

Abstract
β-Catenin, the central component of the canonical Wnt pathway, plays important roles in the processes of liver regeneration, growth, and cancer. Previously, we identified temporal expression of β-catenin during liver development. Here, we characterize the hepatic phenotype, resulting from the successful deletion of β-catenin in the developing hepatoblasts utilizing Foxa3-cyclization recombination and floxed-β-catenin (exons 2 through 6) transgenic mice. β-Catenin loss in developing livers resulted in significantly underdeveloped livers after embryonic day 12 (E12) with lethality occurring at around E17 stages. Histology revealed an overall deficient hepatocyte compartment due to (1) increased cell death due to oxidative stress and apoptosis, and (2) diminished expansion secondary to decreased cyclin-D1 and impaired proliferation. Also, the remnant hepatocytes demonstrated an immature phenotype as indicated by high nuclear to cytoplasmic ratio, poor cell polarity, absent glycogen, and decreased expression of key liver-enriched transcription factors: CCAAT-enhancer binding protein-α and hepatocyte nuclear factor-4α. A paucity of primitive bile ducts was also observed. While the stem cell assays demonstrated no intrinsic defect in hematopoiesis, distorted hepatic architecture and deficient hepatocyte compartments resulted in defective endothelial cell organization leading to overall fetal pallor.
Conclusion
β-Catenin regulates multiple, critical events during the process of hepatic morphogenesis, including hepatoblast maturation, expansion, and survival, making it indispensable to survival.
β-Catenin, a central component of the canonical Wnt pathway, is critical for normal development.1,2 Aberrant activation of this pathway is implicated in cancers of multiple tissues including liver, colon, breast, and skin.3,4 In canonical signaling, Wnt binding to receptor-frizzled and coreceptor low-density lipoprotein reactive protein 5/6, induces dishevelled activation, which inactivates glycogen synthase kinase 3β (GSK3β). The resulting dephosphorylation and dissociation of β-catenin from the cytoplasmic complex with GSK3β, axin and adenomatous polyposis coli gene product leads to its binding to lymphoid enhancer-binding factor/T-cell factor and target gene expression in the nucleus.5 In the absence of Wnt, β-catenin is phosphorylated at specific serine/threonine residues and targeted for ubiquitination.
In the liver, aberrant activation of the Wnt pathway is associated with tumors of varying histology ranging from hepatic adenomas to hepatocellular cancers.6,7 β-Catenin has also been shown to play an important role in hepatic physiology, including liver growth and regeneration.8-12
Previously, we identified a temporal expression of β-catenin during liver development and its antisense-mediated knockdown or exogenous Wnt-3a supplementation, resulting in specific effects on resident cell proliferation and maturation, in the ex vivo embryonic liver cultures.13-15
To conclusively address the role of β-catenin during liver development, we utilized the floxed β-catenin (Ctnnb1flox/flox), in which loxP sites flank exons 2 through 6.16 Since albumin-cyclization recombination (Cre) and α-Fetoprotein-Cre both enabled efficient Cre-mediated recombination of β-catenin only at the postnatal stages, we utilized Foxa3-Cre transgenic mice.17-19 Foxa3 (formerly hepatocyte nuclear factor [HNF]3γ or Hnf3γ)-Cre mice induce efficient excision of loxP-flanked genes in the foregut endoderm and in developing liver as early as E8 to E8.5 in mice.20 Here we characterize the hepatic phenotype only, ensuing from the Foxa3-Cre driven deletion of β-catenin in developing hepatoblasts and hepatocytes to demonstrate the importance of Wnt/β-catenin signaling in multiple key processes during liver development.
Materials and Methods
Generation of β-Catenin Conditional Knockout or Hep-Ctnnb1−//− Mice
Homozygous floxed β-catenin mice and Foxa3-Cre mice, both described previously, were mated as described by Brault et al.16 and Lee et al.20 Briefly, these mice were bred to Foxa3-Cre mice and the offspring carrying a floxed β-catenin allele and Foxa3-Cre were bred to homozygous floxed β-catenin mice. These resulted in floxed and floxdel alleles of Ctnnb1 and are referred to as Hep-Ctnnb1−/− mice. For all experimentation and analysis, the following genotypes were utilized as controls: Ctnnb1loxp/loxp;Foxa3-Cre−/−, Ctnnb1loxp/Wt; Foxa3-Cre+/−, and Ctnnb1loxp/Wt;Foxa3-Cre−/−, and are referred to as Con. No phenotype was observed in the Con mice in any part of the study.
Collection of Embryos and Tissues
We obtained the embryos from pregnant mice at stages E9.5 to E19. For immunohistochemistry (IHC), we fixed the isolated embryos (E9.5 to E14) or livers (E15 to E19) in 10% buffered formaldehyde.
Extraction of RNA, Reverse Transcription, Reverse Transcription Polymerase Chain Reaction, and Affymetrix Microarray
We extracted total RNA from pooled, whole, or E-cadherin-sorted (see below) E16 and E17 Hep-Ctnnb1−/− or Con livers (n = 3) using 1 mL Trizol reagent (Invitrogen, Carlsbad, CA). Following RNase-free DNase (Promega, Madison, WI) treatment, we performed reverse transcription using the First Strand cDNA Synthesis Kit (Fermentas, Hanover, MD). We carried out polymerase chain reaction (PCR) using a standard PCR kit, and a 1-μL aliquot of complementary DNA, Taq DNA polymerase (Invitrogen) with specific primer pairs for mouse albumin and glutathione S-transferases (GSTs). For all samples, we carried out PCR at least three times, as follows: initial denaturation at 94°C for 2 minutes, followed by 35 cycles or 28 cycles of denaturation at 94°C for 1 minute, annealing at 55°C for 1 minute; 72°C for 1 minute, and finally 10 minutes of final extension at 72°C. We analyzed the product by gel electrophoresis.
We utilized gene array Hep-Ctnnb1−/− or Con livers (n = 5) from stage ED16.5 for isolating and purifying RNA by Qiagen RNeasy kit (Qiagen, San Diego, CA). We pooled equivalent amounts of RNA from each liver and used them for the subsequent Affymetrix gene array as per their protocols, as described.10 We also used this probe for Affymetrix chip (set 430) hybridization as per the manufacturer's instructions. We performed final analysis was performed using the Affymetrix Microarray Suite 5.0 software and exported and organized the data in an Excel spreadsheet (Microsoft Office application). We normalized the signals from Hep-Ctnnb1−/− and Con livers to average albumin and α-fetoprotein gene expression to correct for lower epithelial numbers in the Hep-Ctnnb1−/− livers; final analysis is presented as a fold-change (Table 1).
Table 1. Change in Albumin and α-Fetoprotein Gene Expression in Embryonic Livers in the Presence of Normal or Absent β-Catenin at E16.5.
| Gene | Expression in WT (E16.5) | Expression in KO | Fold Change |
|---|---|---|---|
| Albumin | 49280 | 16335 | 3 |
| α-Fetoprotein | 42894 | 11250 | 3.8 |
| Average 3.4* |
Abbreviations: WT, wild-type; KO, knockout.
This average was used in subsequent analysis for normalizing gene expression in the β-catenin-conditional null livers at E16.5 due to observed cell disparity; that is, decreased numbers of hepatocyte and hepatoblasts in β-catenin-deficient livers.
