

Abstract
Three distinct neurodevelopmental disorders arise primarily from deletions or duplications that occur at the 15q11-q13 locus: Prader-Willi syndrome (PWS), Angelman syndrome (AS), and 15q11-q13 duplication syndrome (Dup15q syndrome). Each of these disorders results from the loss of function or over-expression of at least one imprinted gene. Here we discuss the clinical background, genetic etiology, diagnostic strategy, and management for each of these three disorders.
Keywords: Prader-Willi syndrome, Angelman syndrome, chromosome 15q11-q13 duplication, genomic imprinting, copy number variation, DNA methylation, UBE3A, SNRPN
Introduction
Chromosome 15q11-q13 is a region that harbors several genes regulated by genomic imprinting, a phenomenon in which genes are expressed preferentially from one parental allele. As a result, genes subject to regulation by genomic imprinting are functionally haploid, having only a single functional copy. Three distinct neurodevelopmental disorders arise primarily from deletions or duplications that occur at the 15q11-q13 locus: Prader-Willi syndrome (PWS), Angelman syndrome (AS), and 15q11-q13 duplication syndrome (Dup15q syndrome). Each of these disorders results from the loss of function or over-expression of at least one imprinted gene. They each occur with a frequency of approximately 1/15,000 to 1/30,000 live births.
The deletions and duplications of chromosome 15q11-q13 that cause PWS, AS or Dup15q syndrome are mediated by local DNA repeats that occur at the common breakpoints. There are five such elements, which comprise breakpoints 1 through 5 (BP1-BP5). Deletions involving either BP1 or BP2 and BP3 are most common, while the duplications are more complicated, but can frequently involve BPs 4 and 5 and are discussed below. A map of the 15q11-q13 genetic region is shown in Figure 1. Here we discuss the clinical background, genetic etiology, diagnostic strategy, and management for each of these three disorders.
Figure 1. Map of 15q11-q13 region.
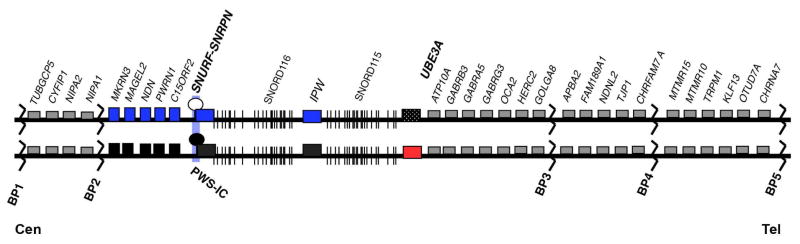
Individual genes are depicted as boxes with their respective names above them. Genes shown in blue and red are imprinted and expressed from the paternal and maternal allelles, respectively. Black boxes mark the silenced allele. Gray boxes indicate genes expressed from both parental alleles. BP1-4 indicate the common breakpoints 1–4. PWS-IC indicates the Prader-Willi imprinting center. Cen and Tel mark the centromere and telomere, respectively to indicate orientation relative to the rest of the chromosome.
Prader-Willi syndrome
1. Clinical background of the disease
Prader-Willi syndrome (PWS) is characterized by hypotonia; failure to thrive with poor suck; hypogonadism; short stature with small hands and feet; hyperphagia leading to morbid obesity, beginning during early childhood; developmental delay/intellectual disability; and behavioral issues, including obsessive compulsive disorder (Table 1). Characteristic facial features are also evident.
Table 1.
Features of Prader-Willi syndrome
| Consistent (100%) | Frequent (80%) | Associated (20–80%) |
|---|---|---|
| Hypotonia | Hypopigmentation | Speech articulation defects |
| Failure to thrive | Behavioral problems | Autism |
| Feeding difficulty | Developmental delay | |
| Hypogonadism | Short stature, if untreated with growth hormone | |
| Obesity, in absence of intervention | Distinctive facial features | |
| Hyperphagia | Sleep disturbances | |
| Small hands/feet |
Hypotonia
Hypotonia becomes evident during pregnancy as decreased fetal movement and atypical presentation at delivery. Assisted delivery and caesarean section are increased with PWS births1. Hypotonia is nearly universal in infants with PWS, thus, a molecular test for PWS should be performed whenever neonatal hypotonia is observed. Infants with PWS are lethargic with reduced movement, often having a weak cry, poor suck, and failure-to-thrive2. The infantile hypotonia improves, but mild to moderate hypotonia persists throughout life in PWS children and adults.
Hypogonadism
Hypogonadism is often evident at birth as gonadal hypoplasia in both males and females. Males typically have a small scrotum and may have a small penis. Unilateral or bilateral cryptorchidism is frequent. Females often have a small labia minoris and clitoris. Puberty can be delayed or disorganized in adolescents with PWS, and most adults are infertile. Hypogonadism is thought to result from both hypothalamic dysfunction resulting in low levels of gonadotropins (and therefore gonadal hormones), as well as primary gonadal deficiency3.
Growth deficits
Growth deficits associated with PWS likely begin in utero. Infants with PWS are typically 15–20% smaller than their siblings1,4. Short stature is often apparent in childhood and persists through adulthood. The lack of a pubertal growth spurt may exacerbate the growth deficit5. Hands and feet are often particularly small, usually averaging below the 5th percentile. Growth deficits are caused, at least in part, by growth hormone deficiency6, which appears to result from hypothalamic-pituitary dysfunction. Indeed, growth hormone replacement therapy improves body mass index and muscle mass in children with PWS, and may even improve body habitus in adults with PWS7–9.
Hyperphagia and obesity
Individuals with PWS pass through a series of seven nutritional phases relating to appetite and weight gain10. The first phase, phase 0, occurs from prenatal to birth with reduced fetal movement and lower birth weight than siblings. The second phase, phase 1a, occurs from birth to approximately 9 months and is characterized by failure-to-thrive with difficulty feeding and decreased appetite. The third phase, phase 1b, occurs from approximately 9 months to 2 years and is marked by improved feeding and appetite and appropriate growth. The fourth phase, phase 2a, occurs between approximately 2 and 4.5 years. This phase involves weight gain without increased appetite or excess calories. The fifth phase, phase 2b, occurs between approximately 4.5 and 8 years. This phase involves continued weight gain with increased appetite and calories, however, individuals in this phase can still feel full. The sixth phase, phase 3, lasts from approximately age 8 to adulthood. This phase is characterized by extreme hyperphagia, and individuals rarely feel full. If food consumption is not limited, obesity is inevitable. The seventh and final phase, phase 4, occurs throughout adulthood. During this phase, appetite is no longer insatiable.
The etiology of hyperphagia in PWS is poorly understood, although it is likely to result from hypothalamic dysfunction5. It has been suggested that increased ghrelin levels may underlie hyperphagia in individuals with PWS11, however, there are no consistently identified hormonal abnormalities that explain the hyperphagic behavior in PWS. Nonetheless, it is not unusual for individuals in phase 3 to exhibit extreme food seeking behaviors, including consumption of non-food items, hoarding of food, or stealing money to buy food5.
