

Abstract
Cryo electron microscopy (cryoEM) has emerged as an excellent tool for resolving high-resolution three-dimensional structures of membrane proteins in a lipid-containing environment with interacting partners. The near atomic resolution structures of Venezuelan equine encephalitis virus and dengue virus revealed transmembrane helices in lipid bilayers, receptor-binding glycosylation moieties, and functionally important interactions between their fusion protein and membrane-anchored chaperone protein. For pleomorphic enveloped viruses, such as human immunodeficiency virus, glycoprotein complexes can be imaged in isolation to reveal molecular interactions at different states. These high-resolution cryoEM structures have clarified important domains not previously resolved by crystallography and illustrate exciting opportunities to visualize viral membrane proteins in their native and possibly transiently stable functional states, thus uncovering mechanisms of action and informing anti-viral strategies.
Introduction
Membrane proteins constitute about 30% of all proteins and are involved in vital functions such as energy metabolism, signal transduction and immune protection. Most current drugs on the market target membrane proteins, and high-resolution three-dimensional (3D) structures of membrane proteins are highly sought after for rational design of improved therapeutic interventions.
Viral membrane proteins typically form the external layer of enveloped viruses. Historically, these viruses are the culprits behind some of the most medically significant human diseases. Examples include the human immunodeficiency virus (HIV — the AIDS virus) — and the various types of influenza viruses — viruses that cause seasonal and pandemic flu. Other enveloped viruses, such as herpes simplex viruses and cytomegaloviruses, can cause chronic or dormant life-long human infections that can be re-activated by environmental factors or in individuals with weakened immune systems, to cause recurring, sometimes life-threatening infections. In tropical and sub-tropical regions, enveloped viruses can be spread rapidly via insect vectors (such as mosquitos) to cause acute, life-threatening infections. Prominent examples include dengue, Yellow fever, West Nile, Ebola.
Recent technology advancement allows for atomic modeling and 3D structure determination of large complexes by high-resolution cryo electron microscopy (cryoEM). These cryoEM structures have revealed protein structures and protein–lipid interactions in or close to their native membrane environment, an advantage over the conventional structural method of X-ray crystallography. When applied to viral membrane proteins that function as large molecular complexes and/or have multiple, transiently stable states, cryoEM is particularly advantageous, as these qualities make them unsuitable for either X-ray crystallography or NMR. This review aims to provide an overview of recent progress of high-resolution cryoEM in structural studies of membrane proteins of enveloped viruses.
High-resolution cryoEM and membrane protein structures
Membrane proteins are difficult to study for a number of reasons. First, the presence of trans-membrane (TM) domains in membrane proteins presents technical difficulties for the isolation of membrane proteins. TM domains are hydrophobic and have a tendency either to form large aggregates or to stick to hydrophobic surfaces when removed from their native lipid-bilayer environment. Second, for viral membrane proteins, it is technically challenging to ensure structural homogeneity and stability, which are pre-requisites of high-resolution structural studies. Drastic conformational changes occur when they are exposed to the low pH environment in the late endosome and/or upon binding to their host receptor molecules during the endocytosis pathway of cell entry. As a result, isolation of membrane proteins in large quantity, at high concentration and in soluble form — conditions required for X-ray crystallography or nuclear magnetic resonance — has always been quite difficult and remains a rate-limiting step of structure determination.
Single-particle cryoEM is a technique for determining 3D structures from projection images of molecular complexes suspended in physiological buffer solution, thus in a non-crystalline form. Images are recorded while molecular complexes are kept within vitreous ice in the vacuum environment of the column of a transmission electron microscope. Recent hardware improvements in microscope optics, sample holders and electron detection devices have made recording of cryoEM images of biological samples a routine practice. Automation in both image acquisition and data processing has significantly reduced the amount of sample required for cryoEM imaging to just a few micrograms, and time for data processing to a few days. When conventional methods (such as photographic films or charge-coupled devices, CCD) are used for recording images, the image contrast is relatively low and high-resolution cryoEM reconstructions are limited to large complexes, such as an entire virus particle. Indeed, atomic or near-atomic resolution structures of several viruses and protein assemblies have been determined by single-particle cryoEM, allowing ab initio atomic model building by following the amino acid side chains or nucleic acid bases identifiable in their cryoEM density maps. In particular, these cryoEM structures have revealed protein structures and protein–lipid interactions in or close to their native membrane environment, an advantage over the conventional virus structure determination method of X-ray crystallography.
