

Abstract
Background and Objectives
Fidaxomicin treatment of Clostridium difficile infection is known to produce minimal systemic exposure, as the antibacterial (antibiotic) remains primarily in the gut. In this randomized, double-blind, placebo-controlled study, the safety, tolerability, and pharmacokinetics of single and multiple ascending doses of fidaxomicin were evaluated in healthy Japanese and Caucasian subjects.
Methods
Thirty-six healthy subjects were randomly assigned in a 3:1 ratio to receive either fidaxomicin or placebo. Cohort 1 (100 mg) and Cohort 2 (200 mg) comprised 12 Japanese subjects each and Cohort 3 (200 mg) comprised 12 Caucasian subjects. Subjects received a single dose of the study drug on Day 1 and received multiple doses for 10 days after a wash-out period.
Results
After multiple 200 mg dosing of fidaxomicin, both mean maximum plasma concentrations (Cmax) in Japanese (8.7 ± 5.3 ng/mL) and Caucasian (7.0 ± 3.7 ng/mL) subjects and the area under the concentration–time curve (AUC) were higher in Japanese subjects (58.5 ± 36.7 ng·h/mL) than in Caucasian subjects (37.6 ± 15.7 ng·h/mL), although variation in both groups was large. The mean fecal concentrations of fidaxomicin in Japanese and Caucasian subjects were 2669 and 2181 μg/g, respectively. The possibly study drug-related adverse events were diarrhea (n = 1), feeling hot (n = 1), and hypersomnia (n = 2), which were mild in severity.
Conclusions
In both Japanese and Caucasian subjects, fidaxomicin demonstrated similarly minimal systemic absorption, and was mainly excreted in feces. Fidaxomicin was safe and well-tolerated in all subjects.
Key Points
| In both Japanese and Caucasian subjects, fidaxomicin showed minimal systemic absorption following oral administration. |
| Plasma concentrations of fidaxomicin and its main metabolite OP-1118 were in the ng/mL range, with the maximum plasma concentration and area under the concentration–time curve values higher in Japanese subjects than in Caucasian subjects. |
| The drug was mainly excreted in feces. Fidaxomicin was safe and well-tolerated in both subject groups. |
Introduction
Clostridium difficile is a Gram-positive, anaerobic, spore-forming bacillus [1]. It is the major infectious cause of nosocomial diarrhea and can lead to prolonged hospital stays, renal failure, toxic megacolon, and occasionally death [2, 3]. C. difficile infection (CDI) is primarily associated with exposure to broad-spectrum antibacterials (antibiotics) [4, 5]. Over the past several decades, CDI has dramatically increased in both frequency and severity in the USA and European countries, owing to an increase in the use of third-generation cephalosporins [6, 7] and fluoroquinolones [8–10]. Honda et al. [11] reported that the incidence of healthcare-onset CDI in Japan was 3.11 cases per 10,000 patient-days. The incidence was lower than that reported in the USA and European countries, but the rates of patients requiring surgical therapy and dying within 30 days in non-outbreak conditions were higher than those in the USA [11]. A nationwide multicenter study in Japan indicated that intravenous cephems and carbapenems were important risk factors for CDI in 1025 Japanese patients [12]. Use of penicillin and proton pump inhibitors were not identified as risk factors, although these are well-known as risk factors in the other countries [4].
Metronidazole is the first-line treatment for a first non-severe episode of CDI, and vancomycin is recommended for a first severe episode [13, 14]. However, relapse occurs in approximately 20 % of patients within 1–3 months [15]. According to the guidelines [13, 14], the first recurrence should be treated with the same antibacterials as those for the first episode, but metronidazole is not recommended for the second and any subsequent recurrence. Second and subsequent recurrences are seen in approximately 20–25 % of patients following their first recurrence, and some of them endure as many as ten or more episodes [16].
Fidaxomicin was approved in 2011 by the USA and European countries for the treatment of C. difficile-associated diarrhea (CDAD), but has not yet been approved in Japan. The label of fidaxomicin indicates that fidaxomicin should not be used for systemic infections and should only be used for infections proven or strongly suspected to be caused by C. difficile. Fidaxomicin is a macrocyclic antibacterial with potent inhibiting activity against bacterial RNA polymerase [17, 18]. It possesses a narrow spectrum of antibacterial activity against Gram-positive aerobic and anaerobic bacteria, including C. difficile, without discernible activity against Gram-negative gut organisms [19, 20]. The metabolism of fidaxomicin occurs through hydrolysis at the isobutyryl ester to form the major active metabolite OP-1118 [21]. It is noteworthy that systemic absorption of fidaxomicin is minimal after oral administration and that high fecal concentrations are observed [22]. Approval of fidaxomicin in the USA and European countries was based on two phase III clinical trials involving 1164 patients with CDI [23, 24] in which the drug showed efficacy and safety similar to vancomycin. Importantly, fidaxomicin significantly reduced overall recurrence rates, as defined by the reappearance of more than three diarrheal stools per 24-h period with a positive toxin test for C. difficile within 4 weeks after cessation of therapy, compared with the vancomycin group [23].
The objective of this study was to evaluate the safety, tolerability, and pharmacokinetics of fidaxomicin in healthy Japanese and Caucasian subjects and to compare these between the two ethnic groups.
Subjects and Methods
Study Design
This was a phase I randomized, double-blind, placebo-controlled dose-escalation study. The study was performed at California Clinical Trials Medical Group, Glendale, California, USA in 2013. The protocol and the informed consent form were approved by the institutional review board. All subjects gave written informed consent before the initiation of any study-specific procedure. The study was conducted in accordance with the ethical principles originating in or derived from the Declaration of Helsinki, International Conference on Harmonization Good Clinical Practice Guidelines, and locally applicable laws and regulations.
Subjects
Healthy Japanese or Caucasian male subjects aged 20–55 years whose clinical laboratory test results at screening and Day −2 were within normal limits or who had been determined to be clinically insignificant by the investigator were eligible to participate in the study. Healthy Japanese subjects living in the USA were required to have a body mass index (BMI) of 18.0–28.0 kg/m2, been born in Japan with two parents and four grandparents of Japanese descent, and resided outside of Japan for not more than 5 years, maintaining the Japanese lifestyle including diet before the start of the study. Although the Japanese diet was not defined clearly, the maintenance of a Japanese lifestyle including diet was confirmed by questionnaire. Healthy Caucasian subjects with a BMI of 18.0–32.0 kg/m2 were included.