Protein Extraction and Western Blots
We prepared whole-cell lysates using Hep-Ctnnb1−/− and Con livers (n > 4) in radioimmunoprecipitation assay buffer containing fresh protease and phosphatase inhibitor cocktails (Sigma, St. Louis, MO) as described.10 We resolved a total of 20 or 50 μg of the proteins by sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis using the mini-PROTEIN 3-electrophoresis module assembly (Bio-Rad Laboratories, Hercules, CA) and transferred them to Immobilon polyvinylidene fluoride membranes; we detected proteins by Super-Signal West Pico Chemi-luminescent Substrate (Pierce, Rockford, IL) and visualized them by autoradiography. We used primary antibodies against β-catenin, cyclin-D1, and CCAAT-enhancer binding protein-α (C/EBPα; Santa Cruz Biotechnology, Santa Cruz, CA) and β-actin (Chemicon, Temecula, CA). We purchased horseradish peroxidase-conjugated secondary antibodies from Chemicon.
Histology, IHC, Immunofluorescence, and Special Stains
We analyzed 4-μ-thick section from at least three Hep-Ctnnb1−/− and control whole embryos or isolated livers (7 gt; E14 stages) by hematoxylin and eosin and indirect immunoperoxidase IHC. We examined immunolocalization of β-catenin, hepatocyte nuclear factor-4α, cytokeratin-19 and platelet endothelial cell adhesion molecule (PECAM; Santa Cruz Biotechnology).21 For negative control, we incubated the sections with secondary antibodies (Chemicon) only.
For proliferation assay, we performed IHC for proliferating cell nuclear antigen (PCNA; Dako, Carpinteria, CA). We detected apoptotic nuclei by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining using the ApopTag Peroxidase kit (Intergen Company, Purchase, NY). We counted the numbers of PCNA-positive or TUNEL-positive cells at ×400 in five random fields from three individually stained E12, E14, and E17 Hep-Ctnnb1−/− or control livers. We compared the average number of positive cells at each stage for statistical significance by Student t test; a two-tailed P value of <0.05 was considered significant. We performed Isolectin-B4 staining for endothelial cells (Vector Laboratories, Burlingame, CA). We performed periodic acid-Schiff staining was performed using Schiff reagent, periodic acid, and Gill No. 3 Hematoxylin (Sigma). We used anti-4-hydroxynonenal (4-HNE; Calbiochem, San Diego, CA) as a marker of oxidative stress by IHC.
For immunofluorescence (IF), the protocol has been described elsewhere.8 We used antibodies against ZO-1 (Zymed, San Francisco, CA) and C/EBPα and vascular endothelial growth factor (VEGF; Santa Cruz Biotechnology). We used secondary antibodies Cy2-conjugated or Cy3-conjugated (Molecular Probes, Eugene, OR) and counterstained the samples with 4′,6′-diamino-2-phenylindole or Hoechst dye.
We viewed all slides under a Zeiss Axioskop 40 upright research microscope and we obtained digital images by a Nikon Coolpix camera. We prepared collages using Adobe PhotoShop 5.0 software.
Malondialdehyde Assay
We assessed lipid peroxidation by malondialdehyde (MDA) assay using a commercially available kit (BIOXYTECH MDA-586; Oxis International, Foster City, CA). Briefly, we prepared total cell lysates from E17 Hep-Ctnnb1−/− and Con livers (n = 4) in phosphate-buffered saline (PBS) with 5 mM butylated hydroxytoluene to prevent sample oxidation during homogenization. We measured MDA and 4-HNE content in pooled liver samples according to the manufacturer's protocol.
Flow Cytometry Analysis and Cell Sorting
We stained suspended single fetal liver cells with anti-E-cadherin-fluorescein isothiocyanate (BD Biosciences). We used the FACSAria Flow Cytometer (BD Biosciences) for cell sorting for messenger RNA and protein isolation.
Colony Forming Unit Cells Assay
We performed the in vitro assay of colony-forming unit cells (CFCs) using E15.5 to E16.5 Hep-Ctnnb1−/− and Con liver cells. We mixed suspended single cells in MethoCult M3434 medium (StemCell Technologies Inc., Canada) and cultured them for 10 days. We scored and counted the different types of colonies under a microscope.
Electron Microscopy
We fixed freshly isolated Hep-Ctnnb1−/− and Con livers from stages E13 to E19 in 2.5% glutaraldehyde for 2 days at 4°C. We washed several 1-mm3 cubes in PBS and postfixed them in 1% OsO4, 1% K3Fe(CN)6 for 1 hour. After additional washes in PBS, we dehydrated the tissue through a graded series of 30% to 100% ethanol, 100% propylene oxide, and then infiltrated it in a 1:1 mixture of propylene oxide:Polybed 812 epoxy resin (Polysciences, Warrington, PA) for 1 hour. After several changes of 100% resin over 24 hours, we embedded the tissue in molds and cured it at 37°C overnight, followed by additional hardening at 65°C for 2 more days. We collected ultrathin (60-nm) sections on 200-mesh copper grids and stained them with 2% uranyl acetate in 50% methanol for 10 minutes, followed by 1% lead citrate for 7 minutes. We viewed the sections with a JEOL JEM 1210 transmission electron microscope at 80 or 60 kV.
Results
Successful Deletion of β-Catenin in the Epithelial Cells During Liver Development Results in Mid to Late Gestational Lethality and Decrease in Epithelial (Hepatoblast, Hepatocyte, and Biliary) Compartment
To induce excision of loxP flanked exons 2 to 6 genes of β-catenin gene in the hepatoblasts, we bred transgenic mice homozygous for floxed β-catenin allele (Ctnnb1flox/flox) and Foxa3-Cre transgenic mice as described in Materials and Methods and elsewhere.16,20 We confirmed the genotype Ctnnb1flox/flox;Foxa3-Cre (referred to as Hep-Ctnnb1−/− from here on) by genomic DNA analysis, which we confirmed by the simultaneous presence of homozygous β-catenin floxed alleles and Cre-recombinase at stages E9.5, E12, E14, and E17 (Fig. 1A). We identified a dramatic decrease in total hepatic β-catenin protein by western blot, which is shown at stages E12, E14, and E17 (Fig. 1B). This coincided with a decrease in cyclin-D1, a known target of the Wnt/β-catenin pathway, in the Hep-Ctnnb1−/− livers (Fig. 1B).
Fig. 1.