The etiology of obesity in PWS manifold. The onset of weight gain in phase 2a, before increased appetite occurs suggests that individuals with PWS have lower caloric requirement10. This is due, at least in part, to decreased resting energy expenditure caused by decreased activity and decreased lean muscle mass compared with neurotypical individuals12. Increased appetite and hyperphagia also contributes to obesity. The obesity in PWS primarily occurs in the abdomen, buttocks, and thighs; there is less visceral fat than would be expected13. Obesity is the major cause of morbidity and mortality in PWS.
Developmental delay/intellectual disability
Motor development and language skills are delayed in most PWS individuals, although nearly all individuals with PWS walk and can effectively communicate verbally. Individuals with PWS have mild to moderate intellectual disability14,15 and poor academic achievement is typical and may be exacerbated by behavioral difficulties in addition to developmental and intellectual disability14.
Behavioral difficulties
Up to 90% of individuals with PWS have characteristic behaviors including stubbornness, temper tantrums, manipulative behaviors, compulsivity, and difficulty with change in routine16. Features similar to obsessive compulsive disorder and skin picking are common. Some of the behaviors are consistent with autism, and indeed some individuals with PWS meet diagnostic criteria for autism. This may be more common in individuals with maternal uniparental disomy14. Psychosis is also prevalent in approximately 10–20% of adults with PWS17,18.
Other features
Facial features associated with PWS include almond shaped eyes; narrow, but prominent nasal bridge; high, narrow forehead; thin upper lip; and downturned mouth. Other features associated with PWS include sleep disorders including sleep apnea19, strabismus (60–70%)5, scoliosis (40–80%)5, light colored hair and skin, striae, and tapering of fingers.
2. Recent advances in genetics and pathomechanism of diseases
Prader-Willi syndrome is caused by the loss of paternally-inherited chromosome 15q11.2-q13. The loss of expression from this chromosomal region typically occurs by one of three mechanisms: 1.) approximately 70% of individuals with PWS have a large deletion of the entire 15q11-q13 imprinted region; 2.) approximately 25% of individuals with PWS have maternal uniparental disomy (matUPD) in which both copies of chromosome 15 have been inherited from the mother; and 3.) less than 5% of individuals with PWS have an imprinting defect that causes paternal 15q11-q13 to behave as though it were inherited from the mother5. Notably, there are no instances of a point mutation in any gene causing PWS, suggesting that PWS is a true contiguous gene syndrome, resulting from the loss of more than one gene.
In the vast majority of PWS individuals, approximately 20 paternally-expressed genes are missing, including Necdin (NDN), Makorin ring 3 (MKRN3), Mage-like 2 (MAGEL2), PWRN2, PWRN1, Nuclear pore associated protein 2 (NPAP2), SNURF, SNRPN, SNORD109A, SNORD116, SNORD115, SNORD109B, and SNHG14 (formerly known as UBE3A-ATS). Typical individuals with deletions of paternal NDN, MKRN3, and MAGEL2 and with deletions of paternal SNORD115 and distal portions of SNHG14 have been identified, suggesting that these genes are not causative for PWS. Conversely, individuals with PWS have been identified with smaller, atypical deletions that have narrowed down the PWS critical region to a 91 kb region that includes SNORD109A, SNORD116, and IPW20–23. Mouse models further suggest that deletions of Snord116 are sufficient to cause PWS, however, no human patient with PWS caused by deletion of SNORD116 alone has been reported to date.
The SNORD genes that are located in 15q11.2-q13 are orphan CD box snoRNA genes that have been reported to modify mRNAs24,25,26 and rRNAs27, as well as act as small interfering RNAs (siRNAs)28. SNORD116 is actually a cluster of 29 similar, but not identical snoRNA genes that although evolutionarily related may affect different mRNAs and rRNAs. It is not known how loss of SNORD116 results in the phenotypic manifestations of PWS.
3. Diagnostic Strategy
Methylation analysis
DNA methylation analysis using southern blot or methylation-specific PCR will diagnose PWS in 99 % of cases, including all three classes; paternal deletion, maternal UPD, and imprinting defect5 (Figure 2). The most widely used assays target the 5’ CpG island of the SNURF-SNRPN (SNRPN) locus, a region known as the imprinting center. The promoter, exon 1 and intron 1 of SNRPN are unmethylated on the paternal allele and thus expressed, and methylated on the maternal, non-expressed allele. A normal individual will have both a methylated and unmethylated SNRPN allele, whereas individuals with PWS will have only the maternally methylated allele5.
Figure 2. Diagnostic strategy for Prader-Willi syndrome.

Blue boxes indicate the diagnostic test, pink boxes indicate the diagnosic decision.
Methylation analysis allows for identification of patients with PWS, but provides no information about the molecular class of the disease. As discussed, 65–75 % of cases results from a deletion at 15q11.2–q 13, 20–30 % result from uniparental disomy in which both copies of chromosome 15 are maternally inherited and less than 5% are caused by some form of imprinting defect5. Differentiation of the molecular class of PWS allows the physician to provide more accurate prognostic information and is crucial for accurate recurrence risk counseling. Testing should proceed in the order outlined below, from most to least common cause.
Search for a deletion. FISH with a SNRPN probe is the most cost efficient means of identifying a deletion. If done with simultaneous chromosomal analysis, rare cases caused by translocation or inversion will be identified. Increasingly, CMA (chromosomal microarray analysis) is sent instead of FISH, and allows for accurate measurement of deletion size in addition to providing information about other genomic changes, if present. The extent of the deletion is expected to become increasingly important as our understanding of genotype-phenotype correlations in PWS grows. CMA is more expensive than FISH, but is increasingly available, and typically is the first test done when a patient with a suspected genetic condition is evaluated, particularly if PWS was not initially considered. In this case, methylation testing is important as a confirmatory test, as identification of a deletion does not distinguish between Prader-Willi and Angelman Syndromes, which may have considerable clinical overlap in the young child. Additionally, CMA may identify rare patients that have deletions that do not include the imprinting center.
UPD testing. DNA polymorphism analysis of chromosome 15 loci on the proband and parents’ DNA will identify cases of maternal uniparental disomy in which both copies of chromosome 15 are maternally inherited29.
Imprinting Defect. Sequencing of the imprinting center can be done in specialized laboratories. Mutations are found in 15 % of those with imprinting defects, with the remainder caused by epimutations. Epimutations carry a low recurrence risk whereas an IC mutation on the paternal allele may be associated with a 50% recurrence risk, thus searching for a mutation provides important information to families about future risk.
Additional Options
MPLA testing is another increasingly popular option as a first line test for diagnosis of PWS, particularly in Europe. MPLA testing has the ability to assess methylation at 5 sites in the PWS region, as opposed to one site in the standard methylation assay. It also will detect a deletion, if present, but cannot distinguish between UPD and imprinting defects.