Recently, technologies for direct electron detection (DED) and electron counting became commercially available for cryoEM imaging (Figure 1a). Such DED cameras have changed how cryoEM is performed in three significantly ways. First, movie stacks of dose-fractionating image frames are recorded, allowing motion correction to minimize image blurring incurred by specimen motion and charging. Second, when such cameras are operated in counting mode, electron events are distinguished from background noise, essentially achieving optimal quantum detection efficiency in imaging. Third, in rare cases where the subject of interest are over 100–200 nm in size (such as an entire herpesvirus or mimivirus virion), ‘super-resolution’ counting mode available on some of these cameras can be used to image such large objects at atomic resolution by subpixel sampling. For all these cameras, image processing programs have been developed to correct motion-induced image by either per-frame or per-particle alignment [1•,2•].
Figure 1.
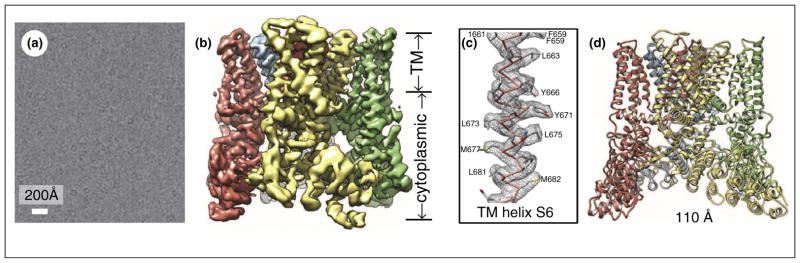
High-resolution cryoEM structure of membrane protein complex TRPV1. (a) Image of TRPV1 embedded in vitreous ice. The image was obtained by motion correction from image stacks acquired on a Gatan K2 direction electron detection camera using ‘super-resolution’ counting mode. (b) Shaded surface representation of the cryoEM density map of TRPV1 channel complex. The overall resolution is 3.4 Å for the entire map. The transmembrane (TM) domain is more stable and better resolved than the cytoplasmic domain. Each complex contains four identical subunits, which are color-coded differently. (c) CryoEM density map (gray wire) and atomic model (sticks) of TM helix S6. Amino acid residues are labeled revealing prominent side chain densities projecting out from the helix. (d) Ribbon diagram of TRPV1 atomic model with each of the four identical subunits color-coded, showing views from side. Adapted from Liao et al. [3••] with permissions from the authors and the publisher.
Indeed, DED cameras have made it possible to derive atomic models for even small membrane protein complexes, such as the ~0.4 MDa truncated mammalian transient receptor potential channel (TRPV1) [3••,4] (Figure 1b). Using motion-corrected, super-resolution counting mode direct electron detection images, the structure of the truncated TRPV1 has been determined to an overall resolution of 3.4 Å. Comparison with voltage-gated channels revealed structural similarities and differences that account for its unique functionalities. Like voltage-gated channels, TRPV1 exhibits four-fold symmetry around a central ion pathway formed by transmembrane (TM) segments 5–6 (S5–S6) and the intervening pore loop, which is flanked by S1–S4 voltage-sensor-like domains (Figure 1c). This cryoEM structure of TRPV1 also shows a wide extracellular ‘mouth’ with a short selectivity filter and facilitation of subunit organization by interactions among cytoplasmic domains, including amino-terminal ankyrin repeats. The same approach can be readily applied to viral membrane protein complexes of similar sizes, such as the herpesvirus gB/gH-gL complexes and HIV env trimers.