Subjects who had any of the following were excluded: clinically significant disease history of the pulmonary, gastrointestinal, cardiovascular [including a history of clinically significant arrhythmia or conduction delays on electrocardiogram (ECG)], hepatic, neurological, psychiatric, renal, genitourinary, endocrine, metabolic, dermatological, immunological, and hematological systems; who had a history of other major disease or malignancy; a history of or current CDI or history of stomach or intestinal surgery or resection that would potentially alter absorption and/or excretion of orally administered drugs; or any clinically significant laboratory abnormalities (e.g., hematology, chemistry, and urinalysis).
Treatment
Subjects who met the eligibility criteria were enrolled in one of three cohorts: Cohort 1–100 mg dose in Japanese subjects; Cohort 2–200 mg dose in Japanese subjects; and Cohort 3–200 mg dose in Caucasian subjects in parallel with Cohort 2. Each cohort comprised 12 subjects, making a total of 36 subjects. After a screening period of up to 28 days, subjects entered the study unit on Day −2 and were hospitalized until Day 17. On the morning of Day 1, subjects of each 12-subject cohort were randomly assigned in a 3:1 ratio to receive either fidaxomicin or placebo (nine subjects for fidaxomicin, three subjects for placebo) in accordance with the randomization codes generated by the sponsor’s designee. The study staff and investigator did not have access to the treatment received by each subject. Only the unblinded pharmacist had access to the randomization code. The placebo matched the fidaxomicin tablets; therefore, there was no unblinding risk by the subjects or clinic staff. Subjects received a single dose of the study drug 30 min after breakfast on the morning of Day 1. After a wash-out period of 5 days, subjects received the study drug twice a day, 30 min after a meal approximately every 12 h on Day 6 to Day 14. On Day 15, subjects received the final dose of the study drug in the morning 30 min after a meal.
After the completion of Cohort 1, a safety review team reviewed the available safety data prior to the enrollment of Cohort 2 and 3, which were tested in parallel.
Sample Collection
Blood samples for the measurement of fidaxomicin and OP-1118 concentration in plasma after single oral administration on Day 1 were taken immediately pre-dose and then 0.5, 1, 2, 3, 4, 6, 8, 12, 24 (Day 2), 48 (Day 3), 72 (Day 4), 96 (Day 5), and 120 (Day 6) h post-dose. Blood samples were also taken pre-dose on Days 6, 7, 10 and 12, and then pre-dose, 0.5, 1, 2, 3, 4, 6, 8, 12, 24 (Day 16), and 48 (Day 17) h post-dose on Day 15.
Twelve-hour urine samples were collected on Day −1, −12 to 0 h; on Day 1, 0–12, 12–24, 24–48, 48–72, 72–96, and 96–120 h post-dose; and on Day 15, 0–12 h post-dose. Feces were collected on Day −1, −24 to 0 h pre-dose; on Day 1, 0–24, 24–48, 48–72, 72–96, and 96–120 h post-dose; and on Days 15–17, the first bowel movement following the last dose was to be collected.
Pharmacokinetic Analysis
Plasma, urine, and feces concentrations of fidaxomicin and OP-1118 were measured by validated liquid chromatography–tandem mass spectrometry methods. The plasma assay method was validated over a range of 0.2–100.0 ng/mL for fidaxomicin and OP-1118. Intra- and inter-precision values for fidaxomicin and OP-1118 were <8.7 and <9.3 %, respectively. Intra- and inter-accuracy values were from −10.0 to 7.2 % and from −5.0 to 12.8 %, respectively. The lower limit of quantification (LLOQ) was 0.2 ng/mL for fidaxomicin and OP-1118 in plasma. The urine assay method was validated over a range of 5–1000 ng/mL for fidaxomicin and OP-1118. Intra- and inter-precision values for fidaxomicin and OP-1118 were <7.3 and <9.3 %, respectively. Intra- and inter-accuracy values for fidaxomicin and OP-1118 were from −6.4 to 8.4 % and from −8.0 to 9.3 %, respectively. LLOQ was set at 5.0 ng/mL for fidaxomicin and OP-1118 in urine. The fecal assay method was validated over a range of 2–400 μg/g for fidaxomicin and 10–2000 μg/g for OP-1118. Intra- and inter-precision values for fidaxomicin and OP-1118 were <8.9 and <5.5 %, respectively. Intra- and inter-accuracy values were from −3.3 to 3.7 % and from −10.1 to 4.9 %, respectively. LLOQ was set at 2.0 μg/g for fidaxomicin and at 10.0 μg/g for OP-1118 in feces.
Pharmacokinetic parameters for fidaxomicin and OP-1118 included maximum plasma concentration (Cmax), time to Cmax (tmax), area under the concentration–time curve (AUC) from time zero to time of last measurable concentration (AUClast), AUC from time zero to infinity (AUC∞), AUC from time zero to 12 h (AUC12), AUC over the dosing interval at steady state (AUCτ), and terminal elimination half-life (t½) after first dose. AUC was calculated using the linear up/log down trapezoidal rule. Apparent total clearance (CL/F) was evaluated for fidaxomicin. The accumulation ratio (Racc) was calculated as AUCτ/AUC12. Urine pharmacokinetic parameters including the cumulative amount of drug excreted (Ae) in the urine from time zero to the time of last measurable concentration (Aelast) and percentage Aelast (%Aelast) were calculated for both fidaxomicin and OP-1118 urine concentrations. Fecal concentrations of fidaxomicin and OP-1118 were determined, and derived pharmacokinetic parameters included cumulative Ae and percentage Ae (%Ae) for a single dose (Days 1–5) and Ae and %Ae for last dose (Days 15–16).
Safety and Tolerability
The safety and tolerability of fidaxomicin were assessed with adverse events (AEs), clinical laboratory evaluations (hematology, chemistry, and urinalysis), 12-lead ECG measurements, vital sign measurements [oral temperature, pulse, and sitting systolic (SBP) and diastolic blood pressure (DBP)], and physical examinations. The investigators asked the subjects non-leading questions to determine how they felt at daily physical examinations and twice-daily rounds. The subjects were also asked to self-report AEs at any point in time to any staff member, including the investigator and research nurse.