4. Current Management
Diet and Nutrition
Failure to thrive in infancy results from poor suck in the setting of hypotonia. Special nipples or gavage feeding are often required. Close monitoring of growth parameters is required in the first year of life 2. If failure to thrive is noted, despite adequate caloric intake, testing for hypothyroidism is indicated, as this is not uncommon in infancy30.
As weight gain begins to increase from the age of 2 onwards, careful supervision of caloric intake is necessary. Weight gain often begins after the age of 2, though appetite increase is not typical until after age 4. It is important to monitor food intake, before the onset of obesity. Nutritional supervision to assess appropriate intake and supplementation of vitamin D, calcium and other nutrients is recommended. Locking of cupboards and refrigerator is often necessary as appetite increases. Evidence suggests that early dietary intervention with a controlled prescribed diet as early as 14 months of age may result in a normal BMI 31. Continued monitoring of diet and weight is central to long-term health including avoidance of diabetes mellitus and other obesity related complications.
Hormonal / Endocrine
Treatment with Growth Hormone (GH) is now recommended as standard of care for children with PWS. Early treatment appears beneficial with normalization of height, increase in muscle mass and decrease in fat mass. Treatment should begin between 4 months and two years of age as benefits in head circumference, gross motor and language development and cognition have been demonstrated with early treatment 32,33. Due to concern about the possibility of an increase in the rate of sudden death from upper airway obstruction in the first months of treatment, a sleep study is recommended prior to initiating treatment, 6 to 12 weeks after initiation of treatment and on an annual basis thereafter34. Children with PWS are at increased risk of obstructive and central apnea and this risk may rise with GH treatment, possibly due to lymphoid hyperplasia. While receiving GH treatment, close monitoring for scoliosis, hypothyroidism, diabetes and elevation of IGF-1 is suggested32. Treatment with GH may need to continue into adulthood, as recent studies suggest that BMI may increase significantly following cessation of GH therapy35.
Cryptorchidism should be identified with referral to urology and treatment if noted in infancy. Treatment of hypogonadism should be considered, with human chorionic gonadotrophin (hCG) or testosterone, to assist with testicular descent, as well as scrotal and phallic development and growth2. In early adolescence, replacement of sex hormones may be appropriate; low dose estrogen or combined estrogen/progestin for girls beginning at age 11 to 12, particularly if there is emenorrhea/oligomenrrhea or low bone mineral density in the setting of low estradiol levels. Testosterone or hCG (increases endogenous testosterone production) in the setting of hypogonadism in boys beginning at 12 to 13 years36.
Monitoring of free thyroxine in addition to TSH should be done annually in childhood36. Awareness of elevated risk for adrenal insufficiency is important as it may be present in up to 60% of children. Central adrenal insufficiency has been observed in PWS, though frequency is unclear. Consideration of measuring ACTH and Cortisol levels with illness is appropriate and some have advocated stress dose steroids with illness or before surgery36. Monitoring for Type 2 Diabetes Mellitus is essential in adults.
Behavioral and Educational
Physical therapy beginning in infancy assists with motor skills development with speech therapy often warranted by 2 years of age. Requirement for educational support should be anticipated including personal classroom aides and behavior management given the frequency of challenging behaviors such as tantrums, compulsive and stubborn behaviors. Serotonin reuptake inhibitors can be helpful for severe behavioral issues, including psychosis, which may emerge in adolescence37. Adolescents and adults often are successful residing in a group home where attention to daily exercise and diet can be emphasized.
Other
Annual assessment for scoliosis should begin in early childhood. Opthalmological evaluation for strabismus and impaired acuity should be done in the first year of life and continue thereafter. Given the increased risk for osteoporosis, bone density studies (DEXA studies) are recommended beginning in adolescence and continuing to adulthood38.
Future Potential Therapies
Several medication trials are ongoing, aimed at addressing the hyperphagia and associated symptoms of PWS. Oxytocin nasal spray is being studied, as there is a reduction in oxytocin producing neurons in the hypothalamic periventricular nucleus in individuals with PWS. A recent publication, however, did not demonstrate benefit in 30 individuals in an 18-week double-blind, placebo-controlled crossover trial39. Other trials are ongoing. A trial of a candidate obesity drug called Beloranib is also underway. Belonarib is an inhibitor of methionine aminopeptidase-2 and works to reduce fatty acid synthesis, insulin levels, and food consumption. It also increases mobilization of fats and energy expenditure40.
Recommendation for family counseling
Family counseling is recommended. Most deletions, UPD, and epimutations are associated with a low recurrence risk. However, IC mutations and some translocations may be associated with a 50% recurrence risk.
Angelman syndrome
1. Clinical background of the disease
Angelman syndrome (AS) is characterized by developmental delay, intellectual disability, absent speech, seizures, ataxic gait, easily excitable happy demeanor, and characteristic facies (Table 2). This disorder has been referred to as “happy puppet syndrome” due to the ataxic gait and disposition of children with AS.
Table 2.
Features of Angelman syndrome
| Consistent (100%) | Frequent (80%) | Associated (20–80%) |
|---|---|---|
| Developmental delay | Seizures | Hypotonia |
| Ataxia and/or tremors | Microcephaly | Strabismus |
| Absent speech | Frequent drooling, mouthing behaviors | |
| Happy demeanor: including hand flapping, frequent laughter/smiling | Protruding tongue, tongue thrusting | |
| Wide mouth, wide spaced teeth Sleep disturbances | ||
| Sleep disturbances | ||
| Fascination with water | ||
| Anxiety |
Developmental delay/intellectual disability
Most infants with AS do not show any signs of the disorder at birth. However, delayed attainment of gross and fine motor skills, language, and social skills are usually evident within the first year of life41. Motor skill delays can be severe, and many individuals with AS are not able to walk. Tremors can further complicate fine motor skill development. Toilet training is typically severely delayed, but is achieved in many with adults with AS. Functionally, individuals with AS only reach a developmental level of approximately 24 to 30 months42. Cognitive ability is severely impaired, however, cognition is difficult to ascertain due to the profound lack of speech, hyperactivity, and inability to pay attention in individuals with AS. Adults with AS are not capable of independent living, although many can perform tasks with supervision, can dress themselves, and can use feeding utensils43,44.
Speech
Language development in individuals with AS is severely impaired. Most individuals with AS are entirely non-verbal, some will speak a few words, and a rare few have some phrase speech. Augmentative communication devices and sign language can be successfully used to communicate with individuals with AS. Their receptive communication exceeds expressive communication45.
