

Abstract
Background
4-chloro-2-methylphenoxy acetic acid (MCPA) is one of the most important pesticides which is extensively used to control weeds in arable farmland. Exposure to this compound occurs in general population and persons who occupationally handle it. The aim of this present work was the preparation of MCPA imprinting polymer and its application as a selective sample preparation technique for trace determination of MCPA in biological and environmental samples.
Methods
In this study, MCPA imprinting polymer was obtained by precipitation polymerization using methacrylic acid (the functional monomer), ethylene glycol dimethacrylate (the cross-linker), 2, 2’-azobisisobutyronitrile (the initiator) and MCPA (the template molecule) in acetonitrile solution. The MIP-NPs were characterized by thermogravimetric analysis and scanning electron microscopy. The optimization process was carried out applying batch method. After optimization of the parameters, affecting the adsorption and desorption of analyte, urine and different water samples were used to determine MCPA.
Results
Imprinted MCPA molecules were removed from the polymeric structure using acetic acid in methanol (20:80 v/v %) as the eluting solvent. Both sorption and desorption process occur within 10 min. The maximum sorbent capacity of the molecular imprinted polymer is 87.4 mg g-1. The relative standard deviation and limit of detection for water samples by introduced selective solid phase extraction were 4.8% and 0.9 μg L-1, and these data for urine samples were 4.5% and 1.60 μg L-1, respectively.
Conclusion
The developed method was successfully applied to determine MCPA in urine and different water samples.
Keywords: Molecular imprinted polymer nanoparticles, 4-chloro-2-methylphenoxy acetic acid, Selective preconcentration, Urine and water samples.
Introduction
The chlorophenoxy herbicides (CPHs) and their derivatives are widely used throughout the world to control weeds in arable farmland, as well as non crop land (1). Among these herbicides, 4-chloro-2-methylphenoxy acetic acid (MCPA) is an aryloxyalkanoic acid, systemic, polar herbicide, which is widely used for post-emergence control of annual and perennial broad-leaved weeds in a range of crops, in grasslands and on roadside verges. It is also used to control aquatic broad-leaved weeds (2).
Exposures to MCPA occurs not only in general population groups through trace residues in the diet, from drinking water and direct contact with lawns recently treated with MCPA (3) but also in persons who occupationally handle it during manufacture, formulation and its application (4, 5). MCPA is poorly metabolized and is excreted largely unchanged in urine (6) This herbicide represent a potential risk for water sources as they are poorly biodegradable and quite mobile in aqueous systems because of their acidiccarboxyl group. Due to the presence of the carboxylate group in its structure (7), this compound can react with metal ions to form complexes which are sparingly soluble in water.
The World Health Organization (WHO), U.S. Environmental Protection Agency (EPA), and national governments such as New Zealand and United State of America (USA) regulation set the maximum contaminant level (MCL) of chlorophenoxy acid herbicides in drinking water in the range of 10-70 ng mL-1(8, 9). MCPA has been found in well water in some countries and is classified by the US EPA as a potential groundwater contaminant and systemic toxicant (10). Because of its high water solubility and toxicological risk, monitoring of ground and drinking water and urine for its presence has great importance with respect to estimation of the future risk of the human health and the ecosystem.
There are various methods for analysis of MCPA in different matrixes. solid phase micro extraction gas chromatography-mass spectrometry (SPME-GC-MS)(11), solid phase extraction high-performance liquid chromatographic with UV detector (SPE-HPLC-UV) (12), liquid liquid extraction gas chromatography-mass spectrometry (LLE-GC-MASS) (13) solid phase extraction liquid chromatography-mass spectrometry-mass spectrometry (SPE- LC-MS-MS)(14) and solid phase extraction-gas chromatography (SPE- GC-ECD) (15) method have been reported in the literature. The above mentioned methods are not only time consuming and require expensive instrumentation but also affected by matrix effects and have low selectivity and recoveries. Because of the complexity of matrix and low content of MCPA in real samples, high efficiency clean-up and enrichment procedures are required prior to analysis. Therefore, it is important to develop techniques for the rapid and selective extraction of MCPA from environmental and biological matrices.
Among different sample preparation techniques, SPE is the most popularly used technique for its advantages like time and cost saving application and minimal consumption of organic solvents (1620) Nevertheless, the major disadvantage of conventional SPE sorbents, such as C18, ion-exchange and size-exclusion phases, is the lack of selectivity, leading to co-extraction of matrix interference components with the target analytes.
In recent years, molecularly imprinted polymers (MIPs) and ion imprinted polymers (IIPs) have been increasingly used (e.g. SPE sorbent) for preconcentration and high efficient separation of various trace analytes in complex matrices (21–30). Molecular imprinting is a technique that creates specific recognition sites for a target molecule within a synthetic polymer(31–36). MIPs exhibit high potential for sample clean-up of very complicated matrix(31), solid-phase extraction(37–40), and solid-phase micro extraction fibers(41), Traditionally, MIPs have been synthesized in bulk polymerization followed by a grinding and sieving process to acquire the desired particle size. Because of some disadvantages of bulk polymerization such as the heterogeneous distribution of the binding sites, embedding of most binding sites, and poor site accessibility for template molecule (42) the consideration of researchers has moved toward achieving highly uniform spherical imprinted particles, particularly on the nanoscale(43). Unlike the bulk monoliths, MIPs possess nano structure and higher surface-to-area ratio; therefore, imprinted cavities are more easily accessible to templates and the binding kinetic is en hanced(44). Nano MIPs particles are obtained by different methods, namely suspension, multistep swelling and precipitation polymerization(45). Among mentioned methods, the precipitation technique is the most convenient one, since it is a homogeneous, one-step synthesis and does not require the use of a surfactant or stabilizer that can remain adsorbed to the surface of the polymer, interfering with the selective binding of the target to the imprinted materials. MIPs using precipitation polymerization were first prepared by Ye et al. (46).