Membrane protein structures in alphaviruses
Viruses in the Togaviridae and Flaviviridae are icosahedral enveloped viruses and exhibit drastic structural transformation during viral maturation and cell entry. These viruses were among the first to have their viral membrane proteins resolved by cryoEM [5–12]. Mature virions enter cells through an endocytic pathway and sense the low pH environment inside the late endosome, leading to fusion of the viral membrane envelop with host membrane and the release of a viral capsid into the host cytoplasm. The fusion pathway has been postulated based on the pre-fusion and post-fusion crystal structures of the viral fusion protein E1 in conjunction with many cryoEM structures and biochemical studies. These postulated fusion intermediates are now subjected to direct observation by cryo electron microscopy and tomography at molecular resolution, employing reconstituted systems such as liposome and virus incubation [13].
The first high-resolution cryoEM structure for enveloped viruses is also that of an alphavirus, an attenuated vaccine strain (TC-83) of Venezuelan equine encephalitis virus (VEEV) at 4.4 Å resolution [14••] (Figure 2). VEEV has been developed into a biological weapon and is classified as an NIAID Category B priority pathogen. The cryoEM density map contained strong mass densities of viral proteins E1, E2 and weak density for E3 (Figure 2a). Though earlier subnanometer resolution cryoEM structure already resolved TM helices [15], amino acid residues with bulky side chains on the TM helix of both E1 and E2 began to be identifiable in the 4.4 Å cryoEM map (Figure 2b). Using these amino acid residues as constraints, atomic models of the full-length VEEV membrane proteins E1 and E2 were built by combining de novo models of E1/E2 TM helices and atomic models derived from homologous ectodomains of Chikungunya virus [16] (Figure 2c). The TM domains of E1 and E2 were absent in the available X-ray models of alphaviruses [16,17]. Derived from the cryoEM densities of the VEEV virion, these atomic models represent the full-length E1 and E2 molecules in their native lipid-containing environment with interacting partners. In addition, the cryoEM structure also revealed the cytoplasmic tails of the membrane proteins that extend beyond their TM helices to interact with the nucleocapsid protein (Figure 2c).
Figure 2.

In situ structures of alphavirus membrane proteins. (a) Density slice of the cryoEM reconstruction of the VEEV TC-83 virion. (b) CryoEM densities of an asymmetric unit segmented out from the virion map. Each unit contains four copies of membrane proteins E1, E2, as well as E3 and the capsid protein. (c) Enlarged view of the boxed region of (b) showing the TM domains (endodomains) of E1 and E2 and their interactions with the capsid protein. The cryoEM densities are shown as semitransparent shades and the models are shown as ribbon (homology model) or as sticks (de novo model). The C terminal helix of both E1 and E2 interacts with the capsid protein. Adapted from Zhang et al. [14••] with permissions from the authors and the publisher.
The in situ molecular interactions among E1, E2 and E3 clarify mechanisms for the initial stage of alphavirus nucleocapsid core formation, and shed light on the virulence attenuation, host recognition and neutralizing activities of VEEV and other alphavirus pathogens. Notably, no human vaccines or antiviral drugs thus far have been licensed for general use and the vaccine strain reconstructed by Zhang et al. is one of the few experimental vaccines that have been used to protect laboratory workers and military personnel [14••]. Although derived from a map of the still limited 4.4 Å resolution, the atomic model of this experimental VEEV vaccine reveals new molecular interactions not available from X-ray models of the ectodomains of E1 and E2 and offers valuable information for bio-engineering efforts to develop vaccines and antiviral strategies.
Protein–lipid interactions in dengue virus
Dengue virus is a prevalent mosquito-borne flavivirus that is endemic across tropical and some subtropical regions and is listed as a potential bio-threat agent [18]. Dengue virus infection can cause diseases ranging from self-limiting dengue fever to potentially lethal dengue shock syndrome and dengue hemorrhagic fever (DSS/DHF). With an estimate of more than 50 million people infected annually, dengue virus spread is recognized as a major urban public health concern by the World Health Organization [19]. Currently, there are neither licensed vaccines nor specific antiviral therapies against dengue infections.