Statistical Analyses
Approximately 12 subjects per cohort (a total of 36 subjects) were considered sufficient for achieving the study objectives. All safety analyses were conducted for all randomized subjects who received at least one dose of the study drug. All pharmacokinetic analyses were performed for subjects for whom sufficient plasma concentration data were available to facilitate the derivation of at least one pharmacokinetic parameter.
The pharmacokinetic parameters calculated based on fidaxomicin and OP-1118 concentrations in plasma, urine, and feces were used to study the pharmacokinetics, steady state, and accumulation with multiple-dose administration and ethnic differences in pharmacokinetics between Japanese and Caucasian subjects. Data points below LLOQ were substituted by zero for the calculation of mean values. To evaluate ethnic differences between Japanese and Caucasian subjects, AUClast, AUC∞, and Cmax after 200 mg of single-dose fidaxomicin administration in Japanese subjects and Caucasian subjects were fitted using an analysis of variance (ANOVA) model with a factor for ethnic group. Geometric mean ratio (Japanese/Caucasian) and the corresponding 90 % confidence intervals (CIs) were presented. The AUCτ and Cmax at steady state (Cmax,ss) from the last dose after multiple-dose administration were analyzed in the same manner.
AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA®), version 15.1.
Results
Subject Demographics
Thirty-six healthy subjects were enrolled in this study and placed into the three specific cohorts mentioned previously. Each cohort consisted of 12 subjects, and they were randomly assigned in a 3:1 ratio to receive either fidaxomicin or placebo (nine subjects for fidaxomicin, three subjects for placebo). Thirty-five subjects completed the study. Six Japanese subjects and three Caucasian subjects were randomized to receive placebo. A total of 27 subjects were randomized to receive fidaxomicin (n = 9 at 100 mg for Japanese, 200 mg for Japanese, and 200 mg for Caucasian subjects). One Japanese subject receiving 200 mg doses of fidaxomicin withdrew from the study on Day 9 because of consent withdrawal due to a personal scheduling issue. This subject reported no treatment-emergent AE (TEAE). All subjects were males and were either Caucasian or Japanese according to their cohort. The mean height and weight of Caucasian subjects were greater than those of Japanese subjects. The mean ages and BMIs of the groups were similar, as shown in Table 1.
Table 1.
Summary of demographics and baseline characteristics
| Category | Placebo | Fidaxomicin | ||||
|---|---|---|---|---|---|---|
| Japanese (n = 6) | Caucasian (n = 3) | Japanese 100 mg (n = 9) | Japanese 200 mg (n = 9) | Caucasian 200 mg (n = 9) | Total (n = 27) | |
| Sex [n (%)] | ||||||
| Male | 6 (100.0) | 3 (100.0) | 9 (100.0) | 9 (100.0) | 9 (100.0) | 27 (100.0) |
| Ethnicity [n (%)] | ||||||
| Not Hispanic or Latino | 6 (100.0) | 3 (100.0) | 9 (100.0) | 9 (100.0) | 9 (100.0) | 27 (100.0) |
| Asian | 6 (100.0) | 0 | 9 (100.0) | 9 (100.0) | 0 | 18 (66.7) |
| Race [n (%)] | ||||||
| White | 0 | 3 (100.0) | 0 | 0 | 9 (100.0) | 9 (33.3) |
| Age (years) [mean ± SD] | 34.3 ± 3.7 | 36.7 ± 13.8 | 36.6 ± 10.6 | 34.2 ± 10.3 | 32.6 ± 8.8 | 34.4 ± 9.7 |
| Height (cm) [mean ± SD] | 169.6 ± 6.6 | 177.7 ± 10.7 | 170.8 ± 7.3 | 171.0 ± 6.6 | 176.1 ± 10.1 | 172.6 ± 8.2 |
| Weight (kg) [mean ± SD] | 71.7 ± 8.9 | 78.4 ± 15.8 | 67.9 ± 8.9 | 70.8 ± 9.2 | 78.7 ± 10.8 | 72.5 ± 10.4 |
| BMI (kg/m2) [mean ± SD] | 24.9 ± 2.1 | 24.6 ± 2.5 | 23.2 ± 2.0 | 24.2 ± 2.6 | 25.5 ± 3.8 | 24.3 ± 2.9 |
BMI body mass index, SD standard deviation
Pharmacokinetics
Plasma concentration–time profiles of fidaxomicin and OP-1118 after a single dose and multiple doses of fidaxomicin are shown in Fig. 1; their pharmacokinetic parameters are summarized in Table 2. Because of insufficient positive plasma concentrations available in the terminal phase, t½ of fidaxomicin or OP-1118 could not be evaluated in 12 or 7 out of 53 cases, respectively. In addition, tmax could not be obtained in one case because all concentrations were below LLOQ. After a single dose of fidaxomicin, absorption led to a median tmax ranging from 2 to 3 h post-dose with maximum individual tmax values of up to 6 h. The Cmax value for fidaxomicin after a single dose of fidaxomicin 200 mg in Caucasian subjects was 4.5 ± 3.4 ng/mL (range 0–11.3 ng/mL), which was closer to the 100 mg dose Cmax of 4.5 ± 2.4 ng/mL (range 2.0–7.7 ng/mL) in Japanese subjects than to the 200 mg dose Cmax of 8.3 ± 4.5 ng/mL (range 3.3–17.9 ng/mL). The Cmax values for fidaxomicin on Day 15 after multiple dosing of 100 mg in Japanese subjects, 200 mg in Japanese subjects, and 200 mg in Caucasian subjects were 5.3 ± 2.9 ng/mL (range 2.3–11.3 ng/mL), 8.7 ± 5.3 ng/mL (range 2.7–18.9 ng/mL), and 7.0 ± 3.7 ng/mL (range 2.1–14.1 ng/mL), respectively. The AUC∞ values for fidaxomicin after a single 100 mg dose in Japanese subjects, 200 mg in Japanese subjects, and 200 mg in Caucasian subjects were 40.6 ± 15.8, 55.6 ± 26.4, and 57.5 ± 30.0 ng·h/mL, respectively. The AUC12 values for fidaxomicin after multiple dosing of 100 mg in Japanese subjects, 200 mg in Japanese subjects, and 200 mg in Caucasian subjects were 32.8 ± 20.5, 58.5 ± 36.7, and 37.6 ± 15.7 ng·h/mL, respectively. The AUC showed minor accumulation upon multiple dosing, with an Racc of 1.3 and 1.4 for the 100 and 200 mg Japanese subjects, respectively. The Racc of 1.6 for the 200 mg Caucasian group was higher than those for the Japanese groups. The percentage coefficient of variation (%CV) of the Cmax and AUC of single and multiple dosing of fidaxomicin ranged from 52.6 to 76.1 % and 39.0 to 62.7 %, respectively. Statistical assessment of the ethnicity effect for single-dose AUClast and steady-state AUCτ showed evidence of higher exposure in Japanese than in Caucasian subjects: the AUClast ratio was 1.3 (90 % CI 0.8–2.1) and the AUCτ ratio was 1.4 (90 % CI 0.9–2.2) (Table 3).