The polymer particles were obtained according to this procedure, have more uniform particles size and require no crushing and sieving steps in comparison with the polymers obtained by bulk polymerization.
Several methods have been described for the determination of MCPA in complex matrix. Biesaga et al. and Lagana et al. used different SPE sorbents coupled with HPLC-UV and LC-MS-MS (12, 14) for determination of MCPA from environmental samples in separated studies and the limit of detections in the range of 2—3 μgL-1 and 0.3 and reproducibilities of 5 and 8 were reported respectively. In another study conducted by Catalina et al. liquid liquid extraction (LLE) coupled with GC-MS (13) was used for determination of MCPA in water samples. Based on the results, the limit of detection in the range of 0.02-0.04 μgL-1 and reproducibility in the range of 9-11 were reported.
These mentioned methods require one or more preconcentration steps for trace level detection and quantification of herbicides due to insufficient sensitivity.
The aim of this present work was the preparation of MCPA imprinting polymer via non-covalent precipitation polymerization. Also, its application as a selective sample preparation technique for trace determination of MCPA in biological and environmental samples was studied in order to eliminate the matrix effects. In addition, HPLC-UV was used for determination of MCPA after preconcentration by MIP-NPs in water and urine samples.
Materials and Methods
Reagents and chemicals
MCPA was purchased from SUPELCO (Sigma-Aldrich, USA). Azobisisobutyronitrile (AIBN) as the initiator was purchased from Acros Organic (NJ, USA). Methacrylic acid (MAA), ethylene glycol dimethacrylare (EGDMA), acetic acid (HOAC), sodium hydroxide (NaOH), acetonitrile (ACN) and methanol (MeOH) were purchased from Merck (Darmstadt, Germany). HPLC grade water was obtained through a Milli-Q® system (Millipore, Milford, MA, USA). Stock standard solution of MCPA (1000 mg L-1) was prepared in methanol and stored at 4 °C in refrigerator to avoid exposure to light.
Instrumentation
Analysis of the standard and test samples were performed by a reversed-phase HPLC system from the Knauer Company (Germany), consisting of a K-1001 series high-pressure pump, a K-2006 photo diode-array detector and a VS injection valve, equipped with a 20-μL loop.
The analytes were separated on a Chromolith performance RR-C18 100 mm×4.6 mm i.d. (Merch KGa A, Germany) and column guards (Chromolith Guard Cartridge Kit RP-C18 and 5 cm ×4.6 mm i.d., 5 μm). The mobile phase was 79.5 % ace-tonitrile, 19.5 % purified water and 1 % acetic acid at the flow rate of 1 mL min-1 in isocratic mode with the detector’s wavelength set at 280 nm.
A digital pH meter, WTW Metrohm 827 Ion analyzer (Herisau, Switzerland), equipped with a combined glass calomel electrode was used for the pH adjustments at 25 ± 1 °C temperature.
The prepared nanosized imprinted polymers were characterized by scanning electron microscopy (SEM), and thermogravimetric analysis (TGA). Thermal stability of the synthesized imprinted polymer nanoparticles was evaluated by TGA. Scanning electron microscopy (SEM) was performed by gently distributing the powder sample on the stainless steel stubs, using SEM (Philips, XL-30, Almelo, the Netherlands) instrument. The thermal properties of synthesized polymers were determined using a BAHR-Thermoanalyse GmbH (Germany) with employing, heating and cooling rates of 10 °C min-1 and using a condenser as the coolant. The samples were weighed as a thin film and carefully packed into a clean aluminum pan (11.5-12.5 mg), and sealed by crimping an aluminum lid on the pan (Shimadzu universal crimper). An Al2O3 empty pan sealed with a cover pan was used as a reference sample. A scanning range of 10 to 800 °C was used for samples at 10 °C min-1 in nitrogen gas.
Synthesis of molecular imprinted and non imprinted polymer nanoparticles
MIP-NPs were prepared according to the following procedure (Fig. 1): 1 mmol of MCPA as the template, and 5 mmol of MAA as the functional monomer were dissolved in 40 mL of acetonitrile as a progen solvent as a medium for polymerization process. The solution was mixed by ultrasonic wave for 10 min and then 1 mmol of AIBN as the initiator and 25 mmol of EGDMA as a cross-linker agent were added to the solution. The solution was purged with a gentle flow of nitrogen gas for 6 min, then; polymerization was performed in an oil bath at 60 °C for 24 h in the presence of nitrogen under magnetic stirring at 400 rpm.
Fig. 1.