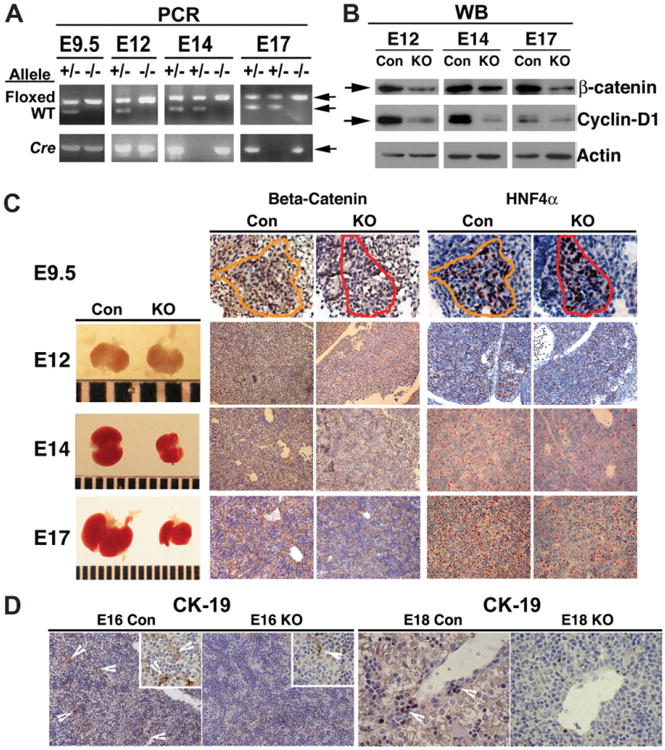
β-Catenin deletion in Hep-Ctnnb1−/− livers with resulting effect on hepatocyte and biliary epithelial cells. (A) PCR analysis of the genomic DNA identifies β-catenin-null (knockout [KO]) or the Hep-Ctnnb1−/− embryos at E9.5 to E17 by the concomitant presence of floxed alleles and Cre-recombinase transgene, whereas the littermate controls (Con) show floxed and wild-type alleles with or without Cre-recombinase transgenes. (B) Western blot (WB) analysis using whole cell lysate of the developing livers at E12 to E17 identified a dramatic decrease in total β-catenin protein along its target, cyclin-D1 in the KO. Successful deletion of β-catenin in KO livers was also verified by IHC for β-catenin at E9.5 (×600) and E12 to E17 (×200). A dramatic decrease in overall liver size was detectable after E12. No difference in HN4α-positive hepatoblasts was apparent at E9.5 (×600) in the KO, however a dramatic difference was observed at E12 to E17 (×200) as compared to the respective controls.(D) E16 control livers show several CK-19-positive primitive bile ducts with a remarkable decrease in KO (×200). Higher magnification (600×) in the inset also shows CK-19-positive cells in KO and Con. A similar decrease in CK-19-positive cells in primitive portal triads are observed in E18 KO livers as compared to the controls (×600).
We obtained no viable Hep-Ctnnb1−/− offspring, with lethality occurring at stage E17 (range, E16 to E19). The Hep-Ctnnb1−/− fetuses were marginally small and pale, with disproportionately smaller livers readily observed after the E12 stage (Fig. 1C).
We employed IHC to verify the stage of successful deletion of β-catenin by Foxa3-Cre driven recombination. A clear decrease in nuclear and cytoplasmic β-catenin was evident at E9.5 in hepatoblasts in the Hep-Ctnnb1−/− livers as compared to the controls (Con) with genotypes Ctnnb1flox/+;Foxa3-Cre or Ctnnb1flox/flox (Fig. 1C). There was no effect of β-catenin deletion on the hepatoblast compartment at this stage as evident by IHC for HNF4α in a consecutive section (Fig. 1C). β-Catenin loss continued throughout development, as shown at stages E12, E14, and E17, which resulted in grossly smaller livers only after E12 (Fig. 1C). A dramatic decrease in the numbers of HNF4α-positive hepatoblasts and hepatocytes was evident at E12 and all later stages (Fig. 1C).
Next, we examined Con and Hep-Ctnnb1−/− livers for primitive bile ducts, which are poorly defined but strongly positive for creatine kinase (CK)-19 after the E16 stage. Several CK-19-positive primitive ductular structures were visible in E16 and E18 Con livers (Fig. 1D). Such CK-19-positive cells were markedly decreased in the primitive ductal plates in Hep-Ctnnb1−/− livers at E16 or E18 stages (Fig. 1D).
Impaired Proliferation and Survival of Hepatoblasts and Hepatocytes in Hep-Ctnnb1−/− Livers
Histological analysis of E12, E14, and E17 livers also showed relatively unaffected hematopoietic cells, with a decrease in the numbers of epithelial cells in the Hep-Ctnnb1−/− livers, especially after E14 (Fig. 2A). The sinusoidal and vascular spaces were dilated in the mutants only.
Fig. 2.

Decreased proliferation and survival of hepatocytes is evident in the Hep-Ctnnb1−/− livers. (A) Hematoxylin and eosin (H&E) staining of the developing livers reveal several hepatoblasts and hepatocytes (arrowheads) in the control livers at E12 to E17, with decreasing numbers of hematopoietic cells (arrows) with progressing fetal age. Knockout (KO) livers reveal a lower number of hepatoblasts and hepatocytes than controls at all ages after E12, with continued presence of hematopoietic cells even at E17. Several PCNA-positive cells are observed in E12 to E17 control livers with a dramatic decrease in the respective KO livers. A few TUNEL-positive apoptotic nuclei are observed in E12 Con and KO livers, whereas increased apoptotic nuclei were observed in E14 and E17 KO livers as compared to their controls (×400). (B) A 3-fold to 6-fold decrease in the numbers of PCNA-positive cells was identified in β-catenin KO livers at various stages of liver development, which was significant (P < 0.05). (C) A 3-fold to 4-fold increase in apoptosis was observed at stages E14 and E17 in β-catenin KO livers, which was significant (P < 0.05).
Because β-catenin has been shown to regulate proliferation and survival of resident cells during ex vivo liver development,14,15 we next investigated any alterations in these events as a possible mechanism of diminished epithelial cell compartment in the Hep-Ctnnb1−/− livers. We observed a dramatic decrease in the number of PCNA-positive cells in the E12, E14, and E17 Hep-Ctnnb1−/− livers, as compared to the Con livers (Fig. 2A). This decrease ranged from 3-fold to 6-fold and was statistically significant at all stages (P < 0.05) (Fig. 2B). As shown before and elsewhere, cyclin-D1, a downstream β-catenin target crucial in G1 to S transition was downregulated in the Hep-Ctnnb1−/− livers19 (Fig. 1B).
Next, we investigated apoptosis by IHC for TUNEL in the Hep-Ctnnb1−/− and Con livers at stages E12, E14, and E17. Comparable and low numbers of apoptotic nuclei were observed at E12 in the Hep-Ctnnb1−/− and control livers (Fig. 2A). However, increased numbers of apoptotic nuclei were observed in Hep-Ctnnb1−/− livers at stages E14 and E17 (Fig. 2A). This difference was around 3-fold to 4-fold and significant (P < 0.05) (Fig. 2B).
Utrastructural Morphology of β-Catenin-Null Livers
Next, we examined Hep-Ctnnb1−/− and Con livers for ultrastructural differences by electron microscopy. Con livers at E15 and E17 showed tightly arranged hepatocytes and hematopoietic and endothelial cells (Fig. 3A). In addition, hepatocytes contained appreciable numbers of mitochondria, endoplasmic reticulum, and glycogen. In contrast, the hepatocytes in the E15 and E17 Hep-Ctnnb1−/− livers depicted 0.25 to 0.5 μ wide vesicular structures that progressed to blebbing and loss of hepatocyte ultrastructure (Fig. 3A), whereas the hematopoietic cells were normal. This morphology was reminiscent of oncotic cell death as has also been reported in the biopsies of patients with nonalcoholic fatty liver disease due to oxidative stress22 and led us to examine oxidative stress as a potential cause of hepatocyte death.
Fig. 3.