Fig. 1.
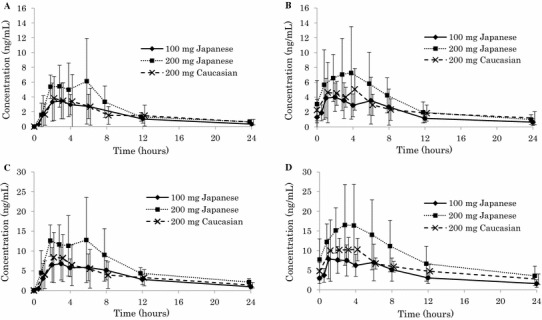
Mean ± standard deviation plasma concentration–time curve of fidaxomicin and OP-1118 after single and multiple doses of fidaxomicin in Japanese subjects and Caucasian subjects: a fidaxomicin plasma concentration after single-dose administration of fidaxomicin; b fidaxomicin plasma concentration after multiple-dose administration of fidaxomicin (Day 15); c OP-1118 plasma concentration after single-dose administration of fidaxomicin; d OP-1118 plasma concentration after multiple-dose administration of fidaxomicin (Day 15)
Table 2.
Pharmacokinetic parameters for fidaxomicin and its metabolite OP-1118
| Compound | Pharmacokinetic parameter | Japanese 100 mg single dose | Japanese 200 mg single dose | Caucasian 200 mg single dose | Japanese 100 mg multiple dose | Japanese 200 mg multiple dose | Caucasian 200 mg multiple dose | |
|---|---|---|---|---|---|---|---|---|
| Fidaxomicin | C max (ng/mL) | n | 9 | 9 | 9 | 9 | 8 | 9 |
| Mean ± SD | 4.5 ± 2.4 | 8.3 ± 4.5 | 4.5 ± 3.4 | 5.3 ± 2.9 | 8.7 ± 5.3 | 7.0 ± 3.7 | ||
| AUCa (ng·h/mL) | n | 8 | 7 | 8 | 9 | 8 | 9 | |
| Mean ± SD | 40.6 ± 15.8 | 55.6 ± 26.4 | 57.5 ± 30.0 | 32.8 ± 20.5 | 58.5 ± 36.7 | 37.6 ± 15.7 | ||
| t ½ (h) | n | 8 | 7 | 8 | 5 | 6 | 7 | |
| Mean ± SD | 7.0 ± 3.5 | 10.6 ± 8.1 | 13.9 ± 5.8 | 11.7 ± 6.1 | 7.8 ± 1.8 | 13.9 ± 3.5 | ||
| CL/F (L/h) | n | 8 | 7 | 8 | 9 | 8 | 9 | |
| Mean ± SD | 2791 ± 964 | 4464 ± 2291 | 4459 ± 2288 | 3877 ± 1699 | 4734 ± 2636 | 6265 ± 2896 | ||
| t max (h) | n | 9 | 9 | 8 | 9 | 8 | 9 | |
| Median | 3.00 | 2.00 | 2.00 | 2.00 | 3.00 | 4.00 | ||
| Min–Max | 1.00–4.07 | 1.00–6.00 | 1.00–4.00 | 1.00–6.00 | 1.00–8.00 | 1.00–4.00 | ||
| R acc | n | NA | NA | NA | 9 | 8 | 8 | |
| Mean ± SD | NA | NA | NA | 1.30 ± 0.44 | 1.37 ± 0.48 | 1.61 ± 0.79 | ||
| OP-1118 | C max (ng/mL) | n | 9 | 9 | 9 | 9 | 8 | 9 |
| Mean ± SD | 8.6 ± 4.0 | 18.0 ± 7.6 | 10.1 ± 5.9 | 10.2 ± 4.8 | 19.8 ± 8.9 | 14.7 ± 5.5 | ||
| AUCa (ng·h/mL) | n | 8 | 7 | 8 | 9 | 8 | 9 | |
| Mean ± SD | 92.7 ± 40.3 | 154.2 ± 48.5 | 129.9 ± 49.6 | 67.6 ± 31.9 | 144.2 ± 74.9 | 86.8 ± 18.9 | ||
| t ½ (h) | n | 8 | 7 | 8 | 8 | 6 | 9 | |
| Mean ± SD | 8.8 ± 3.1 | 11.5 ± 7.6 | 16.7 ± 7.0 | 10.4 ± 4.6 | 8.3 ± 1.5 | 13.7 ± 6.3 | ||
| t max (h) | n | 9 | 9 | 8 | 9 | 8 | 9 | |
| Median | 3.00 | 3.00 | 2.00 | 2.00 | 3.00 | 3.00 | ||
| Min–Max | 1.00–6.00 | 1.00–6.00 | 1.00–4.00 | 1.00–6.00 | 1.00–8.00 | 1.00–4.00 | ||
| R acc | n | NA | NA | NA | 9 | 8 | 8 | |
| Mean ± SD | NA | NA | NA | 1.26 ± 0.33 | 1.37 ± 0.50 | 1.67 ± 0.71 | ||
AUC area under the concentration–time curve, AUC ∞ AUC from time zero to infinity, AUC 12 AUC from time zero to 12 h, CL/F apparent total clearance, C max maximum plasma concentration, min minimum, max maximum, NA not analyzed, R acc accumulation ratio, SD standard deviation, t ½ terminal elimination half-life, t max time to C max
aSingle dose: AUC∞, multiple dose: AUC12
Table 3.