A scheme for the synthesis of MCPA imprinted polymer nanoparticles
The polymer nanoparticles were collected by centrifugation and washed with acetone and methanol, respectively for removing additional reagents and solvent. The template was removed from the cavity of MIP-NP using Soxhelt extraction (acetic acid-methanol (80:20 v/v)), then the eluent solvent was analyzed by HPLC. This procedure was repeated several times until MCPA could not be detected in the eluent solvent by HPLC analysis. Afterwards, the polymer was washed with deionized water until neutralization was obtained. Finally, the prepared MIP-NPs were dried at 60 oC and used for the subsequent studies. The corresponding non imprinted polymers (NIP) were prepared by the same procedure, without the presence of the template (MCPA), to evaluate the prepared MIP-NPs.
Preparation of working solutions and real samples
Stock standard solution of the MCPA (1000 mg L-1) was prepared in methanol and stored in a refrigerator at 4 °C and brought to ambient temperature just prior to use. The working solutions were prepared daily by diluting the standard solutions prior to use. For analysis of urine sample, 2 mL of spiked urine was diluted to 7.0 mL with ultrapure water. The water samples, including distilled water, tap water (Tehran, Iran), river waters obtained from Siahrood river and Derka river (Ghaemshahr, Iran) and sea water (Caspian Sea, Sari, Iran) were collected in cleaned polyethylene bottles and filtered through a 0.45 mm pore size nylon filter (Millipore) immediately after sampling. It should be mentioned that, the sampling period was in March 2013. The amounts of MCPA were successfully determined by the present method.
Extraction procedure
Batch experiments were conducted to investigation of effective factors in sorption and desorption steps. Extraction of MCPA molecules from modeling solutions and real samples is followed by two steps: sorption and desorption. In sorption step, pH and adsorption time of the analyte on the polymer, according to following procedures, were optimized.
To investigate the effect of pH on the extraction efficiency, the pH of 10 mL of sample solutions containing 0.02 mg L-1 of MCPA were adjusted in the range of 2-9. The pH of the suspensions was adjusted to desired values by drop wise addition either sodium hydroxide or hydrochloric acid.
In order to optimize the desorption time, in a typical uptake kinetics test, 50 mg of the sorbent was added to 10 mL of 0.02 mg L-1 MCPA aqueous solution at optimum pH. The mixture was then stirred in different periods of times (i.e., from 5 to 20 min) under magnetic stirring. Then, 50 mg of dried polymer was suspended in aqueous solution (10 mL) containing 0.02 mg L-1 concentration of MCPA and stirred for 10 min at pH 5.0 with a magnetic stirrer, after that the polymer nanoparticles were separated from the solution by centrifugation. In washing step, in order to remove the potential interfering compounds and loosely retained molecules, the MIP-NPs were washed several times with double distilled water, then in desorption step, effective parameters such as desorption times of the analyte from the polymer cavities and type and volume of eluent were optimized.
To determine effective eluent type and composition for removing MCPA out of MIPs, several solvents were examined, while, other parameters were kept constant. Also, in order to investigate the optimum desorption time, various period of times from 5 to 25 min were examined, while other parameters were kept in optimum conditions. The elution of MCPA from MIP-NPs was performed by 1.5mL of HOAC in methanol (20:80 v/v %) for 10 min. The eluate (separated by centrifuge) evaporated to almost dry under a gentle stream of nitrogen and the residue was dissolved in 0.3 mL mobile phase under sonication. Finally, 20 μL of the separated phase was drawn out by a Hamilton syringe and directly injected into the HPLC-Uv for analysis. Figure 2 provides a scheme for illustration of the mentioned extraction procedure.
Fig. 2.

A schematic illustration of extraction procedure
Extraction percent of MCPA was calculated by the following equation:
Adsorption efficiency % = CA- CB / CA × 100
CA and CB are the concentrations of MCPA before and after extraction in the solution.
In order to evaluate the analytical applicability of the proposed method, determination of MCPA in urine and different water samples such as distilled water, tap water (Tehran, Iran), river waters obtained from Siahrood river and Derka river (Ghaemshahr, Iran) and sea water (Caspian Sea, Sari, Iran) was carried out.
Statistical analysis
In order to validate the optimized method, day-today and within-day reproducibility were performed and mean value as well as the relative standard deviation were calculated.
Results
Characterization of the synthesized molecular imprinted polymer nanoparticles
Figure 3 shows TGA plot for the imprinted polymer. The obtained result by TGA can prove the thermal stability of the synthesized MIP-NPs. The morphology of the MIP-NPs was assessed by scanning electron microscopy (Fig. 4).
Fig. 3.
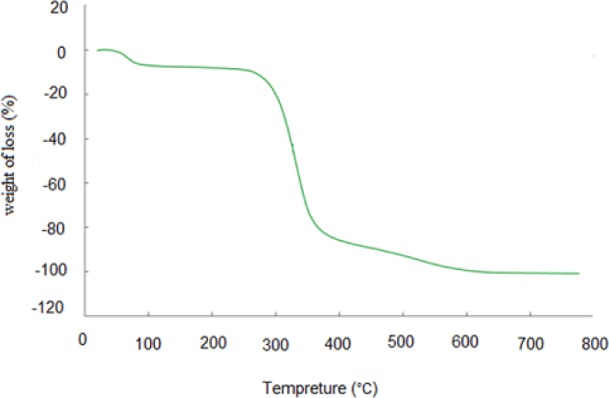
The thermogram of synthesized molecular imprinted polymer nanoparticles
Fig. 4.