Hep-Ctnnb1−/− livers reveal aberrant ultrastructure and elevated oxidative stress. (A) Electron microscopy (EM) of control E15 and E17 livers shows normal hepatocytes (1) containing normal nuclei (n), endoplasmic reticulum cells (e), and mitochondria (m), normal erythroblasts (2), and normal endothelial cells (3). EM of the Hep-Ctnnb1−/− livers at E15 and E17 shows normal blood cells (2), no endothelial cells, and the hepatocytes (1) that display blebbing (bl) and loss of ultrastructural integrity. (B) IHC for 4-HNE reveals smaller hematopoietic cells staining-positive (arrowhead, upper panel) at E17 in control livers, whereas many larger hepatocytes were 4-HNE-positive (arrowhead, lower panel) in the knockout (KO). (×400) (C) Around 2-fold higher MDA and 4-HNE adducts were identified in the Hep-Ctnnb1−/− livers (n = 4) as compared to age-matched controls (n = 4). Also, the baseline lipid peroxidation was around 30% higher in pooled E17 control livers as compared to normal adult livers. (D) Normalized gene array (to the expression of hepatocyte genes-albumin and α-fetoprotein) at E16.5 shows several-fold lower expression of variousGSTs in the Hep-Ctnnb1−/− livers as compared to littermate controls. (E) The decrease in key GSTs in KO was validated by RT-PCR.
Elevated Oxidative Stress in the Absence of β-Catenin during Liver Development
We examined lipid peroxidation due to oxidative stress by IHC for 4-HNE. Smaller cells with hematopoietic morphology were positive for 4-HNE in the Con livers at E17, while hepatocytes were predominantly negative (Fig. 3B). On the other hand, the E17 Hep-Ctnnb1−/− livers displayed several 4-HNE–positive hepatocytes (Fig. 3B), indicating elevated oxidative stress in β-catenin-deficient hepatocytes. Interestingly, smaller and more abundant hematopoietic cells in the E17 Hep-Ctnnb1−/− livers did not show positive staining for 4-HNE (Fig. 3B).
We confirmed these findings further by a biochemical assay for MDA and 4-HNE adducts. Around 2-fold higher adduct content was evident in β-catenin-deficient fetal livers as compared to both controls at E17 (Fig. 3C). Interestingly, the levels of MDA and 4-HNE adducts in the fetal control livers, although dramatically lower than the Hep-Ctnnb1−/− livers, were still around 30% higher than the normal adult livers, suggesting an increased basal oxidative stress (Fig. 3C).
Decreased Expression of Multiple GSTs in Hep-Ctnnb1−/− Livers
GSTs play a protective role against oxidative stress.23,24 We studied the expression of GSTs in the Hep-Ctnnb1−/− and Con livers as a mechanism of continued oxidative stress and loss of cell viability. A dramatic decrease in the expression of several GSTs was evident in the Hep-Ctnnb1−/− livers in the normalized gene array analysis at E16.5 (Fig. 3D) (discussed in Materials and Methods and in Table 1). We used RT-PCR of pooled E-cadherin-sorted cells from Hep-Ctnnb1−/− and Con livers at E16 and E17 stages to verify the decrease, as shown for GST-alpha3, GST-omega1, and GST-mu1 (Fig. 3E).
Impaired Hepatocyte Maturation in the β-Catenin-Deficient Livers
A more dramatic difference in the cellular composition, organization, and architecture was apparent at later stages of liver development in the β-catenin-conditional null livers. At E17, the Con livers were composed of confluent sheets of cells composed of polarized, differentiated hepatocytes with clear cytoplasm, with only a few interspersed hematopoietic cells (Fig. 4A). The Hep-Ctnnb1−/− livers contained predominant hematopoietic cells with only a few hepatocytes, which appeared undifferentiated, with high nuclear to cytoplasmic ratio and lacked polarity (Fig. 4A). To verify the maturation status of the remnant hepatocytes at E17, the livers were examined for glycogen accumulation. Extensive glycogen accumulation by periodic acid-Schiff staining was evident in Con livers and not in Hep-Ctnnb1−/− livers (Fig. 4A). In addition, we assessed tight junctions (TJs) by IF for ZO-1, as an indicator of mature hepatocytes. Several TJs were evident in the Con livers, whereas Hep-Ctnnb1−/− livers showed dramatically lower numbers of TJs as shown at E17 (Fig. 3A).
Fig. 4.

Inadequate maturation of remnant hepatocytes in β-catenin null livers during development is attributable to loss of C/EBPα. (A) Hematoxylin and eosin (H&E) staining (×600) of the E17 control liver (Con) shows predominantly mature hepatocytes with polarity and clear cytoplasm (bold arrow) and a few hematopoietic cells (arrow), whereas the β-catenin-deficient livers (knockout [KO]) show predominant hematopoietic cells (arrow) with a few immature hepatocytes that have high nuclear to cytoplasmic ratio and lack polarity (bold arrow). Periodic acid-Schiff (PAS) staining (×400) shows E17 control livers with extensive glycogen in hepatocytes and E17 KO hepatocytes devoid of glycogen. IF for ZO-1 (×400) shows several tight junctions in the E17 control livers, whereas the KO livers show a dramatic decrease in the numbers of tight junctions. (B) Western blot using the total cell lysates of the E-cadherin-sorted fractions of E17 Con and KO livers show a dramatic decrease in C/EBPα levels in KO. Ponceau Red verifies equal loading in the lower panel. (C) E17 control livers show E-cadherin-positive cells (green) with nuclear C/EBPα (red) as identified by white arrowheads. The KO livers show a few E-cadherin-positive cells (green) that lack nuclear C/EBPα (red) as identified by white arrows (×400). Nuclei were counterstained with Hoesch stain. Smaller panels show E-cadherin (green) and C/EBPα (red) IF without counterstain clearly identifying lack of C/EBPα in β-catenin deficient livers in E-cadherin-positive cells.
Next, we utilized the Affymetrix gene array analysis performed on the pooled whole livers at the E16.5 stages. Table 1 shows differences in the albumin and α-fetoprotein gene expression between the Hep-Ctnnb1−/− and Con livers. While both these genes were markedly lower in the Hep-Ctnnb1−/− livers, we utilized the average fold-change in these two hepatocyte-specific genes to normalize the gene expression data for the differences in cell population; that is, decreased numbers of hepatocyte and hepatoblasts in β-catenin-deficient livers (Table 1). With this normalization, we identified a significantly lower expression of β-catenin gene as well as of its known targets in the liver, such as cyclin-D1 and lect2 (Table 2).11,19,25 The expression of other β-catenin target genes, such as glutamine synthetase, epidermal growth factor receptor, ornithine aminotransferase, CYP2E1, and CYP1A2, remained low overall at E16.5 in both Hep-Ctnnb1−/− and Con livers, or their decrease in Hep-Ctnnb1−/− was less than 3.4-fold and hence was not identified after normalization (data not shown).10,12,19,26,27 A more comprehensive list of all genes down-regulated two-fold or higher is included in the Supplementary Material.
Table 2. Microarray Data from the Affymetrix Gene Array of the Pooled Control and Pooled β-Catenin-Conditional Null Livers at E16.5.
| Gene | Expression in WT (E16.5) | Expression in KO (E16.5) (Normalized to Average Albumin and α-Fetoprotein Expression 3.4*) | Fold Change | References in liver (if any) |
|---|---|---|---|---|
| β-Catenin | 1335.2 | 172.72 | −8 | |
| Regucalcin | 2153.5 | 195.84 | −5 | 19 |
| Cyclin-D1 | 2516 | 480.08 | −5 | 11,19 |
| Leukocyte cell-derived chemotaxin 2 | 1773 | 265.88 | −7 | 25 |
β-Catenin and some of its known gene targets show significant decrease. The expression of genes in the β-catenin conditional null livers was normalized to the average fold change of albumin and α-fetoprotein expression (which is present exclusively in hepatoblasts and hepatocytes) to standardize for differences in cell composition between the two groups; that is, decreased hepatoblast and hepatocyte numbers in the β-catenin-conditional null livers at E16.5. WT, wild-type; KO, knockout
This average was used in subsequent analysis for normalizing gene expression in the β-catenin-conditional null livers at E16.5 due to observed cell disparity; that is, decreased numbers of hepatocyte and hepatoblasts in β-catenin-deficient livers.
