cases of JPS due to a deletion in BMPR1A by Jacoby et al. [1997]. The patient was born at 36 weeks with a club foot deformity to healthy parents. At 10 months, he was referred to our Genetics Clinic where he was noted to have mild cranio-facial abnormalities, mild tricuspid insufficiency, an umbilical hernia, hypoplastic abdominal oblique muscles, short and broad hands and feet, and developmental delay. At 4 years, he presented to the Pediatric Gastroenterology Clinic for further evaluation of a 1 year history of hematochezia. Colonoscopy showed multiple polyps scattered throughout the colon up to 2.5 cm in size. Histopathology revealed juvenile polyps and he was diagnosed with JPS. Metaphase cultures from peripheral leukocytes were analyzed with standard Giemsa method at the time of this diagnosis and showed a large interstitial deletion in the long arm of chromosome 10 [46,XY,del(10)(q22.3q24.1)] [Jacoby et al., 1997]. He was determined to have a de novo deletion as his parents’ karyotypes were normal. Due to continued bleeding he eventually underwent a total colectomy with ileoanal pouch anastomosis.
He did well for a number of years, but presented to our adult Gastroenterology Genetics Clinic at age 23 with recurrent hematochezia. Pouch endoscopy showed multiple polyps (Fig. 1, A and B). Histopathology again confirmed juvenile polyps (Fig. 2, A and B). Further inspection of all pathology specimens showed that he has only had juvenile polyps removed from his gastrointestinal tract; adenomas, hyperplastic polyps, and non-juvenile hamartomas have not been found. Significant discoveries in the genetics of hamartomatous polyposis syndromes had been made since his initial diagnosis [Dahdaleh et al., 2012; Delnatte et al., 2006; Howe et al., 2001; Manfredi, 2010; Sayed et al., 2002]. A review indicated that his chromosome 10 deletion involves contiguous portions of BMPR1A and PTEN which could result in an overlap syndrome of both JPS and CS. Subsequent upper endoscopy was normal, pouch endoscopies continue to show a large polyp burden, and computed tomographic enterography revealed additional smaller polyps just proximal to the pouch. Our patient does not have any clinical features of CS besides developmental delay, however, his chromosome 10 deletion involving PTEN places him at risk for CS associated malignancies, thus altering our surveillance plan.
Figure 1.
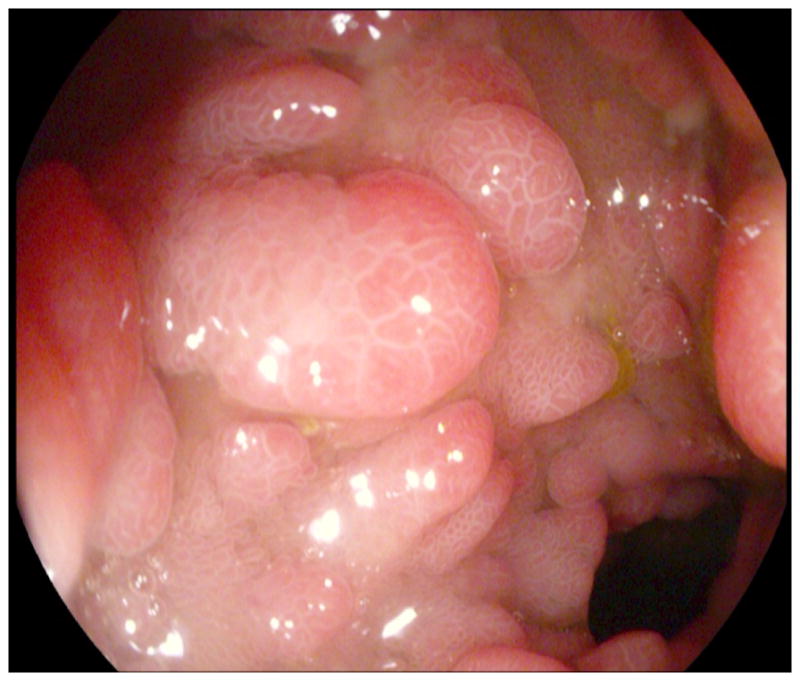

Images (A and B) from pouch endoscopy illustrate multiple spherical pedunculated polyps with smooth surfaces.