Epilepsy
Epilepsy occurs in 80 to 95% of those with Angelman Syndrome, typically with onset before 3 years of age44,46.. Angelman Syndrome is typically associated with generalized epilepsy, though focal seizures occur in up to one third, often in combination with other seizure types 44,46,47. Myoclonic, atypical absence, generalized tonic clonic and atonic seizures are most common, and status epilepticus, frequently myoclonic or non-convulsive, has been reported to occur in up to 90%48. Epilepsy tends to be more severe in early childhood, often easing as children reach puberty, though epilepsy risk appears to persist in to adulthood44. Epilepsy tends to be more severe in those with a maternal deletion, as does disease severity in general47,49. EEG often shows a characteristic pattern, most classically with posterior predominant spike and sharp waves mixed with high amplitude sharply contoured 3 to4 Hz activity.
Movement disorder
Ataxic gait and/or tremulous movement of the limbs is a consistent finding in AS50. Individuals with AS have a slow, stiff legged gait. Typical posture includes raised arms, flexed at the elbows and wrists. Hand flapping is common when walking or excited. Movements are generally jerky and abrupt. The specific brain region responsible for the movement disorder is not known.
Happy demeanor
Individuals with AS have a characteristic happy demeanor and are easily excitable. They have frequent, sometimes inappropriate laughter. Hyperactivity and hypermotoric activity often accompany the happy disposition. Individuals with AS are typically highly social, with social interest beginning in infancy. Most children with AS are eager to communicate, despite difficulty in doing so41. Social disinhibition is common and there is little fear of strangers.
Other features
Other behavioral features associated with AS include stereotypic movements (such as hand flapping), difficulty sleeping, and anxiety. Individuals with AS are frequently fascinated with water. Facial features associated with AS include lightly pigmented skin, hair, and eyes; strabismus; tongue protrusion; prognathia; and widely spaced teeth.
2. Recent advances in genetics and pathomechanism of diseases
AS is caused by the lack of function of maternal UBE3A51,52. This arises due to one of four mechanisms: 1.) deletion of maternal 15q11.2-q13 is found in approximately 74% of individuals with AS, 2.) loss of function mutation of maternal UBE3A is found in approximately 11% of individuals with AS, 3.) paternal uniparental disomy (UPD) is found in approximately 8% of individuals with AS, and 4.) imprinting defect is found in approximately 7% of individuals with AS53. In contrast to PWS, the presence of individuals with AS due to point mutations in the maternal copy of the UBE3A gene demonstrate that AS is a single gene disorder, although other genes in the deletion region can contribute to the severity of AS.
The mechanism by which loss of UBE3A causes AS is still not completely understood. The UBE3A protein, also known as E6AP, is an E3 ubiquitin ligase, which transfers an activated ubiquitin from an E2 ubiquitin ligase to its target protein54. UBE3A/E6AP typically adds lysine 48 (K48)-linked polyubiquitin chains to its substrates, thus targeting them for degradation by the proteasome55. Some substrates of UBE3A/E6AP, such as HHR23A56, RPN1057, and EPHEXIN558 have been identified. HHR23A (also known as RAD23A) stimulates nucleotide excision repair, RPN10 is a subunit of the proteasome and EPHEXIN5 is a rho-GEF involved in regulating dendritic spine density.
Mouse models of AS have suggested some underlying neuronal pathologies associated with the disorder. Deficits in long-term potentiation59,60, inhibitory CAMKII phosphorylation60, presynaptic release probability in inhibitory neurons61, and golgi acidification and protein surface sialylation62 have been reported. Full connections between UBE3A/E6AP substrates and the neuronal pathologies have not yet been made.
3. Diagnosis
Methyation analysis
Methylation analysis using southern blot or methylation-specific PCR of the promoter region of the SNRPN gene/imprinting center will identify roughly 75 to 80 % of cases of AS (Figure 3). Absence of a maternal methylation pattern is indicative of AS, but typically will not distinguish between deletion, UPD or imprinting defect as the cause. If methylation testing is consistent with AS, testing should proceed as follows:
Figure 3. Diagnostic strategy for Angelman syndrome.

Blue boxes indicate the diagnostic test, pink boxes indicate the diagnosic decision.
Deletion testing. The majority of AS cases (65%) are caused by de novo microdeletion of the 15q11.2-13.1 region, which can be identified by FISH or microarray, and are associated with very low recurrence risk.
UPD testing. If a deletion is not found, UPD testing will identify the presence of two paternal copies of chromosome 15 in 8% of cases, though parental samples are required. Paternal UPD is also generally associated with a low recurrence risk.
Imprinting center sequencing. If methylation testing is positive, but microdeletion and UPD testing are negative, an imprinting center defect is suspected. Imprinting defects account for roughly 3 % of AS cases. Molecular testing to look for deletion in the imprinting center will be successful in roughly 10 to 20 % of these cases and carry up to a 50% recurrence risk. If an imprinting center deletion is not found, an epimutation is presumed and recurrence risk again appears low.
UBE3A sequencing
For those patients with clinically suspected AS, but a negative methylation test, UBE3A sequencing should be sought. Mutations in UBE3A are the second most common molecular cause of AS, found in 12% of cases, and can carry a 50% recurrence risk.
If methylation testing and UBE3A sequencing are negative, AS is unlikely to be the diagnosis, and diagnosis of “Angelman-like” syndromes should be considered including Pitt Hopkins syndrome, CDKL5 mutation, and Kleefstra syndrome, among others.
4. Management of Angelman Syndrome
Epilepsy
Seizures are often difficult to control. Patients may require polypharmacy and status epilepticus is not uncommon47,63. Seizures typically respond best to medications traditionally effective for generalized epilepsy including valproic acid, leviteractem, lamotrigine, clonazepam and clobazam. Seizures also may respond well to the ketogenic diet. Medications most likely to exacerbate seizures include vigabatrin, tiagabine, oxcarbazepine and carbamazepine64.
Sleep Disturbance
Children with AS frequently have disrupted sleep with difficulty both in falling asleep and maintaining sleep and possibly a reduced requirement for sleep65,66. Sleep disturbance can be a tremendous strain on caregivers and families. Treatment with melatonin one hour before bedtime has been shown to decrease sleep latency and nighttime awakening67. Doses as low as 0.3 mg can be effective, with typical doses ranging from 0.3 mg to 5 mg nightly68. Additional medical management may be needed as well as providing a safe and restricted environment for children when they do wake at night.
Diet and Nutrition
In infancy and early childhood feeding issues may require special nipples and gastroesophageal reflux can lead to poor weight gain and vomiting, typically managed with positioning and/or medication.
Muscle tone and gait
Hypotonia is frequent in young children though spasticity of limbs often develops over time. Gait is typically ataxic50. Children should receive physical and occupational therapy services. Orthotics and other adaptive devices are often helpful and orthopedic consultation may be warranted for issues including scoliosis.
Speech
Early intervention with speech therapy is important to maximize communication, with emphasis on non-verbal methods of communication such as picture boards and devices recommended.
Other
Children with AS should have regular opthalmolgic assessment for management of strabismus, and hyperopia69.