Similar analysis revealed a multifold decrease in the expression of several genes associated with hepatocyte maturation and function (Table 3). Of significant relevance include genes encoding for apolipoprotein M and A-1, coagulation factors XI, XII, and XIII, transthyretin, α2-macroglobulin, transferrin, and haptoglobin.
Table 3. Microarray Data from the Affymetrix Gene Array of the Pooled Control and Pooled β-Catenin-Conditional Null Livers at E16.5 Show Differences in Hepatocyte Maturation.
| Gene | Expression in WT (E16.5) | Expression in KO (E16.5) (Normalized to Average Albumin and α-Fetoprotein Expression 3.4*) | Fold Change |
|---|---|---|---|
| Apolipoprotein M | 6445.7 | 345.44 | −19 |
| Apolipoprotein A-I | 34804 | 3779.4 | −9 |
| Coagulation factor XI | 787.7 | 102.34 | −8 |
| C/EBPα | 4259.4 | 589.56 | −7 |
| Transthyretin | 41128 | 5684.8 | −7 |
| Hydroxysteroid 17-beta dehydrogenase 2 | 2479.5 | 340 | −7 |
| Aldehyde dehydrogenase 2 | 4501 | 840.14 | −5 |
| Glutamate dehydrogenase | 17825.2 | 3615.9 | −5 |
| Haptoglobin | 1732.3 | 352.24 | −5 |
| Hepatic lipase | 1036.2 | 199.24 | −5 |
| Coagulation factor | 1921.4 | 433.16 | −4 |
| Coagulation factor XII | 1106 | 307.02 | −4 |
| α-2-macroglobulin | 9700.4 | 2567 | −4 |
| Fatty acid binding protein, liver | 7099.4 | 1779.56 | −4 |
| Transferrin | 43850 | 16454.3 | −3 |
As in Table 2, the expression of genes in KO livers was normalized to the average fold change of albumin and α-fetoprotein expression to standardize for cell disparity between the control and β-catenin-conditional null livers at E16.5. WT, wild-type; KO, knockout
This average was used in subsequent analysis for normalizing gene expression in the β-catenin-conditional null livers at E16.5 due to observed cell disparity; that is, decreased numbers of hepatocyte and hepatoblasts in β-catenin-deficient livers.
β-Catenin-Deficient Livers Reveal a Diminished Expression of C/EBPα
As shown in Table 3, expression of C/EBPα was 7-fold lower in the absence of β-catenin in E16.5 livers. C/EBPα is a fundamental regulator of hepatocyte differentiation and maturation that controls expression of multiple liver-specific transcriptional genes, several of which were observed to be significantly decreased in Hep-Ctnnb1−/− livers at E16.5 (Table 3).28-30 To verify the decrease in C/EBPα identified in the gene array, we subjected E-cadherin-sorted cells from E17 Hep-Ctnnb1−/− and Con livers to radioimmunoprecipitation assay buffer whole-cell protein isolation. A dramatic decrease in C/EBPα protein was evident in β-catenin-deficient hepatocytes (Fig. 4B). Dual IF for E-cadherin and C/EBPα also identified a noteworthy decrease in nuclear C/EBPα in E-cadherin-positive cells, which were fewer overall in the Hep-Ctnnb1−/− livers at E17 (Fig. 4C). Thus a significant decrease in C/EBPα was evident in the absence of β-catenin in hepatocytes, which appears to be a major mechanism of their compromised maturation.
Changes in the Adherens Junctions in β-Catenin-Deficient Livers
Because β-catenin is a normal component of the adherens junctions (AJs) along with E-cadherin, p120, and γ-catenin, we investigated this complex in β-catenin-null livers. Western blots utilizing whole cell lysates showed comparable levels of p120 and γ-catenin at E14, followed by lower levels in the Hep-Ctnnb1−/− livers at E15 to E16 (Fig. 5A). However, decreased E-cadherin levels were seen at all stages (Fig. 5A). Coprecipitation studies showed increased association of p120 and E-cadherin in E14 Hep-Ctnnb1−/− livers followed by a gradual decrease (Fig. 5B). This was consistent with decreasing levels of both proteins in the Hep-Ctnnb1−/− livers. The association of γ-catenin to E-cadherin appeared to increase at all stages in the Hep-Ctnnb1−/− livers (Fig. 5B). This was especially prominent in light of low levels of both proteins in the Hep-Ctnnb1−/− livers.
Fig. 5.

Changes in AJs in absence of β-catenin during liver development. (A) Decreased total protein levels of E-cadherin are observed in Hep-Ctnnb1−/− livers at E14 to E16, whereas decrease in p120 and plakoglobin or γ-catenin was observed after the E15 stage in the Hep-Ctnnb1−/− livers, in a representative western blot (WB). Actin confirms equal loading. (B) A relative increase in association of p120 and γ-catenin to E-cadherin at E14 and of γ-catenin and E-cadherin at E15 to E16 stages in the Hep-Ctnnb1−/− livers by coprecipitation. This increase is more pronounced when the low levels of total E-cadherin and γ-catenin levels in the Hep-Ctnnb1−/− livers at these stages is taken into account.
No Intrinsic Defect in Hematopoiesis in the Absence of β-Catenin in Epithelial Compartment
As the embryos lacking β-catenin appeared pale, we investigated any changes in intrinsic hematopoiesis in the Hep-Ctnnb1−/− livers. Hep-Ctnnb1−/− livers were smaller than controls due to deficient hepatocyte compartment but displayed comparable numbers of hematopoietic cells, as shown at the E16 stage (Fig. 6A). Next, we examined cells from these livers for hematopoiesis, utilizing the CFCs assay. Cells from E15.5 to E16.5 Hep-Ctnnb1−/− and Con livers showed comparable numbers of CFCs by this assay as shown in a representative study at E16 stage (Fig. 6B), suggesting absence of any intrinsic defect in hematopoiesis in the Hep-Ctnnb1−/− livers.
Fig. 6.

Normal intrinsic hematopoiesis in β-catenin-deficient livers. (A) Fluorescence-activated cell sorting (FACS) analysis shows comparable numbers of C-kit-positive and Ter-119-positive cells in the Con and knockout (KO) livers at E16 stage. Respective isotype controls are included in the left panel. (B) CFC-assay identified equivalent numbers of colonies using 5 × 105 cells from E16 Hep-Ctnnb1−/− or control livers. The different types of colonies are labeled s follows: Mix, colony-forming unit granulocyte-erythrocyte-megakaryocyte-monocyte (CFU-GEMM) multipotential; GM, CFU granulocyte-monocyte; M, CFU monocyte; G, CFU granulocyte; E, burst-forming unit erythroid (BFU-E); Endo, endothelial.
