

Abstract
Transforming growth factor-beta (TGF-β) is a pleiotropic cytokine, secreted by a variety of cells including immune cells, tumor cells, and stromal cells. TGF-β signaling is dysregulated in cancer patients, and this aberrant signaling at least in part contributes to initiation and progression of many cancers including glioma. The dysregulated signaling components provide molecular targets for the treatment of glioma. In this article, we review TGF-β signaling and its targeting in glioma.
Keywords: TGF-β, TGF-β signaling, glioma, gliomagenesis, glioma treatment
Introduction
Transforming growth factor-beta (TGF-β) is a multifunctional cytokine that regulates cell proliferation, differentiation and tissue homeostasis [1]. The TGF-β superfamily includes various TGF-βs (TGF-β1, -β2, and -β3, which are highly homologous), Activin, Nodal, growth and differentiation factors (GDFs), bone morphogenetic proteins (BMPs), and anti-mullerian hormone (AMH) [2].
In the latent form, TGF-β binds with latent TGF-β binding protein (LTBP) and latency-associated peptide (LAP) to form a latent complex [3]. Many kinds of proteases such as plasmin can catalyze the latent complex of TGF-β to release the active TGF-β. Active TGF-β signals via a heteromeric complex of type I and type II transmembrane serine/threonine kinase receptors and activates different intracellular signaling pathways. TGF-β first binds to TGF-β receptor II (TGFβRII) and alters its conformation, and then TGFβRII phosphorylates TGF-β receptor I [TGFβRI, also termed activin receptor-like kinase (ALK) 5] (Figure 1). Subsequently, TGFβRI phosphorylates receptor-regulated (R-)Smad proteins (Smad 2, 3) on the C-terminal Ser-Ser-X-Ser motif. Activated R-Smads form heteromeric complexes with the Co-Smad, Smad-4, and translocate to the nucleus (Figure 1), where they cooperate with other transcriptional regulators to regulate the expression of target genes such as plasminogen activator inhibitor-1, fibronectin, and collagen type I [4]. Smads consist of conserved Mad homology 1 (MH1), intermediate linker (L) and MH2 domains [5]. In most cell types, TGF-β transduces signals through TGFβRI. However, in endothelial cells, TGF-β signals through two distinct type I receptors, TGFβRI and ALK1, which lead to phosphorylation of Smad2/3 and Smad1/5, respectively [6]. Besides Smad-dependent pathways, various Smad-independent pathways have been identified, including MAPK, PI3K/Akt, JNK/p38, and Rho-like GTPase signaling in a cell type-specific and context-dependent manner [7].
Figure 1.
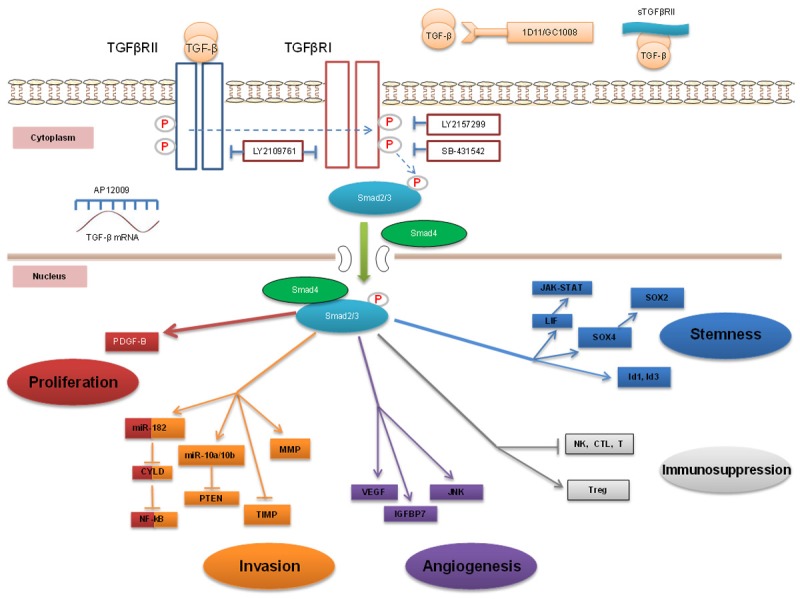
TGF-β signaling in gliomagenesis and its targeting. The TGF-β/Smads signaling pathway contributes to glioma development through induction of multiple carcinogenic processes. This pathway promotes glioma proliferation via PDGF-B and miR-182, invasiveness via miR-182, miR-10 and MMP, as well as angiogenesis via VEGF, IGFBP7, and JNK. The TGF-β/Smads signaling pathway induces immunosuppression by inhibiting NK cells, cytotoxic T lymphocytes (CTL), dendritic cells (DC), and by upregulating T regulatory (Treg) cells. The TGF-β/Smads signaling pathway also drives GSC stemness via LIF, Sox4-Sox2, and Id1-Id3. Aberant TGF-β signaling can be targeted by multiple apporaches, including blockade of TGF-β mRNA translation using antisense oligonucleotides (AON) (AP12009), sequestering TGF-β with soluble receptors (sTGFβRII) or neutralizing antibodies (GC1008), and suppressing TGF-β receptors activity with kinase inhibitors (LY2157299, LY2109761 and SB-431542).
In normal epithelial cells, TGF-β acts as a strong tumor suppressor at least in part due to its inhibitory effect on cell proliferation. This is at least in part attributed to TGF-β mediated pSmad3C (C-terminally phosphorylated Smad3) signaling that results in resistance to proliferative responses induced by mitogenic signals [8]. Even in premalignant stages of cancer, TGF-β can still act as a tumor suppressor by inhibiting epithelial cell proliferation and inducing apoptosis. However, during malignant transformation, epithelial cells or lymphoid cells, which constitute the basis of the majority of cancers in humans, become resistant to the growth inhibitory properties of TGF-β by reductions and/or mutations in TGF-β receptors or intracellular Smads [9,10]. At this malignant transformation stage, Smad3 signaling may shift from a tumor-suppressive pSmad3C to a tumorigenic pSmad3L (linker-phosphorylated Smad 3) pathway [8]. Finally, TGF-β signaling changes to more invasive and proliferative pSmad2L/C and pSmad3L/C signaling (dually phosphorylated at linker and C-terminal regions of Smad2 and Smad3) [8], resulting in tumor metastasis.