The SEM micrograph of synthesized molecular imprinted polymer nanoparticles
Removal step Effect of solution’s pH
As shown in Fig. 5, the adsorption of MCPA by MIP-NPs was approximately constant from pH 2.0 to 5.0 but decreased from pH 6.0 to 9.0. Therefore, for quantitative adsorption of MCPA on the synthesized MIP-NPs the pH of solution was adjusted in the pH of 5.0 in the subsequent experiments.
Fig. 5.
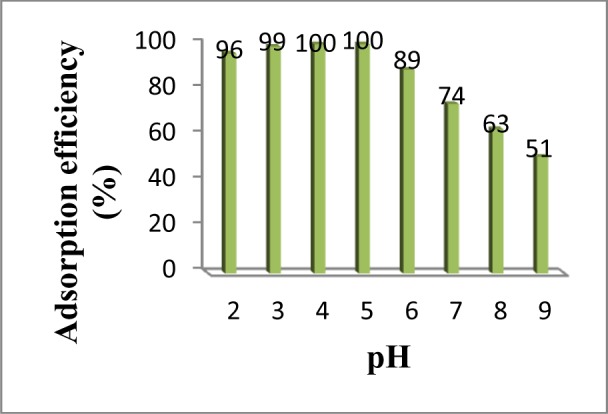
The effect of solution’s pH on the adsorption efficiency of MCPA on MIP-NPs (Conditions: MCPA concentration: 0.02 mgL-1, Sample volume: 10 mL, Adsorption time: 10 min)
Equilibrium sorption time
It was observed that after 10 min, further extraction time had no serious effect. Thus, an optimum equilibration time of 10 minutes was obtained for quantitative removal of MCPA from solution into the solid phase (Fig. 6a).
Fig. 6.
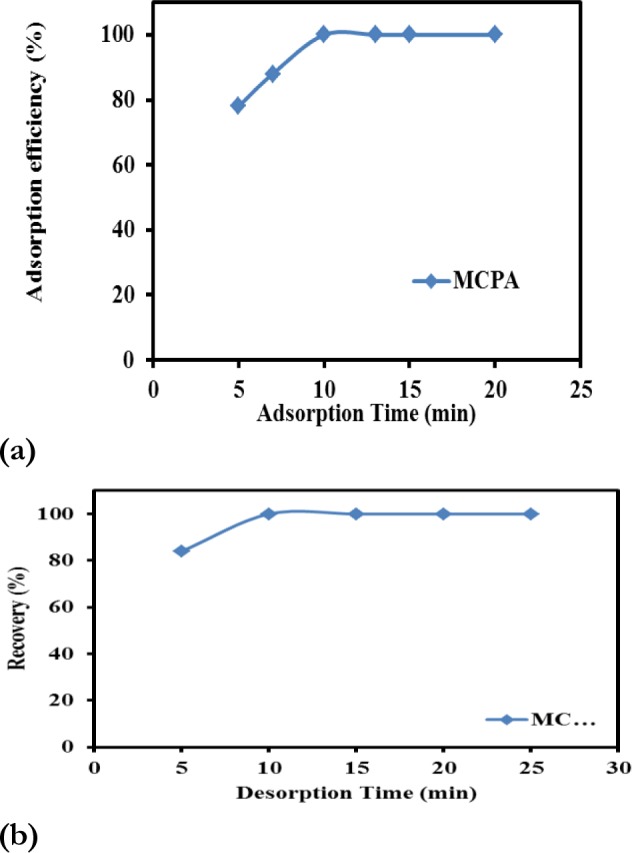
The effect of adsorption time (a) and desorption time (b) on the adsorption efficiency and recovery of MCPA from MIP-NPs
Desorption step Choice of eluent and desorption time
Complete removal of template from synthesized polymer structure is a very crucial step to guarantee the absence of memory effect. It is obvious that HOAC in methanol (20:80 v/v %) shows better recovery compared with the other solvents (Table 1). Acetic acid can affect the hydrogen bonding between template and functional monomer, so that the removal of the template would be easier. In subsequent experiments (Table 2), 1.5mL of HOAC in methanol (20:80 v/v %) was used as optimum eluent.
Table 1.
The effect of elution solvent type (The volume for each elution solvent was 5mL) on the recovery of the target molecule from MIP-NPs (The obtained results are the mean of three measurements)
| Elution solvent | Concentration (V/V (%)) | Ra ± Sb |
|---|---|---|
| Methanol | 100 | 68.0 ± 1.5 |
| Water | 100 | 50.0 ± 1.4 |
| Methanol: Acetic acid | 95:5 | 81.0 ± 1.2 |
| Water: Acetic acid | 95:5 | 66.0 ± 1.3 |
| Methanol: Acetic acid | 90:10 | 92.0 ± 1.4 |
| Methanol: Acetic acid | 85:15 | 96.0 ± 0.8 |
| Methanol: Acetic acid | 80:20 | 99.0 ± 1.0 |
| Methanol: Acetic acid | 75:25 | 89.0 ± 0.9 |
a Recovery (%) / b Standard deviation
Table 2.