Perturbation in Endothelium in the β-Catenin-Deficient Livers
Based on no observable differences in intrinsic hematopoiesis, we next sought to investigate whether endothelial cell organization was perturbed in the Hep-Ctnnb1−/− livers. A similar scenario had been previously attributed to fetal pallor and lethality.31 Isolectin-B4-positive endothelial cells were observed lining the primitive sinusoidal spaces in developing livers (Fig. 7A). However, endothelial cells in the Hep-Ctnnb1−/− livers showed disorganized endothelia within the hepatic architecture (Fig. 7A). This haphazard arrangement of endothelial cells was also verified by IHC for platelet endothelial cell adhesion molecules (Fig. 7B) and Flk-1 (not shown) in Hep-Ctnnb1−/− livers as compared to their controls. While this could be secondary to the overall distorted architecture due to perturbed hepatocyte compartment in the absence of β-catenin, epithelial cells are also a source of VEGF, which is a critical player in endothelial cell homeostasis. Indeed, a noteworthy diminution of VEGF was readily identified by IF in the Hep-Ctnnb1−/− livers as compared to the controls, as shown at E16 (Fig. 7C). Thus defective blood flow secondary to endothelial cell disorganization might also be contributing to the overall phenotype.
Fig. 7.

Disrupted endothelial cell organization in the Hep-Ctnnb1−/− livers. (A) Isolectin B4-positive endothelial cells (arrowheads) line primitive sinusoidal space in E17 control livers, whereas several sinusoidal spaces (*) showed discontinuous isolectin-B4-positive cells in the knockout (KO) livers shown in two representative right panels. (B) Platelet endothelial cell adhesion molecule (PECAM) IHC also identified endothelial cells in control (WT) and KO livers, with a haphazard arrangement of endothelial cells in the latter (arrowhead). (C) Several hepatocytes in the E16 control livers are VEGF-positive (red), whereas only a few VEGF-expressing cells were observed in the E16 Hep-Ct-nnb1−/− livers.
Discussion
Wnt/β-catenin signaling plays multiple fundamental roles in many processes during normal growth and development.1,16 β-Catenin is the critical mediator of the canonical Wnt pathway, which has been implicated in many physiological processes inherent to the liver. The role of Wnt/β-catenin signaling has been demonstrated in liver growth and regeneration, hepatic zonation, and metabolism.8,9,12,19,26,32 Previously, we and others have reported temporal expression during normal liver development, which was validated in vitro for functional significance.13-15,33 While repression of Wnt signaling in the foregut endoderm has been recently demonstrated to be critical for hepatic specification, it was shown to be necessary for liver growth of the bud in Xenopus.34 Interestingly, Wnt2bb has been recently been shown to be inductive for hepatic specification during development in zebrafish.35 While this might be due to mechanistic disparity due to species difference, it might also be due to difference in the time at which the analysis was performed in the two species.
We previously reported high β-catenin protein and gene expression during mouse liver development.14,15 This is observed during hepatic morphogenesis at the time when liver bud components are undergoing expansion, organization, and differentiation.36 In this study, we identified successful deletion of β-catenin during early liver development, resulting in a profound defect in hepatoblast expansion, maturation, and hepatocyte function, leading to lethality at E17.
Proliferation is severely impaired in the absence of β-catenin, secondary to decreased expression of downstream targets such as cyclin-D1, which are critical in proliferation.37 This has also been shown in liver during the processes of regeneration, postnatal liver development, ex vivo embryonic liver development, and more recently in facultative liver stem cells or oval cells.11,15,19,38-40 In this study we observed a significant decrease in cyclin-D1 secondary to β-catenin loss, which leads to failure of expansion of hepatoblasts and eventually hepatocytes into cords and plates to form confluent sheet-like structures. In addition, hepatocytes undergo increased apoptotic death, which has also been identified.15 We also identified a novel role of β-catenin in regulating oxidative stress during liver development via several GSTs, which were repressed in the absence of β-catenin and caused elevated oncotic and apoptotic hepatocytic death. Indeed, low expression of GSTs is associated with elevated oxidative stress.23 Whether this observation is a direct transcriptional consequence of β-catenin loss or an indirect event will need further investigation.
In addition to the limited numbers of hepatoblasts and hepatocytes in β-catenin-deficient livers, we also noted their inadequate maturation. This was indicated by diminished glycogen accumulation and significantly lower expression of factors associated with hepatocyte function such as apolipoproteins, transthyretin, transferrin, haptoglobin, glutamate dehydrogenase, aldehyde dehydrogenase 2, and several coagulation factors.41,42 Another unique observation was a dramatic decrease in the RNA and protein levels of C/EBPα, a master regulator of hepatocyte function via transcriptional control of multiple aforementioned factors.28-30 This pronounced affect of loss of β-catenin in hepatoblasts on C/EBPα and to a lesser extent on HNF4α, which is another key regulator of hepatocyte differentiation and maturation,31 appear to dictate the overall hepatic phenotype and lethality. It is also important to note that GSTs that are protective against oxidative stress are also regulated by HNFs and C/EBP.41,43-45 Whether β-catenin directly controls the expression of the key transcriptional of hepatocyte function such as C/EBPα and HNF4α would need additional experiments. We also suspect that most of the hepatocyte function associated genes mentioned above are not direct transcriptional targets of β-catenin and might be secondary to effects on key regulators such as C/EBPα.28-30,41,43,46
Other evidence of compromise in hepatocyte maturation in the β-catenin-deficient livers was from the histology. The epithelial cells continued to show high nuclear to cytoplasmic ratio and lacked cell polarity, which is dictated by the presence of normal cell-cell junctions consisting of TJs and AJs. During this process, β-catenin redistribution to the hepatocyte membrane has been reported, where it plays a role in AJs, which in turn impact TJs.47,48 Absence of β-catenin led to a decrease in the numbers of TJs as evidenced by the ZO-1 IF. There appeared to be a compensatory increase in total and E-cadherin-associated fractions of p120 and γ-catenin during earlier stages, followed by continued increase in E-cadherin-γ-catenin association at stages E15 to E16. This is of relevance since functional redundancy between β-catenin and γ-catenin (or plakoglobin) has been reported.49,50 It should be noted that while γ-catenin does have transcriptional activation capability, it is distinct and contrary to β-catenin.51 Thus, while γ-catenin might be partially maintaining AJs in the absence of β-catenin, its transcriptional activation was not anticipated to play a vital role. Overall, these compensations were inadequate to sustain hepatic or fetal viability. It is also relevant to point out that absence of beta-catenin leading to poor expansion of hepatoblasts and hepatocytes, with resulting lack of cell-cell contact, might be the primary event dictating lack of hepatocyte maturation, which might in turn affect the expression of master regulators of hepatocyte function such as C/EBPα, rather than it being a direct transcriptional target of Wnt/β-catenin signaling.41,48 Additional studies would be necessary to examine this interaction in greater depth.
Insignificant gross differences between the control and β-catenin conditional null livers were evident before the E12 stage, despite successful deletion of β-catenin. This appears to differ from the observed phenotype in zebrafish, in which Wnt2b/β-catenin stabilization was essential for hepatic induction.35 This could be due to species difference or redundant signaling during early stages. However, more recently, suppression of Wnt signaling has been identified to be important in hepatic specification.34 While additional analysis of Hep-Ctnnb−/− embryos for successful β-catenin deletion at E8:5 in hepatic bud would be necessary to address the role in hepatic specification in murine liver development. β-catenin was imperative for the expansion of hepatoblasts and their maturation, and its deletion finally took a toll at around stage E17, when hepatocyte function became indispensable for survival. Also, discussions of nonhepatic effects of β-catenin loss due to Foxa3-Cre-induced recombination are not within the scope of the present study.