Beyond the tumor suppressor and oncogenic functions of TGF-β, it is also implicated in several other aspects of the tumor microenvironment. In later stages of oncogenesis, tumor cells as well as tumor stromal cells frequently secrete high levels of TGF-β resulting in a favorable microenvironment for angiogenesis [11] and immunoevasion [12]. Furthermore, TGF-β also acts on tumor cells directly by stimulating an epithelial to mesenchymal transition (EMT), allowing migration, extravasation and metastatic dissemination [13].
TGF-β signaling and glioma
Gliomas represent 80% of primary malignant brain tumors [14]. Gliomas are categorized into four grades according to the 2007 World Health Organization (WHO) classification criteria: grade I, grade II, grade III (anaplastic) and grade IV (glioblastoma, GB). GB is the most devastating malignant form of primary brain tumors and is characterized by high invasiveness, aberrant proliferation, chemo- and radiation therapy resistance, and relapse after surgical resection with a median overall survival of 14.6 months [15-17]. Given this poor prognosis, it is critical to understand the fundamental molecular pathways leading to glioma formation in order to develop novel therapeutic strategies for this disease. The TGF-β pathway has been identified as a mediator in glioma initiation and progression due to its effects on cell proliferation [18], tumor invasion [19], angiogenesis [20], immunosuppression [21] and the maintenance of stemness of glioma stem cells (GSCs) [22]. Additionally, human studies have demonstrated that TGF-β is overexpressed in malignant glioma tissues but undetectable in normal brain tissues, further suggesting that TGF-β contributes to glioma development. In particular, TGF-β2 is strongly upregulated in GB [23-25]. Here, we will write a review on the tumor promoting role of the TGF-β signaling pathway and the potential to target its signaling components for the treatment of GB.
TGF-β signaling in glioma cell proliferation
In most normal cells, TGF-βs act as the cell growth inhibitor. This cytostatic effect is dependent on its repression of c-myc and Id1-Id3 as well as its activation of cyclin-dependent kinase inhibitors p21WAF1/CIP1 and p15Ink4b [18,26]. In contrast, aberrant signaling in GB cells, such as alterations to the Smad, PI3K and FoxG1 signaling pathways mediate resistance to TGF-β-induced cell growth inhibition [26]. In addition, in GB cells, it is demonstrated that high TGF-β/Smad signing induces induction of platelet derived growth factor-B (PDGF-B), thus resulting in tumor cell proliferation (Figure 1) [27]. In contrast, in gliomas that are not aggressive with a low proliferation index, TGF-β/Smad signaling is inactive and the induction of PDGF-B by TGF-β is impaired as the PDGF-B promoter remains hypermethylated [27].
TGF-β signaling in glioma invasion and migration
TGF-β is a key player in tumor invasion and metastasis [2]. Macrophages/microglia that constitute the major tumor-infiltrating immune cells in GB are recruited by tumor-secreted cytokines such as TGF-β1 and are induced to become immunosuppressive and adopt a tumor supportive/immune-suppressive phenotype (M2) [28]. TGF-β1 produced by glioma-infiltrating microglia/macrophages itself further enhances glioma invasion in vitro and in vivo [19]. TGFβRII downregulation with small hairpin RNAs (shRNAs) impairs TGF-β-induced GB invasiveness and migration in vitro in human T98G glioblastoma and rat C6 glioma cells. Moreover, C6 glioma cells stably expressing TGFβRII shRNAs in nude mice exhibit 50% less tumorigenicity. Microglia enhance glioma invasiveness when co-cultured with unmodified glioma cells, but this capability is lost when co-cultured with glioma cells stably expressing TGFβRII shRNA [19]. The invasiveness of GSCs is also crucial for the migration of glioma. In a recent study, glioma-associated macrophages/microglia with high expression of TGF-β1 could recruit CD133(+) GSCs. Furthermore, neutralization of TGF-β1 or knockdown of TGFβRII in GSCs inhibits their invasiveness [29]. Proteases such as the matrix metalloproteinases (MMPs) and cathepsins degrade the extracellular matrix, facilitating tumor cells to spread and invade [20,29]. TGF-β is able to enhance MMPs expression and suppress tissue inhibitors of metalloproteinase (TIMP) (Figure 1), thus promoting invasiveness of U87 and LN-229 in matrigel invasion assays [30]. Additionally, TGF-β has been demonstrated to induce miR-10a/10b expression, which enhances glioma cell migration through suppression of PTEN (Figure 1) [31].
Radiation is considered an effective way to prolong survival of GB patients; however, tumor progression with enhanced invasiveness frequently occurs at or close to the original radiation treatment site [32]. Previous studies have demonstrated that irradiation increases the tumor cell invasion in malignant gliomas, but the mechanisms underlying this process are largely unknown. A study shows that, after irradiation, it is observed that both TGF-β and β1-integrin are increased and the invasion capability of U87 cells is enhanced in matrigel invasion assays [33], suggesting that increased TGF-beta may be associated with enhanced invasiveness of GB cells after irradiation.
Recently, TGF-β was also found to induce the expression of miR-182, a microRNA that directly suppresses cylindromatosis (CYLD). CYLD negatively regulates NF-κB activity by ubiquitin deconjugation. TGF-β-mediated suppression of CYLD leads to NF-κB activation, thus promoting glioma invasion and increasing its aggressiveness (Figure 1) [34].