Future potential therapies
Strategies to augment DNA methylation through administration of supplements such as betaine, folate, or other supplements are being investigated with the goal of increasing expression of the dormant allele, though the results of trials have been disappointing. Recent studies are focusing on methods of unsilencing the paternal UBE3A allele. Studies by Huang et al and others, using topoisomerase inhibitors or other approaches to “unsilence” the paternal UBE3A allele are ongoing in animal models70. A recent report of restoration of paternal Ube3a expression in the AS mouse model by genetically terminating transcription of the antisense RNA also provides hope for improving disease treatment in the future71.
Recommendation for family counseling
Family counseling is recommended. Most deletions, UPD, and epimutations are associated with a low recurrence risk. However, IC mutations, UBE3A mutations, and some translocations may be associated with a 50% recurrence risk.
Dup15q syndrome
1. Clinical background of the disease
Individuals with 15q11.2-q13 duplication (Dup15q) syndrome have features of both PWS and AS, as well as some features unique to the disorder. Dup15q syndrome is characterized by central hypotonia, developmental delay, intellectual disability, seizures, and autism (Table 3). There is remarkable diversity in the severity of these symptoms, even in individuals with exactly the same genotype. Duplications involving 15q11.2 or 15q13.3 alone are distinct from 15q11.2-q13 duplications and are not discussed here.
Table 3.
Features of Dup15q syndrome
| Consistent (100%) | Frequent (80%) | Associated (20–80%) |
|---|---|---|
| Hypotonia | Characteristic facial features | |
| Speech/language disorder | Reduced growth | |
| Developmental delay | Autism | |
| Behavior challenges | Sensory processing disorders | |
| Abnormal EEG | Seizures | |
| Hyperpigmentation | ||
| Autism |
Hypotonia
Muscle hypotonia is observed in almost all individuals with Dup15q syndrome, and can be severe, prompting testing for PWS72. Feeding difficulties are common. Joint hyperextensibility and drooling accompanies the hypotonia in most individuals. Major motor milestones such as rolling over, sitting and walking are delayed. Weak cry is often reported. While hypotonia can persist in some adults, it also can subside or progress to hypertonia in the limbs72.
Developmental delay/intellectual disability
Gross and fine motor skill delays are common in individuals with Dup15q syndrome. Hypotonia contributes to these delays. Sitting is reportedly achieved between 10 and 20 months, with walking typical between 2 and 3 years. Although some children with Dup15q syndrome do not walk, the vast majority do walk independently. Fine motor delays include non-functional use of objects and immature exploration of objects. Cognitive and social/emotional delays are apparent in all children with Dup15q. Comprehension is very limited. Cognitive impairment/intellectual disability is frequently in the severe to profound range.
Epilepsy
Seizures are a major medical issue for Dup15q syndrome. Seizures affect approximately 60% of children with Dup15q syndrome, with the typical onset occurring before age 573. Seizures often first present as infantile spasms and later progress to a Lennox-Gastaut type syndrome. Seizures are more common in children with isodicentric chromosome 15 (idic(15)) than in those with interstitial duplications. Most affected children reportedly have multiple seizure types, including infantile spasms, tonic, atonic, tonic-clonic, myoclonic, complex partial, and atypical absence73. Seizures can be intractable. There is an increased risk of sudden unexpected death in epilepsy (SUDEP) in individuals with Dup15q syndrome. Many individuals without overt seizures, have abnormal electroencephalogram (EEG) activity74.
Autism
A majority of individuals with Dup15q syndrome meet the diagnostic criteria for autism74. Speech and language delays occur in most individuals. While some individuals are completely non-verbal, a few are highly verbal. Expressive language is typically severely impacted, and may even be absent. Language is often echolalic with pronoun reversal72. Intent to communicate is also absent or very poor in many individuals. Inappropriate social interactions are typical in individuals with Dup15q. Gaze and bodily contact avoidance is common. Symbolic play is almost never acquired, and individuals with Dup15q syndrome usually do not show interest in their peers. Difficult behaviors such as tantrums, shouting, and aggressiveness often occur, as do stereotypies. Hand flapping, clapping, or wringing are frequently seen, as well as finger biting, head turning, and repeated spinning. Despite a frequent diagnosis of Autism, many individuals with Dup15q syndrome score well on the Autism Diagnostic Observation Scale-general (ADOS-G) test.
Other
Subtle facial features that are characteristic for Dup15q syndrome can be seen in most affected individuals. These include a small button nose, down-slanting palpebral fissures, and low-set and/or posteriorly rotated ears. Increased pigmentation can be observed. Growth retardation occurs in approximately 20–30% of individuals72. Hypogonadism occurs in approximately 20% of affected individuals, although precocious puberty sometimes occurs72.
2. Recent advances in genetics and pathomechanism of diseases
Dup15q syndrome usually occurs in one of two forms. Isodicentric chromosome 15q (idic(15)) and maternal interstitial duplication74. Idic(15) is the most common presentation. In addition to the two normal chromosomes 15, individuals with idic(15) have a small supernumerary chromosome harboring two extra copies of the maternal 15q11.2-q13 region in a tail-to-tail orientation. As the name implies, the idic(15) chromosome also contains two centromeres, but is apparently stable despite this, possibly owing to the inactivation of one of the centromeres. Individuals with idic(15) are tetrasomic for the 15q11.2-q13 region, having three maternal copies and one paternal copy of the locus. Maternal interstitial duplications consist of one extra copy of the maternal 15q11.2-q13 region inserted inverted and in tandem with another copy of the region. Thus, these individuals are triploid for the 15q11.2-13 region; two maternal copies and one paternal copy of the locus are present. Individuals with paternal interstitial duplications do exist in the population, but appear to have a distinct, milder phenotype compared to individuals with maternal interstitial duplications.
Due to the dependence of the phenotypes of individuals on the parent-of-origin of the duplicated allele(s), it is assumed that UBE3A, the gene whose loss of function causes AS, contributes significantly to the Dup15q syndrome. A mouse model carrying two extra copies of murine Ube3a has autistic features and supports this hypothesis75, although mice with only a single extra copy of maternal Ube3a do not have an autistic phenotype75,76. Other, biallelically expressed genes could also play important roles. A cluster of GABA receptor subunit genes (GABRB3, GABRA5, and GABRG3), and a gene encoding another ubiquitin ligase, HERC2, are duplicated in individuals with both maternal interstitial duplication and idic(15). These genes may also contribute to the disorder.
3. Diagnosis
Duplications in the 15q11q13 region, which is prone to genomic rearrangement due to the presence of repeated DNA elements, are most often detected through array CGH 77, which has become a standard screening tool when evaluating children with hypotonia, autism or developmental delay. Prior to the widespread use of this technology, standard high resolution karyotype along with FISH analysis identified cases of inverted duplication 15 (idic 15) syndrome, visible as a marker chromosome 72. Cases of idic (15) may still be identified in this manner, though interstitial duplications, would typically not be detectable by chromosomal analysis. Array CGH also allows for the precise delineation of break points and size of the duplicated material.