The effect of elution solvent volume on the recovery of the target molecule from MIP-NPs (The obtained results are the mean of three measurements)
| Elution solvent (V/V (%)) | Volume (mL) | Ra ± Sb |
|---|---|---|
| Methanol: Acetic acid (80:20) | 5 | 99.0 ± 0.8 |
| Methanol: Acetic acid (80:20) | 4 | 99.0 ± 0.9 |
| Methanol: Acetic acid (80:20) | 3 | 99.0 ± 1.6 |
| Methanol: Acetic acid (80:20) | 2 | 99.0 ± 1.0 |
| Methanol: Acetic acid (80:20) | 1.5 | 99.0 ± 1.0 |
| Methanol: Acetic acid (80:20) | 1.0 | 78.0 ± 1.4 |
a Recovery (%)/ b Standard deviation
As shown in Fig. 6b, extraction recovery was increased up to 10 min and it was constant in longer times. Therefore, 10 minutes can be considered to be the best quantitative time for the elution of MCPA from the imprinted polymer.
Comparison of MIP and NIP
In order to compare MIP and NIP, two extractions by both MIP and NIP in water sample were assessed under the optimal conditions. The results from binding experiments showed that all imprinted polymers had much higher MCPA binding than corresponding NIPs (Table 3). It is because of the high capacity of MIP for extraction of MCPA in comparison with NIP. Template binding by NIP can be explained with nonspecific recognition because of physical adsorption and random interaction of the template molecule with functional groups in the polymer chains (47).
Table 3.
The comparison between extraction recovery and adsorption capacity of MIP and NIP
| Sorbent | Ra ± Sb | Adsorption capacity (mg g-1) |
|---|---|---|
| Molecular imprinted polymer (MIP) | 99.0 ± 1.0 | 87.4 |
| Non imprinted polymer (NIP) | 31.0 ± 1.6 | 11.6 |
a Recovery (%) / b Standard deviation
Statistical and calibration parameters
Under optimum conditions that have been described, the MIP-NPs-SPE showed a linear calibration curve within concentration ranging from 3 to 600 μg L-1. The least square equations at above linear dynamic range for water and urine samples were as follows:
Peak area = 0.296 C a (μg L-1) + 0.336, (R2 = 0.997)/a Concentration of MCPA (For water)
Peak area = 0.341 C a (μg L-1) + 0.469, (R2 = 0.994) / a Concentration of MCPA (For urine)
The limits of detections, defined as CLOD = 3Sb/m, where Sb is the standard deviation of seven replicate blank signals and m is the slope of the linear section of calibration curve after preconcentration, for a sample volume of 10 mL, were found to be 0.9 μg L-1 and 1.60 μg L-1 for MCPA in water and urine samples, respectively. The relative standard deviations for eight separate batch experiments with 50 mg of sorbent for determination of 0.1 μg MCPA in 10 mL of water were calculated to be 4.8% and 4.5% in water and urine samples, respectively.
Determination of MCPA in different water and urine samples
Table 4 shows that the results of three replicate analyses of each real sample obtained by the method are in good agreement with the spiking amounts. The repeatability of the method was demonstrated by the mean relative standard deviation (R.S.D.). Also, the relative recovery is defined as following equation:
Table 4.
Determination of MCPA in urine and different water samples
| Sample | Analyte | Cadded | Cfounded | RSD %Intra-day (N=3) | RSD % Inter-day (N=3) | Relative recovery (%) |
|---|---|---|---|---|---|---|
| Distilled water | MCPA | --- | --- | --- | --- | --- |
| 50.0 µg L-1 | 49.4 µg L- 1 | 3.2 | 3.7 | 98.8 | ||
| Tap water | MCPA | --- | --- | --- | --- | --- |
| 50.0 µg L-1 | 49.4 µg L- 1 | 3.3 | 3.6 | 98.8 | ||
| Siahrood river water | MCPA | --- | --- | --- | --- | --- |
| 50.0 µg L-1 | 49.4 µg L- 1 | 4.1 | 4.4 | 98.9 | ||
| Derka river water | MCPA | --- | 0.9 µg L-1 | --- | --- | --- |
| 50.0 µg L-1 | 50.4 µg L- 1 | 3.4 | 4.2 | 99.0 | ||
| Sea water | MCPA | --- | --- | --- | --- | --- |
| 50.0 µg L-1 | 49.3 µg L- 1 | 4.1 | 4.6 | 98.6 | ||
| Urine | MCPA | --- | --- | --- | --- | --- |
| 50.0 µg L-1 | 48.7 µg L- 1 | 4.2 | 4.5 | 97.4 |
RR = (Cfound - Creal / Cadded) × 100
where Cfound, Creal, and Cadded are the concentrations of analyte after addition of known amount of standard in the real sample, the concentration of analyte in real sample and the concentration of known amount of standard which was spiked to the real sample, respectively.