Two membrane proteins M (also prM, its precursor) and E play multiple roles in viral maturation and subsequent low pH-induced fusion with host endosomal membrane; as such, they are critical to the viral life cycle. These membrane proteins are derived from a polyprotein that is first cleaved to yield prM and E. prM initially binds to E in the ‘spiky immature’ form [20] in the neutral pH of the endoplasmic reticulum. They undergo a maturation process that includes the formation of E dimers in the low pH environment of the trans-Golgi network (TGN) [20] (to form the low pH immature, or ‘smooth immature’ virus), the cleavage of the pr portion from prM (to produce M), and the shedding of pr when the virus is finally released in the neutral pH in the extracellular space, yielding the ‘smooth mature’ virus. In an effort to illustrate this process, a structure of an engineered montage protein that contains pr, the N-terminal 20-amino acid segment of M (M1–20) and the ectodomain of E (the portion outside the membrane) was solved crystallographically [21], along with the low resolution structures of both the spiky and the smooth immature viruses (12.5 Å and 25 Å, respectively) by cryoEM [20]. This crystal structure was then fit into the 25 Å resolution cryoEM structure of the ‘smooth immature’ virus [20] to explain how pr stabilizes E dimer at the low pH encountered in TGN. The pre-fusion dimer of the ectodomain of E at neutral pH, which was thought to resemble that in the smooth mature virus, was also determined by X-ray crystallography [22,23]. Following endocytosis and exposure to low pH in the late endosome, these E dimers dissociate to form fusogenic trimers that mediate fusion with endosomal membrane. The structure of the ectodomain of E as a trimer (after fusion) was captured in the crystal structure in its low pH post-fusion form [24]. The crystals used for these structural determinations [21–24] were obtained outside the context of the whole virion and only contained the ectodomain of E. CryoEM has been used to obtain the 3D structures of the immature viruses [9,25••], and the structure of the mature dengue virions first at 24 Å [10], then to subnanometer [11], and recently to near atomic resolutions [26••,27•].
The 3.5 Å resolution cryoEM structure of the mature virion of dengue virus serotype 2 reveals the in situ molecular interactions among the lipid bilayer, the full-length membrane proteins M and E (Figure 3a–d) [26••]. The structure shows that interactions between the N-terminal 20-amino acid segment of M (M1–20) and E are critical for sensing the pH changes during dengue virus cell entry. Thus, M is a chaperone membrane protein and acts as a latch to keep E in its prefusion, relatively higher energy state. In particular, M–E interactions involve three hydrophobic pockets with three histidine residues that constitute a putative low-pH sensor controlling release of the latch (Figure 3e). Pocket #2 contains two juxtaposed histidine residues, one each from M and E (Figure 3f). Protonation of these histidine residues in the low pH environment in the late endosome during viral entry would disrupt E-M interactions, allowing rising of E and exposure of its fusogenic peptide. The spatial relationship between the TM domain of M, its loop region (amino acids 1–20) and a histidine ‘double-door’, together with the inferred position of pr segment [22], also explains how the virus prevents E from popping outward throughout the low pH environment through trans-Golgi network during maturation. This earlier maturation of dengue virus in the trans-Golgi network, which involves another pH-sensitive event, is to load the latch [26••].
Figure 3.

Atomic model of the dengue virus mature virion. (a) Overview of the E:M:M:E heterotetramer with associated membrane. Transmembrane and perimembrane helices are labeled. The density map prior to Fourier amplitude rescaling and B-factor correction is displayed in gray by volume rendering. (b) Shaded surface representation of the density map of the heterotetramer, composed of two E:M dimers, one at right and also behind in the center, the other at left and also in front in the center. (c) Ribbon diagram of the atomic model of M. (d) Ribbon diagrams of the E:M:M:E heterotetramer. (e–f) Pockets involved in the E:M interaction. Adapted from Zhang et al. [26••] with permissions from the authors and the publisher.
HIV Env protein structures and prospect of high-resolution cryoEM for smaller complexes
Human immunodeficiency virus 1 (HIV-1) is an enveloped virus that causes acquired immunodeficiency syndrome (AIDS). HIV-1 uses a trimeric complex of its envelope glycoprotein (Env) to gain entry into host cells. During its synthesis and assembly in the host cell, the Env precursor is highly glycosylated, forms a trimeric complex, and is subsequently cleaved to yield a heterotrimeric fusion complex of gp120 and gp41 subunits. At the initial stage of HIV infection, binding of gp120 to CD4 and the che-mokine receptors on target cells triggers conformational changes that drive fusion of the viral and cell membranes. Env trimer contains the receptor binding sites and membrane fusion machinery and is the only target for broadly neutralizing antibodies (bnAbs). Atomic structures of the soluble gp120 core fragments in the CD4-bound state [28–31] and the trimeric gp41 ectodomain fragments in the post-fusion state [32–35] have been solved by X-ray crystallography. However, these gp120 and gp41 structures were determined in isolation and in the absence of functionally important components, such as the V1/V2/V3 regions of gp120 and the fusion peptide of gp41.