TGF-β and angiogenesis
The growth of solid tumors including glioma requires neovascularization for nutrient delivery and debris management [35,36]. The correlation between TGF-β and angiogenesis was reported in Chinese hamster ovary (CHO) cells which overexpress recombinant TGF-β1 [20]. After the subcutaneous injection of the modified CHO cells into nude mice, enhanced tumor proliferation and angiogenesis were observed compared to parental CHO cells. Treatment with TGF-β1 neutralizing antibody inhibited tumor growth and angiogenesis, confirming the role of TGF-β1 in angiogenesis [20]. TGF-β, especially TGF-β1, mediates this effect by up-regulation and activation of various angiogenic factors including vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF) and plasminogen activator inhibitor (PAI-1) [35]. A transcriptional profiling study in human GB vessels further suggested that VEGF-A and TGF-β2 played key roles in GB angiogenesis [37]. Some studies have demonstrated that TGF-β signaling pathways and hypoxia synergize in VEGF gene regulation at the transcriptional level (Figure 1). Consistent with this observation, the human VEGF gene promoter region at -1006 to -954 is demonstrated containing functional DNA binding sites for both Smads and HIF-1 (hypoxia-inducible factor) [38].
In a zebrafish glioma model study, glioma U87 cells expressing red fluorescent protein (RFP) were transplanted in green fluorescent protein (GFP) transgenic zebrafish embryos as a model for studying angiogenesis [39]. TGF-β1 increased glioma-induced angiogenesis; however, this was abrogated by the c-Jun N-terminal kinase (JNK) inhibitor SP600125 but not by the ERK inhibitor PD98059, PI3K inhibitor LY294002, or p38 MAPK inhibitor SB202190. These findings demonstrated the critical role of TGF-β1 and JNK pathways in mediating angiogenesis (Figure 1) [39].
Insulin-like growth factor-binding protein 7 (IGFBP7) is highly expressed in tumor endothelial cells and vascular basement membrane, which makes it a biomarker of tumor vessels in GB [40]. Human brain endothelial cells (HBECs) treated with U87-conditioned media (CM) up-regulated IGFBP7 mRNA and protein in comparison to untreated HBECs [40]. ELISA assay showed that U87-CM contained sufficient TGF-β1 (5 pM) to stimulate IGFBP7 in HBEC. U87-CM-induced IGFBP7 expression in HBECs can be blocked by both TGFβR1 antagonist SB431542 and pan-TGF-β neutralizing antibody (1D11), indicating that TGF-β1 may be able to induce IGFBP7-dependent angiogenesis in brain endothelial cells (Figure 1) [40].
TGF-β signaling in tumor-mediated immunosuppression
Gliomas mediate an immunosuppressive tumor microenvironment through a variety of mechanisms (Figure 1). The immunosuppressive cytokines such as interleukin (IL)-10, TGF-β2, cyclooxygenase-2 (COX2) and prostaglandin E2 (PGE2) secreted by gliomas have been proven to play a major role in impeding anti-tumor immune responses in the microenvironment [41]. TGF-β2 down-regulates HLA-DR antigen expression on human malignant glioma cells, facilitating their immune escape from T lymphocytes [42]. Moreover, TGF-β has been reported to specifically inhibit the expression of perforin, granzyme A, granzyme B, IFN-γ, and Fas ligand, which are co-responsible for cytotoxic T lymphocyte (CTL)-mediated tumor cytotoxicity [43]. TGF-β also promotes the generation of immunosuppressive regulatory T (Treg) cells (Figure 1) [44] and TGF-β1 increases macrophage capacity to produce immunosuppressive cytokine IL-10 [45]. Furthermore, TGF-β1 suppresses the activating receptor NKG2D on the surface of CD8+ T cells and NK cells in glioma patients, rendering them less effective at cytotoxicity against glioma cells [46]. TGF-β is upregulated during malignant glioma progression, and this correlates with MICA (NKG2D ligand) downregulation. Consequently, NKG2D ligand-dependent NK cell-mediated lysis is decreased [47]. NKG2D down-regulation is prevented on the NK cell line, NKL, when incubated with supernatant from LN-229 glioma cells with TGF-β1/β2 knock-down by siRNA compared with supernatant from control LN-229 cells [48]. The TGF-β1/β2 knock-down in LN-229 cells also strongly up-regulates expression of MICA and renders the tumor cells more susceptible to killing by NK cells. LN-229 glioma cells deficient in TGF-β show less subcutaneous and orthotopic tumorigenicity after implantation in nude mice, and isolated NK cells from these mice show an activated phenotype [48]. Additionally, TGF-β inhibits NK cell activity through Smad2, Smad3, and Smad4 (Figure 1) to suppress IFN-γ production by NK cells at least in part through inhibition of T-BET, a positive regulator of human NK cell function [49]. Additional work demonstrates that TGF-β/Smad signaling inhibits CD16-mediated human NK cell IFN-γ production and ADCC [50]. These negative modulations of T or NK cell activity by TGF-β in the tumor microenvironment are likely to be important for the immunosuppression observed in glioma patients, as T and NK cells have been identified as important immune cells for eradicating glioma cells [51-53].
There are a variety of antibodies and targeted inhibitors of TGF-β that aim to subvert TGF-β-mediated immunosuppression. Examples including the TGFβR1 kinase inhibitor SD-208 and the anti-TGF-β neutralizing monoclonal antibody (1D11) have been reported useful for enhancing the tumor-directed immune response and improving the therapeutic efficacy in the treatment of gliomas [36,54]. Together, these findings demonstrate that immunotherapeutic strategies to suppress TGF-β signaling may be promising for improving the prognosis of patients with malignant gliomas.