Statistical assessment of ethnic differences of pharmacokinetic parameters in fidaxomicin (200 mg) and its metabolite OP-1118
| Compound | Dose schedule | Pharmacokinetic parameter | Japanese (Test) | Caucasian (reference) | Geometric LS mean ratio (test/reference)a | 90 % CI for the ratio | ||
|---|---|---|---|---|---|---|---|---|
| n | Geometric LS mean | n | Geometric LS mean | |||||
| Fidaxomicin | Single dose | AUC∞ (ng·h/mL) | 7 | 50.06 | 8 | 50.70 | 0.99 | 0.61–1.60 |
| AUClast (ng·h/mL) | 9 | 51.69 | 8 | 40.32 | 1.28 | 0.79–2.09 | ||
| C max (ng/mL) | 9 | 7.28 | 8 | 4.29 | 1.70 | 1.05–2.75 | ||
| Multiple dose | AUCτ (ng·h/mL) | 8 | 49.36 | 9 | 34.76 | 1.42 | 0.91–2.23 | |
| C max,ss (ng/mL) | 8 | 7.42 | 9 | 6.11 | 1.21 | 0.74–2.00 | ||
| OP-1118 | Single dose | AUC∞ (ng·h/mL) | 7 | 148.03 | 8 | 123.24 | 1.20 | 0.90–1.61 |
| AUClast (ng·h/mL) | 9 | 146.91 | 8 | 112.52 | 1.31 | 0.95–1.80 | ||
| C max (ng/mL) | 9 | 16.92 | 8 | 10.45 | 1.62 | 1.16–2.27 | ||
| Multiple dose | AUCτ (ng·h/mL) | 8 | 127.91 | 9 | 84.85 | 1.51 | 1.08–2.11 | |
| C max,ss (ng/mL) | 8 | 18.10 | 9 | 13.79 | 1.31 | 0.92–1.87 | ||
A one-way ANOVA was utilized to analyze the effect of ethnic group differences (Japanese vs. Caucasian) on pharmacokinetic response. Pharmacokinetic parameters were analyzed on the logarithmic scale
ANOVA analysis of variance, AUC area under the concentration–time curve, AUC ∞ AUC from time zero to infinity, AUC last AUC from time zero to time of last measurable concentration, AUC τ AUC over the dosing interval at steady state, CI confidence interval, C max maximum plasma concentration, C max,ss C max at steady state, LS least squares
aRatio of log-transformed pharmacokinetic parameters between Japanese and Caucasian subjects and its 90 % CI are back-transformed to the raw scale
The pharmacokinetic profile of OP-1118 was similar to that of fidaxomicin, but plasma concentrations and AUC values were approximately double compared with those of fidaxomicin (Table 2). The Cmax values for OP-1118 after a single fidaxomicin 200 mg dose in Caucasian subjects was 10.1 ± 5.9 ng/mL (range 0–18.3 ng/mL), which was closer to the 100 mg dose Cmax of 8.6 ± 4.0 ng/mL (range 4.1–13.9 ng/mL) than to the 200 mg dose Cmax of 18.0 ± 7.6 ng/mL (range 10.9–35.2 ng/mL) in Japanese subjects. The Cmax values for OP-1118 after multiple dosing of 100 mg in Japanese subjects, 200 mg in Japanese subjects, and 200 mg in Caucasian subjects were 10.2 ± 4.8 ng/mL (range 4.1–18.4 ng/mL), 19.8 ± 8.9 ng/mL (range 9.8–36.3 ng/mL), and 14.7 ± 5.5 ng/mL (range 7.4–24.4 ng/mL), respectively. The AUC∞ values for OP-1118 after a single 100 mg dose in Japanese subjects, 200 mg in Japanese subjects, and 200 mg in Caucasian subjects were 92.7 ± 40.3, 154.2 ± 48.5, and 129.9 ± 49.6 ng·h/mL, respectively. The AUC12 values for OP-1118 after multiple dosing of 100 mg in Japanese subjects, 200 mg in Japanese subjects, and 200 mg in Caucasian subjects were 67.6 ± 31.9, 144.2 ± 74.9, and 86.8 ± 18.9 ng·h/mL, respectively. For OP-1118, the ratios for Japanese versus Caucasian subjects were higher for all parameters with a ratio of 1.6 (90 % CI 1.2–2.3) for Day 1 Cmax, 1.3 (90 % CI 0.9–1.9) for Day 15 Cmax,ss, 1.2 (90 % CI 0.9–1.6) for Day 1 AUC∞, 1.3 (90 % CI 1.0–1.8) for Day 1 AUClast, and 1.5 (90 % CI 1.1–2.1) for Day 15 AUCτ (Table 3).
Fidaxomicin could not be detected in urine with an LLOQ of 5.0 ng/mL, and amounts excreted in the urine as OP-1118 were ≤0.0104 % of the administered dose. The fecal pharmacokinetic parameters of fidaxomicin and OP-1118 after single-dose administration are summarized in Table 4. Fidaxomicin and OP-1118 were mainly excreted in the feces, with average recoveries from 28.7 to 38.9 % for fidaxomicin and from 18.6 to 24.8 % for OP-1118 (Fig. 2). The mean fecal concentrations of fidaxomicin after multiple dosing of fidaxomicin 200 mg on Day 15 were 2669 μg/g in Japanese subjects and 2181 μg/g in Caucasian subjects. The mean fecal concentrations of OP-1118 in Japanese subjects and Caucasian subjects were 961 and 1163 μg/g, respectively (Table 5).
Table 4.