Identification of an interstitial duplication or idic (15) should be followed by a test to clarify parent of origin, which impacts phenotype74. This could be done either by methylation analysis of the proband sample, or targeted array of parental samples.
4. Treatment/Management
Several studies have confirmed that phenotype is more severe for those children with idic (15) than interstitial duplications78,79. In addition, maternally inherited interstitial duplications appear to be more consistently expressed than paternally inherited ones, and are associated with significantly higher risk of autism spectrum disorder74. The impact of paternally inherited duplications remains unclear, though some pathogenicity is clear.
Muscle tone/growth and nutrition
Hypotonia, particularly impacting oro-facial musculature can lead to feeding difficulties in infancy, more often in idic (15) children, and may require special attention to feeding and growth. Delay in acquisition of motor milestones is also most pronounced in those with idic (15) than interstitial duplications, and early intervention with physical therapy may be beneficial.
Autism
Given the high risk of autism with idic (15) and maternally inherited duplication, evaluation by a Developmental Pediatrician is recommended. This should be considered for paternally inherited duplications as well, particularly if concerns about behavior and social relatedness exist. Children with Dup15q syndrome will also benefit from early intervention with speech and educational therapies.
Epilepsy
Seizures are significantly more common and severe in children with idic (15), affecting roughly two thirds, as opposed to 25 % of those with interstitial duplications 73. Risk for infantile spasms is high in idic (15) and parents should be advised to watch for early signs of this seizure type, as early treatment is beneficial. Screening EEG may be warranted. Treatment with ACTH/steroids for infantile spasms appears to be more effective in this group than vigabatrin (75 % versus 29 %)73, thus should be considered as initial therapy. Seizures often evolve into Lennox Gastaut and appear to respond to medications typically beneficial for those with generalized epilepsy including valproic acid, rufinamide, lamotrigine and zonisamide, though formal studies remain limited. Carbamazepine was also reported to be effective in many, suggesting epilepsy may be multifocal as opposed to primary generalized. Children with idic(15) do not respond well to benzodiazepines and seizure exacerbation was also reported in almost half of those treated with Leviteracetam73. There is little information available regarding the efficacy of the ketogenic diet or vagal nerve stimulator in this population. Unfortunately, epilepsy risk appears persistent in idic (15) with seizures continuing into adulthood in two thirds73. Refractory epilepsy does appear to associated with risk of early death secondary to status epilepticus or SUDEP (sudden unexplained death in epilepsy), reported in 8 % of idic (15) patients with seizures73.
Behavior and sleep
Behavioral difficulties with defiant and aggressive behaviors, particularly in idic (15) patients, can be challenging and may require medical management80. In addition, sleep difficulties may be noted, possibly secondary to epileptic discharges, thus overnight EEG and sleep study may be helpful in directing management.
Future potential therapies
Current work toward future therapies may focus on reducing UBE3A expression levels or activity in Dup15q patients81, although no specific therapies have been reported.
Recommendation for family counseling
Family counseling is recommended. Idic (15) is associated with a low recurrence risk. However, interstitial duplication may be associated with a 50% recurrence risk.
Conclusion
The chromosome 15q11-13 region contains several genes that are regulated by genomic imprinting and impact neurodevelopment. Despite being caused by variation of the same genetic region, Prader-Willi, Angelman, and Dup15q syndromes are distinct disorders with different phenotypic manifestations and diagnostic strategies. Since imprinted genes are involved in these disorders, potential therapies may involve modulating expression of the implicated genes and provide optimism for improved outcomes in the future.
Key Points.
Three distinct neurodevelopmental disorders are caused by copy number variation at human chromosome 15q11-q13: Prader-Willi syndrome, Angelman syndrome, and 15q11-q13 duplication syndrome.
Prader-Willi and Angelman syndromes can also be caused by uniparental disomy, microdeletions and/or single gene mutations, and imprinting defects. An organized diagnostic strategy is required in order to confirm or full exclude the diagnosis.
Prader-Willi syndrome is characterized by infantile hypotonia and failure to thrive followed by obesity, hyperphagia, small stature, and behavioral issues. Early growth hormone treatment improves body habitus and stature.
Angelman syndrome is characterized by severe intellectual disability, absent speech, epilepsy, and characteristic happy affect. It is caused by the loss of function from the maternal UBE3A gene.
15q11-q13 duplication syndrome is characterized by developmental delay, epilepsy, and autism. Duplications that lead to this syndrome are almost always of maternal origin.
Footnotes
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Louisa Kalsner, Email: Lkalsner@connecticutchildrens.org, Departments of Pediatrics and Neurology, University of Connecticut School of Medicine and Connecticut Children’s Medical Center, 505 Farmington Avenue, Farmington, CT 06032.
Stormy J. Chamberlain, Email: chamberlain@uchc.edu, Department of Genetics and Genome Sciences, University of Connecticut Health Center, 400 Farmington Ave., Farmington, CT 06030-6403
References
- 1.Butler MG, et al. Is gestation in Prader-Willi syndrome affected by the genetic subtype? J Assist Reprod Genet. 2009;26:461–6. doi: 10.1007/s10815-009-9341-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCandless SE Committee on G. Clinical report-health supervision for children with Prader-Willi syndrome. Pediatrics. 2011;127:195–204. doi: 10.1542/peds.2010-2820. [DOI] [PubMed] [Google Scholar]
- 3.Eldar-Geva T, Hirsch HJ, Benarroch F, Rubinstein O, Gross-Tsur V. Hypogonadism in females with Prader-Willi syndrome from infancy to adulthood: variable combinations of a primary gonadal defect and hypothalamic dysfunction. Eur J Endocrinol. 2010;162:377–84. doi: 10.1530/EJE-09-0901. [DOI] [PubMed] [Google Scholar]
- 4.Butler MG, et al. Growth standards of infants with Prader-Willi syndrome. Pediatrics. 2011;127:687–95. doi: 10.1542/peds.2010-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14:10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 6.Burman P, Ritzen EM, Lindgren AC. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocr Rev. 2001;22:787–99. doi: 10.1210/edrv.22.6.0447. [DOI] [PubMed] [Google Scholar]
- 7.Carrel AL, et al. Growth hormone improves mobility and body composition in infants and toddlers with Prader-Willi syndrome. J Pediatr. 2004;145:744–9. doi: 10.1016/j.jpeds.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Carrel AL, Myers SE, Whitman BY, Eickhoff J, Allen DB. Long-term growth hormone therapy changes the natural history of body composition and motor function in children with prader-willi syndrome. J Clin Endocrinol Metab. 2010;95:1131–6. doi: 10.1210/jc.2009-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitman B, et al. Growth hormone improves body composition and motor development in infants with Prader-Willi syndrome after six months. J Pediatr Endocrinol Metab. 2004;17:591–600. doi: 10.1515/jpem.2004.17.4.591. [DOI] [PubMed] [Google Scholar]