TGF-β and glioma-initiating cells
Cancer stem cells or tumor-initiating cells are a subpopulation of tumor cells with the capability to undergo self-renewal and multi-lineage differentiation and to recapitulate the entire tumor population [55]. Autocrine TGF-β signaling has been shown to play an essential role in the maintenance of tumorigenicity of glioma stem cells (GSC) [22]. It has been demonstrated that human GSC express higher amounts of TGF-β2 than differentiated glioma cells, and the secreted TGF-β2 appears to correlate with the pathological grade of the glioma [56]. TGF-β produced by GSC enhances effective DNA damage response and self-renewal capacity, leading to microenvironment-mediated resistance to ionizing radiation (IR), while these effects are reversed by TGF-β inhibition through LY364947 (TGFβR1 kinase inhibitor) [57]. The Sox family of proteins have been demonstrated to maintain the GSC population. TGF-β is implicated in this pathway by directly inducing expression of Sox4, which promotes Sox2 expression by associating with the Sox2 enhancer region (Figure 1). Sox2 is an essential factor for maintenance of GSC stemness [22]. Sox2 knockdown by siRNA results in a significant decrease of GSC sphere-forming ability and self-renewal capacity. Inhibition of TGF-β signaling leads to Sox2 downregulation, depriving GSC of stemness, promoting GSC differentiation, and reducing their tumorigenicity in orthotopic immune compromised mice. There are also other GSC-related signaling pathways in which TGF-β is involved. Penuelas et al. demonstrated that TGF-β increases GSC self-renewal capacity through the Smad-dependent induction of leukemia inhibitory factor (LIF) and the subsequent activation of the JAK-STAT signaling pathway (Figure 1) [58]. Furthermore, GB neurospheres that are pretreated with TGF-β or LIF generate tumors earlier and decrease survival significantly in mice compared with untreated GB neurospheres [58].
A GSC-enriched cell population has been found to express high levels of CD44 and inhibitor of DNA-binding protein1 (Id1) and tend to be located in a perivascular niche [59], a place suitable for GSC expansion and tumor development [60]. High CD44 and Id1 levels correlate with poor prognosis in GB patients [59]. Inhibition of the TGF-β signaling cascade by blockade of TGFβRI decreases the CD44high/Id1high GSC population through reduction of Id1 and Id3 transcription factors levels, resulting in an inhibition of their ability to initiate tumors (Figure 1) [59].
GSC not only often locate in perivascular niches, but also generate vascular pericytes to promote vessel function and tumor growth [61]. CXCR4-expressing GSC are recruited toward endothelial cells in brain and GB through SDF-1/CXCR4 axis, and are induced to become pericytes mainly by TGF-β [61].
Preclinical and clinical studies on TGF-β inhibition in glioma
Therapeutic inhibition of TGF-β signaling can be accomplished by inhibiting translation of TGF-β mRNA using antisense oligonucleotides (AON), sequestering the ligands using soluble receptors or their ectodomain constructs (ligand traps) and antibodies, and suppressing TGF-β receptor kinase activity (Figure 1) [62]. Below, we show a summary of pre-clinical studies and clinical trials exploring TGF-β inhibition as a therapeutic approach in glioma. Several therapeutic approaches explored to inhibit oncogenic TGF-β signaling involved in glioma, which are documented in the clinical study database (www.clinicaltrials.gov), are summarized in Table 1.
Table 1.
Anti-TGF-β compounds currently under clinical development for glioma treatment (summarized from www.clinicaltrials.gov)
| Drug | Type | Target | Clinical trial identifier and status |
|---|---|---|---|
| AP12009 | AON | TGF-β2 | NCT00431561, Phase IIb completed [65] |
| NCT00761280, Phase III terminated | |||
| LY2157299 | Kinase inhibitor | TGFβR1 (ALK-5) | NCT01682187, Phase I recruiting [71,72] |
| NCT01220271, Phase I/II recruiting | |||
| NCT01582269, Phase II recruiting | |||
| GC1008 | Antibodies | TGF-β | NCT01472731, Phase II completed [74] |
Antisense TGF-β oligonucleotides
The most effective AON so far for high-grade glioma therapy is a phosphorothioate-modified AON, AP12009 (trabedersen), which is complementary to the human TGF-β2 mRNA sequence (Figure 1, Table 1). Based on three early-passage primary tumor cell cultures isolated from high-grade glioma patients, AP12009 treatment significantly reduced TGF-β2 protein secretion by 49% to 73% compared to untreated and nonsense controls [63]. AP12009 also reduces glioma cell proliferation and migration and reverses the immunosuppressive effects resulted from TGF-β2 [64]. In a phase IIb study, AP12009 was infused intratumorally by a convection-enhanced delivery, which bypassed the blood-brain-barrier and achieved a homogeneous distribution throughout the tumor. In anaplastic astrocytoma (AA, grade III glioma) patients, the median survivals were 39.1 months in the 10 µM AP12009 arm (n = 12) and 35.2 months in the 80 µM AP12009 arm (n = 15), compared with 21.7 months in the chemotherapy arm (n = 12). In this study with limited numbers of patients, AP12009 shows the trend of treatment benefits, but the trend is not statistically significant. However, the benefit was not observed in the patients with GB [65]. In addition, the frequency of glioma patients (with AA or GB) experiencing adverse events was higher with standard chemotherapy (64%) than with 10 or 80 µM AP12009 (27% and 43%, respectively) [65]. Information from the clinical study database (Table 1) indicated that a phase III clinical trial with AP12009 has been temporarily terminated due to insufficient number of suitable participants recruited within the planned period.