Fidaxomicin and OP-1118 fecal pharmacokinetic parameters at Day 1 after single dose
| Compound | Parameter | Japanese 100 mg | Japanese 200 mg | Caucasian 200 mg | |||
|---|---|---|---|---|---|---|---|
| Aelast (mg) | %Aelast (%) | Aelast (mg) | %Aelast (%) | Aelast (mg) | %Aelast (%) | ||
| Fidaxomicin | n | 9 | 9 | 9 | 9 | 9 | 9 |
| Mean ± SD | 28.72 ± 17.72 | 28.72 ± 17.72 | 72.66 ± 58.38 | 36.33 ± 29.19 | 77.86 ± 47.05 | 38.93 ± 23.53 | |
| Median | 28.85 | 28.85 | 103.5 | 51.76 | 86.86 | 43.43 | |
| Min–max | 0a–67.4 | 0a–67.4 | 0a–149 | 0a–74.7 | 0a–143 | 0a–71.3 | |
| OP-1118 | n | 9 | 9 | 9 | 9 | 9 | 9 |
| Mean ± SD | 23.14 ± 13.18 | 24.78 ± 14.11 | 34.64 ± 28.29 | 18.55 ± 15.15 | 43.56 ± 20.84 | 23.32 ± 11.16 | |
| Median | 23.89 | 25.59 | 41.98 | 22.48 | 43.08 | 23.07 | |
| Min–max | 0b–40.8 | 0b–43.7 | 0b–75.3 | 0b–40.3 | 0b–73.9 | 0b–39.6 | |
Ae last cumulative amount of drug excreted in the feces from time zero to the time of last measurable concentration, LLOQ lower limit of quantification, max maximum, min minimum, SD standard deviation
aSubjects with value less than LLOQ (10 ng/mL) or missing during Days 1–5
bSubject with all values less than LLOQ (50 ng/mL) during Days 1–5
Fig. 2.
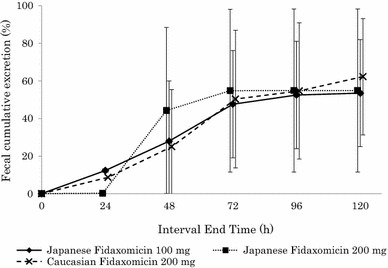
Mean total percentage (±standard deviation) recovery of fecal excretion of fidaxomicin plus OP-1118 versus nominal time at Day 1 after single-dose administration of fidaxomicin in Japanese subjects and Caucasian subjects. The percentage fecal cumulative excretion of OP-1118 was adjusted by multiplying by the ratio: [fidaxomicin molecular weight (1058.04)/OP-1118 molecular weight (987.95) × dose]
Table 5.
Fecal concentration (μg/g) of fidaxomicin and OP-1118 after the multiple dose at Day 15
| Compound | Concentration (μg/g) | ||
|---|---|---|---|
| Japanese 100 mg | Japanese 200 mg | Caucasian 200 mg | |
| Fidaxomicin | |||
| n | 9 | 8 | 9 |
| Mean ± SD | 906.8 ± 312.8 | 2669.2 ± 1023.5 | 2180.9 ± 688.9 |
| Median | 857.28 | 3015.03 | 2342.79 |
| Min–max | 240.96–1272.73 | 1126.99–3952.51 | 1039.61–3113.80 |
| OP-1118 | |||
| n | 9 | 8 | 9 |
| Mean ± SD | 746.0 ± 357.4 | 960.9 ± 388.6 | 1163.3 ± 569.7 |
| Median | 754.80 | 1053.28 | 1360.33 |
| Min–max | 200.83–1266.80 | 397.41–1482.88 | 368.13–1976.66 |
Max maximum, min minimum, SD standard deviation
Safety
In the single-dose cohort, three TEAEs were reported in three subjects: one each in the 100 mg Japanese group, 200 mg Caucasian group, and placebo Japanese group. In the multiple-dose period, there were three TEAEs: one each in the 100 mg Japanese group, 200 mg Japanese group, and placebo Japanese group. No subject died, experienced a serious AE, or discontinued owing to an AE. Possibly study drug-related AEs were diarrhea (n = 1, single dose in the placebo Japanese group), feeling hot (n = 1, multiple dose in the 200 mg Japanese group), and hypersomnia (n = 2, multiple dose in the 100 mg Japanese group and placebo Japanese group), as shown in Table 6. All reported TEAEs were mild in severity.
Table 6.
Subjects with treatment-emergent adverse events by single- and multiple-dose period
| Dose schedule | AE (MedDRA® preferred term) | Placebo [n (%)] | Fidaxomicin [n (%)] | |||
|---|---|---|---|---|---|---|
| Japanese (n = 6) | Caucasian (n = 3) | Japanese 100 mg (n = 9) | Japanese 200 mg (n = 9) | Caucasian 200 mg (n = 9) | ||
| Single dosea | Overall | 1 (16.7) | 0 | 1 (11.1) | 0 | 1 (11.1) |
| Constipation | 0 | 0 | 1 (11.1) | 0 | 0 | |
| Diarrhea | 1 (16.7) | 0 | 0 | 0 | 0 | |
| Dermatitis contact | 0 | 0 | 0 | 0 | 1 (11.1) | |
| Multiple doseb | Overall | 1 (16.7) | 0 | 1 (11.1) | 1 (11.1) | 0 |
| Feeling hot | 0 | 0 | 0 | 1 (11.1) | 0 | |
| Hypersomnia | 1 (16.7) | 0 | 1 (11.1) | 0 | 0 | |
AE adverse event, MedDRA ® Medical Dictionary for Regulatory Activities
aSingle-dose period includes any AEs observed on or after the time of the Day 1 dose up until prior to the first dose in the multiple-dose period starting on Day 6
bMultiple-dose period includes any AEs observed on or after the time of the first dose on Day 6 until end of follow-up
Throughout the single- and multiple-dose periods, no clinically significant changes in hematology and chemistry laboratory test results were observed. No clinical laboratory abnormalities were reported as a TEAE by the investigator.
In the single- and multiple-dose periods, mean vital sign values (DBP, SBP, pulse, oral body temperature) at baseline were similar to those after dosing throughout the study. Throughout both dose periods, such abnormal vital signs were not reported as a TEAE by the investigator.
With regard to ECG results, no subject experienced Fridericia’s formula-corrected QT interval (QTcF) >450 ms at any timepoint during the single- and multiple-dose period, and no subject experienced a clinically significant 12-lead ECG abnormality during the study. No 12-lead ECG abnormality was reported as a TEAE by the investigator. One subject in the Japanese placebo group experienced an increase in QTcF from baseline of ≥30 and <60 ms at one timepoint during the single-dose period (1 h after dosing on Day 1). All other QTcF changes from baseline were either <0 or ≥ 0 to <30 ms.