Figure 2.
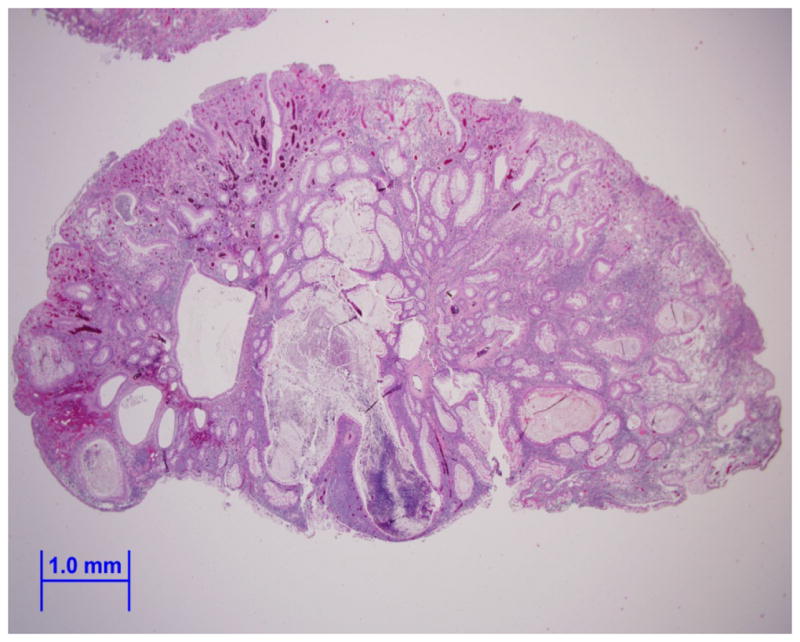

Image (A) is an endoscopically removed juvenile polyp from our patient showing a smooth surface with dilated mucus-filled cystic glands in the lamina propria (hematoxylin-eosin, x10). Image (B) shows a view of the dense stroma, inflammatory infiltrate, and fibrovascular core of the juvenile polyp (hematoxylin-eosin, x20).
DISCUSSION
JPS and CS are two rare autosomal dominant hamartomatous polyposis syndromes. Cases of overlapping syndromes due to a contiguous deletion of BMPR1A and PTEN on chromosome band 10q23, as in our patient, are rare. Our patient presented at a young age with a significant juvenile polyp burden in his colon causing bleeding and anemia; he continues with multiple, large juvenile polyps in his pouch and distal small bowel. Previous investigators who have described similar patients to ours (some with even more severe JPS phenotypes and more clinical features of CS) have hypothesized that deletion of both genes (BMPR1A and PTEN) may lead to a more aggressive clinical picture and that deletion of both genes might have a greater than additive effect on the predisposition to gastrointestinal and extra-intestinal malignancies [Dahdaleh et al., 2012; Delnatte et al., 2006; Arch et al., 1997; Zigman et al., 1997; Tsuchiya et al., 1998; Salviati et al., 2006; Menko et al., 2008]. According to the NCBI Genome database, our patient’s deletion includes approximately 270 genes. Apart from BMPR1A and PTEN, none have a known association with gastrointestinal polyposis. Nonetheless, his severe polyposis phenotype may be influenced by other genes in the deleted region. While isolated juvenile polyps are conventionally presumed to be benign, in patients with JPS and CS the risk of malignant transformation is thought to be due to adenomatous foci in the polyps which later become dysplastic and finally progress to carcinoma [Howe et al., 1998a; Jacoby et al., 1997; Jass et al., 1988; Manfredi, 2010; Schreibman et al., 2005]. Due to this increased risk of malignant transformation, the general recommendation is to remove polyps when encountered in these patients.