Therefore, MIP-NPs-SPE can be used as a reliable sample treatment technique for trace determination of MCPA in water and urine samples. Figure 7a and Fig. 7b, provides chromatograms for determination of MCPA in spiked urine and sea water sample.
Fig. 7.

The chromatograms of the (A) non-spiked and (B) spiked (100 μg L-1) sea water sample after extraction under optimum conditions (a) and the chromatograms of the (A) non-spiked and (B) spiked (100 μg L-1) urine sample after extraction under optimum conditions (b)
Discussion
According to the results, MIP-NPs can be used as a nano-size selective solid phase for very fast extraction of ultra trace amounts of MCPA in complex matrices. The obtained results of the Characterization can prove the successful engineering of cavity for MCPA in the structure of synthesized MIP-NPs.
Another important characteristic of the MIP-NPs, is the short sorption and desorption time for quantitative extraction of the analyte from aqueous sample into solid phase. That is because of nano structure and higher surface-to-area ratio of particles. The prepared MIP in form of nanosize particles have also many advantages such as: rapid extraction of analyte from the real samples, high adsorption capacity, high porosity and surface area, greater recognition binding ability in the sensing of MCPA without a time-consuming process of grinding and sieving compared with traditional MIPs, so there is better accessibility of the active site (48).
Conclusion
In this work, The MIP-NPs has been synthesized by synthesized by precipitation polymerization technique. The developed method was successfully applied to determine MCPA in urine and different water samples. The analytical characteristics such as good precision and preconcentration factor, high quantiities of recoveries, wide dynamic linear range, and low detection of limit were achieved due to the powerful efficiency of the MIP-NPs-SPE method. Other advantages of suggested method are: lack of matrix effect, low consumption of organic solvent, environmentally friendly, safe, simplicity and selectivity. Due to relatively high preconcentration factor, trace amount of MCPA at μg L-1 levels can be determined and separated by MIP-NPs.
Ethical considerations
Ethical issues (Including plagiarism, Informed Consent, misconduct, data fabrication and/or falsification, double publication and/or submission, redundancy, etc) have been completely observed by the authors.
Acknowledgements
The authors would like to appreciate Tehran University of Medical Sciences for funding and supporting this project. The authors also thank Mr Mirghani Seyed-Someh and Mrs Safora Arefian for their kind technical assistance through this study. The authors declare that there is no conflict of interests.
Reference
- Caux PY, Kent R, Bergeron V, Fan G, Macdonald D (1995). Environmental fate and effects of MCPA: A Canadian perspective. Crit Rev Env Sci Technol, 25: 313–376. [Google Scholar]
- Roberts T, Hutson D, Lee P, Nicholls P (1998). Pt. 1: Herbicides and plant growth regulators. ed. Cambridge: Royal Society of Chemistry. [Google Scholar]
- Burns CJ, Swaen GM (2012). Review of 2, 4-dichlorophenoxyacetic acid (2, 4-D) biomonitoring and epidemiology. Crit Rev Toxicol, 42: 768–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heederik D, Hooiveld M, Bas Bueno-de-Mesquita H (1998). Modelling of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin levels in a cohort of workers with exposure to phenoxy herbicides and chlorophenols. Chemosphere, 37: 1743–1754. [DOI] [PubMed] [Google Scholar]
- Kogevinas M, Becher H, Benn T, Bertazzi PA, Boffetta P, Bueno-de-Mesqurta HB, Coggon D, Colin D, Flesch-Janys D, Fingerhut M (1997). Cancer mortality in workers exposed to phenoxy herbicides, chlorophenols, and dioxins an expanded and updated international cohort study. Am J Epidemiol, 145: 1061–1075. [DOI] [PubMed] [Google Scholar]
- de Mesquita H, Doornbos G, van der Kuip DA, Kogevinas M, Winkelmann R (1993). Occupational exposure to phenoxy herbicides and chlorophenols and cancer mortality in the Netherlands. Am J Ind Med, 23: 289–300. [DOI] [PubMed] [Google Scholar]
- Addorisio V, Esposito S, Sannino F (2010). Sorption capacity of mesoporous metal oxides for the removal of MCPA from polluted waters. J Agric Food Chem, 58: 5011–5016. [DOI] [PubMed] [Google Scholar]
- Gleick PH (1998). Water in crisis: paths to sustainable water use. Ecological applications, 8: 571–579. [Google Scholar]
- Hamilton D, Ambrus A, Dieterle R, Felsot A, Harris C, Holland P, Katayama A, Kurihara N, Linders J, Unsworth J (2003). Regulatory limits for pesticide residues in water (IUPAC Technical Report). Pure Appl Chem, 75: 1123–1155. [Google Scholar]