Discussion
Fidaxomicin was approved in 2011 by the USA and European countries for the treatment of CDAD but has not yet been approved in Japan. This randomized, double-blind, placebo-controlled dose-escalation study to evaluate the safety, tolerability, and pharmacokinetics of single and multiple doses of fidaxomicin in healthy Japanese and Caucasian subjects was conducted in the USA. This was the first fidaxomicin pharmacokinetics study conducted among subjects of Japanese ethnicity. A double-blind placebo design was utilized in each cohort consisting of 12 subjects; the subjects were randomly assigned in a 3:1 ratio to receive either fidaxomicin or placebo. The aim of this double-blind, placebo-controlled design was to minimize bias and provide reference data, aiding in the interpretation of results. Fidaxomicin was tested in Japanese subjects to support its development in Japan, and Caucasian subjects were used as a reference cohort. Japanese subjects living in the USA were considered to be the same as native Japanese because the maintenance of a Japanese lifestyle, including diet, in addition to ethnicity was confirmed. A population of healthy subjects helped to avoid confounding effects such as disease and/or concomitant medication. The recommended dose of fidaxomicin in many regions, including the USA and Europe, for treating CDI is a 200 mg tablet twice daily for 10 days. However, fidaxomicin was studied for the first time in Japanese subjects. Therefore, after confirming the safety, tolerability, and pharmacokinetic parameters of 100 mg of fidaxomicin in Japanese subjects as a starting dose, we conducted a parallel comparison study of fidaxomicin 200 mg between Japanese and Caucasian subjects.
Fidaxomicin was shown to have a minimal systemic absorption and high fecal concentrations after oral administration to healthy Caucasians in phase I studies in the USA, where the mean Cmax values following a single dose of 200 mg were 5.2 ± 2.8 and 12.0 ± 6.1 ng/mL, respectively [25, 26].
Our results confirmed the minimal systemic absorption of fidaxomicin following oral administration, with plasma concentrations of fidaxomicin and OP-1118 in the low ng/mL range at the therapeutic dose in both Japanese and Caucasian healthy subjects. The exposure in Japanese subjects was relatively higher that than in Caucasian subjects, although variation in both groups was large.
In our study, 29–39 % of fidaxomicin and 19–25 % of OP-1118 were excreted in feces after single-dose administration. The total percentage of fecal cumulative excretion of fidaxomicin and OP-1118 after single-dose administration was similar among the doses and ethnic groups. No ethnic differences were found in the mean fecal concentrations of fidaxomicin and OP-1118 at 24 h after the last dose of twice-daily 200 mg administration for 10 days: 2669 and 961 μg/g for Japanese subjects, and 2181 and 1163 μg/g for Caucasian subjects, respectively. These results were similar to the US phase I study (OPT-80 1B-MD), in which the fecal recovery of fidaxomicin and OP-1118 after multiple dosing of once daily fidaxomicin 450 mg were 2983 ± 1774 and 610 ± 241 µg/g, respectively [25]. The mean fecal levels of fidaxomicin and OP-1118 were three orders of magnitude higher than the minimum inhibitory concentration for inhibition of 90 % (MIC90) of 0.5 μg/mL for C. difficile [19]. Thus, a high potential efficacy in Japanese patients with CDI could be expected by the maintenance of high fidaxomicin concentration in the gut.
Overall, the incidence of subjects reporting TEAEs and the number of TEAEs were low across treatments in Japanese and Caucasian subjects, in agreement with the favorable safety and tolerability profiles of fidaxomicin found in the US phase I study [25]. The possibly study drug-related AEs were diarrhea (n = 1), feeling hot (n = 1), and hypersomnia (n = 2). All TEAEs were mild in severity. No subjects withdrew because of an AE. The TEAEs in this study were independent of ethnicity and fidaxomicin dose.
Conclusion
In both single-dose and multiple-dose settings, fidaxomicin had minimal systemic absorption following 100 and 200 mg oral administration, with plasma concentrations of fidaxomicin and OP-1118 in the low ng/mL range. Fidaxomicin was mainly excreted in feces. The mean fecal levels of fidaxomicin and OP-1118 were three orders of magnitude higher than the MIC90 of C. difficile isolates. Single and multiple 100 and 200 mg doses of fidaxomicin were safe and well-tolerated in healthy male Japanese and Caucasian subjects.
Acknowledgments
This study was sponsored by Astellas Pharma Inc. The authors thank Drs. Pam Sears and Yoshi Ichikawa (Cubist Pharmaceuticals, Inc., USA) for supporting and assisting in the development of fidaxomicin and Dr. Tetsuji Asao (SunFlare Co., Ltd.) for scientific writing support, for which he received financial compensation from Astellas. Drs. Oshima, Yamazaki, Benner, Miki, Michon, Wojtkowski, Kaibara, and Mujais played a substantial role in the design and overall management of this study and also in the collection, analysis, and interpretation of data. All authors listed as authors were involved in the critical review and revision of the manuscript, and all provided final approval of the content.
Conflict of interest
All authors are paid employees of Astellas Pharma Inc. or Astellas Pharma Global Development, Inc.