Intrahepatic bile ducts arise from the hepatic progenitors or hepatoblasts, the bipotential stem cells that mark liver development. The hepatoblasts in contact with the portal mesenchyme differentiate into biliary epithelial cells, which form a ductal plate comprised of a single layer of cells.36 Such primitive bile ducts are poorly defined but are strongly positive for CK-19 and are abundant after the E16 stage of gestational development. In line with our previous findings, lack of β-catenin also led to paucity or complete absence of CK-19-positive primitive intrahepatic bile ducts, suggesting a critical role of β-catenin in the biliary differentiation of hepatoblasts.13,15
A relevant and interesting observation was persistence of normal hematopoiesis in the β-catenin-deficient livers despite the presence of “ineffective hepatopoiesis.” However, based on the obvious fetal pallor there appeared to be ongoing defects relevant to hematopoiesis. However, while comparable hematopoietic cells, as well as their ability to form colonies, were observed in the absence of β-catenin in hepatocytes, inadequate endothelial cell organization was also observed, secondary to distorted hepatic architecture as well as lack of hepatocytes, which are the source of endothelial cell growth factors such as VEGF. This disorganization contributed to overall fetal pallor and perhaps to demise. A similar scenario has also been attributed to fetal loss in the HNF-4α-null phenotype.31
Mechanisms by which hematopoiesis ceases in the liver during late fetal stages remain obscure. Some evidence suggests that factors such as Oncostatin M might be playing a role in this event.52 Continued presence of the hematopoietic cells in the β-catenin-null livers at stages later than E17 suggested that lack of competition for physical space due to the failure of hepatocyte expansion maybe playing a role in the persisting hematopoietic compartment. Additionally, presence of oxidative stress during normal liver development, which was detected in late fetal stages, might be an important mechanism, adversely affecting normal hematopoiesis, stimulating their relocation. Absence of the protective GSTs in hepatocytes in the absence of β-catenin might be allowing for these cells to succumb to the elevated oxidative stress. Such a route of hematopoietic ablation during normal fetal liver development would need to be explored further.
Supplementary Material
Acknowledgments
Supported by the National Institutes of Health (NIH) grants 1R01DK62277 and R01CA124414 to S.P.S.M.; Rango's Fund for Enhancement of Pathology Research; and the Cleveland Foundation.
Abbreviations
- AJ
adherens junction
- C/EBPα
CCAAT-enhancer binding protein-α
- CFC
colony forming unit cell
- CK
creatine kinase
- Cre
cyclization recombination
- E1
embryonic day 1
- GSK3β
glycogen synthase kinase 3β
- GST
glutathione S-transferase
- HNE
hydroxynonenal
- HNF
hepatocyte nuclear factor
- IF
immunofluorescence
- IHC
immunohistochemistry
- MDA
malondialdehyde
- PCNA
proliferating cell nuclear antigen
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- TJ
tight junction
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- VEGF
vascular endothelial growth factor
Footnotes
Potential conflict of interest: Nothing to report.
Supplementary material for this article can be found on the Hepatology Web site (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html).
References
- 1.Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- 2.Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science. 2000;287:1606–1609. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- 3.Morin PJ. beta-Catenin signaling and cancer. Bioessays. 1999;21:1021–1030. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 4.Pennisi E. How a growth control path takes a wrong turn to cancer. Science. 1998;281:1438–1439. doi: 10.1126/science.281.5382.1438. News; Comment. erratum appears in Science 1998;281:1809. [DOI] [PubMed] [Google Scholar]
- 5.Barker N, Morin PJ, Clevers H. The Yin-Yang of TCF/beta-catenin signaling. Adv Cancer Res. 2000;77:1–24. doi: 10.1016/s0065-230x(08)60783-6. [DOI] [PubMed] [Google Scholar]
- 6.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C, Rebouissou S, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43:515–524. doi: 10.1002/hep.21068. [DOI] [PubMed] [Google Scholar]
- 8.Monga SP, Pediaditakis P, Mule K, Stolz DB, Michalopoulos GK. Changes in WNT/beta-catenin pathway during regulated growth in rat liver regeneration. Hepatology. 2001;33:1098–1109. doi: 10.1053/jhep.2001.23786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sodhi D, Micsenyi A, Bowen WC, Monga DK, Talavera JC, Monga SP. Morpholino oligonucleotide-triggered beta-catenin knockdown compromises normal liver regeneration. J Hepatol. 2005;43:132–141. doi: 10.1016/j.jhep.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 10.Tan X, Apte U, Micsenyi A, Kotsagrelos E, Luo JH, Ranganathan S, et al. Epidermal growth factor receptor: a novel target of the Wnt/beta-catenin pathway in liver. Gastroenterology. 2005;129:285–302. doi: 10.1053/j.gastro.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sekine S, Gutierrez PJ, Lan BY, Feng S, Hebrok M. Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology. 2007;45:361–368. doi: 10.1002/hep.21523. [DOI] [PubMed] [Google Scholar]
- 12.Sekine S, Lan BY, Bedolli M, Feng S, Hebrok M. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology. 2006;43:817–825. doi: 10.1002/hep.21131. [DOI] [PubMed] [Google Scholar]
- 13.Hussain SZ, Sneddon T, Tan X, Micsenyi A, Michalopoulos GK, Monga SP. Wnt impacts growth and differentiation in ex vivo liver development. Exp Cell Res. 2004;292:157–169. doi: 10.1016/j.yexcr.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 14.Micsenyi A, Tan X, Sneddon T, Luo JH, Michalopoulos GK, Monga SP. Beta-catenin is temporally regulated during normal liver development. Gastroenterology. 2004;126:1134–1146. doi: 10.1053/j.gastro.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 15.Monga SP, Monga HK, Tan X, Mule K, Pediaditakis P, Michalopoulos GK. Beta-catenin antisense studies in embryonic liver cultures: role in proliferation, apoptosis, and lineage specification. Gastroenterology. 2003;124:202–216. doi: 10.1053/gast.2003.50000. [DOI] [PubMed] [Google Scholar]
- 16.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, et al. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 17.Apte U, Zeng G, Muller P, Tan X, Micsenyi A, Cieply B, et al. Activation of Wnt/beta-catenin pathway during hepatocyte growth factor-induced hepatomegaly in mice. Hepatology. 2006;44:992–1002. doi: 10.1002/hep.21317. [DOI] [PubMed] [Google Scholar]
- 18.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26:149–150. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 19.Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology. 2006;131:1561–1572. doi: 10.1053/j.gastro.2006.08.042. [DOI] [PubMed] [Google Scholar]