Guidelines exist for screening and surveillance of JPS and CS; however, there are no comparative studies to demonstrate the benefit of aggressive screening for gastrointestinal or extra-intestinal malignancies. The National Comprehensive Cancer Network (NCCN) has published recommendations for both syndromes (www.nccn.org) but indicates that all are considered category 2A (based on lower-level evidence with uniform NCCN consensus that the intervention is appropriate). For JPS, regular surveillance with colonoscopy and upper endoscopy are recommended starting at age 15. If the polyp burden cannot be managed endoscopically, surgery may be necessary. Upper endoscopies may be more important in JPS patients with a SMAD4 mutation. Sayed et al. [2002] compared patients with JPS due to a mutation in BMPR1A to those with a mutation in SMAD4 and found no significant differences in clinical findings besides a preponderance of upper gastrointestinal tract polyps in families with SMAD4 mutations. Surveillance recommendations for CS include regular colonoscopy, thyroid ultrasound, mammography and breast MRI, and consideration of regular screening for endometrial and renal cell cancers. There is also general agreement that patients with both of these syndromes should be referred to a specialized multidisciplinary team involving gastroenterology, dermatology, surgery, oncology, and genetics due to the rarity and complexities of these conditions [Jasperson et al., 2010; Schreibman et al., 2005].
Review of the surveillance recommendations illustrates the need for an individualized algorithm for our patient. It also raises the potential need for further testing in patients with JPS due to BMPR1A deletions for concurrent deletions in PTEN and vice versa to assure comprehensive screening protocols are followed. Our patient underwent a total proctocolectomy with ileoanal pouch anastomosis due to a polyp burden in his colon that could not be managed endoscopically. With the continued polyp burden in his pouch and just proximal to the pouch, further surgical management with revision of the pouch and an end-ileostomy was discussed. However, due to his developmental delay there was concern for how he would adjust to the ileostomy. We have elected to pursue annual pouch endoscopies with continued removal of the largest polyps, as well as upper endoscopies every 2–3 years and we are starting a program of annual thyroid ultrasound. This case illustrates that the surveillance algorithm should be influenced by the severity of associated symptoms, the feasibility of achieving endoscopic clearance of polyps, and the genetic mutation(s). It also highlights the importance of transitional care between pediatric and adult gastroenterology and establishing longitudinal follow-up, due to ongoing research in hereditary polyposis syndromes leading to new knowledge and revised surveillance recommendations.
Acknowledgments
FINANCIAL SUPPORT
Support was provided by a Mentored Research Scholar Grant in Applied and Clinical Research from the American Cancer Society (MRSG-13-144-01 – CPHPS), a Clinical and Translational Science Award through the National Institutes of Health Center for Advancing Translational Sciences (UL1TR000427), the National Cancer Institute (grant P30CA014520-34), and the UW Health Innovation Program.
Footnotes
DISCLOSURE
None of the authors have relevant financial interests or ethical conflicts to disclose.