References
- 1.Carroll KC, Bartlett JG. Biology of Clostridium difficile: implications for epidemiology and diagnosis. Annu Rev Microbiol. 2011;65:501–521. doi: 10.1146/annurev-micro-090110-102824. [DOI] [PubMed] [Google Scholar]
- 2.Kelly CP, LaMont JT. Clostridium difficile–more difficult than ever. N Engl J Med. 2008;359:1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 3.Forster AJ, Taljaard M, Oake N, Wilson K, Roth V, van Walraven C. The effect of hospital-acquired infection with Clostridium difficile on length of stay in hospital. CMAJ. 2012;184:37–42. doi: 10.1503/cmaj.110543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freeman J, Bauer MP, Baines SD, Corver J, Fawley WN, Goorhuis B, Kuijper EJ, Wilcox MH. The changing epidemiology of Clostridium difficile infections. Clin Microbiol Rev. 2010;23:529–549. doi: 10.1128/CMR.00082-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Owens RC, Jr, Donskey CJ, Gaynes RP, Loo VG, Muto CA. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin Infect Dis. 2008;46(Suppl 1):S19–S31. doi: 10.1086/521859. [DOI] [PubMed] [Google Scholar]
- 6.Thomas C, Riley TV. Restriction of third generation cephalosporin use reduces the incidence of Clostridium difficile-associated diarrhoea in hospitalised patients. Commun Dis Intell Q Rep. 2003;27(Suppl):S28–S31. doi: 10.33321/cdi.2003.27.17. [DOI] [PubMed] [Google Scholar]
- 7.Slimings C, Riley TV. Antibiotics and hospital-acquired Clostridium difficile infection: update of systematic review and meta-analysis. J Antimicrob Chemother. 2014;69:881–891. doi: 10.1093/jac/dkt477. [DOI] [PubMed] [Google Scholar]
- 8.Pépin J, Saheb N, Coulombe MA, Alary ME, Corriveau MP, Authier S, et al. Emergence of fluoroquinolones as the predominant risk factor for Clostridium difficile-associated diarrhea: a cohort study during an epidemic in Quebec. Clin Infect Dis. 2005;41:1254–1260. doi: 10.1086/496986. [DOI] [PubMed] [Google Scholar]
- 9.Kallen AJ, Thompson A, Ristaino P, Chapman L, Nicholson A, Sim BT, et al. Complete restriction of fluoroquinolone use to control an outbreak of Clostridium difficile infection at a community hospital. Infect Control Hosp Epidemiol. 2009;30:264–272. doi: 10.1086/595694. [DOI] [PubMed] [Google Scholar]
- 10.McDonald LC, Killgore GE, Thompson A, Owens RC, Jr, Kazakova SV, Sambol SP, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 11.Honda H, Yamazaki A, Sato Y, Dubberke ER. Incidence and mortality associated with Clostridium difficile infection at a Japanese tertiary care center. Anaerobe. 2014;25:5–10. doi: 10.1016/j.anaerobe.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi M, Mori N, Bito S. Multi-institution case-control and cohort study of risk factors for the development and mortality of Clostridium difficile infections in Japan. BMJ Open. 2014;4:e005665. doi: 10.1136/bmjopen-2014-005665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen SH, Gerding DN, Johnson S, Kelly CP, Loo VG, McDonald LC, Society for Healthcare Epidemiology of America; Infectious Diseases Society of America et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA) Infect Control Hosp Epidemiol. 2010;31:431–455. doi: 10.1086/651706. [DOI] [PubMed] [Google Scholar]
- 14.Debast SB, Bauer MP, Kuijper EJ, European Society of Clinical Microbiology and Infectious Diseases European Society of Clinical Microbiology and Infectious Diseases: update of the treatment guidance document for Clostridium difficile infection. Clin Microbiol Infect. 2014;20(Suppl 2):1–26. doi: 10.1111/1469-0691.12418. [DOI] [PubMed] [Google Scholar]
- 15.Eyre DW, Walker AS, Wyllie D, Dingle KE, Griffiths D, Finney J, et al. Infections in Oxfordshire Research Database. Predictors of first recurrence of Clostridium difficile infection: implications for initial management. Clin Infect Dis. 2012;55(Suppl 2):S77–S87. doi: 10.1093/cid/cis356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pepin J, Alary ME, Valiquette L, Raiche E, Ruel J, Fulop K, et al. Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin Infect Dis. 2005;40:1591–1597. doi: 10.1086/430315. [DOI] [PubMed] [Google Scholar]
- 17.Cavalleri B, Arnone A, Di Modugno E, Nasini G, Goldstein BP. Structure and biological activity of lipiarmycin B. J Antibiot (Tokyo). 1988;41:308–315. doi: 10.7164/antibiotics.41.308. [DOI] [PubMed] [Google Scholar]
- 18.Kurabachew M, Lu SHJ, Krastel P, Schmitt EK, Suresh BL, Goh A, et al. Lipiarmycin targets RNA polymerase and has good activity against multidrug-resistant strains of Mycobacterium tuberculosis. J Antimicrob Chemother. 2008;62:713–719. doi: 10.1093/jac/dkn269. [DOI] [PubMed] [Google Scholar]
- 19.Goldstein EJC, Babakhani F, Citron DM. Antimicrobial activities of fidaxomicin. Clin Infect Dis. 2012;55(Suppl 2):S143–S148. doi: 10.1093/cid/cis339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louie TJ, Cannon K, Byrne B, Emery J, Ward L, Eyben M, et al. Fidaxomicin preserves the intestinal microbiome during and after treatment of Clostridium difficile infection (CDI) and reduces both toxin reexpression and recurrence of CDI. Clin Infect Dis. 2012;55(Suppl 2):S132–S142. doi: 10.1093/cid/cis338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babakhani F, Gomez A, Robert N, Sears P. Killing kinetics of fidaxomicin and its major metabolite, OP-1118, against Clostridium difficile. J Med Microbiol. 2011;60:1213–1217. doi: 10.1099/jmm.0.029470-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sears P, Crook DW, Louie TJ, Miller MA, Weiss K. Fidaxomicin attains high fecal concentrations with minimal plasma concentrations following oral administration in patients with Clostridium difficile infection. Clin Infect Dis. 2012;55(Suppl 2):S116–S120. doi: 10.1093/cid/cis337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louie TJ, Miller MA, Mullane KM, Weiss K, Lentnek A, Golan Y, OPT-80-003 Clinical Study Group et al. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med. 2011;364:422–431. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 24.Cornley OA, Crook DW, Esposito R, Poirier A, Somero MS, Weiss K, OPT-80-004 Clinical Study Group et al. Fidaxomicin versus vancomycin for infection with Clostridium difficile in Europe, Canada, and the USA: a double-blind, non-inferiority, randomized control trial. Lancet Infect Dis. 2012;12:281–289. doi: 10.1016/S1473-3099(11)70374-7. [DOI] [PubMed] [Google Scholar]
- 25.Shue YK, Sears PS, Shangle S, Walsh RB, Lee C, Gorbach SL, et al. Safety, tolerance, and pharmacokinetic studies of OPT-80 in healthy volunteers following single and multiple oral doses. Antimicrob Agents Chemother. 2008;52:1391–1395. doi: 10.1128/AAC.01045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.FDA label and approval history of Dificid, supplement number 004. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Label_ApprovalHistory#labelinfo. Accessed 14 Oct 2014.