Modulation of TGF-β receptors
Soluble receptors can bind to TGF-β, thereby preventing it from binding to its cell surface receptors (Figure 1) [66]. Naumann et al. used adenoviral gene transfer to express secreted TGFβRII in the human glioma cell line LN-229, leading to reduced Smad2 phosphorylation and enhanced NK cell cytotoxicity against glioma cells. LN-308 glioma cells expressing the secreted TGFβRII had significantly delayed growth compared to control cells in an intracerebral xenograft nude mouse model [66]. Another therapeutic method to inhibit TGF-β signaling is to block TGFβRI or TGFβRII kinase activity, thereby preventing phosphorylation of downstream effectors such as R-Smads. SB-431542 is a novel, small molecule kinase inhibitor of type I TGF-β receptor (Figure 1) [67]. It is not surprising that SB-431542 treatment results in blockade of Smad phosphorylation and nuclear translocation, and inhibition of expression of TGF-β downstream targets, VEGF and PAI-1. Consequently, SB-431542 treatment inhibits in vitro glioma proliferation and migration [67]. LY2109761, a novel TGF-β receptor type I and type II dual inhibitor (Figure 1), has been shown to inhibit tumor development in a variety of murine tumor models including pancreatic cancer and hepatocellular carcinoma [68,69]. LY2109761 reduces in vitro survival of U87 and T98 glioma cell lines along with an anti-migratory and anti-angiogenic effects. Using a subcutaneous xenograft U87 or T98 model, LY2109761 impedes tumor growth alone or in combination with radiation and temozolomide (TMZ). LY2109761 also decreases tumor blood perfusion as measured by noninvasive dynamic contrast-enhanced magnetic resonance imaging [70].
In a phase 1 dose-escalation study regarding TGFβRI inhibitor LY2157299 monohydrate (Figure 1, Table 1), 16.6% (5/30) and 7.7% (2/26) of patients with glioma had either a complete response (CR) or a partial response (PR) in the LY2157299 monotherapy arm and the LY2157299-lomustine combination arm, respectively [71,72]. In both groups, 15 patients with glioma had stable disease (SD), among whom 5 had SD ≥ 6 cycles of treatment. In total, 12/56 (21.4%) of glioma patients had a clinical benefit (CR, PR, or SD ≥ 6 cycles), which correlated with low expression of pSmad2 (normalized to total Smad2) in their tumors. LY2157299 was safe on intermittent administration (14 days on/14 days off) of 300 mg/day for 28 days without cardiac adverse effects [71,72].
Antibodies
The pan-TGF-β neutralizing antibody 1D11 (Figure 1) enters both subcutaneous and intracranial implanted gliomas after intravenous injection and remains detectable within the tumor for several days, while only minimal amounts of 1D11 are found in other organs and tissues [73]. However, 1D11 shows different effects on the treatment of gliomas in immunocompetent and immunodeficient mice [73]. Treatment of immunocompetent mice bearing subcutaneous GL261 tumors with 1D11 results in complete remission, but for unknown reasons, repetition of the treatment in immune deficient mice with subcutaneous GL261 tumors shows an opposite effect [73]. In addition, while intracranially implanted GL261 glioma cells in immunocompetent C57BL/6J mice show no tumor reduction after the 1D11 treatment, the glioma cells show less invasion into the adjacent areas of the brain [73]. A Phase II study of the human analog of the 1D11 antibody, GC1008 (Figure 1, Table 1), for the treatment of glioma has been completed [74]. 89Zirkonium (Zr)-GC1008 showed excellent and specific uptake by recurrent gliomas, determined by positron emission tomography (PET) scan. No major toxicity was observed in GC1008 treatment, but all patients showed clinical and/or radiological disease progression after 1 to 3 rounds of the treatment. Thus, in this study with 12 patients, clinical benefit of GC1008 was not achieved [74].
Concluding remarks
Gliomas are characterized by aggressive proliferation, diffuse infiltration and resistance to radio- and chemotherapy. As TGF-β plays a major role in glioma progression, targeting of TGF-β or its downstream signaling in combination with radio-chemotherapy might be a promising therapeutic approach. Inhibitors of the TGF-β pathway developed so far comprise several classes, as listed above. Some of these classes have entered clinical trials for the treatment of glioma as well as other types of cancer. Although various studies have demonstrated the potential benefits of targeting the TGF-β signaling pathway in glioma, data are not consistently encouraging. The reason could be that inhibition of either the TGF-β receptor binding or kinase activity may also result in alternative compensatory pathways mediated by other activators of the Smad pathway or Smad independent factors [2]. Thus, in addition to targeting the TGF-β signaling pathway alone, a synergistic response may be achieved by simultaneously targeting other aberrant signaling pathways such as EGFR, PI3K/Akt, NF-κB, JAK/STAT, etc. Indeed, in preclinical studies in pancreatic tumors, targeting the EGFR and TGF-β signaling pathways concurrently shows better efficacy than targeting either signaling pathway alone [75]. Several clinical trials evaluating the effects of the TGFβRI kinase inhibitor LY-2157299 in combination with Sorafenib (a small molecular inhibitor of multiple tyrosine protein kinases and Raf kinases) in hepatocellular carcinoma are also ongoing (NCT02240433, NCT02178358, NCT01246986). Importantly, the combination of TGF-β signaling inhibitors with U.S. FDA-approved immune checkpoint blockade agents, such as anti-PD1, anti-PD-L1, and anti-CTLA4 antibodies, most likely would improve clinical outcomes over targeting a single pathway, especially as these antibodies have recently been shown to have efficacy in murine models of glioma [76-78].
Acknowledgements
This review was supported by the grant CA155521 from the National Institutes of Health. The authors sincerely thank David M. Lucas for his careful and critical reading of this manuscript.
Disclosure of conflict of interest
The authors confirm that this article content has no conflicts of interest.