Recently, 3D structures of engineered variants of cleaved, soluble Env trimers [i.e. SOSIP trimers, which are cleaved at residues 664 or 681 recombinant trimers from the BG505 genotype, stabilized by specific substitutions, including a disulfide bond (termed SOS) between residue 501 (HXB2 numbering) and 605, and an Ile-to-Pro mutation at position 559] in complex with a CD4 binding site bnAb’s, PGV04 and VRC03, both were determined by single-particle cryoEM at ~6 Å resolution [36••,37••] (Figure 4a,b). These stabilized SOSIP trimer structures are similar to one another and recapitulate the ‘closed’, prefusion form of the Env trimer. The structures reveal shielding glycans and the spatial arrangement of Env components, including the V1/V2, V3, HR1, and HR2 domains. The cryoEM density maps also show that the stem, coiled coil helices of gp41 forms the core of the Env trimer and provide insights into trimer assembly and gp120-gp41 interactions (Figure 4). The cryoEM structure by Lyumkis et al. [36••] correlates nicely with an X-ray structure solved in parallel by the same group to about 5 Å resolution [38] and maps out the CD4bs epitope cluster for bnAbs, which covers a more extensive area and defines a more complex site of vulnerability than previously described. Comparison with the structure of Env trimer in its open, CD4-bound state [39] suggests that the gp41 stem helix bundle of gp41 forms the core of the trimer and acts as the anchor for structural reorganization from the closed, unliganded form to the ‘open’ conformation upon CD4 receptor binding [37••]. This use of a coiled coil helix bundle as an anchor for structural transition from the closed, prefusion state to an activated intermediate state is a feature common to class 1 viral fusion proteins, such as the influenza hemagglutinin trimers [40].
Figure 4.

Prefusion structure of the ectodomain of HIV Env trimer determined at ~6 Å resolution by cryoEM [36••]. (a) CryoEM image (left, 2.6 μm underfocus) and the side (middle) and top (right) views of the 6 Å resolution cryoEM structure of Env–VRC03 complex. Key structure components, as well as the fitted X-ray models (ribbons) of the gp120, gp41 and antibody subunits are indicated. (b) CryoEM structure of Env-pgv04 and models. (c) Model of the ectodomain of Env monomer shown as ribbons. Panel a and panels b–c are adapted from Lyumkis et al. [36••] and Bartesaghi et al. [37••], respectively, with permissions from the authors and the publishers.
It is of note that structures of the full-length HIV-1 Env trimer complex in its unliganded, prefusion state have also appeared in recent years [41,42••], purportedly at progressively improved resolutions from 11 to 6 Å. These structures posited the presence of a cavity at the center of the spike, thus differ drastically from those described above. One could argue that this situation might reflect possible flexibility of Env trimer and the possibility that the different structures are simply different incarnation of the same Env trimer in different functional states. Others opined that some of these structures suffered from model bias or what is known as the ‘Einstein out of noise’ problem [43].
The predisposing factors of model bias include image data with very noisy images, huge number of noisy images, and use of models with high-resolution information (such as atomic models derived from homology modeling) or strong features (such as aggressively masked intermediate maps) to drive refinement. With just one of these factors, the very choice of initial model (such as a portrait of Einstein) has been shown to bias or even dictate the outcome of structure refinement [44,45]; that is, you get what you seek.