References
- Arch EM, Goodman BK, Van Wesep RA, Liaw D, Clarke K, Parsons R, McKusick VA, Geraghty MT. Deletion of PTEN in a patient with Bannayan-Riley-Ruvalcaba syndrome suggests allelism with Cowden disease. Am J Med Genet. 1997;71(4):489–493. [PubMed] [Google Scholar]
- Dahdaleh FS, Carr JC, Calva D, Howe JR. Juvenile polyposis and other intestinal polyposis syndromes with microdeletions of chromosome 10q22–23. Clin Genet. 2012;81(2):110–116. doi: 10.1111/j.1399-0004.2011.01763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delnatte C, Sanlaville D, Mougenot JF, Vermeesch JR, Houdayer C, de Blois MC, Genevieve D, Goulet O, Fryns JP, Jaubert F, Vekemans M, Lyonnet S, Romana S, Eng C, Stoppa-Lyonnet D. Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am J Hum Genet. 2006;78(6):1066–1074. doi: 10.1086/504301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM, Velculescu VE, Traverso G, Vogelstein B. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28(2):184–187. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998a;5(8):751–756. doi: 10.1007/BF02303487. [DOI] [PubMed] [Google Scholar]
- Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, Stone EM, Aaltonen LA. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998b;280(5366):1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- Jacoby RF, Schlack S, Sekhon G, Laxova R. Del(10)(q22.3q24.1) associated with juvenile polyposis. Am J Med Genet. 1997;70(4):361–364. doi: 10.1002/(sici)1096-8628(19970627)70:4<361::aid-ajmg6>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138(6):2044–2058. doi: 10.1053/j.gastro.2010.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jass JR, Williams CB, Bussey HJR, Morson BC. Juvenile polyposis - a precancerous condition. Histopathology. 1988;13(6):619–630. doi: 10.1111/j.1365-2559.1988.tb02093.x. [DOI] [PubMed] [Google Scholar]
- Kato M, Mizuki A, Hayashi T, Kunihiro T, Nagata H, Tsukada N, Orikasa H, Morinaga S. Cowden’s disease diagnosed through mucocutaneous lesions and gastrointestinal polyposis with recurrent hematochezia, unrevealed by initial diagnosis. Intern Med. 2000;39(7):559–563. doi: 10.2169/internalmedicine.39.559. [DOI] [PubMed] [Google Scholar]
- Manfredi M. Hereditary hamartomatous polyposis syndromes: understanding the disease risks as children reach adulthood. Gastroenterology & hepatology. 2010;6(3):185–196. [PMC free article] [PubMed] [Google Scholar]
- Menko FH, Kneepkens CM, de Leeuw N, Peeters EA, Van Maldergem L, Kamsteeq EJ, Davidson R, Rozendaal L, Lasham CA, Peeters-Scholte CM, Jansweijer MC, Hilhorst-Hoftsee Y, Gille JJ, Heins YM, Nieuwint AW, Sistermans EA. Variable phenotypes associated with 10q23 microdeletions involving the PTEN and BMPR1A genes. Clin Genet. 2008;74(2):145–154. doi: 10.1111/j.1399-0004.2008.01026.x. [DOI] [PubMed] [Google Scholar]
- Salviati L, Patricelli M, Guariso G, Sturniolo GC, Alaggio R, Bernardi F, Zuffardi O, Tenconi R. Deletion of PTEN and BMPR1A on chromosome 10q23 is not always associated with juvenile polyposis of infancy. Am J Hum Genet. 2006;79(3):593–596. doi: 10.1086/507151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed MG, Ahmed AF, Ringold JR, Anderson ME, Bair JL, Mitros FA, Lynch HT, Tinley ST, Petersen GM, Giardiello FM, Vogelstein B, Howe JR. Germline SMAD4 or BMPR1A mutations and phenotype of juvenile polyposis. Ann Surg Oncol. 2002;9(9):901–906. doi: 10.1007/BF02557528. [DOI] [PubMed] [Google Scholar]
- Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005;100(2):476–490. doi: 10.1111/j.1572-0241.2005.40237.x. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Stanich PP, Owens VL, Sweetser S, Khambatta S, Smyrk TC, Richardson RL, Goetz MP, Patnaik MM. Colonic polyposis and neoplasia in Cowden syndrome. Mayo Clin Proc. 2011;86(6):489–492. doi: 10.4065/mcp.2010.0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya KD, Wiesner G, Cassidy SB, Limwonqse C, Boyle JT, Schwartz S. Deletion 10q23.2-q23.33 in a patient with gastrointestinal juvenile polyposis and other features of Cowden-like syndrome. Genes Chromosomes Cancer. 1998;21(2):113–118. doi: 10.1002/(sici)1098-2264(199802)21:2<113::aid-gcc6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Zigman AF, Lavine JE, Jones MC, Boland CR, Carethers JM. Localization of the Bannayan-Riley-Ruvalcaba syndrome gene to chromosome 10q23. Gastroenterology. 1997;113(5):1433–1437. doi: 10.1053/gast.1997.v113.pm9352843. [DOI] [PubMed] [Google Scholar]