References
- 1.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–215. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 2.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hyytiainen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41:233–264. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- 4.Hawinkels LJ, Ten Dijke P. Exploring anti-TGF-beta therapies in cancer and fibrosis. Growth Factors. 2011;29:140–152. doi: 10.3109/08977194.2011.595411. [DOI] [PubMed] [Google Scholar]
- 5.Chen YG, Hata A, Lo RS, Wotton D, Shi Y, Pavletich N, Massague J. Determinants of specificity in TGF-beta signal transduction. Genes Dev. 1998;12:2144–2152. doi: 10.1101/gad.12.14.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 7.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuzaki K. Smad phosphoisoform signaling specificity: the right place at the right time. Carcinogenesis. 2011;32:1578–1588. doi: 10.1093/carcin/bgr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfraim LA, Fernandez TM, Mamura M, Fuller WL, Kumar R, Cole DE, Byfield S, Felici A, Flanders KC, Walz TM, Roberts AB, Aplan PD, Balis FM, Letterio JJ. Loss of Smad3 in acute T-cell lymphoblastic leukemia. N Engl J Med. 2004;351:552–559. doi: 10.1056/NEJMoa031197. [DOI] [PubMed] [Google Scholar]
- 11.ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11:79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 12.Beck C, Schreiber H, Rowley D. Role of TGF-beta in immune-evasion of cancer. Microsc Res Tech. 2001;52:387–395. doi: 10.1002/1097-0029(20010215)52:4<387::AID-JEMT1023>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 13.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol. 2013;15(Suppl 2):ii1–56. doi: 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 16.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 18.Alexandrow MG, Moses HL. Transforming growth factor beta and cell cycle regulation. Cancer Res. 1995;55:1452–1457. [PubMed] [Google Scholar]
- 19.Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, Franciszkiewicz K, Chouaib S, Kaminska B. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion--an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene. 2008;27:918–930. doi: 10.1038/sj.onc.1210683. [DOI] [PubMed] [Google Scholar]
- 20.Ueki N, Nakazato M, Ohkawa T, Ikeda T, Amuro Y, Hada T, Higashino K. Excessive production of transforming growth-factor beta 1 can play an important role in the development of tumorigenesis by its action for angiogenesis: validity of neutralizing antibodies to block tumor growth. Biochim Biophys Acta. 1992;1137:189–196. doi: 10.1016/0167-4889(92)90201-l. [DOI] [PubMed] [Google Scholar]
- 21.Platten M, Wick W, Weller M. Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc Res Tech. 2001;52:401–410. doi: 10.1002/1097-0029(20010215)52:4<401::AID-JEMT1025>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 22.Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5:504–514. doi: 10.1016/j.stem.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 23.Maxwell M, Galanopoulos T, Neville-Golden J, Antoniades HN. Effect of the expression of transforming growth factor-beta 2 in primary human glioblastomas on immunosuppression and loss of immune surveillance. J Neurosurg. 1992;76:799–804. doi: 10.3171/jns.1992.76.5.0799. [DOI] [PubMed] [Google Scholar]
- 24.Samuels V, Barrett JM, Bockman S, Pantazis CG, Allen MB Jr. Immunocytochemical study of transforming growth factor expression in benign and malignant gliomas. Am J Pathol. 1989;134:894–902. [PMC free article] [PubMed] [Google Scholar]
- 25.Yamada N, Kato M, Yamashita H, Nister M, Miyazono K, Heldin CH, Funa K. Enhanced expression of transforming growth factor-beta and its type-I and type-II receptors in human glioblastoma. Int J Cancer. 1995;62:386–392. doi: 10.1002/ijc.2910620405. [DOI] [PubMed] [Google Scholar]
- 26.Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 27.Bruna A, Darken RS, Rojo F, Ocana A, Penuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, Seoane J. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11:147–160. doi: 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 28.Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12:1113–1125. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, Xiao HL, Wang B, Yi L, Wang QL, Jiang XF, Yang L, Zhang P, Qian C, Cui YH, Zhang X, Bian XW. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol. 2012;189:444–453. doi: 10.4049/jimmunol.1103248. [DOI] [PubMed] [Google Scholar]
- 30.Wick W, Platten M, Weller M. Glioma cell invasion: regulation of metalloproteinase activity by TGF-beta. J Neurooncol. 2001;53:177–185. doi: 10.1023/a:1012209518843. [DOI] [PubMed] [Google Scholar]
- 31.Liu S, Sun J, Lan Q. TGF-beta-induced miR10a/b expression promotes human glioma cell migration by targeting PTEN. Mol Med Rep. 2013;8:1741–1746. doi: 10.3892/mmr.2013.1709. [DOI] [PubMed] [Google Scholar]
- 32.Zhai GG, Malhotra R, Delaney M, Latham D, Nestler U, Zhang M, Mukherjee N, Song Q, Robe P, Chakravarti A. Radiation enhances the invasive potential of primary glioblastoma cells via activation of the Rho signaling pathway. J Neurooncol. 2006;76:227–237. doi: 10.1007/s11060-005-6499-4. [DOI] [PubMed] [Google Scholar]
- 33.Canazza A, Calatozzolo C, Fumagalli L, Bergantin A, Ghielmetti F, Fariselli L, Croci D, Salmaggi A, Ciusani E. Increased migration of a human glioma cell line after in vitro CyberKnife irradiation. Cancer Biol Ther. 2011;12:629–633. doi: 10.4161/cbt.12.7.16862. [DOI] [PubMed] [Google Scholar]