- Waldner G, Friesl-Hanl W, Haberhauer G, Gerzabek MH (2012). Differences in sorption behavior of the herbicide 4-chloro-2-methylphenoxyacetic acid on artificial soils as a function of soil pre-aging. J soil sediment, 12: 1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez I, Rubi E, Gonzalez R, Quintana J, Cela R (2005). On-fibre silylation following solid-phase microextraction for the determination of acidic herbicides in water samples by gas chromatography. Anal Chim Acta, 537: 259–266. [Google Scholar]
- Biesaga M, Jankowska A, Pyrzyńska K (2005). Comparison of different sorbents for solid-phase extraction of phenoxyalkanoic acid herbicides. Microchimica Acta, 150: 317–322. [Google Scholar]
- Catalina MI, Dallüge J, Vreuls RJJ, Brinkman UAT (2000). Determination of chlorophenoxy acid herbicides in water by in situ esterification followed by in-vial liquid-liquid extraction combined with large-volume on-column injection and gas chromatography-mass spectrometry. J Chromatogr A, 877: 153–166. [DOI] [PubMed] [Google Scholar]
- Laganà A, Bacaloni A, De Leva I, Faberi A, Fago G, Marino A (2002). Occurrence and determination of herbicides and their major transformation products in environmental waters. Anal Chim Acta, 462: 187–198. [Google Scholar]
- Vink M, Van der Poll J (1996). Gas chromatographic determination of acid herbicides in surface water samples with electron-capture detection and mass spectrometric confirmation. J Chromatogr A, 733: 361–366. [Google Scholar]
- Shahtaheri SJ, Ghamari F, Golbabaei F, Rahimi-Froushani A, Abdollahi M (2005). Sample preparation followed by high performance liquid chromatographic (HPLC) analysis for monitoring of muconic acid as a biomarker of occupational exposure to benzene. Int J Occup Saf Ergon, 11: 337–388. [DOI] [PubMed] [Google Scholar]
- Shahtaheri SJ, Ibrahimi L, Golbabaei F, Hosseini M (2007). Solid phase extraction for 1-hydroxypyrene as a biomarker of exposure to PAHs prior to high performance liquid chromatography, Iranian Journal of Chemistry and Chemical Engineering, 26: 75–81. [Google Scholar]
- Shahtaheri SJ, Khadem M, Golbabaei F, Rahimi-Froushani A (2007). Optimization of sample preparation procedure for evaluation of occupational and environmental exposure to nickel, Iranian J Publ Health, 36: 73–81. [Google Scholar]
- Shahtaheri SJ, Abdollahi M, Golbabaei F, Rahimi-Froushani A (2008). Monitoring of mandelic acid as a biomarker of environmental and occupational exposures to styrene. Int J Environl Res, 2: 169–176. [Google Scholar]
- Heidari HR, Shahtaheri SJ, Golbabaei F, Alimohamadi M, Rahimi-Froushani A (2009). Trace analysis of xylene in occupational exposures monitoring. Iranian J Publ Health, 38: 89–99. [Google Scholar]
- Jing T, Wang Y, Dai Q, Xia H, Niu J, Hao Q, Mei S, Zhou Y (2010). Preparation of mixed-templates molecularly imprinted polymers and investigation of the recognition ability for tetracycline antibiotics. Biosens Bioelectron, 25: 2218–2224. [DOI] [PubMed] [Google Scholar]
- Ebrahimzadeh H, Behbahani M, Yamini Y, Adlnasab L, Asgharinezhad AA (2013). Optimization of Cu(II)-ion imprinted nanoparticles for trace monitoring of copper in water and fish samples using a Box-Behnken design. React funct polym Polymers, 73: 23–29. [Google Scholar]
- Hoshina K, Horiyama S, Matsunaga H, Haginaka J (2009). Molecularly imprinted polymers for simultaneous determination of antiepileptics in river water samples by liquid chromatography-tandem mass spectrometry. J Chromatogr A, 1216: 4957–4962. [DOI] [PubMed] [Google Scholar]
- Behbahani M, Barati M, Bojdi MK, Pourali AR, Bagheri A, Tapeh NAG (2013). A nanosized cadmium (II)-imprinted polymer for use in selective trace determination of cadmium in complex matrices. Microchim Acta, 180: 1117–1125. [Google Scholar]
- Prieto A, Schrader S, Bauer C, Möder M (2011). Synthesis of a molecularly imprinted polymer and its application for microextraction by packed sorbent for the determination of fluoroquinolone related compounds in water. Anal Chim Acta, 685: 146–152. [DOI] [PubMed] [Google Scholar]
- Lucci P, Núñez O, Galceran MT (2011). Solid-phase extraction using molecularly imprinted polymer for selective extraction of natural and synthetic estrogens from aqueous samples. J Chromatogr A, 1218: 4828–4833. [DOI] [PubMed] [Google Scholar]
- Behbahani M, Bagheri A, Taghizadeh M, Salarian M, Sadeghi O, Adlnasab L, Jalali K (2013). Synthesis and characterisation of nano structure lead (II) ion-imprinted polymer as a new sorbent for selective extraction and preconcentration of ultra trace amounts of lead ions from vegetables, rice, and fish samples. Food Chem, 138: 2050–2056. [DOI] [PubMed] [Google Scholar]
- Behbahani M, Taghizadeh M, Bagheri A, Hosseini H, Salarian M, Tootoonchi A (2012). A nanostructured ion-imprinted polymer for the selective extraction and preconcentration of ultra-trace quantities of nickel ions. Microchim Acta, 178: 429–437. [Google Scholar]