- 20.Lee CS, Friedman JR, Fulmer JT, Kaestner KH. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944–947. doi: 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- 21.Monga SP, Hout MS, Baun MJ, Micsenyi A, Muller P, Tummalapalli L, et al. Mouse fetal liver cells in artificial capillary beds in three-dimensional four-compartment bioreactors. Am J Pathol. 2005;167:1279–1292. doi: 10.1016/S0002-9440(10)61215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roskams T, Yang SQ, Koteish A, Durnez A, DeVos R, Huang X, et al. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. AmJ Pathol. 2003;163:1301–1311. doi: 10.1016/S0002-9440(10)63489-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallagher EP, Gardner JL, Barber DS. Several glutathione S-transferase isozymes that protect against oxidative injury are expressed in human liver mitochondria. Biochem Pharmacol. 2006;71:1619–1628. doi: 10.1016/j.bcp.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 24.Knight TR, Choudhuri S, Klaassen CD. Constitutive mRNA expression of various glutathione s-transferase isoforms in different tissues of mice. Toxicol Sci. 2007;100:513–524. doi: 10.1093/toxsci/kfm233. [DOI] [PubMed] [Google Scholar]
- 25.Ovejero C, Cavard C, Perianin A, Hakvoort T, Vermeulen J, Godard C, et al. Identification of the leukocyte cell-derived chemotaxin 2 as a direct target gene of beta-catenin in the liver. Hepatology. 2004;40:167–176. doi: 10.1002/hep.20286. [DOI] [PubMed] [Google Scholar]
- 26.Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, et al. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002;21:8293–8301. doi: 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- 27.Loeppen S, Koehle C, Buchmann A, Schwarz M. A beta-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis. 2005;26:239–248. doi: 10.1093/carcin/bgh298. [DOI] [PubMed] [Google Scholar]
- 28.Darlington GJ, Wang N, Hanson RW. C/EBP alpha: a critical regulator of genes governing integrative metabolic processes. Curr Opin Genet Dev. 1995;5:565–570. doi: 10.1016/0959-437x(95)80024-7. [DOI] [PubMed] [Google Scholar]
- 29.Dinic S, Bogojevic D, Petrovic M, Poznanovic G, Ivanovic-Matic S, Mihailovic M. C/EBP alpha and C/EBP beta regulate haptoglobin gene expression during rat liver development and the acute-phase response. Mol Biol Rep. 2005;32:141–147. doi: 10.1007/s11033-005-0750-0. [DOI] [PubMed] [Google Scholar]
- 30.Zannis VI, Kan HY, Kritis A, Zanni E, Kardassis D. Transcriptional regulation of the human apolipoprotein genes. Front Biosci. 2001;6:D456–D504. doi: 10.2741/zannis. [DOI] [PubMed] [Google Scholar]
- 31.Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet. 2003;34:292–296. doi: 10.1038/ng1175. [DOI] [PubMed] [Google Scholar]
- 32.Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, et al. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev Cell. 2006;10:759–770. doi: 10.1016/j.devcel.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 33.Suksaweang S, Lin CM, Jiang TX, Hughes MW, Widelitz RB, Chuong CM. Morphogenesis of chicken liver: identification of localized growth zones and the role of beta-catenin/Wnt in size regulation. Dev Biol. 2004;266:109–122. doi: 10.1016/j.ydbio.2003.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLin VA, Rankin SA, Zorn AM. Repression of Wnt/beta-catenin signaling in the anterior endoderm is essential for liver and pancreas development. Development. 2007;134:2207–2217. doi: 10.1242/dev.001230. [DOI] [PubMed] [Google Scholar]
- 35.Ober EA, Verkade H, Field HA, Stainier DY. Mesodermal Wnt2b signalling positively regulates liver specification. Nature. 2006;442:688–691. doi: 10.1038/nature04888. [DOI] [PubMed] [Google Scholar]
- 36.Lemaigre F, Zaret KS. Liver development update: new embryo models, cell lineage control, and morphogenesis. Curr Opin Genet Dev. 2004;14:582–590. doi: 10.1016/j.gde.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 37.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 38.Apte U, Thompson MD, Cui S, Liu B, Cieply B, Monga SP. Wnt/beta-catenin signaling mediates oval cell response in rodents. Hepatology. 2008;47:288–295. doi: 10.1002/hep.21973. [DOI] [PubMed] [Google Scholar]
- 39.Apte U, Zeng G, Thompson MD, Muller P, Micsenyi A, Cieply B, et al. beta-Catenin is critical for early postnatal liver growth. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1578–G1585. doi: 10.1152/ajpgi.00359.2006. [DOI] [PubMed] [Google Scholar]
- 40.Hu M, Kurobe M, Jeong YJ, Fuerer C, Ghole S, Nusse R, et al. Wnt/beta-catenin signaling in murine hepatic transit amplifying progenitor cells. Gastroenterology. 2007;133:1579–1591. doi: 10.1053/j.gastro.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 41.Block GD, Locker J, Bowen WC, Petersen BE, Katyal S, Strom SC, et al. Population expansion, clonal growth, and specific differentiation patterns in primary cultures of hepatocytes induced by HGF/SF, EGF and TGF alpha in a chemically defined (HGM) medium. J Cell Biol. 1996;132:1133–1149. doi: 10.1083/jcb.132.6.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spath GF, Weiss MC. Hepatocyte nuclear factor 4 provokes expression of epithelial marker genes, acting as a morphogen in dedifferentiated hepatoma cells. J Cell Biol. 1998;140:935–946. doi: 10.1083/jcb.140.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomez-Lechon MJ, Jover R, Donato T, Ponsoda X, Rodriguez C, Stenzel KG, et al. Long-term expression of differentiated functions in hepatocytes cultured in three-dimensional collagen matrix. J Cell Physiol. 1998;177:553–562. doi: 10.1002/(SICI)1097-4652(199812)177:4<553::AID-JCP6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 44.Paulson KE, Darnell JE, Jr, Rushmore T, Pickett CB. Analysis of the upstream elements of the xenobiotic compound-inducible and positionally regulated glutathione S-transferase Ya gene. Mol Cell Biol. 1990;10:1841–1852. doi: 10.1128/mcb.10.5.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Romero L, Higgins MA, Gilmore J, Boudreau K, Maslen A, Barker HJ, et al. Down-regulation of alpha class glutathione S-transferase by interleukin-1beta in human intestinal epithelial cells (Caco-2) in culture. Drug Metab Dispos. 2002;30:1186–1193. doi: 10.1124/dmd.30.11.1186. [DOI] [PubMed] [Google Scholar]
- 46.Timchenko NA, Wilde M, Nakanishi M, Smith JR, Darlington GJ. CCAAT/enhancer-binding protein alpha (C/EBP alpha) inhibits cell proliferation through the p21 (WAF-1/CIP-1/SDI-1) protein. Genes Dev. 1996;10:804–815. doi: 10.1101/gad.10.7.804. [DOI] [PubMed] [Google Scholar]
- 47.Matsui T, Kinoshita T, Morikawa Y, Tohya K, Katsuki M, Ito Y, et al. K-Ras mediates cytokine-induced formation of E-cadherin-based adherens junctions during liver development. EMBO J. 2002;21:1021–1030. doi: 10.1093/emboj/21.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monga SP, Micsenyi A, Germinaro M, Apte U, Bell A. beta-Catenin regulation during matrigel-induced rat hepatocyte differentiation. Cell Tissue Res. 2006;323:71–79. doi: 10.1007/s00441-005-0045-8. [DOI] [PubMed] [Google Scholar]
- 49.Gallicano GI, Bauer C, Fuchs E. Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development. 2001;128:929–941. doi: 10.1242/dev.128.6.929. [DOI] [PubMed] [Google Scholar]
- 50.Zhou J, Qu J, Yi XP, Graber K, Huber L, Wang X, et al. Upregulation of gamma-catenin compensates for the loss of beta-catenin in adult cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;292:H270–H276. doi: 10.1152/ajpheart.00576.2006. [DOI] [PubMed] [Google Scholar]
- 51.Solanas G, Miravet S, Casagolda D, Castano J, Raurell I, Corrionero A, et al. beta-Catenin and plakoglobin N- and C-tails determine ligand specificity. J Biol Chem. 2004;279:49849–49856. doi: 10.1074/jbc.M408685200. [DOI] [PubMed] [Google Scholar]
- 52.Kinoshita T, Sekiguchi T, Xu MJ, Ito Y, Kamiya A, Tsuji K, et al. Hepatic differentiation induced by oncostatin M attenuates fetal liver hematopoiesis. Proc Natl Acad Sci U S A. 1999;96:7265–7270. doi: 10.1073/pnas.96.13.7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.