To prevent model bias, the obvious strategy is simply to avoid these predisposing factors. Mass of large complexes, such as entire virion particles of the alpha virus and dengue virus described above, provides sufficient electron scattering power to yield images of high contrast and reduces the number of particles needed in refinement. When operated with the revolutionary electron counting mode, novel DED cameras can capture all individual electron events, thus achieving quantum efficiency and essentially eliminating background noise in the cryoEM images. Images recorded with counting mode on a DED camera have greatly improved contrast, thus reducing the number of images required and enabling atomic resolution reconstruction for small complexes such as the HIV Env trimers.
In addition to the above practices aimed at avoiding model bias, one must also rely on other internal controls or external measures for structural validation. For structures better than 4.5 Å resolution, chemical information from other independent sources, such as the identities of amino acid residues and nucleic acid bases, can be utilized as powerful internal controls, provided that this sequence information is not used for refinement. One way to utilize such controls is to intentionally omit a sequence segment (e.g. 10%) in initial model building and test for the emergence of densities that match the omitted sequence in the refined structure. The emerged densities should match the chemical information omitted in terms of identity of amino acid or base residues. Of note, carboxyl groups in acidic amino acids (Glu and Asp) are particularly susceptible to electron damage. Carboxyl side chains in Glu and Asp could be simply knocked off by high-energy electrons and become invisible in cryoEM maps, whereas side chain densities of other amino acids are resolved and remain visible [46,47,48,49•]. This is not the case for electron densities maps in X-ray crystallography where carboxyl groups of Glu and Asp are not damaged by X-ray photons and are visible. Relying on these powerful internal controls, high-resolution cryoEM can finally forgo the tedious Fourier shell correlation calculations, which, tagged ‘gold-standard’ or not, measure merely the reproducibility of a structure but do not eliminate systematic errors of software packages.
Conclusions
In 1975, Henderson and Unwin demonstrated the potential of electron microscopy for studying membrane proteins through their pioneering effort of imaging purple membrane crystalline protein arrays and resolving trans-membrane helices of bacteriorhodopsin [50]. Since then, much of the progress in high-resolution cryoEM has come from non-crystalline samples directly imaged in vitrified water without the use of embedding materials, thanks to improvements in both electron microscope instrumentation and image processing methods. Inherently low contrast of cryoEM images recorded on photographic films had limited realization of near atomic resolution to large complexes, exemplified by icosahedral and helical viruses. Atomic models of several membrane-containing viruses by high-resolution cryoEM revealed the trans-membrane domains of their membrane proteins and critically important molecular interactions among viral proteins in situ. These earlier advancements, together with the improved image contrast afforded by direct electron detection or counting, high-resolution cryoEM now offers exciting opportunities to reveal molecular interactions at atomic details of viral membrane protein complexes, either in situ on structurally homogeneous viruses or off site after being isolated from structural heterogeneous/pleomorphic viruses.
Acknowledgments
Research in the author’s group is supported in part by NIH grants (R01 AI/069015/094386 and GM071940).
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1•.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. The first paper documenting use of electron counting with a DED camera for high-resolution cryoEM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2•.Campbell MG, Cheng A, Brilot AF, Moeller A, Lyumkis D, Veesler D, Pan J, Harrison SC, Potter CS, Carragher B, et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 2012;20:1823–1828. doi: 10.1016/j.str.2012.08.026. The first paper documenting use of movie-mode direct electron detection with a DED camera for high-resolution cryoEM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3••.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. Atomic models built for a small membrane protein complex by electron counting with a DED camera. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moiseenkova-Bell VY, Stanciu LA, Serysheva II, Tobe BJ, Wensel TG. Structure of TRPV1 channel revealed by electron cryomicroscopy. Proc Natl Acad Sci U S A. 2008;105:7451–7455. doi: 10.1073/pnas.0711835105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuller SD. The T = 4 envelope of Sindbis virus is organized by interactions with a complementary T = 3 capsid. Cell. 1987;48 :923–934. doi: 10.1016/0092-8674(87)90701-x. [DOI] [PubMed] [Google Scholar]