- Koohpaei AR, Shahtaheri SJ, Ganjali MR, Forushani AR, Golbabaei F (2008). Application of multivariate analysis to the screening of molecularly imprinted polymers (MIPs) for ametryn. Talanta, 75: 978–986. [DOI] [PubMed] [Google Scholar]
- Koohpaei AR, Shahtaheri SJ, Ganjali MR, Forushani AR, Golbabaei F (2009). Optimization of solid-phase extraction using developed modern sorbent for trace determination of ametryn in environmental matrices. J Hazard Mater, 170: 1247–1255. [DOI] [PubMed] [Google Scholar]
- Alexander C, Andersson HS, Andersson LI, Ansell RJ, Kirsch N, Nicholls IA, O’Mahony J, Whitcombe MJ (2006). Molecular imprinting science and technology: a survey of the literature for the years up to and including 2003. J Mol Recognit, 19: 106–180. [DOI] [PubMed] [Google Scholar]
- Haupt K (2003). Peer reviewed: molecularly imprinted polymers: the next generation. Anal Chem, 75: 376 A-383 A. [DOI] [PubMed] [Google Scholar]
- Zimmerman SC, Lemcoff NG (2004). Synthetic hosts via molecular imprinting-are universal synthetic antibodies realistically possible? Chem Commun: 5–14. [DOI] [PubMed] [Google Scholar]
- Maeda M, Bartsch RA (1998) Molecular and ionic recognition with imprinted polymers: a brief overview. ACS symposium series, ACS Publications, pp. 1–9. [Google Scholar]
- Le Moullec S, Truong L, Montauban C, Begos A, Pichon V, Bellier B (2007). Extraction of alkyl methylphosphonic acids from aqueous samples using a conventional polymeric solid-phase extraction sorbent and a molecularly imprinted polymer. J Chromatogr A, 1139: 171–177. [DOI] [PubMed] [Google Scholar]
- Tamayo FG, Casillas JL, Martin-Esteban A (2003). Highly selective fenuron-imprinted polymer with a homogeneous binding site distribution prepared by precipitation polymerisation and its application to the clean-up of fenuron in plant samples. Anal Chim Acta, 482: 165–173. [Google Scholar]
- Haginaka J, Sanbe H (2000). Uniform-sized molecularly imprinted polymers for 2-arylpropionic acid derivatives selectively modified with hydrophilic external layer and their applications to direct serum injection analysis. Anal Chem, 72: 5206–5210. [DOI] [PubMed] [Google Scholar]
- Turiel E, Martín-Esteban A, Tadeo JL (2007). Molecular imprinting-based separation methods for selective analysis of fluoroquinolones in soils. J Chromatogr A, 1172: 97–104. [DOI] [PubMed] [Google Scholar]
- He J, Lv R, Zhu J, Lu K (2010). Selective solid-phase extraction of dibutyl phthalate from soybean milk using molecular imprinted polymers. Anal Chim Acta, 661: 215–221. [DOI] [PubMed] [Google Scholar]
- Yan H, Qiao J, Pei Y, Long T, Ding W, Xie K (2012). Molecularly imprinted solid-phase extraction coupled to liquid chromatography for determination of Sudan dyes in preserved beancurds. food chem, 132: 649–654. [DOI] [PubMed] [Google Scholar]
- Djozan D, Ebrahimi B (2008). Preparation of new solid phase micro extraction fiber on the basis of atrazine-molecular imprinted polymer: Application for GC and GC/MS screening of triazine herbicides in water, rice and onion. Anal Chim Acta, 616: 152–159. [DOI] [PubMed] [Google Scholar]
- Kan X, Zhao Y, Geng Z, Wang Z, Zhu J-J (2008). Composites of multiwalled carbon nanotubes and molecularly imprinted polymers for dopamine recognition. J Phys Chem C, 112: 4849–4854. [Google Scholar]
- Shea KJ, Yan M, Roberts M, Cremer PS, Crooks RM. Materials Research Society Symposium Proceedings. Volume 723 Molecularly Imprinted Materials-Sensors and Other Devices. Symposia Held in San Francisco, California on April 2–5, 2002: DTIC Document2002. [Google Scholar]
- Tokonami S, Shiigi H, Nagaoka T (2009). Review: Micro-and nanosized molecularly imprinted polymers for high-throughput analytical applications. Anal Bioanal Chem, 641: 7–13. [DOI] [PubMed] [Google Scholar]
- Haginaka J (2008). Monodispersed, molecularly imprinted polymers as affinity-based chromatography media. J Chromatogr B, 866: 3–13. [DOI] [PubMed] [Google Scholar]
- Ye L, Cormack PA, Mosbach K (1999). Molecularly imprinted monodisperse micros-pheres for competitive radioassay. Anal Commun, 36: 35–38. [Google Scholar]
- Abouzarzadeh A, Forouzani M, Jahanshahi M, Bahramifar N (2012). Synthesis and evaluation of uniformly sized nalidixic acid-imprinted nanospheres based on precipitation polymerization method for analytical and biomedical applications. J Mol Recognit, 25: 404–413. [DOI] [PubMed] [Google Scholar]
- Say R, Ersoz A, sener I, Atılır A, Diltemiz S, Denizli A (2005). Comparison of adsorption and selectivity characteristics for 4-nitrophenol imprinted polymers prepared via bulk and suspension polymerization. Sep Sci Technol, 39(15): 3471–3484. [Google Scholar]