- 10.Miller JL, et al. Nutritional phases in Prader-Willi syndrome. American journal of medical genetics. Part A. 2011;155A:1040–9. doi: 10.1002/ajmg.a.33951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cummings DE, et al. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med. 2002;8:643–4. doi: 10.1038/nm0702-643. [DOI] [PubMed] [Google Scholar]
- 12.Butler MG, Theodoro MF, Bittel DC, Donnelly JE. Energy expenditure and physical activity in Prader-Willi syndrome: comparison with obese subjects. Am J Med Genet A. 2007;143A:449–59. doi: 10.1002/ajmg.a.31507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstone AP, et al. Visceral adipose tissue and metabolic complications of obesity are reduced in Prader-Willi syndrome female adults: evidence for novel influences on body fat distribution. J Clin Endocrinol Metab. 2001;86:4330–8. doi: 10.1210/jcem.86.9.7814. [DOI] [PubMed] [Google Scholar]
- 14.Whittington J, Holland A. Neurobehavioral phenotype in Prader-Willi syndrome. Am. 2010;154C:438–47. doi: 10.1002/ajmg.c.30283. [DOI] [PubMed] [Google Scholar]
- 15.Whittington J, et al. Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome. J Intellect Disabil Res. 2004;48:172–87. doi: 10.1111/j.1365-2788.2004.00556.x. [DOI] [PubMed] [Google Scholar]
- 16.Dykens EM, Cassidy SB, King BH. Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. Am J Ment Retard. 1999;104:67–77. doi: 10.1352/0895-8017(1999)104<0067:MBDIPS>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 17.Butler JV, et al. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study. Dev Med Child Neurol. 2002;44:248–55. doi: 10.1017/s001216220100202x. [DOI] [PubMed] [Google Scholar]
- 18.Boer H, et al. Psychotic illness in people with Prader Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet. 2002;359:135–6. doi: 10.1016/S0140-6736(02)07340-3. [DOI] [PubMed] [Google Scholar]
- 19.Festen DA, et al. Sleep-related breathing disorders in prepubertal children with Prader-Willi syndrome and effects of growth hormone treatment. J Clin Endocrinol Metab. 2006;91:4911–5. doi: 10.1210/jc.2006-0765. [DOI] [PubMed] [Google Scholar]
- 20.Sahoo T, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719–21. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duker AL, et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. European journal of human genetics : EJHG. 2010;18:1196–201. doi: 10.1038/ejhg.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Smith AJ, et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet. 2009;18:3257–65. doi: 10.1093/hmg/ddp263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bieth E, et al. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cavaille J, et al. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc Natl Acad Sci U S A. 2000;97:14311–6. doi: 10.1073/pnas.250426397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bazeley PS, et al. snoTARGET shows that human orphan snoRNA targets locate close to alternative splice junctions. Gene. 2008;408:172–9. doi: 10.1016/j.gene.2007.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kishore S, et al. The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Human Molecular Genetics. 2010;19:1153–64. doi: 10.1093/hmg/ddp585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bortolin-Cavaille ML, Cavaille J. The SNORD115 (H/MBII-52) and SNORD116 (H/MBII-85) gene clusters at the imprinted Prader-Willi locus generate canonical box C/D snoRNAs. Nucleic Acids Research. 2012 doi: 10.1093/nar/gks321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen M, et al. Direct cloning of double-stranded RNAs from RNase protection analysis reveals processing patterns of C/D box snoRNAs and provides evidence for widespread antisense transcript expression. Nucleic Acids Res. 2011;39:9720–30. doi: 10.1093/nar/gkr684. Epub 2011 Aug 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaffer LG, et al. American College of Medical Genetics statement of diagnostic testing for uniparental disomy. Genet Med. 2001;3:206–11. doi: 10.1097/00125817-200105000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller JL, et al. Pituitary abnormalities in Prader-Willi syndrome and early onset morbid obesity. Am J Med Genet A. 2008;146A:570–7. doi: 10.1002/ajmg.a.31677. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt H, Pozza SB, Bonfig W, Schwarz HP, Dokoupil K. Successful early dietary intervention avoids obesity in patients with Prader-Willi syndrome: a ten-year follow-up. J Pediatr Endocrinol Metab. 2008;21:651–5. doi: 10.1515/JPEM.2008.21.7.651. [DOI] [PubMed] [Google Scholar]
- 32.Deal CL, et al. GrowthHormone Research Society workshop summary: consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome. J Clin Endocrinol Metab. 2013;98:E1072–87. doi: 10.1210/jc.2012-3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Festen DA, et al. Mental and motor development before and during growth hormone treatment in infants and toddlers with Prader-Willi syndrome. Clin Endocrinol (Oxf) 2008;68:919–25. doi: 10.1111/j.1365-2265.2007.03126.x. [DOI] [PubMed] [Google Scholar]
- 34.Berini J, et al. Growth hormone therapy and respiratory disorders: long-term follow-up in PWS children. J Clin Endocrinol Metab. 2013;98:E1516–23. doi: 10.1210/jc.2013-1831. [DOI] [PubMed] [Google Scholar]
- 35.Oto Y, et al. Exacerbation of BMI after cessation of growth hormone therapy in patients with Prader-Willi syndrome. Am J Med Genet A. 2014;164A:671–5. doi: 10.1002/ajmg.a.36355. [DOI] [PubMed] [Google Scholar]
- 36.Emerick JE, Vogt KS. Endocrine manifestations and management of Prader-Willi syndrome. Int J Pediatr Endocrinol. 2013;2013:14. doi: 10.1186/1687-9856-2013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soni S, et al. The course and outcome of psychiatric illness in people with Prader-Willi syndrome: implications for management and treatment. J Intellect Disabil Res. 2007;51:32–42. doi: 10.1111/j.1365-2788.2006.00895.x. [DOI] [PubMed] [Google Scholar]
- 38.Goldstone AP, et al. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab. 2008;93:4183–97. doi: 10.1210/jc.2008-0649. [DOI] [PubMed] [Google Scholar]
- 39.Einfeld SL, et al. A double-blind randomized controlled trial of oxytocin nasal spray in Prader Willi syndrome. Am J Med Genet A. 2014;164A:2232–9. doi: 10.1002/ajmg.a.36653. [DOI] [PubMed] [Google Scholar]