- 6.Paredes AM, Brown DT, Rothnagel R, Chiu W, Schoep RJ, Johnston RE, Prasad BVV. Three-dimensional structure of a membrane-containing virus. Proc Natl Acad Sci U S A. 1993;90 :9095–9099. doi: 10.1073/pnas.90.19.9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pletnev SV, Zhang W, Mukhopadhyay S, Fisher BR, Hernandez R, Brown DT, Baker TS, Rossmann MG, Kuhn RJ. Locations of carbohydrate sites on alphavirus glycoproteins show that E1 forms an icosahedral scaffold. Cell. 2001;105:127–136. doi: 10.1016/s0092-8674(01)00302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Mukhopadhyay S, Pletnev SV, Baker TS, Kuhn RJ, Rossmann MG. Placement of the structural proteins in Sindbis virus. J Virol. 2002;76:11645–11658. doi: 10.1128/JVI.76.22.11645-11658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Corver J, Chipman PR, Zhang W, Pletnev SV, Sedlak D, Baker TS, Strauss JH, Kuhn RJ, Rossmann MG. Structures of immature flavivirus particles. EMBO J. 2003;22:2604–2613. doi: 10.1093/emboj/cdg270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, et al. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108:717–725. doi: 10.1016/s0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003;10:907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Kaufmann B, Chipman PR, Kuhn RJ, Rossmann MG. Structure of immature West Nile virus. J Virol. 2007;81 :6141–6145. doi: 10.1128/JVI.00037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao S, Zhang W. Characterization of an early-stage fusion intermediate of Sindbis virus using cryoelectron microscopy. Proc Natl Acad Sci U S A. 2013;110:13362–13367. doi: 10.1073/pnas.1301911110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Zhang R, Hryc CF, Cong Y, Liu X, Jakana J, Gorchakov R, Baker ML, Weaver SC, Chiu W. 4.4 Å cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 2011;30:3854–3863. doi: 10.1038/emboj.2011.261. The first full-length model of the VEEV membrane proteins by high-resolution cryoEM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukhopadhyay S, Zhang W, Gabler S, Chipman PR, Strauss EG, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Structure. 2006;14:63–73. doi: 10.1016/j.str.2005.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature. 2010;468:709–712. doi: 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. Structural changes of envelope proteins during alphavirus fusion. Nature. 2010;468:705–708. doi: 10.1038/nature09546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borio L, Inglesby T, Peters CJ, Schmaljohn AL, Hughes JM, Jahrling PB, Ksiazek T, Johnson KM, Meyerhoff A, O’Toole T, et al. Hemorrhagic fever viruses as biological weapons: medical and public health management. J Am Med Assoc. 2002;287:2391–2405. doi: 10.1001/jama.287.18.2391. [DOI] [PubMed] [Google Scholar]
- 19.WHO. Dengue: guidelines for diagnosis, treatment, prevention and control. Geneva, Switzerland: WHO; 2009. new edition. [PubMed] [Google Scholar]
- 20.Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science. 2008;319:1834–1837. doi: 10.1126/science.1153264. [DOI] [PubMed] [Google Scholar]
- 21.Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, Rossmann MG. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science. 2008;319:1830–1834. doi: 10.1126/science.1153263. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Zhang W, Ogata S, Clements D, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. Conformational changes of the flavivirus E glycoprotein. Structure. 2004;12:1607–1618. doi: 10.1016/j.str.2004.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 25••.Kostyuchenko VA, Zhang Q, Tan JL, Ng TS, Lok SM. Immature and mature dengue serotype 1 virus structures provide insight into the maturation process. J Virol. 2013;87:7700–7707. doi: 10.1128/JVI.00197-13. A near-atomic resolution structure of the dengue virus serotype 1 by cryoEM in both their mature and immature states. Differences of the surface properties of the membrane protein E were revealed between denguevirus serotype 1 and 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26••.Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, Schein S, Zhou ZH. Cryo-EM structure of the mature dengue virus at 3.5-Å resolution. Nat Struct Mol Biol. 2013;20:105–110. doi: 10.1038/nsmb.2463. The first atomic model of the entire dengue virus virion by high-resolution cryoEM. Comparisons of this structure of the mature virion with earlier structures of immature dengueviruses suggest how the virus sense and respond to different pH during intra-cellular maturation and extra-cellular infection processes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Kostyuchenko VA, Chew PL, Ng TS, Lok SM. Near-atomic resolution cryo-electron microscopic structure of dengue serotype 4 virus. J Virol. 