- 34.Song L, Liu L, Wu Z, Li Y, Ying Z, Lin C, Wu J, Hu B, Cheng SY, Li M, Li J. TGF-beta induces miR-182 to sustain NF-kappaB activation in glioma subsets. J Clin Invest. 2012;122:3563–3578. doi: 10.1172/JCI62339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8:21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 36.Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma JY, Almirez R, Mangadu R, Liu YW, Platten M, Herrlinger U, Murphy A, Wong DH, Wick W, Higgins LS, Weller M. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004;64:7954–7961. doi: 10.1158/0008-5472.CAN-04-1013. [DOI] [PubMed] [Google Scholar]
- 37.Dieterich LC, Mellberg S, Langenkamp E, Zhang L, Zieba A, Salomaki H, Teichert M, Huang H, Edqvist PH, Kraus T, Augustin HG, Olofsson T, Larsson E, Soderberg O, Molema G, Ponten F, Georgii-Hemming P, Alafuzoff I, Dimberg A. Transcriptional profiling of human glioblastoma vessels indicates a key role of VEGF-A and TGFbeta2 in vascular abnormalization. J Pathol. 2012;228:378–390. doi: 10.1002/path.4072. [DOI] [PubMed] [Google Scholar]
- 38.Sanchez-Elsner T, Botella LM, Velasco B, Corbi A, Attisano L, Bernabeu C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001;276:38527–38535. doi: 10.1074/jbc.M104536200. [DOI] [PubMed] [Google Scholar]
- 39.Yang XJ, Chen GL, Yu SC, Xu C, Xin YH, Li TT, Shi Y, Gu A, Duan JJ, Qian C, Cui YH, Zhang X, Bian XW. TGF-beta1 enhances tumor-induced angiogenesis via JNK pathway and macrophage infiltration in an improved zebrafish embryo/xenograft glioma model. Int Immunopharmacol. 2013;15:191–198. doi: 10.1016/j.intimp.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 40.Pen A, Moreno MJ, Durocher Y, Deb-Rinker P, Stanimirovic DB. Glioblastoma-secreted factors induce IGFBP7 and angiogenesis by modulating Smad-2-dependent TGF-beta signaling. Oncogene. 2008;27:6834–6844. doi: 10.1038/onc.2008.287. [DOI] [PubMed] [Google Scholar]
- 41.Zhu VF, Yang J, Lebrun DG, Li M. Understanding the role of cytokines in Glioblastoma Multiforme pathogenesis. Cancer Lett. 2012;316:139–150. doi: 10.1016/j.canlet.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 42.Zuber P, Kuppner MC, De Tribolet N. Transforming growth factor-beta 2 down-regulates HLA-DR antigen expression on human malignant glioma cells. Eur J Immunol. 1988;18:1623–1626. doi: 10.1002/eji.1830181023. [DOI] [PubMed] [Google Scholar]
- 43.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, Khazaie K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U S A. 2005;102:419–424. doi: 10.1073/pnas.0408197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maeda H, Kuwahara H, Ichimura Y, Ohtsuki M, Kurakata S, Shiraishi A. TGF-beta enhances macrophage ability to produce IL-10 in normal and tumor-bearing mice. J Immunol. 1995;155:4926–4932. [PubMed] [Google Scholar]
- 46.Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. 2010;12:7–13. doi: 10.1093/neuonc/nop009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, Weller M, Friese MA. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain. 2006;129:2416–2425. doi: 10.1093/brain/awl205. [DOI] [PubMed] [Google Scholar]
- 48.Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, Weller M. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004;64:7596–7603. doi: 10.1158/0008-5472.CAN-04-1627. [DOI] [PubMed] [Google Scholar]
- 49.Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, Jaung MS, Blaser BW, Sun J, Benson DM Jr, Mao H, Yokohama A, Bhatt D, Shen L, Davuluri R, Weinstein M, Marcucci G, Caligiuri MA. Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity. 2006;24:575–590. doi: 10.1016/j.immuni.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 50.Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, Allard J 2nd, Wei M, Mao H, Byrd JC, Perrotti D, Caligiuri MA. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. 2008;181:3784–3792. doi: 10.4049/jimmunol.181.6.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, Griffero F, Marubbi D, Spaziante R, Bellora F, Moretta L, Moretta A, Corte G, Bottino C. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. 2009;182:3530–3539. doi: 10.4049/jimmunol.0802845. [DOI] [PubMed] [Google Scholar]
- 52.Barcia C Jr, Gomez A, Gallego-Sanchez JM, Perez-Valles A, Castro MG, Lowenstein PR, Barcia C Sr, Herrero MT. Infiltrating CTLs in human glioblastoma establish immunological synapses with tumorigenic cells. Am J Pathol. 2009;175:786–798. doi: 10.2353/ajpath.2009.081034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker GJ, Chockley P, Yadav VN, Doherty R, Ritt M, Sivaramakrishnan S, Castro MG, Lowenstein PR. Natural killer cells eradicate galectin-1-deficient glioma in the absence of adaptive immunity. Cancer Res. 2014;74:5079–5090. doi: 10.1158/0008-5472.CAN-14-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ueda R, Fujita M, Zhu X, Sasaki K, Kastenhuber ER, Kohanbash G, McDonald HA, Harper J, Lonning S, Okada H. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res. 2009;15:6551–6559. doi: 10.1158/1078-0432.CCR-09-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jordan CT. Cancer stem cells: controversial or just misunderstood? Cell Stem Cell. 2009;4:203–205. doi: 10.1016/j.stem.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiu B, Zhang D, Wang C, Tao J, Tie X, Qiao Y, Xu K, Wang Y, Wu A. IL-10 and TGF-beta2 are overexpressed in tumor spheres cultured from human gliomas. Mol Biol Rep. 2011;38:3585–3591. doi: 10.1007/s11033-010-0469-4. [DOI] [PubMed] [Google Scholar]