- 40.Heymsfield SB, et al. Hyperphagia: current concepts and future directions proceedings of the 2nd international conference on hyperphagia. Obesity (Silver Spring) 2014;22 (Suppl 1):S1–S17. doi: 10.1002/oby.20646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bird LM. Angelman syndrome: review of clinical and molecular aspects. Appl Clin Genet. 2014;7:93–104. doi: 10.2147/TACG.S57386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peters SU, Beaudet AL, Madduri N, Bacino CA. Autism in Angelman syndrome: implications for autism research. Clin Genet. 2004;66:530–6. doi: 10.1111/j.1399-0004.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 43.Clayton-Smith J, Pembrey ME. Angelman syndrome. J Med Genet. 1992;29:412–5. doi: 10.1136/jmg.29.6.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laan LA, den Boer AT, Hennekam RC, Renier WO, Brouwer OF. Angelman syndrome in adulthood. Am J Med Genet. 1996;66:356–60. doi: 10.1002/(SICI)1096-8628(19961218)66:3<356::AID-AJMG21>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 45.Gentile JK, et al. A neurodevelopmental survey of Angelman syndrome with genotype-phenotype correlations. J Dev Behav Pediatr. 2010;31:592–601. doi: 10.1097/DBP.0b013e3181ee408e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galvan-Manso M, Campistol J, Conill J, Sanmarti FX. Analysis of the characteristics of epilepsy in 37 patients with the molecular diagnosis of Angelman syndrome. Epileptic Disord. 2005;7:19–25. [PubMed] [Google Scholar]
- 47.Thibert RL, et al. Epilepsy in Angelman syndrome: a questionnaire-based assessment of the natural history and current treatment options. Epilepsia. 2009;50:2369–76. doi: 10.1111/j.1528-1167.2009.02108.x. [DOI] [PubMed] [Google Scholar]
- 48.Valente KD, et al. Epilepsy in patients with angelman syndrome caused by deletion of the chromosome 15q11-13. Arch Neurol. 2006;63:122–8. doi: 10.1001/archneur.63.1.122. [DOI] [PubMed] [Google Scholar]
- 49.Varela MC, Kok F, Otto PA, Koiffmann CP. Phenotypic variability in Angelman syndrome: comparison among different deletion classes and between deletion and UPD subjects. Eur J Hum Genet. 2004;12:987–92. doi: 10.1038/sj.ejhg.5201264. [DOI] [PubMed] [Google Scholar]
- 50.Williams CA. Neurological aspects of the Angelman syndrome. Brain Dev. 2005;27:88–94. doi: 10.1016/j.braindev.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 51.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–3. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 52.Matsuura T, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–7. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 53.Dagli A, Buiting K, Williams CA. Molecular and Clinical Aspects of Angelman Syndrome. Mol Syndromol. 2012;2:100–112. doi: 10.1159/000328837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 55.Wang M, Pickart CM. Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J. 2005;24:4324–33. doi: 10.1038/sj.emboj.7600895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar S, Talis AL, Howley PM. Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J Biol Chem. 1999;274:18785–92. doi: 10.1074/jbc.274.26.18785. [DOI] [PubMed] [Google Scholar]
- 57.Lee SY, et al. Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell Mol Life Sci. 2014;71:2747–58. doi: 10.1007/s00018-013-1526-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Margolis SS, et al. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell. 2010;143:442–55. doi: 10.1016/j.cell.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang YH, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 60.Weeber EJ, et al. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci. 2003;23:2634–44. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wallace ML, Burette AC, Weinberg RJ, Philpot BD. Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron. 2012;74:793–800. doi: 10.1016/j.neuron.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Condon KH, Ho J, Robinson CG, Hanus C, Ehlers MD. The Angelman syndrome protein Ube3a/E6AP is required for Golgi acidification and surface protein sialylation. J Neurosci. 2013;33:3799–814. doi: 10.1523/JNEUROSCI.1930-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pelc K, Boyd SG, Cheron G, Dan B. Epilepsy in Angelman syndrome. Seizure. 2008;17:211–7. doi: 10.1016/j.seizure.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 64.Thibert RL, Larson AM, Hsieh DT, Raby AR, Thiele EA. Neurologic manifestations of Angelman syndrome. Pediatr Neurol. 2013;48:271–9. doi: 10.1016/j.pediatrneurol.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 65.Bruni O, et al. Sleep disturbances in Angelman syndrome: a questionnaire study. Brain Dev. 2004;26:233–40. doi: 10.1016/S0387-7604(03)00160-8. [DOI] [PubMed] [Google Scholar]
- 66.Didden R, Korzilius H, Smits MG, Curfs LM. Sleep problems in individuals with Angelman syndrome. Am J Ment Retard. 2004;109:275–84. doi: 10.1352/0895-8017(2004)109<275:SPIIWS>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 67.Braam W, Didden R, Smits MG, Curfs LM. Melatonin for chronic insomnia in Angelman syndrome: a randomized placebo-controlled trial. J Child Neurol. 2008;23:649–54. doi: 10.1177/0883073808314153. [DOI] [PubMed] [Google Scholar]
- 68.Braam W, et al. Exogenous melatonin for sleep problems in individuals with intellectual disability: a meta-analysis. Dev Med Child Neurol. 2009;51:340–9. doi: 10.1111/j.1469-8749.2008.03244.x. [DOI] [PubMed] [Google Scholar]
- 69.Michieletto P, Bonanni P, Pensiero S. Ophthalmic findings in Angelman syndrome. J AAPOS. 2011;15:158–61. doi: 10.1016/j.jaapos.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 70.Huang HS, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2012;481:185–9. doi: 10.1038/nature10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meng L, et al. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet. 2013;9:e1004039. doi: 10.1371/journal.pgen.1004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Battaglia A. The inv dup (15) or idic (15) syndrome (Tetrasomy 15q) Orphanet J Rare Dis. 2008;3:30. doi: 10.1186/1750-1172-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Conant KD, et al. A survey of seizures and current treatments in 15q duplication syndrome. Epilepsia. 2014;55:396–402. doi: 10.1111/epi.12530. [DOI] [PubMed] [Google Scholar]
- 74.Urraca N, et al. The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism research : official journal of the International Society for Autism Research. 2013;6:268–79. doi: 10.1002/aur.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith SE, et al. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Science translational medicine. 2011;3:103ra97. doi: 10.1126/scitranslmed.3002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakatani J, et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell. 2009;137:1235–46. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang NJ, Liu D, Parokonny AS, Schanen NC. High-resolution molecular characterization of 15q11-q13 rearrangements by array comparative genomic hybridization (array CGH) with detection of gene dosage. Am J Hum Genet. 2004;75:267–81. doi: 10.1086/422854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bolton PF, et al. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am J Med Genet. 2001;105:675–85. doi: 10.1002/ajmg.1551. [DOI] [PubMed] [Google Scholar]
- 79.Browne CE, et al. Inherited interstitial duplications of proximal 15q: genotype-phenotype correlations. Am J Hum Genet. 1997;61:1342–52. doi: 10.1086/301624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Battaglia A, Parrini B, Tancredi R. The behavioral phenotype of the idic(15) syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:448–55. doi: 10.1002/ajmg.c.30281. [DOI] [PubMed] [Google Scholar]
- 81.Germain ND, CP, Plocik AM, Glatt-Deeley H, Brown J, Fink JJ, Bolduc KA, Robinson TM, Levine ES, Reiter LT, Graveley BR, Lalande M, Chamberlain SJ. Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Molecular Autism. 2014;5 doi: 10.1186/2040-2392-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]