2014;88:477–482. doi: 10.1128/JVI.02641-13. The first high-resolution cryoEM structure of the dengue virus serotype 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang CC, Lam SN, Acharya P, Tang M, Xiang SH, Hussan SS, Stanfield RL, Robinson J, Sodroski J, Wilson IA, et al. Structures of the CCR5N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317:1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, et al. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou T, Xu L, Dey B, Hessell AJ, Van Ryk D, Xiang SH, Yang X, Zhang MY, Zwick MB, Arthos J, et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445:732–737. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci U S A. 2010;107:1166–1171. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 34.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 35.Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 2010;6:e1000880. doi: 10.1371/journal.ppat.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36••.Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1484–1490. doi: 10.1126/science.1245627. A 5.5 Å cryoEM structure of the cleaved HIV-1 envelope trimer was determined by cryoEM and used to build a pseudo-atomic model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Bartesaghi A, Merk A, Borgnia MJ, Milne JL, Subramaniam S. Prefusion structure of trimeric HIV-1 envelope glycoprotein determined by cryo-electron microscopy. Nat Struct Mol Biol. 2013;20:1352–1357. doi: 10.1038/nsmb.2711. A 6 Å cryoEM structure of the cleaved HIV-1 envelope trimer in its prefusion state (closed) was determined by cryoEM and compared to its ‘open’ state to suggest a mechanism of cell entry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris A, Borgnia MJ, Shi D, Bartesaghi A, He H, Pejchal R, Kang YK, Depetris R, Marozsan AJ, Sanders RW, et al. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc Natl Acad Sci U S A. 2011;108:11440–11445. doi: 10.1073/pnas.1101414108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15 :690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mao Y, Wang L, Gu C, Herschhorn A, Desormeaux A, Finzi A, Xiang SH, Sodroski JG. Molecular architecture of the uncleaved HIV-1 envelope glycoprotein trimer. Proc Natl Acad Sci U S A. 2013;110:12438–12443. doi: 10.1073/pnas.1307382110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42••.Mao Y, Wang L, Gu C, Herschhorn A, Xiang SH, Haim H, Yang X, Sodroski J. Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat Struct Mol Biol. 2012;19 :893–899. doi: 10.1038/nsmb.2351. The structure of the HIV-1 envelope protein presented in this paper differs drastically from other cryoEM structures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henderson R. Avoiding the pitfalls of single particle cryo-electron microscopy. Einstein from noise. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1314449110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Penczek P, Radermacher M, Frank J. Three-dimensional reconstruction of single particles embedded in ice. Ultramicroscopy. 1992;40:33–53. [PubMed] [Google Scholar]
- 45.van Heel M, Winkler H, Orlova E, Schatz M. Structural analysis of ice-embedded single particles. Scan Microsc. 1992;(Suppl 6):23–42. [Google Scholar]
- 46.Grigorieff N, Ceska TA, Downing KH, Baldwin JM, Henderson R. Electron-crystallographic refinement of the structure of bacteriorhodopsin. J Mol Biol. 1996;259:393–421. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 47.Mitsuoka K, Hirai T, Murata K, Miyazawa A, Kidera A, Kimura Y, Fujiyoshi Y. The structure of bacteriorhodopsin at 3.0 Å resolution based on electron crystallography: implication of the charge distribution. J Mol Biol. 1999;286:861–882. doi: 10.1006/jmbi.1998.2529. [DOI] [PubMed] [Google Scholar]
- 48.Sachse C, Chen JZ, Coureux PD, Stroupe ME, Fandrich M, Grigorieff N. High-resolution electron microscopy of helical specimens: a fresh look at tobacco mosaic virus. J Mol Biol. 2007;371:812–835. doi: 10.1016/j.jmb.2007.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49•.Ge P, Zhou ZH. Hydrogen-bonding networks and RNA bases revealed by cryo electron microscopy suggest a triggering mechanism for calcium switches. Proc Natl Acad Sci U S A. 2011;108:9637–9642. doi: 10.1073/pnas.1018104108. This paper documents identification of hydrogen bonds can be identified to interpret viral molecular interactions by high resolution cryoEM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henderson R, Unwin PN. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]