- 57.Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, Barcellos-Hoff MH. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res. 2012;72:4119–4129. doi: 10.1158/0008-5472.CAN-12-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Penuelas S, Anido J, Prieto-Sanchez RM, Folch G, Barba I, Cuartas I, Garcia-Dorado D, Poca MA, Sahuquillo J, Baselga J, Seoane J. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009;15:315–327. doi: 10.1016/j.ccr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 59.Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, Prieto-Sanchez RM, Barba I, Martinez-Saez E, Prudkin L, Cuartas I, Raventos C, Martinez-Ricarte F, Poca MA, Garcia-Dorado D, Lahn MM, Yingling JM, Rodon J, Sahuquillo J, Baselga J, Seoane J. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell. 2010;18:655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 60.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 61.Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, Min W, McLendon RE, Rich JN, Bao S. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagaraj NS, Datta PK. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs. 2010;19:77–91. doi: 10.1517/13543780903382609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hau P, Jachimczak P, Schlingensiepen R, Schulmeyer F, Jauch T, Steinbrecher A, Brawanski A, Proescholdt M, Schlaier J, Buchroithner J, Pichler J, Wurm G, Mehdorn M, Strege R, Schuierer G, Villarrubia V, Fellner F, Jansen O, Straube T, Nohria V, Goldbrunner M, Kunst M, Schmaus S, Stauder G, Bogdahn U, Schlingensiepen KH. Inhibition of TGF-beta2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides. 2007;17:201–212. doi: 10.1089/oli.2006.0053. [DOI] [PubMed] [Google Scholar]
- 64.Hau P, Jachimczak P, Bogdahn U. Treatment of malignant gliomas with TGF-beta2 antisense oligonucleotides. Expert Rev Anticancer Ther. 2009;9:1663–1674. doi: 10.1586/era.09.138. [DOI] [PubMed] [Google Scholar]
- 65.Bogdahn U, Hau P, Stockhammer G, Venkataramana NK, Mahapatra AK, Suri A, Balasubramaniam A, Nair S, Oliushine V, Parfenov V, Poverennova I, Zaaroor M, Jachimczak P, Ludwig S, Schmaus S, Heinrichs H, Schlingensiepen KH Trabedersen Glioma Study Group. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol. 2011;13:132–142. doi: 10.1093/neuonc/noq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naumann U, Maass P, Gleske AK, Aulwurm S, Weller M, Eisele G. Glioma gene therapy with soluble transforming growth factor-beta receptors II and III. Int J Oncol. 2008;33:759–765. [PubMed] [Google Scholar]
- 67.Hjelmeland MD, Hjelmeland AB, Sathornsumetee S, Reese ED, Herbstreith MH, Laping NJ, Friedman HS, Bigner DD, Wang XF, Rich JN. SB-431542, a small molecule transforming growth factor-beta-receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol Cancer Ther. 2004;3:737–745. [PubMed] [Google Scholar]
- 68.Melisi D, Ishiyama S, Sclabas GM, Fleming JB, Xia Q, Tortora G, Abbruzzese JL, Chiao PJ. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther. 2008;7:829–840. doi: 10.1158/1535-7163.MCT-07-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fransvea E, Angelotti U, Antonaci S, Giannelli G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 2008;47:1557–1566. doi: 10.1002/hep.22201. [DOI] [PubMed] [Google Scholar]
- 70.Zhang M, Herion TW, Timke C, Han N, Hauser K, Weber KJ, Peschke P, Wirkner U, Lahn M, Huber PE. Trimodal glioblastoma treatment consisting of concurrent radiotherapy, temozolomide, and the novel TGF-beta receptor I kinase inhibitor LY2109761. Neoplasia. 2011;13:537–549. doi: 10.1593/neo.11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodon J, Carducci M, Sepulveda-Sanchez JM, Azaro A, Calvo E, Seoane J, Brana I, Sicart E, Gueorguieva I, Cleverly A, Pillay NS, Desaiah D, Estrem ST, Paz-Ares L, Holdhoff M, Blakeley J, Lahn MM, Baselga J. Pharmacokinetic, pharmacodynamic and biomarker evaluation of transforming growth factor-beta receptor I kinase inhibitor, galunisertib, in phase 1 study in patients with advanced cancer. Invest New Drugs. 2015;33:357–70. doi: 10.1007/s10637-014-0192-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodon J, Carducci MA, Sepulveda-Sanchez JM, Azaro A, Calvo E, Seoane J, Brana I, Sicart E, Gueorguieva I, Cleverly AL, Sokalingum Pillay N, Desaiah D, Estrem ST, Paz-Ares L, Holdoff M, Blakeley J, Lahn MM, Baselga J. First-in-Human Dose Study of the Novel Transforming Growth Factor-beta Receptor I Kinase Inhibitor LY2157299 Monohydrate in Patients with Advanced Cancer and Glioma. Clin Cancer Res. 2015;21:553–60. doi: 10.1158/1078-0432.CCR-14-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hulper P, Schulz-Schaeffer W, Dullin C, Hoffmann P, Harper J, Kurtzberg L, Lonning S, Kugler W, Lakomek M, Erdlenbruch B. Tumor localization of an anti-TGF-beta antibody and its effects on gliomas. Int J Oncol. 2011;38:51–59. [PubMed] [Google Scholar]
- 74.den Hollander MW, Bensch F, Glaudemans AWJM, Enting RH, Bunskoek S, Oude Munnink TH, Lub-de Hooge MN, Pearlberg J, Gietema JA, de Vries EGE, Walenkamp AME. 89zr-GC1008 PET imaging and GC1008 treatment of recurrent glioma patients. J. Clin. Oncol. 2013;31(Suppl) abstr 2050. [Google Scholar]
- 75.Deharvengt S, Marmarelis M, Korc M. Concomitant targeting of EGF receptor, TGF-beta and SRC points to a novel therapeutic approach in pancreatic cancer. PLoS One. 2012;7:e39684. doi: 10.1371/journal.pone.0039684. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76.Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, Han Y, Lesniak MS. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290–5301. doi: 10.1158/1078-0432.CCR-14-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vom Berg J, Vrohlings M, Haller S, Haimovici A, Kulig P, Sledzinska A, Weller M, Becher B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J Exp Med. 2013;210:2803–2811. doi: 10.1084/jem.20130678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, Nicholas S, Kellett M, Ruzevick J, Jackson C, Albesiano E, Durham NM, Ye X, Tran PT, Tyler B, Wong JW, Brem H, Pardoll DM, Drake CG, Lim M. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS One. 2014;9:e101764. doi: 10.1371/journal.pone.0101764. [DOI] [PMC free article] [PubMed] [Google Scholar]